

Abstract
Transcription elongation by multisubunit RNA polymerases (RNAPs) is processive, but neither uniform nor continuous. Regulatory events during elongation include pausing, backtracking, arrest, and transcription termination, and it is critical to determine whether the absence of continued synthesis is transient or permanent. Here we describe mechanisms to generate large quantities of stable archaeal elongation complexes on a solid support to permit (1) single-round transcription, (2) walking of RNAP to any defined template position, and (3) discrimination of transcripts that are associated with RNAP from those that are released to solution. This methodology is based on untagged proteins transcribing biotin- and digoxigenin-labeled DNA templates in association with paramagnetic particles.
Keywords: Archaea, Transcription, Walking, Termination, Nascent transcript, Stalled elongation complex, Intrinsic termination, Single-round transcription
1 Introduction
Processive transcription elongation adumbrates, and research findings support that the decision to terminate transcript synthesis is tightly regulated [1–21]. It is therefore imperative that methods are available to discriminate delayed synthesis (paused, backtracked, or arrested complexes) from bona vide transcript release signaling transcription termination. Release of an RNA transcript from a fully formed elongation complex is the definition of transcription termination [22]. This definition discriminates true termination from abortive initiation wherein short transcripts are often repeatedly released from complexes transitioning from initiation to elongation [23–26]. Release of the nascent RNA may or may not occur simultaneously with recycling of RNAP from the DNA for another round of transcription [27].
Studies of elongation and termination often require positioning elongation complexes at discrete template positions in vitro. By noncovalently linking template DNAs to a solid—and often zmagnetic—support, stable transcription complexes can be generated by limiting the NTP substrates provided to RNAP for synthesis [28, 29]. Transcription of a template attached to a solid support permits walking RNAP to discrete positions and also provides a simple technique to distinguish those transcripts associated with stalled but stable elongation complexes from true termination events.
Herein we describe methods to expand this technology to the study of archaeal RNAPs. The simplified transcription system employed by all archaea most closely mimics the eukaryotic RNA polymerase II (Pol II) system rather than the bacterial or eukaryotic Pol I or Pol III machinery [30–37]. Archaeal transcription systems necessitate that at least two additional considerations must be addressed. First, essentially all current in vitro transcription systems with archaeal components are derived from hyperthermophiles [38–43], and transcription at high temperatures often requires two compatible solid-support matrixes to facilitate multiple rounds of walking and to distinguish true termination events. Second, the archaeal transcription apparatus is not sensitive to commonly used RNAP inhibitors (i.e., rifampicin or α-amanitin) [44], and thus transcription of a template linked to a solid support is often necessary to obtain a single elongation complex per DNA template to analyze single-round transcription elongation and termination in vitro. The methods presented here detail the promoter-directed in vitro transcription derived from Thermococcus kodakarensis and the steps necessary to generate stalled elongation complexes and monitor transcription termination.
2 Materials
2.1 Bacterial Cell Growth and Purification of Recombinant TBP, TFB1, and TFB2
LB media (10 g/L peptone, 5 g/L yeast extract, 10 g/L NaCl). 1 L autoclave sterilized within a 4 L baffled Erlenmeyer flask.
Kanamycin.
Chloramphenicol.
Rosetta 2 (DE3) cells (Novagen) (see Note 1).
Isopropyl-β-D-1 thiogalactopyranoside (IPTG).
Sorbitol.
Lysozyme.
TEN-x buffers: 20 mM Tris–HCl pH 8, 0.1 mM EDTA, × mM Nacl. Buffers are named based on [NaCl]. For example, TEN-100 represents a solution with 100 mM NaCl.
Protein storage buffer: 20 mM Tris–HCl pH 8, 0.1 mM EDTA, 100 mM NaCl, 50 % glycerol, 5 mM β-ME.
6× SDS-loading buffer (0.375 M Tris pH 6.8, 12 % SDS, 60 % glycerol, 0.6 M DTT, 0.06 % bromophenol blue).
5× SDS-running buffer (94 g glycine, 15 g Tris-base, and 5 g sodium dodecyl sulfate (SDS) per liter).
Dialysis tubing (10 kDa and 100 kDa molecular weight cut-off).
2.2 Thermococcus Cell Growth and Purification of RNAP
Anaerobic chamber (Coy Laboratories) (see Note 2).
Artificial seawater medium supplemented with 0.5 % (w/v) tryptone, 0.5 % (w/v) yeast extract, 1× trace mineral solution, and 1× vitamin mixture. 1× artificial seawater contains 20 g NaCl, 3 g MgCl2·6H2O, 6 g MgSO4·7H2O, 1 g (NH4)2SO4, 200 mg NaHCO3, 300 mg CaCl2·2H2O, 0.5 g KCl, 420 mg KH2PO4, 50 mg NaBr, 20 mg SrCl2·6H2O, and 10 mg Fe(NH4)2(SO4)2·6H2O per L.
Trace mineral solution (1,000×): 0.5 g MnSO4·H2O, 0.1 g CoCl2·6H2O, 0.1 g ZnSO4·7H2O, 0.01 g CuSO4·5H2O, 0.01 g AlK(SO4)2·12H2O, 0.01 g H3BO3, 0.01 g Na2MoO4·2H2O per L.
Vitamin mixture (200×): 0.2 g niacin, 0.08 g biotin, 0.2 g p antothenate, 0.2 g lipoic acid, 0.08 g folic acid, 0.2 g P-amionbenzoic acid, 0.2 g thiamine, 0.2 g riboflavin, 0.2 g py ridoxine, 0.2 g cobalamin per L.
Elemental sulfur (flowers of sulfur).
2.3 In Vitro Transcription
rNTPs (see Note 3).
10× Transcription Buffer: 155 mM Tris–HCl pH 8.0, 27.5 mM MgCl2.
SA Buffer: 10 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1 mg/ml bovine serum albumin (BSA).
Wash Buffer: 20 mM Tris–HCl, 0.1 mM EDTA, 250 mM KCl, 4 mM MgCl2, 20 μg/ml BSA.
1.2× Stop buffer: 0.6 M Tris–HCl pH 8.0, 12 mM EDTA.
1.0× Stop buffer: 0.5 M Tris–HCl pH 8.0, 10 mM EDTA.
Formamide loading buffer: 95 % formamide, 1× TBE containing 0.1 % bromophenol blue and 0.1 % xylene cyanol.
1 M KCl.
Streptavidin-coated magnetic beads (We use Streptavidin MagneSphere Paramagnetic Particles from Promega).
Anti-digoxigenin magnetic beads (We use Anti-digoxigenin magnetic particles from Roche).
25:24:1 Phenol:Chloroform:Isoamyl alcohol mixture; Tris-saturated at pH 8.0.
Dinucleotide (ApC; see Note 3).
OmniFlex 200 μl Gel-Load pipette tips (Life Science Products).
32P-γ-ATP.
T4 polynulceotide kinase (We use PNK purchased from New England Biolabs).
10× T4 PNK buffer (We use the buffer supplied from New England Biolabs).
2.4 Additional Instrumentation
Top-heated thermocycler (see Note 4).
40 cm vertical gel electrophoresis apparatus and an appropriate power supply.
Sonicator and appropriate horn.
Large capacity high-speed centrifuge. We use an Avanti-J26 XP1 equipped with JA25.50, JLA10.5, and J20/1 rotors; Beckman Coulter.
Liquid chromatography system equipped with fraction collector. We use an AKTA platform; GE Healthcare.
Chromatography columns, including Hi Trap Q-FF, Superdex 200 16/60, Hi Trap SP-FF, Mono Q, and Hi Trap Chelating. We obtain all of our columns from GE Healthcare, although any high-quality columns with similar matrixes will suffice.
Centrifugal concentrators (10, 30, and 100 kDa molecular weight cut-off; we use units from Ambion).
Discontinuous SDS-PAGE system. We generate all gels in-house and resolve gels using a Minimax protein apparatus; Aquebogue Machine Shop.
Coomassie stain (1 g coomassie per liter; 50 % methanol, 10 % acetic acid, 40 % H2O).
Magnetic separation stand (Although any magnet will suffice, we typically rely on commercial stands from Promega).
pH and conductivity meter.
3 Methods
Transcription using archaeal components requires only three protein complexes for accurate promoter-initiated transcription, namely RNAP, TATA-binding protein (TBP), and Transcription Factor B (TFB) [38, 45]. Procedures for the purification of the necessary protein complexes are provided first, followed by details regarding the formation of elongation complexes. Separation of intact transcription elongation complexes from RNAs released into solution is then detailed.
3.1 Purification of Recombinant Archaeal TBP
Grow Rosetta2 (DE3) cells carrying a pET30b-derived vector expressing recombinant TBP (TK0132) from T. kodakarensis in LB supplemented with 34 μg/ml chloramphenicol and 40 μg/ml kanamycin to an A600 of 0.4 at 37 °C. Induce expression by addition of IPTG to a final concentration of 0.25 mM and sorbitol to final concentration of 1 % (w/v) (see Note 5). Allow 12 h at 22 °C for induction (see Note 6).
Harvest biomass (5,000 × g) and discard supernatant. Resuspend biomass (3 ml per gram) in TEN-0 with 0.2 mg lysozyme/ml and lyse by repeated sonication (see Note 7).
Clarify mixture by centrifugation (10,000 × g). Discard pelleted debris.
Load the clarified supernatant over a 5 ml Hi-Trap Q-FF column (pre-equilibrated with TEN-100) attached to a chromatography system. Discard flowthrough.
Flush the column with minimally 20 column volumes TEN-100 (continue to flush with TEN-100 if substantial protein elution can be detected by monitoring the UV absorbance of the eluent). Elute bound proteins, including TBP, with a ~30 column volume linear gradient of NaCl from TEN-100 to TEN-2000.
Collect 1–2 ml fractions during the elution. 10 μl of peak fractions (identified by UV absorbance) are combined with 2 μl 6× SDS-loading buffer and boiled for 3 min.
Fractions containing TBP (molecular weight ~21.5 kDa) should be identified by SDS-PAGE and coomassie staining.
Fractions containing TBP should be pooled and concentrated (at 4 °C) to <1 ml using 10 kDa molecular-weight cut-off centrifugal concentrators following the manufacturers recommendations.
Concentrated fractions containing TBP should be loaded onto and resolved through a Superdex 200 16/60 column equilibrated with TEN-500 at 0.5 ml/min. Collect 1–2 ml fractions during the elution.
Fractions containing TBP should be identified by SDS-PAGE and coomassie staining (as in Subheading 3.1, steps 6 and 7). These fractions should be pooled and concentrated (as in Subheading 3.1, step 7).
Concentrated fractions containing TBP should be reapplied onto a 5 ml HiTrap Q-FF column pre-equilibrated with TEN-200. The column should be washed with TEN-200 until no changes in UV absorbance are noted in the eluent. Bound proteins are eluted with a linear ~30 CV gradient from TEN-200 to TEN-900.
Fractions containing TBP should be identified by SDS-PAGE and coomassie staining. These fractions should be pooled and concentrated (if necessary). Secure samples in 10 kDa dialysis tubing and dialyze samples (twice, for at least 6 h) against minimally 1,000-volumes protein storage buffer. Recover purified protein from the dialysis tube and store at −80 °C.
3.2 Purification of Recombinant Archaeal TFB
T. kodakarensis encodes two isoforms of transcription factor B [(TFB); TFB1 = TK1280; TFB2 = TK2287], both of which are fully functional in vivo and in vitro for all transcription reactions [39]. Details are provided that are generally applicable for purification of either TFB isoform tagged with an N-terminal His6-sequence.
Grow Rosetta2 (DE3) cells carrying a pET28a-derived vector expressing a recombinant TFB from T. kodakarensis in LB supplemented with 34 μg/ml chloramphenicol and 40 μg/ml kanamycin to an A600 of 0.5 at 37 °C. Induce expression by addition of IPTG to a final concentration of 0.25 mM and sorbitol to final concentration of 1 % (w/v). Allow 24 h at 22 °C for induction (see Note 8).
Harvest biomass (5,000 × g) and discard supernatant. Resuspend biomass in TEN-100 (3 ml per gram) containing 0.2 mg lysozyme/ml and lyse by repeated sonication.
Clarify mixture by centrifugation (10,000 × g). Discard pelleted debris.
Load the clarified supernatant over a 5 ml Hi-Trap SP-FF column (pre-equilibrated with TNE-100) attached to a chromatography system. Discard flowthrough.
Flush the column with minimally 20 CV TEN-100 (continue to flush with TEN-100 if substantial protein elution can be detected by monitoring the UV absorbance of the eluent). Elute the bound proteins, including TFB, with a ~30 CV linear gradient of TEN-100 to TEN-2000.
Collect 1–2 ml fractions during the elution. 10 μl of peak fractions (identified by UV absorbance) are combined with 2 μl 6× SDS-loading buffer and boiled for 3 min.
Fractions containing TFB (molecular weight ~34 kDa) should be identified by SDS-PAGE and coomassie staining.
Fractions containing TFB should be pooled, and loaded directly onto a 5 ml Ni2+-charged chelating column (see Note 9). After extensive washing with 20 ml Tris–HCl pH 8.0 containing 500 mM NaCl and 5 mM imidazole, bound proteins should be eluted with a linear gradient of 500 mM NaCl and 5 mM imidazole to 100 mM NaCl and 500 mM imidazole in 20 mM Tris–HCl pH 8.0.
Fractions containing TFB should be identified by SDS-PAGE and coomassie staining. These fractions should be pooled and concentrated (if necessary). Secure samples in 10 kDa dialysis tubing and dialyze samples (twice, for at least 6 h) against minimally 1,000-volumes protein storage buffer. Recover purified protein from the dialysis tube and store at −80 °C.
3.3 Purification of Archaeal RNAP
Many archaeal RNAPs contain Fe-S centers [46, 47], however, there is no evidence for such clusters in T. kodakarensis RNAP and the enzyme can be purified aerobically without concern. Strains wherein a gene encoding a single subunit of RNA polymerase is modified to encode a protein with a His6-tag are now routinely used for purification of RNAP [39].
Prepare sterile artificial seawater media supplemented with yeast extract, tryptone, vitamins, and trace minerals within a Coy anaerobic chamber (see Note 2). Add sulfur to 2 g per L. Media should be inoculated with an appropriate T. kodakarensis culture (1:100) and be placed at 85 °C until growth reaches mid-exponential phase (A600 ~ 0.6).
Harvest biomass (aerobically, 5,000 × g; see Note 10) and discard supernatant. Resuspend biomass (3 ml per gram) in 25 mM Tris–HCl pH 8.0, 1 M NaCl, 10 % (v/v) glycerol and lyse by repeated freeze-thawing employing liquid N2 (see Note 11).
Clarify mixture by centrifugation (15,000 × g). Discard pelleted debris.
Load the clarified supernatant over a 5 ml Ni2+-charged Hi-Trap chelating column (pre-equilibrated with 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol) attached to a chromatography system. Discard flowthrough.
Flush the column with minimally 20 CV 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol (continue to flush with 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol if substantial protein elution can be detected by monitoring the UV absorbance of the eluent). Elute the bound proteins, including RNAP, with a ~30 CV linear gradient from 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol to 25 ml Tris–HCl pH 8.0 containing 0.1 M NaCl, 10 % (v/v) glycerol, and 100 mM imidazole.
Collect 1–2 ml fractions during the elution. 10 μl of peak fractions (identified by UV absorbance) are combined with 2 μl 6× SDS-loading buffer and boiled for 3 min.
Fractions containing RNAP (molecular weight ~380,000 kDa; 12 subunits) should be identified by SDS-PAGE and coomassie staining. The two largest subunits of RNAP run as an easily identifiable doublet near the top of the gel.
Fractions containing RNAP should be pooled and concentrated to <1 ml using 100 kDa molecular-weight cut-off centrifugal concentrators.
Concentrated material containing RNAP should be diluted with 25 ml Tris–HCl pH 8.0 10 % (v/v) glycerol until the conductivity of the sample is below the conductivity of 25 mM Tris–HCl pH 8.0, 10 mM MgCl2, 10 % (v/v) glycerol. The diluted sample should be loaded onto and resolved through a 1 ml Mono Q column. Discard flowthrough. The column should be washed with minimally 20 CV 25 mM Tris–HCl pH 8.0, 10 mM MgCl2, 10 % (v/v) glycerol containing 200 mM KCl, then bound proteins should be eluted with a linear gradient of 200 mM to 400 mM KCl in 25 mM Tris–HCl pH 8.0, 10 mM MgCl2, 10 % (v/v) glycerol.
Collect 1–2 ml fractions during the elution. 10 μl of peak fractions (identified by UV absorbance) are combined with 2 μl 6× SDS-loading buffer and boiled for 3 min.
Fractions containing RNAP (molecular weight ~380,000 kDa; 12 subunits) should be identified by SDS-PAGE and coomassie staining.
Fractions containing RNAP should be pooled and concentrated to <1 ml using 100 kDa molecular-weight cut-off centrifugal concentrators. Concentrated material should be loaded onto and resolved through a Superdex 200 16/60 column equilibrated with 20 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 100 mM NaCl at 0.5 ml/min.
Collect 1–2 ml fractions during the elution. 10 μl of peak fractions (identified by UV absorbance) are combined with 2 μl 6× SDS-loading buffer and boiled for 3 min.
Fractions containing RNAP (molecular weight ~380,000 kDa; 12 subunits) should be identified by SDS-PAGE and coomassie staining. Pool appropriate fractions.
-
Pooled fractions containing RNAP should be reapplied to a 5 ml Ni2+-charged HiTrap chelating column (pre-equilibrated with 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol) attached to a chromatography system. Discard flowthrough.
Flush the column with minimally 20 CV 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol (continue to flush with 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol if substantial protein elution can be detected by monitoring the UV absorbance of the eluent). Elute the bound proteins, including RNAP, with a ~30 CV linear gradient from 25 ml Tris–HCl pH 8.0 containing 1 M NaCl and 10 % (v/v) glycerol to 25 ml Tris–HCl pH 8.0 containing 0.1 M NaCl, 10 % (v/v) glycerol, and 100 mM imidazole.
Collect 1–2 ml fractions during the elution. 10 μl of peak fractions (identified by UV absorbance) are combined with 2 μl 6× SDS-loading buffer and boiled for 3 min.
Fractions containing RNAP should be identified by SDS-PAGE and coomassie staining. These fractions should be pooled and concentrated (if necessary). Secure samples in 100 kDa dialysis tubing and dialyze samples (twice, for at least 6 h) against minimally 1,000-volumes protein storage buffer. Recover purified protein from the dialysis tube and store at −80 °C.
3.4 Design of DNA Template
We employ DNA templates containing the modified promoter from Methanothermobacter sp. used to initiate transcription of histone B (5′-GCGATATATTTATATAGGGATATAGTAATAGATAATATCA-3′) [48]. This short promoter sequence (PhmtB) contains both BRE (underlined) and TATA box (double underlined) sequences to aid transcription factor binding. The transcription start site (bold) is defined and uniform, and >70 % of template DNAs contain elongation complexes when initiation uses equimolar RNAP and template concentrations; TBP and TFB are provided in fourfold molar excess [39]. The sequence downstream from the transcription initiation site can be designed to suit the particular needs of an experiment; for illustrative purposes, we will detail elongation on a DNA template permitting elongation to +116 with the initially transcribed sequence of +1-ACGGTAACCGG (Fig. 1).
Fig. 1.
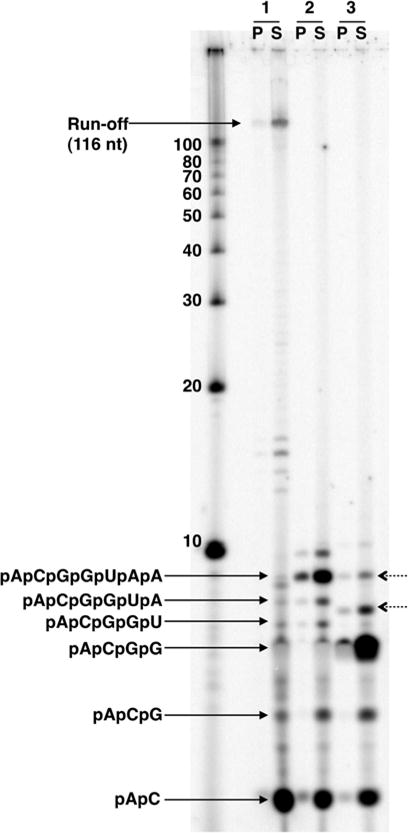
Initial transcription and separation of nascent- versus released-transcripts. Denaturing electrophoresis of 5′-ApC dinucleotide radiolabeled RNAs generated from in vitro transcription reactions with archaeal components permits identification of specific transcripts (identified with solid arrows on the left). Two misincorporation products are visible in reaction #3 (identified with dashed arrows on the right). Reactions were separated into pellet (P) and supernatant (S) fractions to identify transcripts associated with the transcription apparatus (pellet fraction) from those transcripts released to solution (supernatant fraction). +116 nt transcripts represent run-off products. Radiolabeled ssDNA 10-bp markers (far left lane) serve as approximate size standards. Reactions resolved in lanes 1–3 each contain 5′-32P-ApC and were supplemented with 200 μM: (1) ATP, GTP, CTP, and UTP; (2) only ATP, GTP, and UTP; (3) only GTP
It is important to note that the DNA templates employed in transcription assays can be any length and can include the use of even supercoiled plasmid templates. We typically employ templates wherein the 5′ end of the nontemplate strand of DNA is biotinylated or digoxigenin-labeled such that elongation complexes can be captured and washed during the experiment. For experiments requiring multiple walking steps interspersed with heating steps, the template strand should also be labeled at the 5′ end with either digoxigenin or biotin; note that on DNA templates where both 5′ bases are modified, one strand should be labeled with biotin and the complement with digoxigenin. For all of our DNAs used in transcription reactions, nontemplate sequences are fully complementary to template strand sequences. Double-stranded templates can be generated via PCR (with or without 5′-modified primers), or each strand of the DNA can be chemically synthesized and oligonucleotides paired to generate double-stranded templates for transcription.
3.5 Labeling Dinucleotide Primers with32P
For each transcription reaction, combine 1.5 μmole ApC dinucleotide with ~50 μCi32P-γ-ATP, in a 5 μl reaction containing 1× T4 PNK buffer and 10 U T4 PNK.
Incubate the reaction at 37 °C for 1 h.
Incubate the reaction at 85 °C for 10 min to inactivate T4 PNK.
3.6 Transcription Initiation with Subsets of NTPs Permits Walking of RNAP
Combine a double-stranded DNA template (10 nM final) with 40 nM RNAP, 80 nM TFB, 80 nM TBP, in 1× Transcription buffer supplemented with 250 mM KCl, 5 mM DTT, and 5 mM MgCl2. Add the entirety of the reaction wherein the dinucleotide was labeled (from Subheading 3.4). Reactions are typically prepared in sterile, 0.6 ml plastic tubes with flat caps that can be fitted into a thermocycler block equipped with a heated lid. This mixture is heated to 85 °C for 5 min to allow formation of promoter-bound, open complex (see Note 12).
Add a subset of NTPs (each NTP to [200 μM] final, dissolved in 1× transcription buffer, 250 mM KCl, 5 mM MgCl2, 5 mM DTT) that allows for elongation to a discrete, predetermined position in the sequence to the reaction. As one example, addition of GTP alone to a reaction employing a DNA substrate with the nontemplate sequence (+1-ACGGTAACC) permits elongation to generate an RNA that is +4 nucleotides in length. Addition of just ATP, GTP, and UTP would permit elongation to +7, but transcription elongation complexes (TECs) containing 8 or fewer nucleotides are generally not stable and typically do not survive washing steps. Formation of a promoter-proximal stalled complex sterically inhibits the formation of a second TEC on the same template, thus limiting transcription initiation to a single round. Washing the complexes (see below) removes excess RNAP, TBP, and TFB, eliminating any future TEC formation.
The reaction is placed on ice and transferred to pre-equilibrated dry streptavidin-coated paramagnetic particles (see Note 13). The streptavidin-coated paramagnetic particles and the TECs formed on the biotinylated DNA substrates are allowed to incubate on ice for 10 min.
Using the Magnetic Separation Stand, the magnetic particles and the bound TECs are washed three times using 100 μl of wash buffer. These washes remove essentially all unincorporated NTPs. The particles are then resuspended in 1× transcription buffer, supplemented with 5 mM DTT, and 250 mM KCl.
A new subset of NTPs can be added to allow walking to a different position and this process can be repeated until all desired walking steps are performed (see Note 14). To continue the example from above, TECs containing seven nucleotide RNAs can be supplemented with CTP to permit continued elongation to +9, or all NTPs to generate TECs with RNAs that are +116 nucleotides long.
3.7 Stopping of Reaction and Preparation of Samples
Samples are typically removed in 20 μl volumes and added to 100 μl 1.2× stop buffer or 120 μl of 1.0× stop buffer if TECs bound to particles are not resuspended after washing. Additionally, 8 μg of carrier tRNA is added to each sample.
A volume of phenol:chloroform:isoamyl alcohol equal to the total volume of the stopped reaction is added, and each reaction is mixed extensively.
The extracted samples are centrifuged (3 min, 14,000 × g) to separate the aqueous and organic phases. The aqueous layer is removed to a clean tube containing 2.6 volumes of 100 % ethanol. The reactions are vigorously mixed, quickly centrifuged to collect material from the side of the tubes, then placed at −20 °C for minimally 1 h (see Note 15).
The cold samples are centrifuged in a refrigerated benchtop centrifuge (4 °C, 14,000 × g) for 30 min. A small visible pellet should be seen at the end of the centrifugation.
The ethanol is then removed from the samples and appropriately discarded.
Each pellet is then resuspended in 4 μl formamide loading buffer. Samples are heated to 99 °C for 3 min, then immediately transferred to ice prior to loading on denaturing gels.
3.8 Resolving of Samples
The samples are resolved in a 40 cm polyacrylamide gel on an aluminum-backed gel apparatus.
Each total sample (4 μl) is transferred via OmniFlex tips into unique wells in the gel.
The gel is then immediately resolved by applying ~1,500–2,500 V until desired resolution is achieved. For separation of transcripts differing by a few or even a single nucleotide, we resolve samples until the bromophenol blue migrates at least 30 cm in ≤15 % denaturing gels.
3.9 Analysis
When the gel is sufficiently resolved, the apparatus is broken down and the glass plates containing the gel are separated. The specific activity of the nascent transcripts is typically sufficient such that drying of the gels is unnecessary. The gel is covered with a single layer of plastic wrap and is exposed to a phosphorimager screen. Use of a radiolabeled dinucleotide ensures that all transcripts, regardless of length, have the same specific activity permitting immediate and easy quantification of molar ratios of different transcripts present in the reactions.
Acknowledgments
This work was supported by a grant (GM-100329) from the National Institutes of Health to TJS.
Footnotes
Archaeal and bacterial genomes use the same genetic code, but substantial differences in codon bias between the two domains generally necessitates use of an E. coli strain wherein all rare tRNAs are overexpressed to facilitate high-level translation of archaeal transcripts for recombinant protein production.
T. kodakarensis is an obligate anaerobe. Facilities for anaerobic microbiology are critical for the proper passage and growth of Thermococcus strains. We use an anaerobic chamber manufactured by Coy Laboratories, although any unit capable of maintaining an anaerobic environment would be suitable. All media preparations should be carried out within the chamber, sealed within the chamber, and then removed from the chamber for sterilization. Extreme caution is warranted when autoclaving sealed media bottles. 1 L of media is typically prepared in 2 L pyrex bottles with high temperature closures and anaerobic septums.
When employing subsets of NTPs to allow elongation to a specific position, it is critical that the purity of the NTPs be extremely high. The quality and purity of NTP preparations from suppliers differs significantly. We routinely use NTPs purchased from GE Healthcare, although any sufficiently pure NTPs will suffice. Transcription initiation with a dinucleotide is often preferable to initiation using two NTPs, as initiation is more uniform and the dinucleotide permits radiolabeling of the dinucleotide with 32 P-γ-ATP. Use of radiolabeled dinucleotides results in transcripts with a single radiolabel and facilitates rapid quantification of RNA products on a molar ratio.
Small volume transcription reactions (<100 μl) heated to 85 °C in a traditional wet- or dry-bath suffer too great a volumetric loss due to evaporation to control reaction conditions for reproducible results. We rely on top-heated thermocyclers to limit evaporative loss. The reaction tubes used for the experimentation should be sized appropriately to fit into the thermocycler and allow the top of the thermocycler to close tightly. We utilize 0.65 ml tubes with flat caps.
The concentrations of IPTG used here have been empirically determined to maximize protein expression. Alternate expression constructs are likely to require different conditions. The addition of sorbitol aids in expression and limits degradation of recombinant proteins.
TBP expression peaks at 12 h post-induction and solubility is greater when cultures are shifted to 22 °C. Purification is possible from cultures maintained at 37 °C and from cultures induced for as short as 1 h.
Sonication is typically completed with a series of 10 s pulses, on ice, with 30–40 s between pulses to limit heating the sample. Sonication is continued until the mixture has a viscosity approximately equal to water.
Expression of each TFB peaks at least 24 h after induction. Lowering the culture temperature to 22 °C aids in total yield and solubility of each TFB isoform.
Many Ni2+-affinity matrixes are available. We routinely use prepacked chelating columns that we charge with NiSO4 prior to use. Columns are flushed, in order, with 5 CV H2O, 1 CV 0.5 M EDTA (to remove all bound metals), 5 CV H2O, 1 CV 0.1 M NiSO4 (to charge the column with Ni2+), and 5 CV H2O before each use.
The metabolism of T. kodakarensis typically uses elemental sulfur as the terminal electron acceptor, thus generating copious amount of poisonous H2S gas and building substantial pressures within the sealed growth vessel. Extreme care should be taken to properly vent the pressurized gases prior to harvesting biomass. All spent media should be disposed within a fume hood.
Biomass is flash frozen by immersion in liquid N2 then immediately thawed by incubation at 85 °C. Repeated rounds of freezing and thawing effectively lyses T. kodakarensis cultures.
These procedures have been optimized at the normal growth temperature of Thermococcus kodakarensis. Transcription initiation and elongation are possible at lower temperatures. Note that sequences that direct intrinsic termination at physiological temperature only function to direct RNAP to pause at reduced temperatures [6], and thus it is critical to evaluate elongation-termination decisions at the physiological temperature.
Streptavidin MagneSphere Paramagnetic Particles must be equilibrated prior to use. The particles are equilibrated by washing 10 μl of particles (1 mg/ml) with 100 μl of SA buffer three times. To separate the particles from the buffer, a MagneSphere Technology Magnetic Separation Stand (Promega) is used. The buffer is removed from the particles prior to the reaction being added to the particles. For each reaction that particles will be used; 10 μl of particles must be equilibrated.
The streptavidin molecules denature at temperatures above 55 °C, and thus we cool complexes only to ensure capture to the solid support. Complexes must be returned to physiological temperatures before elongation-termination decisions can be properly monitored following NTP addition.
DNAs containing digoxigenin moieties can be employed when coupled with solid supports facilitating capture of digoxigenin-labeled DNAs. We often employ templates with separate digoxigenin and biotin moieties at the 5′ ends of the substrates.
The temperature constraints of the archaeal transcription apparatus necessitate the addition of unheated streptavidin-coated particles after each elongation cycle at high temperature. Elongation at room temperature is possible, but is facilitated by incubation at 85 °C. Room temperature elongation is utilized when the physiological state of the TEC is not under study during the elongation; however elongation at 85° permits analysis at the optimal reaction conditions.
We rely on a high concentration of Tris to aid in the precipitation of in vitro synthesized RNAs. Use of tRNA and Tris as carriers results in samples that do not contain high concentrations of NaCl or KCl that typically lower the resolution and clarity of samples during electrophoresis. It is critical to extract and precipitate the RNAs away from RNAP, as RNAP will nonspecifically associate with transcripts and retard their migration during electrophoresis.
References
- 1.Yarnell WS, Roberts JW. Mechanism of intrinsic transcription termination and antitermination. Science. 1999;284(5414):611–615. doi: 10.1126/science.284.5414.611. [DOI] [PubMed] [Google Scholar]
- 2.Santangelo TJ, Roberts JW. Forward translocation is the natural pathway of RNA release at an intrinsic terminator. Mol Cell. 2004;14(1):117–126. doi: 10.1016/s1097-2765(04)00154-6. [DOI] [PubMed] [Google Scholar]
- 3.Santangelo TJ, Artsimovitch I. Termination and antitermination: RNA polymerase runs a stop sign. Nat Rev Microbiol. 2011;9(5):319–329. doi: 10.1038/nrmicro2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toulokhonov I, Artsimovitch I, Landick R. Allosteric control of RNA polymerase by a site that contacts nascent RNA hairpins. Science. 2001;292(5517):730–733. doi: 10.1126/science.1057738. [DOI] [PubMed] [Google Scholar]
- 5.White E, Kamieniarz-Gdula K, Dye MJ, Proudfoot NJ. AT-rich sequence elements promote nascent transcript cleavage leading to RNA polymerase II termination. Nucleic Acids Res. 2013;41(3):1797–1806. doi: 10.1093/nar/gks1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santangelo TJ, Reeve JN. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J Mol Biol. 2006;355(2):196–210. doi: 10.1016/j.jmb.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 7.Santangelo TJ, Cubonova L, Skinner KM, Reeve JN. Archaeal intrinsic transcription termination in vivo. J Bacteriol. 2009;191(22):7102–7108. doi: 10.1128/JB.00982-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollands K, Sevostiyanova A, Groisman EA. Unusually long-lived pause required for regulation of a Rho-dependent transcription terminator. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1319193111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo J, Turek ME, Price DH. Regulation of RNA polymerase II Termination by phosphorylation of Gdownl. J Biol Chem. 2014;289(18):12657–12665. doi: 10.1074/jbc.M113.537662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb S, Hector RD, Kudla G, Granneman S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biol. 2014;15(1):R8. doi: 10.1186/gb-2014-15-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell. 2013;155(5):1075–1087. doi: 10.1016/j.cell.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 12.Contreras X, Benkirane M, Kiernan R. Premature termination of transcription by RNAP II: the beginning of the end. Transcription. 2013;4(2):72–76. doi: 10.4161/trns.24148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters JM, Mooney RA, Grass JA, Jessen ED, Tran F, Landick R. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 2012;26(23):2621–2633. doi: 10.1101/gad.196741.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conaway RC, Conaway JW. The Mediator complex and transcription elongation. Biochim Biophys Acta. 2013;1829(1):69–75. doi: 10.1016/j.bbagrm.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagschal A, Rousset E, Basavarajaiah P, Contreras X, Harwig A, Laurent-Chabalier S, Nakamura M, Chen X, Zhang K, Meziane O, Boyer F, Parrinello H, Berkhout B, Terzian C, Benkirane M, Kiernan R. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell. 2012;150(6):1147–1157. doi: 10.1016/j.cell.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peters JM, Vangelof f AD, Landick R. Bacterial transcription terminators: the RNA 3′-end chronicles. J Mol Biol. 2011;412(5):793–813. doi: 10.1016/j.jmb.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mischo HE, Proudfoot NJ. Disengaging polymerase: terminating RNA polymerase II transcription in budding yeast. Biochim Biophys Acta. 2013;1829(1):174–185. doi: 10.1016/j.bbagrm.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, Mironov A, Nudler E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505(7483):372–377. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spitalny P, Thomm M. A polymerase III-like reinitiation mechanism is operating in regulation of histone expression in archaea. Mol Microbiol. 2008;67(5):958–970. doi: 10.1111/j.1365-2958.2007.06084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Werner F. Molecular mechanisms of transcription elongation in archaea. Chem Rev. 2013;113(11):8331–8349. doi: 10.1021/cr4002325. [DOI] [PubMed] [Google Scholar]
- 21.Grohmann D, Werner F. Hold on!: RNA polymerase interactions with the nascent RNA modulate transcription elongation and termination. RNA Biol. 2010;7(3):310–315. doi: 10.4161/rna.7.3.11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adhya S, Gottesman M. Control of transcription termination. Annu Rev Biochem. 1978;47:967–996. doi: 10.1146/annurev.bi.47.070178.004535. [DOI] [PubMed] [Google Scholar]
- 23.Goldman SR, Ebright RH, Nickels BE. Direct detection of abortive RNA transcripts in vivo. Science. 2009;324(5929):927–928. doi: 10.1126/science.1169237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu LM. Promoter clearance and escape in prokaryotes. Biochim Biophys Acta. 2002;1577(2):191–207. doi: 10.1016/s0167-4781(02)00452-9. [DOI] [PubMed] [Google Scholar]
- 25.Carpousis AJ, Gralla JD. Cycling of ribonucleic acid polymerase to produce oligonucleotides during initiation in vitro at the lac UV5 promoter. Biochemistry. 1980;19(14):3245–3253. doi: 10.1021/bi00555a023. [DOI] [PubMed] [Google Scholar]
- 26.Gralla JD, Carpousis AJ, Stefano JE. Productive and abortive initiation of transcription in vitro at the lac UV5 promoter. Biochemistry. 1980;19(25):5864–5869. doi: 10.1021/bi00566a031. [DOI] [PubMed] [Google Scholar]
- 27.Epshtein V, Dutta D, Wade J, Nudler E. An allosteric mechanism of Rho-dependent transcription termination. Nature. 2010;463(7278):245–249. doi: 10.1038/nature08669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nudler E, Gusarov I. Analysis of the intrinsic transcription termination mechanism and its control. Methods Enzymol. 2003;371:369–382. doi: 10.1016/s0076-6879(03)71028-3. [DOI] [PubMed] [Google Scholar]
- 29.Nudler E, Gusarov I, Bar-Nahum G. Methods of walking with the RNA polymerase. Methods Enzymol. 2003;371:160–169. doi: 10.1016/s0076-6879(03)71011-8. [DOI] [PubMed] [Google Scholar]
- 30.Hirata A, Murakami KS. Archaeal RNA polymerase. Curr Opin Struct Biol. 2009;19(6):724–731. doi: 10.1016/j.sbi.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reich C, Zeller M, Milkereit P, Hausner W, Cramer P, Tschochner H, Thomm M. The archaeal RNA polymerase subunit P and the eukaryotic polymerase subunit Rpb12 are interchangeable in vivo and in vitro. Mol Microbiol. 2009;71(4):989–1002. doi: 10.1111/j.1365-2958.2008.06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langer D, Hain J, Thuriaux P, Zillig W. Transcription in archaea: similarity to that in eucarya. Proc Natl Acad Sci U S A. 1995;92(13):5768–5772. doi: 10.1073/pnas.92.13.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Werner F. A nexus for gene expression-molecular mechanisms of Spt5 and NusG in the three domains of life. J Mol Biol. 2012;417(1–2):13–27. doi: 10.1016/j.jmb.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grohmann D, Werner F. Recent advances in the understanding of archaeal transcription. Curr Opin Microbiol. 2011;14(3):328–334. doi: 10.1016/j.mib.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Werner F, Grohmann D. Evolution of multisubunit RNA polymerases in the three domains of life. Nat Rev Microbiol. 2011;9(2):85–98. doi: 10.1038/nrmicro2507. [DOI] [PubMed] [Google Scholar]
- 36.Kostrewa D, Zeller ME, Armache KJ, Seizl M, Leike K, Thomm M, Cramer P. RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature. 2009;462(7271):323–330. doi: 10.1038/nature08548. [DOI] [PubMed] [Google Scholar]
- 37.Kusser AG, Bertero MG, Naji S, Becker T, Thomm M, Beckmann R, Cramer P. Structure of an archaeal RNA polymerase. J Mol Biol. 2008;376(2):303–307. doi: 10.1016/j.jmb.2007.08.066. [DOI] [PubMed] [Google Scholar]
- 38.Santangelo TJ, Cubonova L, James CL, Reeve JN. TFB1 or TFB2 is sufficient for Thermococcus kodakaraensis viability and for basal transcription in vitro. J Mol Biol. 2007;367(2):344–357. doi: 10.1016/j.jmb.2006.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomm M, Stetter KO. Transcription in methanogens. Evidence for specific in vitro transcription of the purified DNA-dependent RNA polymerase of Methanococcus thermolithotrophicus. Eur J Biochem. 1985;149(2):345–351. doi: 10.1111/j.1432-1033.1985.tb08932.x. [DOI] [PubMed] [Google Scholar]
- 40.Hudepohl U, Reiter WD, Zillig W. In vitro transcription of two rRNA genes of the archaebacterium Sulfolobus sp. B12 indicates a factor requirement for specific initiation. Proc Natl Acad Sci U S A. 1990;87(15):5851–5855. doi: 10.1073/pnas.87.15.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hethke C, Geerling AC, Hausner W, de Vos WM, Thomm M. A cell-free transcription system for the hyperthermophilic archaeon Pyrococcus furiosus. Nucleic Acids Res. 1996;24(12):2369–2376. doi: 10.1093/nar/24.12.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werner F, Weinzierl RO. A recombinant RNA polymerase II-like enzyme capable of promoter-specific transcription. Mol Cell. 2002;10(3):635–646. doi: 10.1016/s1097-2765(02)00629-9. [DOI] [PubMed] [Google Scholar]
- 43.Qureshi SA, Bell SD, Jackson SP. Factor requirements for transcription in the Archaeon Sulfolobus shibatae. EMBO J. 1997;16(10):2927–2936. doi: 10.1093/emboj/16.10.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madon J, Leser U, Zillig W. DNA-dependent RNA polymerase from the extremely halophilic archaebacterium Halococcus morrhuae. Eur J Biochem. 1983;135(2):279–283. doi: 10.1111/j.1432-1033.1983.tb07649.x. [DOI] [PubMed] [Google Scholar]
- 45.Bell SD, Magill CP, Jackson SP. Basal and regulated transcription in Archaea. Biochem Soc Trans. 2001;29(Pt 4):392–395. doi: 10.1042/bst0290392. [DOI] [PubMed] [Google Scholar]
- 46.Hirata A, Klein BJ, Murakami KS. The X-ray crystal structure of RNA polymerase from Archaea. Nature. 2008;451(7180):851–854. doi: 10.1038/nature06530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lessner FH, Jennings ME, Hirata A, Duin EC, Lessner DJ. Subunit D of RNA polymerase from Methanosarcina acetivorans contains two oxygen-labile [4Fe-4S] clusters: implications for oxidant-dependent regulation of transcription. J Biol Chem. 2012;287(22):18510–18523. doi: 10.1074/jbc.M111.331199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie Y, Reeve JN. In vitro transcription assays using components from Methanothermobacter thermautotrophicus. Methods Enzymol. 2003;370:66–72. doi: 10.1016/s0076-6879(03)70006-8. [DOI] [PubMed] [Google Scholar]