

Summary
Tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) is an adaptor protein that mediates a wide array of protein-protein interactions via its TRAF domain and a RING finger domain that possesses non-conventional E3 ubiquitin ligase activity. First identified nearly two decades ago as a mediator of IL-1 receptor (IL-1R)-mediated activation of NFκB, TRAF6 has since been identified as an actor downstream of multiple receptor families with immunoregulatory functions, including members of the TNFR superfamily, the toll-like receptor (TLR) family, tumor growth factor-β receptors (TGFβR), and T cell receptor (TCR). In addition to NFκB, TRAF6 may also direct activation of mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), and interferon regulatory factor (IRF) pathways. In the context of the immune system, TRAF6-mediated signals have proven critical for the development, homeostasis, and/or activation of B cells, T cells, and myeloid cells, including macrophages, dendritic cells, and osteoclasts, as well as for organogenesis of thymic and secondary lymphoid tissues. In multiple cellular contexts, TRAF6 function is essential not only for proper activation of the immune system, but also for maintaining immune tolerance, and more recent works have begun to identify mechanisms of contextual specificity for TRAF6, involving both regulatory protein interactions, and messenger RNA regulation by microRNAs.
Keywords: TRAF-6, TRAFs, signal transduction, K63-linked, mir-146a, TAK1
Introduction
Tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) was identified through two independent efforts, one using yeast two-hybrid screening to identify proteins that interact with the CD40 cytoplasmic tail (1), and the other by data mining sequences with similarity to other TRAF proteins with characteristics of interleukin-1 receptor (IL-1R) signal transducers (2). Initial characterization showed that TRAF6 was, at the time, the only TRAF family member capable of bridging signaling by the TNFR and TLR/IL-1R superfamilies. TRAF6 is broadly expressed in mammalian tissues and is well conserved across species. The severe phenotype of TRAF6 knockout mice, which experience 100% mortality within two weeks after birth, and are found to exhibit neural tube defects and exencephaly characterized by abnormal regulation of programmed cell death within specific regions of the developing central nervous system is demonstrative of the importance of TRAF6 to critical biological functions (3). To investigate the requirement for TRAF6 specifically in the mammalian immune system, bone marrow chimera mice using TRAF6-deficient donor bone marrow were generated and showed manifold defects, including disorganized lymphoid tissues and inflammatory autoimmune disease (4). The severity of the disruption of the immune system in these chimeric mice suggested important roles for TRAF6 in numerous immune cell types. While many studies have shown the importance of TRAF6 signaling in development (5-7), homeostasis (8), and cancer (9, 10) outside of immune cells, the focus of this review will be on the roles of TRAF6 within the hematopoietic lineage as well as select organogenic processes that regulate the development and anatomy of the immune system.
TRAF6 signaling mechanisms
Our understanding of the mechanisms employed by TRAF6 to effect intracellular signal transduction within immune cells has continued to expand over the past two decades. The apparent adaptability of TRAF6 to various receptor signaling pathways within a given cell, as well as the context- and/or cell type-specific signaling functions of TRAF6 have been made possible by seemingly innumerable TRAF6 interaction partners. At the same time, TRAF6 signaling in both immune and non-immune cells is one of the original proteins identified as containing a type of non-conventional E3 ubiquitin ligase activity. Below, the general TRAF6 signaling mechanisms that have been characterized as operating in immune cells are summarized (Fig. 1), though some mechanisms are context-specific and are addressed in cell type-specific sections.
Figure 1.
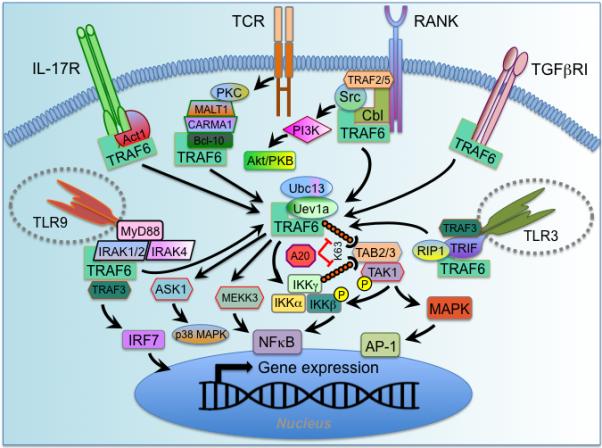
TRAF6 signaling mechanisms. A generalized schema for TRAF6 signal transduction downstream of various receptor families is depicted. TRAF6 is broadly expressed, but most immune cells restrict its activity through selective expression of receptors used to activate it. TRAF6 interacts directly, through TRAF6 binding motifs, with some receptor types, including TNFR superfamily members (represented here by RANK), and TGFβRI. In other cases TRAF6 is linked to the receptor via intermediary proteins, such as Act1 for the IL-17R family, the MALT1-CARMA1-Bcl-10 complex downstream of TCR, the MyD88-IRAK4-IRAK1/2 complex downstream of some TLR/IL-1R superfamily members (e.g., TLR7/8/9, TLR2/4, IL-1/18/33R), and TRIF for others (e.g., TLR3/4). Upon activation, TRAF6 forms a complex with the E2 Ubiquitin conjugating enzyme Ubc13/Uev1a and attaches K63-linked ubiquitin chains to lysine residues on various target proteins, including itself and IKKγ/NEMO. These ubiquitin chains enable the formation of complexes that lead to kinase activation through adaptors, such as TAB2/3, that contain ubiquitin-binding domains. TAB2/3 recruit the kinase TAK1, which phosphorylates downstream kinases, such as IKKβ and MAPK. These events lead to activation of the transcription factors NFκB and AP-1, respectively. In addition to TAK1, TRAF6 lies upstream of additional kinases, such as MEKK3, which leads to NFκB (and possibly AP-1) activation, ASK1, which lies upstream of p38 MAPK, and in the TLR3 complex TRAF6 may lead to RIP1 activation, which may tbe involved in TAK1 activation. Negative regulation of K63-linked ubiquitin chain-mediated TRAF6 signaling is provided by the de-ubiquitinating enzyme A20 and its adaptor TAX1BP1, which lead to signal termination both by removing K63-linked chains, but also by directing K48-linked ubiquitin-mediated proteasomal degradation of Ubc13. In some cases additional scaffolding proteins associated with a receptor engage TRAF6 to activate downstream signaling. In RANK signaling, TRAF6 is recruited to Cbl family scaffolding complexes that recruit Src family kinases, which activate Akt/PKB via PI3 kinase. TRAF6 activity is often coordinated with other TRAFs, such as TRAF2/5 at the RANK receptor, and TRAF3 in some TLR complexes. TRAF3-TRAF6 coordination may be important for activation of the IRF family of transcription factors.
Specific TRAF6 interacting motifs
Orthologs of TRAF6 can be traced back to invertebrates, such a Drosophila where it is called DTRAF2, and plays roles in both dorsal/ventral polarity and anti-microbial immune responses via Toll (11). Mammalian TRAF6 protein is highly conserved, and consists of an N-terminal Zn RING finger domain, a series of five Zn finger domains, a coiled-coil TRAF-N domain, and a C-terminal TRAF-C domain (1, 2). This domain configuration is the same as is found in other mammalian TRAF family members TRAF2, TRAF3, and TRAF5, all of which have also been implicated in various signaling pathways that also utilize TRAF6 (12). Capacity to activate downstream signaling pathways may be similar between these TRAF family members, but TRAF6 likely derives signaling specificity primarily from it TRAF-C domain, which binds to consensus X-X-P-X-E-X-X-aromatic/acidic motifs found in interacting proteins (13). By contrast, TRAFs 2, 3, and 5 are all capable of interacting with distinct overlapping motifs, such as P-X-Q-X-T (14, 15). However, TRAF6 may be recruited to protein complexes involving other TRAFs, such as those downstream of inflammatory pathways that utilize both TRAF6 and TRAF3 (16). Binding to TRAF6 interaction motifs is believed to activate TRAF6 by promoting oligomerization (13, 17), and these motifs can link TRAF6 directly to the intracellular domains of transmembrane receptors, such as TNFR superfamily members (13, 14) and TGFβRI/ALK5 (18, 19), or to signaling receptors indirectly via cytoplasmic intermediaries, such as the kinase IRAK1 for the TLR/IL-1R superfamily (13), the SEFIR domain-containing E3 ubiquitin ligase Act1 for the IL-17 receptor family (20, 21), MAVS/VISA/IPS-1/Cardif for RIG-I family member (22), and the paracaspase MALT1 for T cell receptor (TCR) (23). In addition to proteins containing TRAF6 binding motifs, TRAF6 signaling complexes also include signaling components necessary for recruited kinases and other adaptors. Most notably, downstream of some TLR/IL-1R members, including TLR4, TLR9, and IL-1R, TRAF6 is recruited to the Myddosome, a complex containing the adaptors MyD88, and kinases IRAK1, IRAK2, and IRAK4 (24). In response to TLR3 signaling, TRAF6 is recruited to a complex including the adaptor TRIF, the related family member TRAF3, and the RIP1 kinase, which activates TAK1 in response to TRAF6 activation (16, 25, 26). Even in cases where TRAF6 binds directly to receptors, such as TNFR superfamily members, scaffolding proteins such as c-Cbl, Cbl-b, and RACK1 (unpublished observation) are involved in recruiting downstream kinases, such as c-Src and phosphoinositide 3-kinase (PI3K) (27-29).
TRAF6 as a non-conventional E3 ligase
In all cases investigated thus far, activation of TRAF6 protein requires the Zn RING finger domain, the mechanistic description of which represented a seminal discovery in immunological signal transduction. In 2000, Chen and colleagues described a complex involving TRAF6 and the ubiquitin conjugating enzyme Ubc13 and the Ubc-like protein Uev1A, which is capable of catalyzing the synthesis of unique polyubiquitin chains, in a TRAF6 RING finger domain-dependent manner, that are linked through lysine-63 (K63) of ubiquitin rather than through the conventional K48-linked chains typically associated with proteasomal degradation (30). The same group subsequently showed that these K63-linked ubiquitin chains led to downstream activation of IKK and MAP kinases (MAPK) by first recruiting and activating the kinase TGFβ-activating kinase 1 (TAK1) (31). How K63-linked ubiquitin chains accomplish this task has been a matter of debate, since TRAF6 has been shown to link chains to various target proteins in response to stimulation and it is difficult in situ to parse the specific biological relevance of targets of ubiquitination. In fact, TRAF6 autoubiquitination is often used as a read-out for TRAF6 activation, and in one study has been mapped to the TRAF6 lysine residue K124 (32). While this study reports that K124 is essential for TRAF6-mediated activation of TAK1 and IKK, a separate study showed that while the TRAF6 RING finger domain is essential for IL-1R- and RANK-mediated TRAF6-dependent signaling, ubiquitination of TRAF6 itself is dispensable (33). In both reports, as well as previous others that link TRAF6 to the NOD pathway (34) and to TCR (23), the IKK complex member, IKKγ/NEMO, is reported as an additional target of TRAF6-mediated ubiquitination, and might therefore represents an element of redundancy in TRAF6 ubiquitin targeting. Site-specific ubiquitination of IRAK1 has also been reported as a possible mechanism of TRAF6-mediated signaling downstream in MyD88-dependent signaling pathways of the TLR/IL-1R superfamily (35). It is possible that any one specific anchoring point for K63-linked ubiquitin chains is not required for kinase activation, but rather their mere presence within the signaling complex, as it has been shown that even free, unanchored K63-linked chains were able to activate TAK1 and IKK by first interacting with a ubiquitin-binding domain in the adaptor protein TAB2 (36). This observation does not necessarily mean, however, that unanchored K63-chains mediate signaling in vivo, because although Ubc13/Uev1A can generate free chains, an E3 ligase, such as TRAF6 is required primarily for specifying protein targets and catalyzing transfer of ubiquitin chains to those targets (37), and TRAF6 has been repeatedly demonstrated by genetic approaches to be critical for intracellular (as opposed to cell-free) signal transduction.
While most effort in decoding the canonical TRAF6 signaling mechanism has focused on the formation of protein complexes that lead to activation of TAK1 and subsequently activation of IKK-NFκB and MAPK, activation of other pathways are also associated with TRAF6. For instance, TRAF6-mediated activation of the kinase MEKK3 is required for inflammatory cytokine production in response to some TLR/IL-1R stimuli (38). Evidence suggests that TRAF6-MEKK3 represents a time-delayed mechanism for activating NFκB in TRAF6-dependent signaling pathways in a manner independent of TAK1 (39). TRAF6 has also been shown to activate the p38 MAPK pathway in response to TLR4 and ROS stimulation via activation of the kinase ASK1, though it is unclear whether TAK1 is involved (40). TRAF6 has also been shown to activate the PI3K and downstream Akt/PKB pathway in response to both TNFR and TLR/IL-1R stimulation, but in the former case activation requires recruitment to the receptor of Src family kinases and Cbl family scaffolding proteins (27, 29), while in the latter case IRAK1 is required, though it is unclear whether other bridging molecules are involved (28). Recent work has shown that TRAF6 signaling is also specified by interaction with the scaffolding protein RACK1, which, upon RANK activation leads to recruitment of TRAF6, TAK1, and MKK6 in order to activate the p38 MAPK pathway (unpublished observation). In addition to recruitment of scaffolding and adaptor complexes, specificity of TRAF6 signaling can be determined by quantity of signal. For instance, experiments involving overexpression of mutant versions of CD40 and RANK suggest that differences in downstream signaling results may be attributed to the greater number of TRAF6 binding sites found in RANK (41). By contrast, signaling downstream of TRAF6 is also activated directly through its ubiquitin ligase activity. For instance, it has recently been shown that Akt is a direct target of TRAF6 ubiquitin ligase activity in response to IL-1β stimulation, and that Akt ubiquitination is required for its membrane recruitment and activating phosphorylation (42). Finally, TRAF6 has also been shown to activate the interferon regulatory pathway (IRF) pathway, including members IRFs 5 and 7, downstream of some TLR/IL-1R members in a mechanism that utilizes the MyD88-IRAK complex (43, 44).
Deubiquitinase regulation of TRAF6 signaling
As TRAF6 activates signaling via attachment of K63-linked ubiquitin chains, a class of de-ubiquitinating enzymes has been identified that disassemble these chains in order to negatively modulate signaling. The deubiquitinating enzymes A20, and to a lesser extent CYLD, have both been demonstrated to negatively regulate TRAF6-mediated ubiquitination (45, 46). CYLD has been shown to be recruited to TRAF6 by p62/Sequestosome and to be required for negatively regulating signaling from the TNFR superfamily member RANK in osteoclasts by inhibiting TRAF6 ubiquitination (47). A20 negatively regulates TRAF6 signaling by various mechanisms. In response to TLR/IL-1R signaling A20 de-ubiquitinase activity removes TRAF6 K63-linked ubiquitin chains (45). The zinc finger-containing protein Tax1-binding protein 1 (TAX1BP1) has been shown to regulate A20-mediated inhibition of TRAF6 signaling by recruiting A20 via ubiquitin-binding activity (48, 49). Another mechanism of A20-mediated inhibition of TRAF6 signaling involves disruption of the TRAF6 ubiquitination complex by triggering K48-linked degradative ubiquitination of the E2 ubiquitin-conjugating enzyme Ubc13 (50). A20 regulates IL-17 receptor signaling mediated by TRAF6, first through IL-17 receptor-induced expression, and then by binding directly to IL-17RA within an established inhibitory domain, and disrupting K63-linked ubiquitination of members of the activating complex involving TRAF6 and Act1 (51). Downstream of TGFβRI signaling, TRAF6-dependent MAPK activation is inhibited via Smad6 recruitment of A20, which is associated with decreased TRAF6 K63-linked ubiquitination (52). These and various other studies utilizing genetic approaches demonstrate the importance of negative regulatory mechanisms of TRAF6 signaling in order to maintain proper immune homeostasis (45, 53, 54).
Emerging examples of TRAF6 signaling crosstalk
Just as mechanisms exist to regulate TRAF6 signaling, examples have recently emerged of cases where TRAF6 itself interacts with and modulates signaling in pathways that were previously unrecognized as related. The JAK-STAT pathway activates gene transcription downstream of various cytokine receptors critical to immune cells, including interferons and interleukins. This pathway is often thought to act in parallel with, or sequential to, TRAF6-mediated signaling within a given cell during activation and differentiation events that are triggered by complex stimuli. However, some evidence suggests crosstalk between the two pathways. STAT3 and NFκB transcription factors can be activated downstream of IL-6R and IL-1R, respectively, and IL-1-triggered TRAF6 signaling regulates STAT3 signaling both indirectly by specifying formation of hetero- versus homo-dimers of NFκB members (55), and directly by interacting with STAT3 in a manner that enhanced STAT3 ubiquitination and decreased its transcriptional activity (56). Conversely, it has recently been shown that STAT3 negatively regulates TRAF6 signaling by inhibiting expression and/or function of the E2 conjugating enzyme Ubc13 downstream of TNFR and TLR-IL-1R superfamily members (57). TRAF6 has also recently been shown to positively regulate JAK-STAT signaling through a non-canonical pathway involving activation via serine phosphorylation (as opposed to canonical activation on tyrosine residues), as TRAF6 recruits STAT1 into the TLR signalosome for activation by an as-yet unidentified serine kinase (58). Another area of emerging crosstalk, though its implications for immune processes remain are currently unclear, is between TRAF6 and the Notch signaling pathway. It was first shown that TRAF6-deficiency in myofiber explants results in increased proliferation along with enhanced expression of Notch ligands, demonstrating indirect, but biologically relevant, regulation of the pathway (59). More recently, it has been shown that TGFβ-dependent activation of TRAF6 leads to recruitment of presenilin 1, the protease responsible for activating Notch receptors by enabling their transport from the membrane to the nucleus, to the TGFβRI complex where it serves a similar function (60). TRAF6 apparently then promotes protease activity in a manner associated with K63-linked ubiquitination of presenilin-1, suggesting the possibility that TRAF6 may also play some direct role in Notch activation that will require additional investigation (60). In fact, recent work in Drosophila shows that the TRAF6 ortholog DTRAF2 interacts directly with the Notch intracellular domain (ICD), and that genetic interaction between DTRAF2 and Notch is apparent in related phenotypes (61). As the Notch has been extensively characterized as a critically important and well conserved signaling pathway to cellular development and differentiation fate choices, this work may indicate an important new avenue of study of TRAF6 signaling that will begin to explain how TRAF6 can both directly regulate immediate inflammatory responses, and also subtly control long term differentiation paths of various immune cell types. It is likely that with further advances in proteomic techniques, additional examples of TRAF6 signaling crosstalk will be identified, and as such, these complexities must be considered when studying TRAF6-associated immune phenotypes.
Roles of TRAF6 in thymic and peripheral lymphoid tissues
The immune system is controlled by multiple mechanisms that regulate tolerance and immunity. Self-tolerance protects against autoimmune pathology and is established during both development (central tolerance) (62, 63) and peripheral homeostasis (peripheral tolerance) (64, 65). Humoral and cell-mediated immunity coordinately protect against pathogenic invasion and cancer, and require multiple developmental steps from activation to differentiation to achieve functionally distinct subsets of lymphocytes. TRAF6-deficient mice exhibit severe osteopetrosis, thymic atrophy, defective lymph node organogenesis, splenomegaly, and suffer perinatal death (d13-17) (66), suggesting essential roles for TRAF6 in a wide range of immune compartments. Additionally, studies with fetal liver chimeras further substantiate the role of TRAF6 in controlling development and maintenance of different subsets of hematopoietic cells (4). Below, current understanding of the wide range of roles of TRAF6 in regulating tolerance, activation and differentiation of T cells is reviewed.
TRAF6 regulates thymic selection and central tolerance
Negative selection of self-reactive thymocytes depends on promiscuous expression of tissue-specific antigens (Ag) by medullary thymic epithelial cells (mTECs) (62, 63, 67, 68). The expression of autoimmune regulator (Aire) protein by mTECs provides Ags that are otherwise expressed only in peripheral tissues, eliminating self-reactive cells from the developing T cell repertoire, and playing a crucial role in central T cell tolerance.
Phenotypes of TRAF6−/− mice, along with studies focusing on signaling of the TNFR superfamily members RANK and CD40 during development have suggested critical roles for TRAF6 in the development and functions of mTECs and central tolerance (69, 70). TRAF6−/− mice show compromised mTEC development along with reduced expression of Aire and, consequently, fewer tissue-specific antigens, correlating with their autoimmune phenotype (66). TRAF6−/− fetal thymic stroma tissue fails to mediate negative selection and induces autoimmunity when grafted into athymic nude mice (71). Furthermore, specific deletion of TRAF6 in thymic epithelial cells (TRAF6ΔTEC) results in diminished medullary areas of the thymus along with autoimmune hepatitis and autoantibody development (72), indicating an essential role for TRAF6 in central tolerance through regulation of mTEC development (Fig 2).
Figure 2.
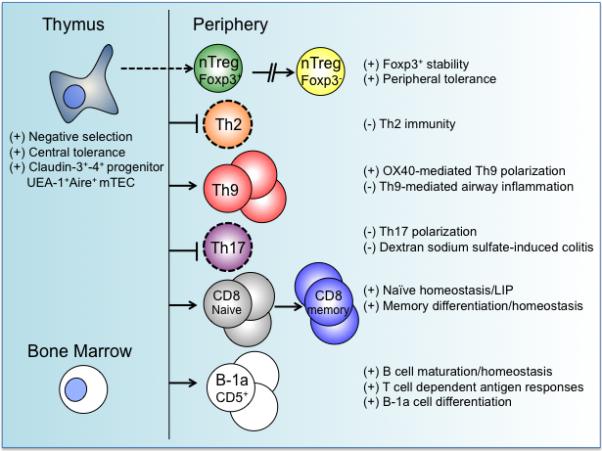
TRAF6 regulation of adaptive immune function and homeostasis. The roles of TRAF6 in lymphocytes are illustrated based on data gathered from studies that employed cell-specific deletion of TRAF6. TRAF6 plays an important role in maintaining central tolerance as well as peripheral immunity. See text for further detail. (+) TRAF6 functions as a positive regulator; (−) TRAF6 functions as a negative regulator.
Several studies suggest mechanistic insights into how TRAF6 regulates development of the thymic medullary microenvironment and expression of Aire. One study showed that claudin-3 and -4 (Cld3,4)+ mTECs, a subset that selectively expresses Aire, are significantly impaired in TRAF6−/− mice (73). This study further suggests the possibility that mTECs arise from a unique progenitor, and that TRAF6 might promote proliferation and/or survival of this cell lineage during embryonic thymic development. TRAF6 also induces expression of RelB, a member of the NFκB family, which is important for mTEC development. RelB-deficient mice show medullary thymic atrophy, impairments of Aire expression and negative selection, and multi-organ inflammation, exhibiting phenotypes similar to TRAF6−/− mice (74). Retroviral introduction of TRAF6 into TRAF6−/− mTEC cell lines restores expression of RelB, suggesting TRAF6 directs RelB induction in mTECs (71). However, it currently remains unclear how TRAF6-mediated RelB induction leads to Aire expression. Additionally, defective mTEC formation observed in RANK/RANKL- (75-77) or CD40/CD40L-deficient mice (70, 78) points to these receptor-ligand pairs as potentially critical activators of TRAF6 during mTEC development. RANK signaling was shown to be important during embryogenesis, and cooperation between CD40 and RANK signals is required to establish the medullary microenvironment upon birth (77). In addition, ligation of RANK or CD40 was shown to activate intracellular signaling pathways necessary for the development of mTECs, including TRAF6- (for canonical NFκB activation) and NFκB inducing kinase (NIK)- (for non-canonical NFκB activation) dependent signaling, by employing TRAF6−/− and aly/aly (NIK mutant) embryonic tissues in fetal thymic organ cultures (FTOC). The development of UEA-1+Aire+ cells is compromised in FTOC of TRAF6−/− and aly/aly mice upon RANK or CD40 ligation, showing the importance of both TRAF6-dependent canonical and NIK-dependent non-canonical NFκB activation pathways for the development of mTECs (77). Further studies into the mechanisms and signals that determine commitment to the mTEC lineage and Aire expression, and spatiotemporal involvement of TRAF6 in this process will be necessary for better understanding the role of TRAF6 in establishing central tolerance.
TRAF6 controls lymphoid tissue organogenesis
TRAF6−/− mice lack peripheral lymph nodes (LN), the development of which require cooperation between RANKL-RANK and lymphotoxin (LTα, LTβ, LTβ) receptor signaling in order to activate both the canonical NFκB and non-canonical NIK-IKKα pathways (79). In this context, TRAF6 appears to be required for the relevant RANK-mediated signaling (80). LN organogenesis first requires RANK engagement on lymphoid tissue inducer (LTi) cells recruited to a rudimentary anlage composed of lymphoid tissue organizer (LTo) progenitor cells. LTi cells subsequently produce lymphotoxin α/β (LTα/β) for LTo progenitors that express LTβR, which induces development of mature LTo cells (81, 82). RANK-mediated positive feedback leads to increased RANKL-RANK expression on mature LTo cells, amplifying LTi growth and tissue organization (83). Finally, it has been shown that LN organogenesis can be reconstituted by intraembryonic injection of IL-7 into TRAF6−/− embryos, suggesting that the effects of RANKL exerted on the LN inducer cell population are functionally similar to those of IL-7 with respect to LTα/β induction (80).
Roles of TRAF6 in T cells
Study of TRAF6 in immune cells initially focused on antigen-presenting cells (APCs) because of the wide array of TRAF6-associated receptors from both the TNFR and TLR/IL-1R superfamilies that they express. While dysregulation of the TRAF6−/− T cell compartment was previously believed to be due to cell non-autonomous effects related to defective APC function, significant upregulation of TRAF6 in response to TCR engagement justified examination of T cell-intrinsic TRAF6 (84). T cell-specific deletion of TRAF6 has revealed a surprisingly broad array of critical cell-intrinsic roles for TRAF6 in the regulation of peripheral T cell tolerance and immunity (Figs. 2 and 3).
Figure 3.
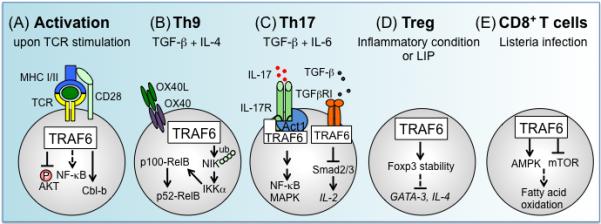
TRAF6 in T lymphocyte activation and differentiation. (A) Upon TCR stimulation, TRAF6 negatively regulates PI3K-Akt activation and restrains IL-2 production and proliferation by enforcing a requirement for costimulation. Absence of TRAF6 expression results in T cells that are refractory to Treg-mediated suppression, and that resist anergy induction in a manner associated with decreased expression of the pro-anergy factor Cbl-b. TCR-dependent activation of NFκB is critical for both T cell activation and differentiation, and currently conflicting evidence exists regarding the requirement for TRAF6 for activation of this pathway. (B) OX40-triggered TRAF6-dependent non-canonical NFκB activation, in combination with TGF-β and IL-4, is important for Th9 cell differentiation. TRAF6 activates NIK, which mediates the processing of p100 to p52, leading to generation of p52-RelB complexes. OX40-OX40L-dependent Th9 polarization is independent of PU.1, a key transcription factor for Th9 cells. (C) TRAF6 is an important regulator of IL-17R signaling as well as Th17 differentiation. TRAF6 is recruited to IL-17 receptor and activated through Act1, an adaptor protein and E3 ubiquitin ligase, and subsequently transduces signals to activates NFκB and MAPK pathways. TRAF6 also functions as an inhibitor of TGF-β-induced Smad2/3 activation, and TRAF6ΔT T cells cultured under Th17 conditions with TGF-β and IL-6 exhibit increased Th17 differentiation due to enhanced TGF-β-mediated inhibition of expression of IL-2. (D) TRAF6 is essential in memory CD8+ T cell differentiation in response to listeria infection. TRAF6ΔT CD8+ T cells fail to differentiate into memory cells following normal primary expansion. This defect can be mitigated by pharmacologically enhancing AMPK activity, or by inhibiting mTOR activity. As a process regulated downstream of AMPK, fatty acid oxidation levels may function as cellular rheostat for determining memory precursor potential.
TCR-mediated T cell activation
TCR stimulation initiates a complex signaling cascade that culminates with activation of transcription factors, including NFκB, NFAT, and AP-1. In the canonical NFκB signaling pathway, phosphorylation and degradation of cytosolic IκB inhibitors by the IκB kinase (IKK) complex result in subsequent nuclear translocation of NFκB. PKCθ, Carma1/CARD11-Bcl10-MALT1 (the CBM complex) and Caspase-8 are key molecules involved in the activation of IKK/NFκB signaling downstream of TCR (85-90). Several studies have suggested an essential role for TRAF6 in TCR-induced IKK activation. For example, IKK/NFκB activation in CD3/CD28-stimulated Jurkat cells was compromised by siRNA knockdown of TRAF6 (23, 91). TRAF6 directly associates with the C-terminus of MALT1 via two conserved binding motifs as shown in overexpression experiments, or is indirectly recruited to MALT1 through association with caspase-8 (91). C-terminal ubiquitination of MALT1 is important for initiating IKK/NFκB signaling and IL-2 induction in response to T cell activation, and TRAF6 has been shown to function as a potential ubiquitin ligase for MALT1 in Jurkat cells (92). Additionally, knockdown experiments using siRNA in Jurkat cells show that association of TRAF6 with activated caspase-8 is crucial for the translocation of caspase-8 to lipid rafts and for subsequent TCR-induced activation of NFκB, suggesting the importance of TRAF6 to TCR signaling (91). However, these findings do not necessarily align with phenotypes observed in T cells from TRAF6ΔT T cells, in which TRAF6 is genetically deleted during thymic development by expression of CD4-Cre (84). In contrast to biochemical and gene knockdown studies in cell lines, TRAF6ΔT T cells exhibit no apparent defects in NFκB activation upon TCR engagement. Additionally, unlike TCR-stimulated T cells deficient in CBM complex members, TRAF6ΔT T cells show no defects in NFκB activation, and exhibit (at least) normal levels of IL-2 production and proliferation (93). These discrepancies may suggest that the requirement for TRAF6 in response to TCR signaling is context-dependent, such that it may be required during some stage of activation or differentiation of T cells not yet tested. It should be noted that TRAF6 expression is strongly upregulated in naïve T cells upon initial TCR-mediated activation (84), possibly suggesting that the requirement for TRAF6 downstream of TCR is specific to activated cells and/or TCR re-stimulation (which may be more similar Jurkat cells). Alternatively, there may be redundancy of TRAFs in primary T cells, such that absence of TRAF6 is compensated by another TRAF during TCR stimulation. Along these lines, siRNA-mediated knockdown of TRAF2 in Jurkat cells suggests its involvement in TCR-dependent NF- κB activation (23). Also, transgenic mice expressing a dominant-negative mutant of TRAF2 in T cells exhibit a defect in IL-2 production (94). Therefore, examining the phenotype of T cells by TRAF2/6 double deletion may be informative for understanding the IKK/NFκB signaling mechanism downstream of TCR, and might resolve seemingly contradictory findings regarding the role of TRAF6 in T cell activation.
In addition to apparently normal TCR-mediated NFκB activation, naïve CD4+ T cells from mice with T cell-specific deletion of TRAF6 (TRAF6ΔT) exhibit hyper-sensitivity TCR engagement, as evidenced by enhanced activation of the PI3K-Akt pathway and diminished requirement for CD28 costimulation to undergo proliferation (84). Uncoupling of CD28 and TCR in TRAF6ΔT CD4+ T cells also results in a defect in anergy induction associated with dysregulation of the E3 ligase Cbl-b (93), and TRAF6ΔT CD4+ T cells are refractory to suppression by CD4+CD25+ Tregs both in vitro and in vivo (84) (Fig. 3).
Differentiation of Th/Treg cells
After initial TCR-mediated stimulation, naive peripheral CD4+ T cells differentiate into distinct T helper (Th) lineages, including Th1, Th2, Th9, Th17, Th22, and iTreg, in the presence of cytokines that drive different types of immune responses. Various factors, including initial cytokine environments, transcription factors, and epigenetic modifications, are involved in CD4+ T cell fate determination and functional stability (95-100). Several lines of evidence show that T cell-intrinsic TRAF6 expression regulates CD4+ T cell lineage differentiation (Fig. 3).
Th9 cells produce IL-9 and IL-10, are believed to contribute to immunopathology in allergy and autoimmunity, and differentiate in response to IL-2, TGF-β and IL-4 (101). Factors known to contribute to Th9 cell differentiation include IL-25, Jagged2, PD-L2, OX40, and thymic stromal lymphopoietin (TSLP) (98, 102). A recent study demonstrated that TRAF6 is required for OX40 signaling-dependent Th9 differentiation (103). Ligation of OX40 activates the ubiquitin ligase activity of TRAF6, which mediates the activation of NIK. NIK mediates the processing of p100 to p52, thus forming the transcriptionally active p52-RelB complex. This noncanonical NFκB pathway is required for differentiation of Th9 cells by OX40 costimulation.
IL-17-producing Th17 cells play an important role in host defense against extracellular bacteria, fungi and other eukaryotic pathogens. Th17 differentiation is induced by IL-6 and TGF- β and further stabilized by IL-23 (104). The transcription factor RORγt regulates the differentiation process (96). TRAF6 also regulates Th17 cell differentiation, as shown in a model of acute colitis, where TRAF6ΔT mice exhibit more severe disease associated with increased Th17 cells in lamina propria tissue (105). Consistent with this finding, naïve TRAF6ΔT CD4+ T cells show increased Th17 differentiation in vitro under Th17 polarizing conditions, suggesting that TRAF6-dependent inhibition of Th17 differentiation is cell-intrinsic. Cells from TRAF6ΔT Th17 cultures were found to be more sensitive to TGF-β stimulation, resulting in enhanced Smad2/3 activation and related inhibition of expression of IL-2, itself an inhibitor of Th17 differentiation (106). As such, TRAF6ΔT cells in Th17 cultures exhibited levels of inhibition of Th17 differentiation comparable with control cells.
Regulatory T cells (Tregs) are responsible for restraining T cell-mediated immunity and come in two basic varieties: thymic (or “natural”) Tregs (nTregs) generated in the thymus during selection, and iTregs, induced in the periphery, both of which require expression of Foxp3 (107). As discussed below, Treg deficiency or impotence may impact in vivo immune tolerance in mice lacking TRAF6 specifically in T cells or Tregs. However, there does not appear to be significant evidence that TRAF6-deficient T cells exhibit defective Treg differentiation or function in vitro. While TRAF6ΔT mice appear to have normal circulating Treg population, it should be noted that since TRAF6 is not deleted until after CD4 is expressed in these mice (as deletion is driven by CD4-Cre), they may not be suitable for determining whether nTreg development is TRAF6-dependent. To address this question, mice in which TRAF6 is deleted upon expression of Foxp3 (TRAF6Δ Foxp3) were generated (108). Like TRAF6ΔT mice, TRAF6Δ Foxp3 mice produce Tregs that exhibit apparently normal suppressive capacity during in vitro suppression assays (108). Using TRAF6−/− mice, it was shown that while TRAF6 is necessary for thymic Treg development, it is dispensable for antigen-dependent iTreg development in the periphery, as TGFβ-driven induction of Foxp3+CD25+CD4+ T cells is not defective in the absence of TRAF6 (109). Similarly, TRAF6ΔT CD4+ T cells also exhibit apparently normal Foxp3+ iTreg differentiation in the presence of TGF-β and IL-2 (105). Together these results suggest that differentiation of the extra-thymic iTreg population does not require T cell-intrinsic TRAF6 expression.
CD8+ T cell homeostasis and memory development
CD8+ T cells are the primary population responsible for protective cell-mediated immunity to infection with intracellular pathogens or cancer. Upon Ag encounter, CD8+ T cells undergo a differentiation program characterized by expansion, effector and contraction phases, and development of a memory population that ensures faster and more efficient immune protection in response to subsequent Ag encounter. T cell-specific deletion of TRAF6 showed a role for TRAF6 in differentiation of memory CD8+ T cells in response to intracellular bacterial infection that also revealed an unexpected link to cellular metabolism. Upon L. monocytogenes infection, TRAF6ΔT CD8+ T cells undergo normal primary expansion, followed by dramatic contraction, and then failure to develop a memory population that could effectively expand upon re-challenge (110). Microarray analysis highlighted abnormal regulation of genes associated with the fatty acid metabolism pathway in TRAF6ΔT CD8+ T cells during the contraction phase. Furthermore, contracting TRAF6ΔT CD8+ T cells were found to exhibit defective activation of AMP kinase (AMPK), an upstream trigger of fatty acid oxidation. Treatment of mice with pharmacological agents that enhance AMPK and/or fatty acid oxidation, rescues the defect in TRAF6ΔT CD8+ T in memory formation, suggesting that TRAF6 integrates signals upstream of AMPK and consequently regulates memory T cell formation. This study shows an important possibility that there could be a direct cause and effect relationship between a metabolic pathway and the CD8+ T cell differentiation process, and that TRAF6 could directly or indirectly modulate AMPK to coordinate signals for energy homeostasis. Currently it remains unclear whether TRAF6 regulates specific metabolic pathways, and if so, whether such regulation determines CD8+ T cell fate. Resolution of these questions may require specific gene targeting of bona fide metabolic pathway components to eliminate possible metabolism-independent effects of TRAF6 deficiency and/or off-target effects of pharmacological agents.
Homeostasis of the naïve T cell compartment is critical for optimal immune responses since maintaining T cells with broad specificity is essential for effective pathogen clearance. Although TRAF6ΔT CD8+ T cells are hyper-proliferative in response to cognate Ag stimulation, it was recently shown that naïve TRAF6ΔT CD8+ T cells exhibit defective homeostatic/lymphopenia-induced proliferation (LIP), and that this defect can be correlated with a novel in vitro model of lymphopenia-induced proliferation (LIP) (111). Specifically, the IL-1 family member IL-18 was found to synergize with IL-7 to support slow LIP-like expansion of naive control CD8+ T cells, whereas cytokine synergy was abrogated in TRAF6ΔT CD8+ T cells, suggesting a TRAF6-dependent pathway required for LIP in naïve CD8+ T cells. Using a model peptide system, it was shown that IL-7/IL-18 cytokine synergy induces naïve CD8+ T cells to proliferate in response to a model self-peptide in vitro, which further correlates with requirements for LIP in vivo. While IL-18R receptor signaling does not appear to be specifically required for in vivo LIP, this could point to the fact that there are various TRAF6-dependent signaling pathways active in a given T cell, and future work may focus on identifying how TRAF6 signaling is coordinated in this context. Additionally, because homeostatic mechanisms are also critical for maintenance of the memory cell compartment, temporal deletion of TRAF6 during the memory phase may also be studied in the context of cytokine-dependent homeostasis (Fig. 3).
Importance of T cell-intrinsic TRAF6 to immune tolerance
As described above, TRAF6 plays significant cell-intrinsic roles in various aspects of T cell activation, differentiation, and homeostasis. However, broader assessment of mice lacking TRAF6 in T cell compartments, TRAF6ΔT and TRAF6ΔFoxp3, suggest both T cell autonomous and non-autonomous effects on immune homeostasis and tolerance. For example, while TRAF6ΔT CD4+ T cells exhibit no apparent propensity to become Th2 cells during in vitro differentiation assays (105), TRAF6ΔT mice exhibit spontaneous multi-organ inflammatory disease with splenomegaly and lymphadenopathy, characterized by elevated Th2 cytokine production and spontaneous Th2 cell differentiation, increased serum IgG1, IgE and IgM levels and autoantibody production, as well as significant expansion of the B cell compartment (84). It may therefore be that TRAF6-deficient T cells trigger initial B cell expansion, but that Th2 polarization occurs as an effect of feedback from B cell expansion. This issue may be addressed in more detail by investigating the fate of naïve wild-type CD4+ T cells transferred to TRAF6ΔT mice. TRAF6ΔFoxp3 mice, in which TRAF6 deletion is restricted to the Treg compartment, also exhibit Th2-associated autoimmunity, and TRAF6ΔFoxp3 Tregs fail to suppress colitis induction in Rag2−/− mice when co-transferred with naïve control T cells, suggesting that T cell-intrinsic TRAF6 expression is required in vivo for Treg-mediated tolerance even if it is dispensable for Treg differentiation and in vitro suppression. By combining TRAF6ΔFoxp3 with a cell-intrinsic reporter for Foxp3 (Foxp3YFP-CreRosa26RFP), it was shown that expansion of Tregs and stable expression of Foxp3 are TRAF6-dependent in the in vivo peripheral immune tissues, and furthermore, that TRAF6ΔFoxp3 T cells that lost Foxp3 acquire GATA-3 and IL-4 expression, suggesting a potential conversion of TRAF6ΔFoxp3 Tregs into Th2 effector cells. These data may imply that Th2 skewing observed in TRAF6ΔT mice is, at least partially, the result of TRAF6 deficiency within the Treg subset, though the source of the non-cell-autonomous loss of tolerance within the B cell compartment would still remain to be determined. It also remains unclear whether in vivo Fox3 stability in TRAF6ΔFoxp3 Tregs is entirely cell-autonomous. This question should probably be first investigated in the context of reported cell-intrinsic increased activation of the PI3K/Akt pathway in TRAFΔT CD4 T cells, as this phenotype may correlate with recent observations relating the PI3K negative regulator PTEN to Foxp3+ Treg lineage stability (Fig 3.) (112). At the same time, additional studies must be undertaken to determine the various contributions of other, more clearly cell-intrinsic effects of TRAF6 on T cell differentiation, including Th9 and Th17, to the overall maintenance of immune tolerance in vivo.
Roles of TRAF6 in B cells
CD40 plays an important role in various B cell functions. Engagement of CD40 by its ligand CD154 recruits TRAF adaptor proteins, triggering signaling cascades leading to activation of NFκB, MAPK/ERK, and PI3K, which promote B cell effector functions such as, proliferation, cytokine and Ig secretion, isotype switching, and development of B cell memory (113, 114). Various studies have shown a critical role for TRAF6 in B cell function as a signal transducer of CD40. The key function of TRAF6 downstream of CD40 was directly examined by expressing a CD40 mutant that fails to bind TRAF6 or a dominant-negative TRAF6 molecule in B cell lines. This study shows that TRAF6 is required for IL-6 and IgM production, and B7 upregulation upon CD40 ligation, but is not essential for NFκB translocation or JNK activation (115). Elsewhere, disruption of the TRAF6 binding sites alone was shown to have little effect on NFκB and JNK activation, and failure of recruitment of TRAF2 and TRAF3 has a more significant effect (116), suggesting the role of TRAF6 in B cells is largely redundant with other TRAF proteins. Additionally, by generating transgenic mice expressing a mutant CD40 receptor in which TRAF6 or TRAF2/3 (and TRAF1/5 recruitment indirectly through interaction with TRAF2/3) binding sites are disrupted (117), it was shown that TRAF6 recruitment is required for generation of long-lived Ig-secreting bone marrow plasma cells. In germinal center (GC) formation, however, TRAF6 exhibits redundancy with TRAF2/3, showing that there exist both TRAF6-dependent and TRAF6-independent pathways shaping antigen-driven humoral responses through CD40 signaling.
Deletion of TRAF6 within the B cell compartment using CD19-Cre mice (TRAF6ΔB) further specifies the contributions of CD40 and/or TRAF6 in signaling, function, and development of B cells (118). B cells from TRAF6ΔB mice fail to activate MAPK and NFκB and to proliferate in response to LPS, CpG-DNA, and anti-CD40 antibody stimulation in vitro. The early development of subsets of B cells in the bone marrow (BM) of TRAF6ΔB mice is not compromised whereas the B220highIgM+IgD+ mature recirculating B cell population in BM is significantly impaired. The percentage of CD19+B220+ cells is significantly reduced with more prominent reduction in mature B cells (IgMlowIgDhighAA4.1−) while no obvious defect is observed in survival, suggesting that TRAF6 regulates a later step in mature B cell development. As expected from the defect in CD40 signaling in vitro, isotype switching to IgG1 and IgG2b in response to T cell-dependent (TD) antigen and generation of long-lived plasma cells are severely impaired in TRAF6ΔB mice. However, other CD40-dependent processes shown in CD40 or CD154 knockout mice, such as affinity maturation and GC formation are not affected, suggesting compensatory mechanisms for TRAF6. TRAF6ΔB mice also show a complete absence of CD5+ B-1a cells, which play an important role in clearing pathogens, such as S. pneumonia by providing natural antibodies, a phenotype observed in neither CD40-deficient nor MyD88/TRIF doubly deficient mice (119). Previously, it was shown that B cell–specific deletion of TAK1 results in B220+CD5+ B-1a population reduction in the peritoneal cavities (120), showing a similar phenotype with TRAF6ΔB mice. Therefore, these results together suggest the TRAF6-TAK1-dependent signaling pathway regulates development of the B-1a population. Revealing upstream stimuli that activate TRAF6, or redundancies in CD40 and TLR signaling that may regulate B-1a cell development and/or homeostasis will require further study. Cumulatively, the TRAF6ΔB phenotype demonstrates the complexity of signaling processes necessary for B cell development and function (Fig. 2).
Epstein–Barr Virus (EBV) infection of B cells results in chronic activation and proliferation through expression of several proteins, including latent infection integral membrane protein 1 (LMP1). Transgenic expression of LMP1 in murine B cells leads to hyperplasia and lymphoma, suggesting an important role for LMP1 in B cell transformation (121, 122). LMP1 resembles a constitutively-activate form of CD40 in B cells. However, the LMP1 signaling complex and its in vivo function in B cells also have some distinctions compared to CD40. Both LMP1 and CD40 induce activation of MAPK and NFκB, cytokine and antibody production, and extrafollicular B cell differentiation. However, LMP1, but not CD40, interacts with TNF receptor 1–associated death domain (TRADD) adapter protein (123). Furthermore, LMP1 expression not only fails to rescue GC formation in CD40−/− mice but also actively suppresses GC formation even in the presence of CD40 as demonstrated with LMP1 transgenic mice (124). Also, LMP1 and CD40 recruit different TRAF complexes for signal transduction. TRAFs 1 and 2 are required for JNK and NFκB activation downstream of CD40 but not LMP1 (125). LMP1 exhibits greater utilization of TRAF5 to activate B cells than does CD40 (126). In another study, transgenic mice that express LMP1 in B cells in the presence or absence of TRAF6 were generated and showed a requirement for TRAF6 in B cells for LMP1-mediated development of GC and anti-dsDNA antibody production, as well as basal immunoglobulin production and isotype switching (127). B cell-specific deletion of TRAF6 has no effect in GC formation or antibody affinity maturation following immunization (118). Therefore, TRAF6 deficiency seems to more severely affect LMP1 functions than CD40 functions, which also rely on TRAF2 recruitment. Decreased requirements for functional LMP1 signals would contribute to its unrestrained signaling during B cell transformation. The importance of TRAF6 is also demonstrated in a study showing the roles of TRAF2 and Nck-interacting kinase (TNIK), a germinal center kinase, in LMP-1 and CD40 signaling (128). Functional proteomics predicted and co-immunoprecipitation assays confirmed association of TNIK and LMP1 in lymphoblastoid cells. siRNA knockdown of TNIK or TRAF6 showed that TNIK is essential to JNK activation and canonical NFκB signaling, and that TRAF6 is required for these processes by mediating TNIK recruitment to LMP1. CD40 stimulation quickly induces the interaction of TNIK and TRAF6 in B cells, and siRNA knockdown of TNIK impairs JNK phosphorylation in human B cells (BL41) following CD40L stimulation, suggesting an important role of TNIK in CD40-mediated JNK activation in B cells. Furthermore, a recent study demonstrates that TRAF6 is required for class switch recombination (CSR) of DNA in B cells, providing additional evidence for TRAF6-dependent B cell function. In another study, MyD88 was shown to increase immunoglobulin diversification and production through the TNFR superfamily member TACI, which activates the MyD88-IRAK1-IRAK4-TRAF6-TAK1 pathway (129). It was further demonstrated that TACI signaling converges with CD40 and TLR signals through MyD88 recruitment and induces class switch recombination (CSR) of DNA in B cells. Expression of a dominant-negative form of signaling transducers downstream of MyD88, or immunoprecipitation assays with 2E2 B cells reveals that IRAK1, IRAK4, TRAF6, TAK1, and IKKβ are involved in TACI signaling. Consistent with these results, MyD88- and IRAK4-deficient patients show a defect in activation-induced cytidine deaminase (AICDA) induction, indicating impaired CSR. Evidence described here collectively represents TRAF6 as an important cell-intrinsic regulator of B cell development and function. As CD40 is a potent immune activator that is involved in the pathogenesis and exacerbation of a variety of human autoimmune diseases, and LMP1 plays a key role in EBV-mediated human B-cell malignancy, it will be of considerable interest to better define the mechanisms by which TRAF6 regulates these signaling pathways.
Roles of TRAF6 in myeloid cells
TRAF6 plays various important roles in myeloid immune cells that regulate immune responses via control of inflammatory responses, recognition of innate immune signals leading to initiation of adaptive immune responses, and homeostasis of microenvironments in which immune responses occur. Many TRAF6-associated signaling receptors and signaling mechanisms are shared between different types of myeloid cells (in addition to common mechanisms shared outside the myeloid lineage), but in this section the focus is on the biological requirements of TRAF6-mediated signaling in the contexts of specific myeloid cell types, and how those requirements are relevant to immune function.
Macrophages
Macrophages are phagocytic activators and regulators of innate inflammatory immunity that play important roles in resolution of microbial infection and wound healing, as well as contributing to conditions like obesity, diabetes, and atherosclerosis (130, 131). In response to soluble Toxoplasma gondii antigen (STAg), TRAF6 expression in macrophages has been shown to be required for induction of the inflammatory cytokine IL-12, which is critical for control of parasite infection, in a manner dependent on the p38 MAPK pathway (132). Toxoplasma gondii has the capacity to enter macrophages by direct penetration in addition to by phagocytosis (133). Another mechanism of Toxoplasma eradication involves vacuole-lysosome fusion in macrophages via autophapy. This process has been found to been triggered by macrophage CD40 activation of the TRAF6 pathway that synergizes with TNFR signaling, demonstrating a novel mechanism of anti-parasite immunity as well as a physiologically relevant example of TRAF6 signaling crosstalk (134). Interestingly, the known anti-viral mediator PKR has recently been shown as a novel target of TRAF6, as it was demonstrated to be activated downstream of CD40-TRAF6 signaling in macrophages in order to trigger autophagic vacuole-lysosome fusion in the service of T. gondii killing (135). Additional study of CD40 signaling mechanisms in macrophages showed that TRAF6, but not TRAF2/3/5, binding sites in CD40 are required for inflammatory cytokine production, and that CD40-TRAF6 mediates downstream activation of IKK-NFκB and ERK MAPK pathways (136). Another study investigated the role of cholesterol depletion by the parasite Leishmania major and its potential role in subverting inflammatory TRAF6 signaling. It was demonstrated that TRAF6 and TRAF2/3/5 form distinct CD40 signalosomes in L. major-infected macrophages, such that in the absence of cholesterol, TRAF6 is prevented from inducing IL-12 and is instead complexed with the kinase Lyn to promote signaling that results in production of the immunomodulatory cytokine IL-10 (137).
In addition to anti-microbial immunity, macrophages are also involved in chronic inflammatory pathology such as atherosclerosis, where macrophages accumulate in a cholesterol-associated manner along arterial walls (131). CD40 signaling in macrophages is an important component of this pathology. In a mouse model of atherosclerosis, CD40L blockade or CD40 deficiency mitigate disease, work to examine the TRAF signaling dependency of atherogenesis showed that TRAF6, not TRAF2/3/5, binding sites in hematopoietically-expressed CD40 on a pro-atherogenic polipoprotein-deficient (ApoE−/−) background are required for monocyte recruitment to the arterial wall and in the absence of CD40-TRAF6 signaling macrophages preferentially differentiate toward the anti-inflammatory M2 fate (138). To directly address the role of TRAF6 in atherogenesis, another group induced specific genetic deletion of TRAF6 in either the endothelial or myeloid lineages on the ApoE−/− background (139). Interestingly, it was found that while endothelial TRAF6 deficiency mitigated disease development, the absence of TRAF6 in myeloid cells resulted in exacerbated disease, characterized by diminished production of the protective anti-inflammatory cytokine IL-10 by macrophages. In contrast to the study focusing only on the role of CD40-TRAF6 signaling, this case encompasses potential TLR-TRAF6 and TGFβR-TRAF6 signaling as well, which may represent potentially important anti-atherogenic mechanisms. Together, these studies show that regulation of TRAF6 signaling within macrophages undergoes complex regulation in order to maintain a balance between protective and pathogenic inflammation.
A newly discovered mechanism of TRAF6 function has begun to clarify how reactive oxygen species produced by mitochondria (mROS) in macrophages contributes to immune responses against intracellular bacteria (140). mROS production is triggered by TLR signaling, and was revealed to involve recruitment of TRAF6 to the mitochondria where it interacts with the mitochondrial respiratory chain assembly mediator protein ECSIT. Apparent ubiquitination of ECSIT by TRAF6 is associated with increased mROS production, and ECSIT- or TRAF6-deficient macrophages exhibit defective TLR-induced ROS production and intracellular bacteria killing. This study not only promotes the importance of mitochondria as platforms for immune signaling, it further expands our understanding of where TRAF6 may function within the cell. Characterization of the mitochondrial anti-viral signaling mediator MAVS/VISA/IPS-1/Cardif and its interaction with TRAF6 has also suggested possible localization of TRAF6 to the mitochondria (22). These findings also may also have implications in other cell types, such as effector-contracting CD8 T cells, where TRAF6 deficiency results in defective mitochondrial fatty acid oxidation in response to unknown (TRAF6-activating) stimuli (110). Future efforts may further focus not only on TRAF6 signaling pathways and interaction partners, but also on TRAF6 subcellular location in response to specific stimuli. Additionally, it will be interesting to determine whether activation of kinase pathways associated with ROS-dependent TRAF6 activity, such as ASK1-p38, is triggered by TRAF6 mitochondrial translocation (40).
Dendritic cells
TRAF6 has multiple important roles in dendritic cell (DC) biology, including regulating DC activation, survival, homeostasis, and tolerance (Fig. 4). The identified roles of TRAF6 in DCs can be divided into those attributed to specific receptor signaling pathways, and those inferred from phenotypic changes in TRAF6-deficient DCs, but which have not necessarily been linked to modulation of specific receptor activity. In the former category, TRAF6 in DCs is a critical mediator of the TNFRSF members CD40 and RANK. The ligand for RANK, RANKL (also called TRANCE, ODF, and OPG-L) was originally identified as a cytokine expressed by T cells that enhanced bone marrow-derived DC (BMDC) survival (141, 142), and TRAF6, which was subsequently shown to bind at multiple sites in the intracellular region of RANK (along with multiple other TRAFs), is shown by dominant-negative TRAF6 overexpression to be required for optimal NFκB activation (143). Similarly, CD40, for which TRAF6 (and various other TRAFs) have been extensively characterized (primarily in B cell models) as mediating downstream activation of NFκB and MAPK pathways, is expressed by DCs and is shown to enhance survival as well as DC-T cell interactions (144). Though CD40-deficient DCs exhibit normal activation of T cell responses, simultaneous blocking of RANK results in defective anti-viral CD4 T cells responses, suggesting redundant roles for RANK and CD40 in DC-mediated immunity (145). TRAF6 also regulates RANK- and CD40-mediated BMDC activation of Akt/PKB, another candidate pathway for promoting survival, in a mechanism that involves c-Src activation and requires the scaffolding protein Cbl-b (27). TRAF6−/− mice and bone marrow chimeras were used in a study to explicitly investigate the role of TRAF6 in development and activation of DCs in the context of acute inflammatory immunity. Results showed first that TRAF6 is required for normal homeostasis of the splenic DC compartment, with TRAF6-deficient mice exhibiting a specific severe defect in the CD4+CD11b+ DC subset that encompasses more than half of total splenic DCs (146). Further, both BMDCs and splenic DCs from TRAF6-deficient mice fail to upregulate MHC and costimulatory surface markers, and fail to elaborate inflammatory cytokines in response to stimulation with either CD40L or various TLR ligands. It should be noted that TRAF6-deficient and MyD88-deficient DCs exhibit similar defects in response to TLR ligand engagement, suggesting a common signaling pathway (147). Not surprisingly, antigen-loaded TRAF6-deficient DCs exhibit defective capacity to activate antigen-specific CD4 T cells in situ (146). However, studying the role of TRAF6 in DCs using complete knockout mice presents a problem first with isolating DC-specific defects in the context of manifold TRAF6-dependent immune processes, and second, with unintentionally activating DC through ex vivo manipulation. In a more recent study mice harboring DC-specific TRAF6 deletion (TRAF6ΔDC) were generated, enabling study of DC biology in situ, and the effects of DC TRAF6 signaling on immune homeostasis. Surprisingly, while splenic DCs in TRAF6ΔDC mice exhibited similar deficiencies in driving acute Th1 immunity, TRAF6ΔDC mice spontaneously developed Th2-associated inflammation and fibrosis specifically in the small intestine (148). This disease phenotype was found to be associated with defective Treg homeostasis, and could be ameliorated with broad-spectrum antibiotics treatment, which initially suggested TRAF6ΔDC-associated inflammation is triggered by gut commensal microbiota. However, when TRAF6ΔDC are re-derived on a germ-free background lacking microbiota the phenotype is exacerbated, though still capable of being ameliorated by antibiotics (unpublished observation). Because TRAF6 signaling in DCs appears to be a critical factor for controlling gut immune homeostasis in a manner that is independent of commensal microbiota, and because this model is relatively unique in its anatomic specificity and spontaneous nature, it is now important to determine what other factors may be triggering this condition in order to draw potential parallels to human autoimmune disease. It is also notable that mice lacking MyD88 specifically in the DC compartment do not exhibit intestinal autoimmune pathology similar to TRAF6ΔDC, suggesting that the upstream receptors involved in this disease are TLR-independent (148, 149). Additional work will be required to identify the relevant stimuli.
Figure 4.
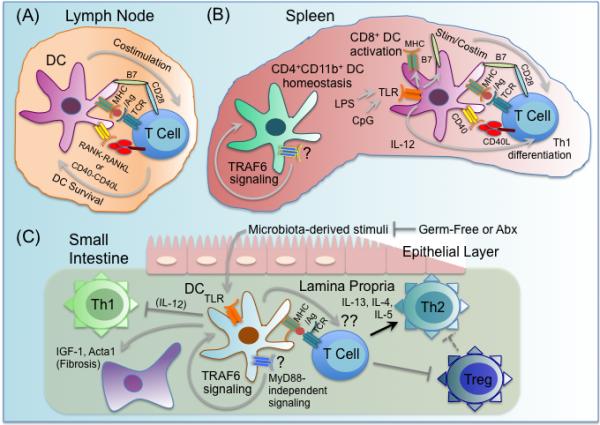
Multiple roles for TRAF6 in dendritic cell function. TRAF6 has multiple important roles in dendritic cell (DC) biology, including regulating DC activation, survival, homeostasis, and tolerance. (A) TRAF6 in DCs is a critical mediator of CD40 and RANK, which function to enhance DC survival and DC-T cell interactions in the lymph nodes. (B) TRAF6 signaling through an undetermined receptor(s) (?) is required for normal homeostasis of the splenic DC compartment, with TRAF6-deficient mice exhibiting a specific severe defect in the CD4+CD11b+ DC subset. Splenic DCs from TRAF6-deficient mice fail to upregulate MHC and costimulatory surface markers like B7-2/CD86, and fail to elaborate inflammatory cytokines like IL-12 in response to stimulation with either CD40L or various TLR ligands, including LPS and CpG DNA, likely due to TRAF6-dependent TLR and CD40 signaling. As a result, TRAF6-deficient DCs exhibit defective capacity to activate antigen-specific CD4 T cells to become IFNγ-producing Th1 cells. (C) TRAF6ΔDC mice, in which TRAF6 is specifically deleted in DCs, spontaneously develop MyD88-independent Th2-associated inflammation and fibrosis specifically in the small intestine, as well as diminished Treg numbers. Broad-spectrum antibiotics (Abx) treatment blocks development of TRAF6ΔDC intestinal phenotypes, while ablation of commensal microbiota via germ-free re-derivation exacerbates TRAF6ΔDC intestinal phenotypes. While microbiota-dependent TLR stimulation is likely TRAF6- and MyD88-dependent and required for lamina propria DC production of inflammatory cytokines like IL-12 that induce Th1 differentiation, TRAF6 signaling (but not MyD88 signaling) through an undetermined receptor(s), (?), is required for regulation of these DC-intrinsic processes. It is also unclear what DC TRAF6-dependent tolerigenic factor(s), (??), produced by lamina propria DCs that both promotes Tregs and inhibits spontaneous Th2 differentiation. It is possible that reduced Tregs leads to unrestrained Th2.
Osteoclasts and osteoimmunology
Bone is a critical component of the immune system, as the bone marrow serves as a reservoir for hematopoietic stem cells and long-lived plasma cells, as well as a key repository for various immunoregulatory factors including TGFβ. Bone is developed and maintained by the competing actions of osteoblasts, which build bone and osteoclasts, which resorb bone (150). Disruption in the balance of these actions can result in developmental defects, such as failure to form a bone marrow cavity, or lead to loss of bone strength via osteoporosis or osteopetrosis. Interestingly, while osteoblasts derive from mesenchymal stem cells, osteoclasts derive from the myeloid lineage and are closely related to DCs (151). In fact, the same cytokine, RANKL that was discovered as promoter of DC survival was also shown to be expressed in the bone by osteoblasts and stromal cells, and to act as a strong inducer of osteoclastogenesis (152). Further, genetic deletion of RANKL or its receptor RANK showed that these factors are absolutely essential to osteoclast development, as mice lacking either gene exhibit severe osteopetrosis, occluded bone marrow cavities, failure of tooth eruption, and near absence of detectable osteoclasts (153, 154). While RANK signaling in osteoclasts is mediated by various TRAFs (143, 155), it was not initially clear which factor was the key activator of the relevant functions downstream of RANK in osteoclasts. The phenotype of TRAF6-deficient mice, which nearly exactly phenocopied all the bone-related phenotypes of RANKL- and RANK-deficient mice, made clear that TRAF6 is the critical downstream mediator of RANK with respect to osteoclast development and maintenance (3). Further studies show that within its role as a regulator of osteoclast differentiation and function, TRAF6 mediates RANK-dependent NF-κB, MAPK, and Akt/PBK signaling (29, 143, 155), osteoclast maturation and cytoskeletal rearrangement (156), as well as induction of reactive oxygen species (ROS) pathways necessary for optimal osteoclastogenesis (157). TRAF6-mediated RANK signaling is also required for expression of the osteoclast master regulatory transcription factor NFATc1 (158, 159). Experimental evidence using overexpression of chimeric receptors further suggests that the specific capacity of RANK, but not the closely related receptor CD40 to drive osteoclastogenesis is due the greater number of TRAF6 binding sites in RANK, and hence stronger potential TRAF6-mediated signaling as compared to CD40 (41). Inhibitory mechanisms specific to osteoclast RANK signaling have been identified that target TRAF6 and prevent excessive osteoclast activity. For instance, in osteoclasts, but not macrophages, RANK induces expression of the dual-function cytoplasmic adaptor/transcriptional co-activator/co-repressor four and half LIM domain 2 (FHL2), which associates with TRAF6 and sequesters it in the nucleus (160). The hyper-resorptive phenotype of FHL2-deficient mice demonstrates its importance as a negative regulator of TRAF6-mediated osteoclastogenesis (161). Further, the de-ubiquitinase CYLD has been shown to inactivate TRAF6 downstream of RANK in osteoclasts, as evidenced by removal of polyubiquitin chains, and the physiologic relevance of this mechanism is supported by the phenotype of CYLD-deficient osteoclasts, which exhibit hyper-responsiveness to RANK (47).
Study of interplay between bone and the immune system is conducted under the banner of a joint field called osteoimmunology (162). One of the definitive examples of osteoimmunology is redirection of RANKL expressed by T cells to prolong interactions with DCs to promoting osteoclastogenesis by engaging immature osteoclasts or osteoclast precursors. In some cases this can result in severe pathology, such as in the case of osteoarthritis, where RANKL-triggered osteoclasts degrade the joints. The inflammatory cytokine TNFα, which can trigger TRAF6 signaling pathways, also promotes osteoclastogenesis when in synergy with RANKL (163), though work with TRAF6-deficient cells show that TRAF6 is dispensable for RANKL-independent osteoclastogenesis driven by TNFα plus TGFβ, so it remains unclear whether TRAF6 contributes to TNFα-mediated osteoclast differentiation or function (164). IL-1 also has dual function in the immune system and bone, signaling in osteoclasts through TRAF6 and induces co-localization of TRAF6 and c-Src to the osteoclast actin ring (165). A mechanism that short-circuits this type of pathology has evolved, in which IFNγ produced by T cells induces proteasomal degradation of TRAF6 in osteoclast precursors, thus preventing RANK-mediated signaling (166). Another potential negative regulatory mechanism that may prevent excessive osteoclast formation in the presence of immune response against infection is the apparent negative regulation of RANK-mediated pre-osteoclast to osteoclast transition when TLRs expressed directly on the pre-osteoclast are triggered (167). TLR ligands are typically microbial products that strongly activate DCs and other inflammatory elements, and while some TLR ligands, such as endotoxin/LPS are known to promote pathogenic osteoclastogenesis in the presence of chronic bacterial infection, this effect may occur through indirect action of TNFα (168). Because osteoclasts share a lineage with DCs, it is not surprising that they also express a wide variety of TLRs, but what is vexing is that TRAF6 is a key signaling mediator of both pro- (RANK) and anti- (TLR) osteoclastogenic receptors within the same cell. This is one example of how diverse the function of TRAF6 is in regulating immunity, and shows that additional effort is required to further define how TRAF6 signaling can be subtly adjusted in such contexts.
MicroRNA-mediated regulation of TRAF6 function
MicroRNAs (miRNA) represent a second order level of negative gene regulation that may function in either or both a cell type- and stimulation-specific manner, often serving within an auto-regulatory feedback loop (169). miRNAs genes products, transcribed independently, as polycistronic clusters, or within intronic regions of coding genes, are sequentially processed by the RNase III Drosha and the endoribonuclease Dicer to form a mature 22-basepair double-stranded RNA, which binds, as part of the cytoplasmic multi-protein RNA-induced silencing complex (RISC) to the 3’ untranslated region (UTR) of a target mRNA, thereby effecting target transcript degradation (170). There are currently hundreds of known miRNAs that regulate various aspects mammalian gene expression, and a smaller group that have been shown to specifically regulate innate and adaptive immune responses (171). One miRNA in particular, miR-146a, has been shown to modulate immune function by directly targeting the transcript for TRAF6 by binding to multiple sites in the TRAF6 3’ UTR (172). While miR-146a-dependent degradation of TRAF6 mRNA prevents production of new TRAF6 protein, existing protein can continue to function. Therefore, miRNA may be described as a gradual means of winding down an inflammatory response. It should be noted that miR-146a, like many miRNAs has additional, functionally related targets, including IRAK family members, so the effects of its expression are difficult in a given context to link directly to TRAF6 inhibition (172). The critical importance of miR-146a to immune homeostasis is demonstrated by miR-146a knockout mice, which exhibit enhanced responsiveness to endotoxin, hyperproliferation of myeloid cells in secondary lymphoid tissues, and eventual development of autoimmune disease and malignant tumors (173, 174). In attempting to understand the basis for these phenotypes, it has been shown that TRAF6 is not only a target of miR-146a, but also a key regulator of its expression. miR-146a expression is induced by a broad array of pattern recognition receptors (PRRs), including TLR4, which utilize TRAF6 for activation of NFκB, a transcription factor family for which the miR-146a promoter contains multiple binding sites (175). Expression of miR-146a and its targeted degradation of TRAF6 mRNA then feeds back to inhibit further NFκB-dependent gene expression, including inflammatory gene targets IL-6, IL-8, IL-1β, and TNFα (172, 173, 175, 176). In response to infection of mouse macrophages with vesicular stomatitis virus (VSV), miR-146a is induced through the RIG-I pathway and production of the anti-viral cytokine type-I interferon is associated with miR-146a-mediated inhibition of TRAF6, as well as IRAK-1 and IRAK-2 expression (176). Importantly, potential relevance of miR-146a inhibition of expression of TRAF6 to human disease has also been shown. In a study of the myelodysplastic 5q- syndrome, loss of expression of miRNAs miR-145 (which targets the TLR adapter TIRAP) and miR-146 was correlated with deletion of chromosome 5q, and experiments using an animal model of disease suggested that resulting TRAF6 over-expression in progenitor cells recapitulates disease phenotypes (177).
While miR-146a has primarily been characterized as a regulator of innate immune responses, recent work has demonstrated that its expression in T cells can be induced by TCR-triggered NFκB activation (178). Further, examination of miR-146a-deficient T cells reveals hyper-responsiveness to both acute and chronic autoimmune antigens that is associated with enhanced NFκB activity, and increased expression of (i.e., failed inhibition of) TRAF6 and IRAK-1 expression (178). Interestingly, because T cell-specific deletion of TRAF6 also leads to spontaneous loss of immune tolerance and hyper-responsiveness to antigen (84), it may be important in this context to determine what the effects are of jointly targeting both TRAF6 and miR-146a in a T cell-specific manner on immune homeostasis. This approach may clarify TRAF6-dependent and – independent roles of miR-146a in T cell regulation, including whether TRAF6-dependent signals are required for miR-146a induction in response to TCR activation. It has also been shown that miR-146a expression in Treg cells is critical for Treg-mediated suppression of Th1 responses in a manner that is believed to be related to failed inhibition of STAT1 expression (179). Because recent studies have identified a requirement for cell-intrinsic TRAF6 signaling for proper Treg function and maintenance of Foxp3 expression (108), it may be interesting to determine whether miR-146a functions with some degree of specificity in Tregs
While miR-146a is the best-characterized miRNA that targets TRAF6 expression, other miRNAs with specificity for TRAF6 have been identified. miR-146b, for instance, shares a very similar sequence with miR-146a, but is expressed from a unique gene locus on a separate chromosome. Additionally, miR-146b expression, at least in monocytes, appears delayed compared to miR-146a, and is induced through the STAT3 pathway (180). Another recent study shows that the miRNA mir-124, which is induced by acetylcholine inhibition in murine bone marrow-derived macrophages directly targets TRAF6, which then attenuates the capacity of those cells to transduce TLR-mediated signals (181). This area of research will require much additional targeted effort in order to better the mechanisms by which TRAF6 signaling is modulated in both cell- and stimuli-dependent manners. In particular, because TRAF6 can potentially be activated through multiple receptor families within a given single cell, it will be of interest to determine whether a form of crosstalk occurs involving miRNA targeting of TRAF6.
Concluding Remarks
Even when only looking within the immune system, the manifold roles of TRAF6 seem ever expanding, which leads to one of the most pressing open questions surrounding TRAF6 biology: how does a single protein affect such various outcomes, downstream of such a variety of signaling receptors, even when sometimes coordinating apparently conflicting signals within the same cell? This issue is even more confounding when considering that virtually all TRAF6-dependent functions appear to rely on the activity of a single RING finger domain. And there remain TRAF6-dependent phenotypes, such as in T cell- or DC-targeted mice, in which it is currently unclear which or how many upstream receptors are active in maintaining TRAF6-regulated homeostasis. In order to begin to better understand the competing functions of TRAF6 within the immune system, additional work will have to be done to not only target TRAF6 within specific cell lineages, but also to inactivate TRAF6 in a temporal manner so that the effects of TRAF6 downstream of a given stimuli can be distinguished from lingering effects of developmental/differentiation defects. Another method by which this issue can be addressed is to further define (and/or target) specific TRAF6-binding motifs so that new studies employing mutant receptors or adaptors, such as those in the TGFβR and TCR pathways, can be initiated to examine stimuli-specific TRAF6-dependent effects in cell types that utilize TRAF6 simultaneously within multiple signaling pathways. Finally, while the RING finger domain and the E3 ligase activity it confers on TRAF6 are clearly critical for active signaling, potential negative regulatory roles of TRAF6, as well as inter-pathway crosstalk, may involve TRAF6 for RING-independent target sequestration, but in a manner that is too subtle to be experimentally resolved using current over-expression approaches. Discovery of potential immunomodulatory effects of RING-deficient TRAF6 may require physiologic expression. With respect to the broader role of TRAF6 in controlling immune homeostasis and function between different cell types, it is interesting that TRAF6 plays a critical role in transducing signals downstream of APC-associated “innate” receptors, such as TLRs, that communicate non-self threats to the adaptive immune system, while at the same time, TRAF6 also acts as a negative of “adaptive” receptor signaling downstream of TCR, raising additional questions about the evolutionary pressures that shaped its functional roles. In particular, the observations that deletion of TRAF6 in either T cells or DCs results not in defective cellular activation, but rather spontaneous induction of Th2 immunity may suggest a broader conserved function for TRAF6 concerned more with favoring inflammation rather than distinguishing self from non-self. By further investigating these questions and others, we can achieve more detailed contextual understanding of the vast biological functions of TRAF6, and hopefully render it a suitable target for therapeutic disease interventions.
Acknowledgements
The authors acknowledge grant support from the NIH (AI64909, AI18627, AR55903, DE19381).
Footnotes
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1.Ishida T, et al. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 2.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 3.Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes & development. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiffoleau E, et al. TNF receptor-associated factor 6 deficiency during hemopoiesis induces Th2-polarized inflammatory disease. J Immunol. 2003;171:5751–5759. doi: 10.4049/jimmunol.171.11.5751. [DOI] [PubMed] [Google Scholar]
- 5.Dickson KM, Bhakar AL, Barker PA. TRAF6-dependent NF-kB transcriptional activity during mouse development. Developmental dynamics : an official publication of the American Association of Anatomists. 2004;231:122–127. doi: 10.1002/dvdy.20110. [DOI] [PubMed] [Google Scholar]
- 6.Fujikawa H, Farooq M, Fujimoto A, Ito M, Shimomura Y. Functional studies for the TRAF6 mutation associated with hypohidrotic ectodermal dysplasia. The British journal of dermatology. 2013;168:629–633. doi: 10.1111/bjd.12018. [DOI] [PubMed] [Google Scholar]
- 7.Ohazama A, et al. Traf6 is essential for murine tooth cusp morphogenesis. Developmental dynamics : an official publication of the American Association of Anatomists. 2004;229:131–135. doi: 10.1002/dvdy.10400. [DOI] [PubMed] [Google Scholar]
- 8.Landstrom M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol. 2010;42:585–589. doi: 10.1016/j.biocel.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 9.Inoue J, Gohda J, Akiyama T, Semba K. NF-kappaB activation in development and progression of cancer. Cancer science. 2007;98:268–274. doi: 10.1111/j.1349-7006.2007.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, et al. TRAF6 activation in multiple myeloma: a potential therapeutic target. Clin Lymphoma Myeloma Leuk. 2012;12:155–163. doi: 10.1016/j.clml.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen B, Liu H, Skolnik EY, Manley JL. Physical and functional interactions between Drosophila TRAF2 and Pelle kinase contribute to Dorsal activation. Proc Natl Acad Sci U S A. 2001;98:8596–8601. doi: 10.1073/pnas.141235698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. 2013;8:7. doi: 10.1186/1750-2187-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye H, et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002;418:443–447. doi: 10.1038/nature00888. [DOI] [PubMed] [Google Scholar]
- 14.Chung JY, Lu M, Yin Q, Wu H. Structural revelations of TRAF2 function in TNF receptor signaling pathway. Advances in experimental medicine and biology. 2007;597:93–113. doi: 10.1007/978-0-387-70630-6_8. [DOI] [PubMed] [Google Scholar]
- 15.Hildebrand JM, et al. A BAFF-R mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-R signaling. J Exp Med. 2010;207:2569–2579. doi: 10.1084/jem.20100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hacker H, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 17.Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes & development. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorrentino A, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulek K, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanamori M, Kai C, Hayashizaki Y, Suzuki H. NF-kappaB activator Act1 associates with IL-1/Toll pathway adaptor molecule TRAF6. FEBS letters. 2002;532:241–246. doi: 10.1016/s0014-5793(02)03688-8. [DOI] [PubMed] [Google Scholar]
- 22.Liu S, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 24.Cohen P. The TLR and IL-1 signalling network at a glance. Journal of cell science. 2014;127:2383–2390. doi: 10.1242/jcs.149831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- 26.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Frontiers in immunology. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arron JR, et al. A positive regulatory role for Cbl family proteins in tumor necrosis factor-related activation-induced cytokine (trance) and CD40L-mediated Akt activation. J Biol Chem. 2001;276:30011–30017. doi: 10.1074/jbc.M100414200. [DOI] [PubMed] [Google Scholar]
- 28.Neumann D, Lienenklaus S, Rosati O, Martin MU. IL-1beta-induced phosphorylation of PKB/Akt depends on the presence of IRAK-1. Eur J Immunol. 2002;32:3689–3698. doi: 10.1002/1521-4141(200212)32:12<3689::AID-IMMU3689>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 29.Wong BR, et al. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999;4:1041–1049. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- 30.Deng L, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 32.Lamothe B, Campos AD, Webster WK, Gopinathan A, Hur L, Darnay BG. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J Biol Chem. 2008;283:24871–24880. doi: 10.1074/jbc.M802749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y. TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS One. 2008;3:e4064. doi: 10.1371/journal.pone.0004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA, Cantley LC. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol. 2007;27:6012–6025. doi: 10.1128/MCB.00270-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol Cell Biol. 2008;28:3538–3547. doi: 10.1128/MCB.02098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia ZP, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nature structural & molecular biology. 2014;21:301–307. doi: 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- 38.Huang Q, et al. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 39.Yamazaki K, et al. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci Signal. 2009;2:ra66. doi: 10.1126/scisignal.2000387. [DOI] [PubMed] [Google Scholar]
- 40.Matsuzawa A, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 41.Kadono Y, et al. Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Rep. 2005;6:171–176. doi: 10.1038/sj.embor.7400345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang WL, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–1138. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawai T, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 44.Takaoka A, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 45.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 46.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 47.Jin W, et al. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–1866. doi: 10.1172/JCI34257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iha H, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008;27:629–641. doi: 10.1038/emboj.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shembade N, Harhaj NS, Liebl DJ, Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007;26:3910–3922. doi: 10.1038/sj.emboj.7601823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garg AV, Ahmed M, Vallejo AN, Ma A, Gaffen SL. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci Signal. 2013;6:ra44. doi: 10.1126/scisignal.2003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung SM, et al. Smad6 inhibits non-canonical TGF-beta1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat Commun. 2013;4:2562. doi: 10.1038/ncomms3562. [DOI] [PubMed] [Google Scholar]
- 53.Shembade N, Harhaj EW. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cellular & molecular immunology. 2012;9:123–130. doi: 10.1038/cmi.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turer EE, et al. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med. 2008;205:451–464. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshida Y, et al. Interleukin 1 activates STAT3/nuclear factor-kappaB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem. 2004;279:1768–1776. doi: 10.1074/jbc.M311498200. [DOI] [PubMed] [Google Scholar]
- 56.Wei J, et al. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS One. 2012;7:e49567. doi: 10.1371/journal.pone.0049567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang H, et al. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13. Nat Commun. 2014;5:5798. doi: 10.1038/ncomms6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunology and cell biology. 2014;92:761–769. doi: 10.1038/icb.2014.51. [DOI] [PubMed] [Google Scholar]
- 59.Hindi SM, et al. Reciprocal interaction between TRAF6 and notch signaling regulates adult myofiber regeneration upon injury. Mol Cell Biol. 2012;32:4833–4845. doi: 10.1128/MCB.00717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gudey SK, et al. TRAF6 stimulates the tumor-promoting effects of TGFbeta type I receptor through polyubiquitination and activation of presenilin 1. Sci Signal. 2014;7:ra2. doi: 10.1126/scisignal.2004207. [DOI] [PubMed] [Google Scholar]
- 61.Mishra AK, Sachan N, Mutsuddi M, Mukherjee A. TRAF6 is a novel regulator of Notch signaling in Drosophila melanogaster. Cell Signal. 2014;26:3016–3026. doi: 10.1016/j.cellsig.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 62.Kyewski B, Klein L. A central role for central tolerance. Annual review of immunology. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 63.Anderson G, Lane PJ, Jenkinson EJ. Generating intrathymic microenvironments to establish T-cell tolerance. Nature reviews Immunology. 2007;7:954–963. doi: 10.1038/nri2187. [DOI] [PubMed] [Google Scholar]
- 64.Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nature reviews Immunology. 2002;2:11–19. doi: 10.1038/nri701. [DOI] [PubMed] [Google Scholar]
- 65.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annual review of immunology. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naito A, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes to cells : devoted to molecular & cellular mechanisms. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 67.Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol. 2001;2:1032–1039. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 68.Anderson MS, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 69.Galy AH, Spits H. CD40 is functionally expressed on human thymic epithelial cells. J Immunol. 1992;149:775–782. [PubMed] [Google Scholar]
- 70.Gray DH, et al. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood. 2006;108:3777–3785. doi: 10.1182/blood-2006-02-004531. [DOI] [PubMed] [Google Scholar]
- 71.Akiyama T, et al. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science. 2005;308:248–251. doi: 10.1126/science.1105677. [DOI] [PubMed] [Google Scholar]
- 72.Bonito AJ, et al. Medullary thymic epithelial cell depletion leads to autoimmune hepatitis. J Clin Invest. 2013;123:3510–3524. doi: 10.1172/JCI65414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hamazaki Y, et al. Medullary thymic epithelial cells expressing Aire represent a unique lineage derived from cells expressing claudin. Nat Immunol. 2007;8:304–311. doi: 10.1038/ni1438. [DOI] [PubMed] [Google Scholar]
- 74.Burkly L, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 75.Hikosaka Y, et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. 2008;29:438–450. doi: 10.1016/j.immuni.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 76.Rossi SW, et al. RANK signals from CD4(+)3(-) inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J Exp Med. 2007;204:1267–1272. doi: 10.1084/jem.20062497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akiyama T, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. doi: 10.1016/j.immuni.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 78.White AJ, et al. Sequential phases in the development of Aire-expressing medullary thymic epithelial cells involve distinct cellular input. Eur J Immunol. 2008;38:942–947. doi: 10.1002/eji.200738052. [DOI] [PubMed] [Google Scholar]
- 79.Mueller CG, Hess E. Emerging Functions of RANKL in Lymphoid Tissues. Frontiers in immunology. 2012;3:261. doi: 10.3389/fimmu.2012.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoshida H, et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer's patches. Immunity. 2002;17:823–833. doi: 10.1016/s1074-7613(02)00479-x. [DOI] [PubMed] [Google Scholar]
- 81.White A, et al. Lymphotoxin a-dependent and -independent signals regulate stromal organizer cell homeostasis during lymph node organogenesis. Blood. 2007;110:1950–1959. doi: 10.1182/blood-2007-01-070003. [DOI] [PubMed] [Google Scholar]
- 82.Benezech C, et al. Ontogeny of stromal organizer cells during lymph node development. J Immunol. 2010;184:4521–4530. doi: 10.4049/jimmunol.0903113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koning JJ, Mebius RE. Interdependence of stromal and immune cells for lymph node function. Trends in immunology. 2012;33:264–270. doi: 10.1016/j.it.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 84.King CG, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. 2006;12:1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- 85.Hara H, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 2003;18:763–775. doi: 10.1016/s1074-7613(03)00148-1. [DOI] [PubMed] [Google Scholar]
- 86.Egawa T, et al. Requirement for CARMA1 in antigen receptor-induced NF-kappa B activation and lymphocyte proliferation. Current biology : CB. 2003;13:1252–1258. doi: 10.1016/s0960-9822(03)00491-3. [DOI] [PubMed] [Google Scholar]
- 87.Ruland J, et al. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell. 2001;104:33–42. doi: 10.1016/s0092-8674(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 88.Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–758. doi: 10.1016/s1074-7613(03)00293-0. [DOI] [PubMed] [Google Scholar]
- 89.Jun JE, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 90.Su H, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 91.Bidere N, Snow AL, Sakai K, Zheng L, Lenardo MJ. Caspase-8 regulation by direct interaction with TRAF6 in T cell receptor-induced NF-kappaB activation. Current biology : CB. 2006;16:1666–1671. doi: 10.1016/j.cub.2006.06.062. [DOI] [PubMed] [Google Scholar]
- 92.Oeckinghaus A, et al. Malt1 ubiquitination triggers NF-kappaB signaling upon T-cell activation. EMBO J. 2007;26:4634–4645. doi: 10.1038/sj.emboj.7601897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.King CG, et al. Cutting edge: requirement for TRAF6 in the induction of T cell anergy. J Immunol. 2008;180:34–38. doi: 10.4049/jimmunol.180.1.34. [DOI] [PubMed] [Google Scholar]
- 94.Cannons JL, Bertram EM, Watts TH. Cutting edge: profound defect in T cell responses in TNF receptor-associated factor 2 dominant negative mice. J Immunol. 2002;169:2828–2831. doi: 10.4049/jimmunol.169.6.2828. [DOI] [PubMed] [Google Scholar]
- 95.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annual review of immunology. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual review of immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 97.Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annual review of immunology. 2012;30:707–731. doi: 10.1146/annurev-immunol-020711-075058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jabeen R, Kaplan MH. The symphony of the ninth: the development and function of Th9 cells. Curr Opin Immunol. 2012;24:303–307. doi: 10.1016/j.coi.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annual review of immunology. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 100.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annual review of immunology. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 101.Dardalhon V, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schmitt E, Klein M, Bopp T. Th9 cells, new players in adaptive immunity. Trends in immunology. 2014;35:61–68. doi: 10.1016/j.it.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 103.Xiao X, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol. 2012;13:981–990. doi: 10.1038/ni.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O'Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21:425–434. doi: 10.1016/j.cytogfr.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cejas PJ, et al. TRAF6 inhibits Th17 differentiation and TGF-beta-mediated suppression of IL-2. Blood. 2010;115:4750–4757. doi: 10.1182/blood-2009-09-242768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 107.Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Frontiers in immunology. 2013;4:152. doi: 10.3389/fimmu.2013.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muto G, et al. TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS One. 2013;8:e74639. doi: 10.1371/journal.pone.0074639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shimo Y, et al. TRAF6 directs commitment to regulatory T cells in thymocytes. Genes to cells : devoted to molecular & cellular mechanisms. 2011;16:437–447. doi: 10.1111/j.1365-2443.2011.01500.x. [DOI] [PubMed] [Google Scholar]
- 110.Pearce EL, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Walsh MC, Pearce EL, Cejas PJ, Lee J, Wang LS, Choi Y. IL-18 Synergizes with IL-7 To Drive Slow Proliferation of Naive CD8 T Cells by Costimulating Self-Peptide-Mediated TCR Signals. J Immunol. 2014;193:3992–4001. doi: 10.4049/jimmunol.1400396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huynh A, et al. Control of PI(3) kinase in T cells maintains homeostasis and lineage stability. Nat Immunol. 2015 doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annual review of immunology. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 114.Banchereau J, et al. The CD40 antigen and its ligand. Annual review of immunology. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 115.Jalukar SV, Hostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6 in CD40-mediated B lymphocyte effector functions. J Immunol. 2000;164:623–630. doi: 10.4049/jimmunol.164.2.623. [DOI] [PubMed] [Google Scholar]
- 116.Manning E, Pullen SS, Souza DJ, Kehry M, Noelle RJ. Cellular responses to murine CD40 in a mouse B cell line may be TRAF dependent or independent. Eur J Immunol. 2002;32:39–49. doi: 10.1002/1521-4141(200201)32:1<39::AID-IMMU39>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 117.Ahonen C, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol. 2002;3:451–456. doi: 10.1038/ni792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kobayashi T, et al. TRAF6 is required for generation of the B-1a B cell compartment as well as T cell-dependent and -independent humoral immune responses. PLoS One. 2009;4:e4736. doi: 10.1371/journal.pone.0004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 120.Sato S, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 121.Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nature reviews Immunology. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 122.Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U S A. 1993;90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci U S A. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Uchida J, et al. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. 1999;286:300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 125.Xie P, Hostager BS, Munroe ME, Moore CR, Bishop GA. Cooperation between TNF receptor-associated factors 1 and 2 in CD40 signaling. J Immunol. 2006;176:5388–5400. doi: 10.4049/jimmunol.176.9.5388. [DOI] [PubMed] [Google Scholar]
- 126.Kraus ZJ, Nakano H, Bishop GA. TRAF5 is a critical mediator of in vitro signals and in vivo functions of LMP1, the viral oncogenic mimic of CD40. Proc Natl Acad Sci U S A. 2009;106:17140–17145. doi: 10.1073/pnas.0903786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Arcipowski KM, Stunz LL, Bishop GA. TRAF6 is a critical regulator of LMP1 functions in vivo. International immunology. 2014;26:149–158. doi: 10.1093/intimm/dxt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shkoda A, et al. The germinal center kinase TNIK is required for canonical NF-kappaB and JNK signaling in B-cells by the EBV oncoprotein LMP1 and the CD40 receptor. PLoS Biol. 2012;10:e1001376. doi: 10.1371/journal.pbio.1001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.He B, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11:836–845. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nature reviews Immunology. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 131.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nature reviews Immunology. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mason NJ, Fiore J, Kobayashi T, Masek KS, Choi Y, Hunter CA. TRAF6-dependent mitogen-activated protein kinase activation differentially regulates the production of interleukin-12 by macrophages in response to Toxoplasma gondii. Infection and immunity. 2004;72:5662–5667. doi: 10.1128/IAI.72.10.5662-5667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Morisaki JH, Heuser JE, Sibley LD. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. Journal of cell science. 1995;108(Pt 6):2457–2464. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- 134.Subauste CS, Andrade RM, Wessendarp M. CD40-TRAF6 and autophagy-dependent anti-microbial activity in macrophages. Autophagy. 2007;3:245–248. doi: 10.4161/auto.3717. [DOI] [PubMed] [Google Scholar]
- 135.Ogolla PS, et al. The protein kinase double-stranded RNA-dependent (PKR) enhances protection against disease cause by a non-viral pathogen. PLoS Pathog. 2013;9:e1003557. doi: 10.1371/journal.ppat.1003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl LM, Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol. 2005;174:1081–1090. doi: 10.4049/jimmunol.174.2.1081. [DOI] [PubMed] [Google Scholar]
- 137.Rub A, et al. Cholesterol depletion associated with Leishmania major infection alters macrophage CD40 signalosome composition and effector function. Nat Immunol. 2009;10:273–280. doi: 10.1038/ni.1705. [DOI] [PubMed] [Google Scholar]
- 138.Lutgens E, et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Polykratis A, van Loo G, Xanthoulea S, Hellmich M, Pasparakis M. Conditional targeting of tumor necrosis factor receptor-associated factor 6 reveals opposing functions of Toll-like receptor signaling in endothelial and myeloid cells in a mouse model of atherosclerosis. Circulation. 2012;126:1739–1751. doi: 10.1161/CIRCULATIONAHA.112.100339. [DOI] [PubMed] [Google Scholar]
- 140.West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Anderson DM, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 142.Wong BR, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186:2075–2080. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J Biol Chem. 1998;273:28355–28359. doi: 10.1074/jbc.273.43.28355. [DOI] [PubMed] [Google Scholar]
- 144.O'Sullivan BJ, Thomas R. CD40 ligation conditions dendritic cell antigen-presenting function through sustained activation of NF-kappaB. J Immunol. 2002;168:5491–5498. doi: 10.4049/jimmunol.168.11.5491. [DOI] [PubMed] [Google Scholar]
- 145.Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, Choi Y. TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med. 1999;189:1025–1031. doi: 10.1084/jem.189.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kobayashi T, et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 2003;19:353–363. doi: 10.1016/s1074-7613(03)00230-9. [DOI] [PubMed] [Google Scholar]
- 147.Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol. 2001;166:5688–5694. doi: 10.4049/jimmunol.166.9.5688. [DOI] [PubMed] [Google Scholar]
- 148.Han D, et al. Dendritic cell expression of the signaling molecule TRAF6 is critical for gut microbiota-dependent immune tolerance. Immunity. 2013;38:1211–1222. doi: 10.1016/j.immuni.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ikeda K, Takeshita S. Factors and mechanisms involved in the coupling from bone resorption to formation: how osteoclasts talk to osteoblasts. Journal of bone metabolism. 2014;21:163–167. doi: 10.11005/jbm.2014.21.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Miyamoto T, et al. Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood. 2001;98:2544–2554. doi: 10.1182/blood.v98.8.2544. [DOI] [PubMed] [Google Scholar]
- 152.Lacey DL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 153.Dougall WC, et al. RANK is essential for osteoclast and lymph node development. Genes & development. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kong YY, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 155.Galibert L, Tometsko ME, Anderson DM, Cosman D, Dougall WC. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-kappaB, a member of the TNFR superfamily. J Biol Chem. 1998;273:34120–34127. doi: 10.1074/jbc.273.51.34120. [DOI] [PubMed] [Google Scholar]
- 156.Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. 2002;277:44347–44356. doi: 10.1074/jbc.M202009200. [DOI] [PubMed] [Google Scholar]
- 157.Lee NK, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- 158.Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J. RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J. 2005;24:790–799. doi: 10.1038/sj.emboj.7600564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Takayanagi H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Developmental cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 160.Bai S, Zha J, Zhao H, Ross FP, Teitelbaum SL. Tumor necrosis factor receptor-associated factor 6 is an intranuclear transcriptional coactivator in osteoclasts. J Biol Chem. 2008;283:30861–30867. doi: 10.1074/jbc.M802525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bai S, et al. FHL2 inhibits the activated osteoclast in a TRAF6-dependent manner. J Clin Invest. 2005;115:2742–2751. doi: 10.1172/JCI24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Walsh MC, et al. Osteoimmunology: interplay between the immune system and bone metabolism. Annual review of immunology. 2006;24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- 163.Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem. 2001;276:563–568. doi: 10.1074/jbc.M008198200. [DOI] [PubMed] [Google Scholar]
- 164.Kim N, et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. 2005;202:589–595. doi: 10.1084/jem.20050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nakamura I, et al. IL-1 regulates cytoskeletal organization in osteoclasts via TNF receptor-associated factor 6/c-Src complex. J Immunol. 2002;168:5103–5109. doi: 10.4049/jimmunol.168.10.5103. [DOI] [PubMed] [Google Scholar]
- 166.Takayanagi H, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 167.Takami M, Kim N, Rho J, Choi Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol. 2002;169:1516–1523. doi: 10.4049/jimmunol.169.3.1516. [DOI] [PubMed] [Google Scholar]
- 168.Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J Clin Invest. 1997;100:1557–1565. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 170.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 171.Gracias DT, Katsikis PD. MicroRNAs: key components of immune regulation. Advances in experimental medicine and biology. 2011;780:15–26. doi: 10.1007/978-1-4419-5632-3_2. [DOI] [PubMed] [Google Scholar]
- 172.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Boldin MP, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhao JL, Rao DS, Boldin MP, Taganov KD, O'Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011;108:9184–9189. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Saba R, Sorensen DL, Booth SA. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Frontiers in immunology. 2014;5:578. doi: 10.3389/fimmu.2014.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hou J, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 177.Starczynowski DT, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010;16:49–58. doi: 10.1038/nm.2054. [DOI] [PubMed] [Google Scholar]
- 178.Yang L, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med. 2012;209:1655–1670. doi: 10.1084/jem.20112218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lu LF, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci U S A. 2013;110:11499–11504. doi: 10.1073/pnas.1219852110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Qiu S, et al. Chronic Morphine-Induced MicroRNA-124 Promotes Microglial Immunosuppression by Modulating P65 and TRAF6. J Immunol. 2014 doi: 10.4049/jimmunol.1400106. [DOI] [PMC free article] [PubMed] [Google Scholar]