

Abstract
Background
Left ventricular non‐compaction (LVNC) is a rare cardiomyopathy. Many genetic variants have been associated with LVNC. However, the number of the previous LVNC‐associated variants that are common in the background population remains unknown. The aim of this study was to provide an updated list of previously reported LVNC‐associated variants with biologic description and investigate the prevalence of LVNC variants in healthy general population to find false‐positive LVNC‐associated variants.
Methods and Results
The Human Gene Mutation Database and PubMed were systematically searched to identify all previously reported LVNC‐associated variants. Thereafter, the Exome Sequencing Project (ESP) and the Exome Aggregation Consortium (ExAC), that both represent the background population, was searched for all variants. Four in silico prediction tools were assessed to determine the functional effects of these variants. The prediction results of those identified in the ESP and ExAC and those not identified in the ESP and ExAC were compared. In 12 genes, 60 LVNC‐associated missense/nonsense variants were identified. MYH7 was the predominant gene, encompassing 24 of the 60 LVNC‐associated variants. The ESP only harbored nine and ExAC harbored 18 of the 60 LVNC‐associated variants. In total, eight out of nine ESP‐positive variants overlapped with the 18 variants identified in ExAC database.
Conclusions
In this article, we identified 9 ESP‐positive and 18 ExAC‐positive variants of 60 previously reported LVNC‐associated variants, suggesting that these variants are not necessarily the monogenic cause of LVNC.
Keywords: Exome aggregation consortium, Exome sequencing project, genes, human gene mutation database, left ventricular non‐compaction, LVNC, LVNC‐associated variants, variants
Introduction
Left ventricular non‐compaction (LVNC) is a rare cardiomyopathy with a prevalence that varies considerably among studies (0.014–14%) (Oechslin et al. 2000; Pignatelli et al. 2003; Stöllberger and Finsterer 2005; Aras et al. 2006; Belanger et al. 2008; Stanton et al. 2009; Tian et al. 2014). Left ventricular non‐compaction is most likely caused by a pathological arrest in the compaction process, which leads to a non‐compacted left ventricular myocardium (Dusek et al. 1975; Chin et al. 1990; Agmon et al. 1999; Zambrano et al. 2002). Left ventricular non‐compaction is characterized by a non‐compacted inner myocardial layer and a compacted outer myocardial layer with deep intratrabecular recesses, that are communicated to the left ventricular chamber (Jenni et al. 2001; Sarma et al. 2010).
There are no single diagnostic criteria for LVNC. The clinical presentation is highly variable from asymptomatic to symptomatic with heart failure (HF), atrial and ventricular arrhythmias, thromboembolic events, and sudden cardiac death (Ichida et al. 1999; Paterick et al. 2010; Bhatia et al. 2011). Left ventricular non‐compaction is associated with congenital cardiac disorders, other cardiomyopathies, and some neuromuscular diseases (Jenni et al. 2001).
Both sporadic and familial LVNC have been reported (Sasse‐Klaassen et al. 2003; Sen‐Chowdhry and McKenna 2008). The pattern of familial LVNC inheritance is mainly autosomal dominant with incomplete penetrance, although autosomal recessive and X‐linked inheritance have been reported (Bleyl et al. 1997a; Sasse‐Klaassen et al. 2003; Xing et al. 2006; Sen‐Chowdhry and McKenna 2008). Left ventricular non‐compaction is associated with variants in mitochondrial, cytoskeletal, Z‐line, and sarcomeric genes (Sen‐Chowdhry and McKenna 2008). The sarcomeric genes include cardiac β‐myosin heavy chain (MYH7), cardiac troponin T (TNNT2), and cardiac α‐actin (ACTC1) (Budde et al. 2007; Hoedemaekers et al. 2007; Monserrat et al. 2007; Klaassen et al. 2008). Furthermore, LVNC has been described with variants in tafazzin (TAZ), alpha‐dystrobrevin (DTNA), tropomyosin 1 (TPM1), and cardiac troponin I (TNNI3) (Ichida et al. 2001; Budde et al. 2007; Klaassen et al. 2008; Probst et al. 2011; Arndt et al. 2013). Carriers of these sarcomeric variants demonstrate a range of phenotypes from no LVNC (asymptomatic) to early onset LVNC or other cardiomyopathies (Moric‐Janiszewska and Markiewicz‐Łoskot 2008; Finsterer 2009).
The first gene associated with LVNC, tafazzin, was reported more than 17 years ago (Bleyl et al. 1997a). Ongoing genetic research has since discovered many novel variants in the genes described above. Despite the known associated genes, the genetic background of this cardiomyopathy is not fully understood.
The aim of this article was to provide an updated list of LVNC‐associated variants previously reported in the literatures with biologic description. In addition, we will investigate the prevalence of LVNC variants in a healthy general population in order to identify false‐positive LVNC‐associated variants.
Methods
Data collection
Genes and variants previously associated with LVNC were identified using the Human Gene Mutation Database (HGMD) and by reviewing published literature in PubMed until July 17th 2015 (HGMD (Human Gene Mutation Database for Human Genetics Research); Exome Variant Server).
The following queries were used: ((Left ventricular non‐compaction), (Left ventricular noncompaction), (left ventricular hypertrabeculation) and (left ventricular hypertrabecular/non‐compaction [Mesh])) and ((Genetic) or (“Genetics” [Mesh])), in order to collect all published data on identified LVNC‐associated variants. Furthermore, we also searched HGMD, Google scholar, and publicly available databases.
All relevant articles were reviewed for data on functional studies and on familial co‐segregation. We defined co‐segregation as at least two genotypically positive family members with the same phenotype. Positive functional data were determined as any in vivo or in vitro model demonstrating results differing from the wild‐type model as described previously.
Exome sequencing project
In the Exome Sequencing Project (ESP), next‐generation sequencing of all protein coding regions in 6503 individuals of African American (n = 2203 individuals) and European American (n = 4300 individuals) descent from different population studies has been carried out. Clinical data were not readily available or available on request.
The ESP database was searched for the missense and nonsense variants previously associated with LVNC.
Left ventricular non‐compaction‐associated variants were subdivided into those that were identified in the ESP database (ESP positive) and those that were not identified in the ESP database (ESP negative).
The exome aggregation consortium
In the exome aggregation consortium, sequenced 60,706 unrelated individuals on the regions of the human genome that encode proteins as part of various disease‐specific and population genetic studies. Individuals with sever pediatric disease have been removed. Thus, the ExAC Browser is a very useful dataset of allele frequencies. Left ventricular non‐compaction‐associated variants were subdivided into those that were identified in the ExAC database (ExAC positive) and those that were not identified in the ExAC database (ExAC negative).
Prediction analysis
The functional effects of all variants were assessed using four prediction tools: (1) Conservation across species; (2) Grantham Score; (3) Sorting Intolerant From Tolerant, v5.1.1 (SIFT); and (4) PolyPhen‐2. Conservation across species was obtained from the HGMD and classified as occurring at a position with no substitutions (conserved/pathogenic) or ≥1 substitutions (not conserved/benign). Grantham physicochemical values were calculated using the Grantham amino acid difference matrix. We defined a value above 100 as radical (pathogenic) and a value under 100 as conservative (benign) (Grantham 1974; Giudicessi et al. 2012). Using PolyPhen‐2, each variant was labeled as “probably damaging,” “possibly damaging,” or “benign” (PolyPhen‐2). Variants labeled “probably damaging” and “possibly damaging” were considered to be “damaging” (pathogenic) in our analysis. Finally, SIFT prediction classified variants as “tolerant” (benign) or “damaging” (pathologic) (SIFT).
In a final analysis using all prediction tools, a variant was considered damaging (pathogenic) if ≥3 in silico prediction tools identified the variant as pathogenic, as previously described (Giudicessi et al. 2012; Andreasen et al. 2013; Jabbari et al. 2013; Risgaard et al. 2013). Variants predicted to be damaging by one or two tools were considered to be variants of uncertain significance (VUS). Each of the four prediction tools predicted separately the pathogenicity of previously published LVNC‐associated variants. We used Giudicessi et al. (2012) agreement of ≥3 in silico tool predictions to assess the pathogenic status of a LVNC‐associated variant. This is an established and robust bioinformatics method, increasing the importance of the assessment using the known online prediction tools. (Giudicessi et al. 2012; Andreasen et al. 2013; Jabbari et al. 2013; Risgaard et al. 2013) This method increases the correlation between genetic findings and clinical practice especially in rare diseases.
Figure 1.
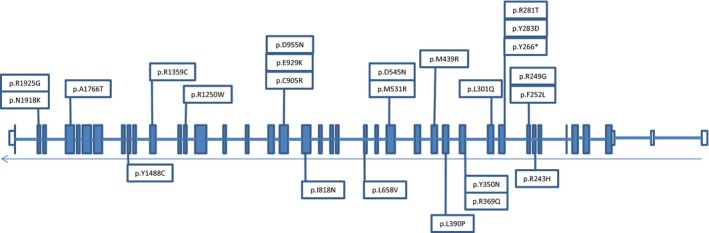
LVNC‐associated variants in the MYH7 exons. White exons are untranslated region and blue exons are translated sequence.
Results
Through a review of the literature, we identified 12 genes previously associated with LVNC (Table S1). Overall, we identified 60 missense/nonsense variants previously associated with LVNC, and most of the variants (40%) were found in MYH7 (24 out of 60).
The ESP database included 9 of these 60 variants, which were found in seven genes: MYBPC3, MYH7B, MIB, TNNT2, CASQ2, TAZ, and LDB3 (Table S1). Notably, no variants in MYH7 were found in the ESP database. In the ExAC database, we identified 18 variants distributed in all genes except TPM1. In total, eight out of nine ESP‐positive variants overlapped with the 18 variants identified in ExAC database (Table S1).
Two variants (p.D545N and p.D955N) coexisted as a heterozygous double mutation in the cis position on the same MYH7 allele segregating with LVNC. Notably, the patients were of various ethnic backgrounds and nationalities from Europe, America, and Asia. Most of the patients were men, and the patient age spectrum was widespread from infancy to adulthood.
Heredity pattern
In total, we found 42 familial forms of LVNC. Familial genetic co‐segregation was found in 30 out of 42 clinical familial cases (Table S1). Some of these variants were also identified in other studies not having a clear heredity pattern, suggesting a sporadic form of LVNC. Therefore, there is some overlap between familial and sporadic cases.
In the familial cases, three heredity patterns, including autosomal dominant, autosomal recessive, and X‐linked, were reported. The majority of the heredity patterns were autosomal dominant. We found only one example of autosomal recessive inheritance pattern among all familial cases (p.R820W/MYBPC3). Some variants showed both sporadic and familial inheritance patterns in different studies (p.G197R/TAZ) (Bleyl et al. 1997b).
Phenotypes of the families
We found a wide clinical spectrum of LVNC phenotypes ranging from asymptomatic to symptomatic (Table S1). Left ventricular non‐compaction was the predominant form of cardiomyopathy in the families (39 of 41). However, in some families, the relatives showed other cardiomyopathies (dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM)) with no sign of LVNC (Table S1). There were also four families with co‐existing LVNC and DCM, and two families showed LVNC and HCM, and one family exhibited LVNC and a congenital heart defect (Ichida et al. 2001). Notably, we found two cases with several types of cardiac diseases in their families (Postma et al. 2011; Ronvelia et al. 2012). In a family with the TAZ/p.G195* variant, LVNC co‐existed with Barth syndrome, DCM, atrial septal defect, and elevated urine 3‐methylglutaconic acid (3‐MGA) levels (Ronvelia et al. 2012). Lastly, LVNC co‐existed with cardiovascular malformation and Ebstein᾿s anomaly in a family with the MYH7/p.Y283D variant (Postma et al. 2011). These results highlighted that multiple factors affect the pathogenicity of variants, but family‐based whole‐genome sequencing makes it possible to identify the association between a genetic variant and a rare disease.
Prediction analysis
Prediction analysis was carried out for 56 of the 60 variants. The remaining four variants were not analyzed because they generated stop codons.
By utilizing the Giudicessi et al. (2012) agreement of ≥3 in silico tools, prediction analysis determined that 51% of missense variants were pathogenic, 41% were VUS, and 5% were benign. The pathogenicity of the p.D626N/LDB3 variant could not be predicted using the agreement of ≥3 in silico tools and it did not match with the amino acids in six isoforms of the protein.
Finally, by using the Giudicessi et al. (2012) agreement of ≥3 in silico tools, 44% (4/9) of the ESP‐positive variants were pathogenic compared with 55% (10/18) of the ExAC‐positive variants (Table S1).
Discussion
Using bioinformatics, this is the first study to identify and investigate, all previous genetic variants associated with LVNC until July 2015. We found 60 variants in 12 genes associated with LVNC. In addition, prediction analyses were carried out for all missense variants.
This study identified 9 LVNC‐associated variants in the ESP database and 18 variants in ExAC database.
In the MYH7 gene, 24 of 60 LVNC‐associated variants were reported. None of those were identified in the ESP, but two SNPs (p.R1359C and p.R243H) were identified in ExAC database (Klaassen et al. 2008). These two variants predicted to be pathogenic with agreement of more than three prediction tools analysis. The assumption is that a pathogenic variant is unlikely to be present in a high number of individuals in the background population. The cut‐off for how many persons, one variant can be present in a cohort like ESP and still be pathogenic, is still unclear. Also, it should be kept in mind that it is of course possible that the geno‐positive individuals in general population can develop LVNC or other cardiomyopathies in late age. The p.R1359C variant in MYH7 was a sporadic case of LVNC, while the p.R243H was identified with family co‐segregation and with low allele frequency (0.000008236) in the ExAC database (Exome Aggregation Consortium (ExAC)). These findings suggest that the p.R243H theoretically develop LVNC in healthy carriers. Family co‐segregation is a valuable tool for determining the pathogenicity of disease‐causing variants (Klaassen et al. 2008). Hoedemaekers et al. (2010) performed a family screening with both molecular and clinical examinations in 58 patients with LVNC and their 194 family members. This screening revealed that 67% (39/58 patients with LVNC) had a genetic background with these variants. Furthermore, nine of 50 families had one of these variants. The findings support that the variants in MYH7 penetrate to the next generation. Budde et al. (2007) assessed the clinical and molecular characterization of a large German family with LVNC. The variant p.R281T in MYH7 was present in affected family members (11/24) and also the p.R281T known as ESP‐negative and ExAC‐negative variant. These results supported the major role of MYH7 variants in the development of LVNC.
Kaneda et al. (2008) screened 99 unrelated probands with DCM and five probands with isolated LVNC and reported a novel p.M531R variant in MYH7 in a patient with isolated LVNC, and we did not find the p.M531R in ESP and ExAC databases. To define the function of the p.M531R variant in MYH7, Kaneda et al. (2008) performed a functional study with transgenic mouse carrying the p.M532R mutant alpha MHC gene, which is identical to the p.M531R variant in human beta MHC. Although none of the mice had the characteristics of LVNC, 50–70% of the transgenic mice demonstrated left ventricular hypertrophy at 2–3 months of age. In addition, dilation of the left ventricle was reported in approximately 25% of transgenic mice by 18 months of age. They showed biphasic changes in the LV wall thickness (HCM developing to DCM). Their results suggested that the p.M531R variant has a malignant or pro cardiomyopathic effect on cardiac function.
By using four prediction analysis, 17 out of 24 (71%) previously reported LVNC‐associated MHY7 variants were predicted pathogenic, and interestingly, none of variants in MYH7 were predicted benign. Due to the effect of LVNC‐associated variants in MYH7 on cardiac development and function, the genetic basis of this gene in LVNC appears to be important. The variants found in MYH7 in patients with isolated LVNC strongly support the importance of this gene in pathogenesis of LVNC. Functional investigations of these variants are important and needed to provide a more comprehensive understanding of the effects of variants on protein function.
We identified LVNC‐associated genes (DTNA, MYH7B and CASQ2) that had only one nonsynonym LVNC‐associated variant (p.P121L, p.R890C, and p.H244R) (Ichida et al. 2001; Hoedemaekers et al. 2010; Esposito et al. 2013). The p.H244R and p.R890C variants present in both ESP and ExAC database, and the p.P121L variant only present in ExAC database. In all of these variants, there was family co‐segregation with the same phenotype (LVNC) (Ichida et al. 2001; Hoedemaekers et al. 2010; Esposito et al. 2013). The genetic screening in the first degree relatives to identify co‐segregation is an important tool to determine the pathogenicity of a variant. These co‐segregation data suggest that these variants are likely to be disease causing in a monogenic pattern, while they are predicted benign, VUS, and pathogenic (Table S1).
MYBPC3 contains seven LVNC‐associated variants, of which three (p.G5R, p.G490R and p.R502W) are found in the ESP database and six variants (p.R820W, p.P873L, p.G5R, p.G490R, p.G148R, and p.R502W) identified in ExAC Browser (Table S1). Interestingly, p.R820W exhibited autosomal recessive inheritance, whereas the rest displayed autosomal dominant inheritance. The p.G490R and the p.R502W predicted as pathogenic variants, while they are present in both ESP database and ExAC database. These variants were also identified in a family co‐segregation study. The family members with the p.R502W developed HCM and the family member with the p.G490R developed LVNC. These data point to the fact that there are many factors (for example modifier genes, age, environment, etc.) that affect the pathogenicity of variants.
Functional studies are often helpful to clarify the function of the protein variants such as rare and low‐frequency LVNC‐associated variants. Functional studies have been performed for eight LVNC‐associated variants (MYH7/p.M531R, MIB1/p.R530*, and p.V943F, PRDM16/p.K702*, TNNT2/p.E96K ,and p.R131W, LDB3/p.D117N, and TPM1/p.D84N). All variants showed loss‐of‐function in mutated proteins (Table S1).
Accurate estimating of rare variants frequency in background population is needed with a widespread ethnic background population with enormous number of healthy individuals. Multiple factors (e.g., modifier genes, age, environment, etc.) affect the penetrance of pathogenic variants in healthy individuals. Thus, it is not possible to determine the possibility of pathogenicity for ExAC‐positive variant. Additional functional study and genetic searching in family members are needed to clarify the pathogenic effect of these ExAC‐positive variants in general population.
Limitations
One limitation of this study is that the variants reported in this manuscript were collected in patients of different nationalities worldwide, while the ESP is restricted to European American and African American patients. Thus, the prevalence of LVNC in individual carrying LVNC‐associated variants in the ESP and ExAC could not been determined.
Due to lack of data regarding variants positioned in promoters, introns, and untranslated regions (UTR) in the ESP, variants found in these regions were not included in our study. The clinical status of variant carriers was not readily available. It is possible that some of individuals suffer from LVNC or other cardiomyopathies in the ExAC population.
Functional investigations have only been carried out for a minority of the variants, and not all studies investigated whether a variant co‐segregated.
Implications
This novel dataset should be used in the evaluation of potential disease‐causing variants. The presence of a potential disease‐causing variant in these large databases implies that the variant is less likely to be disease causing. The presence of a variant in one person in the large databases (more than 5000 persons) does not rule out disease causality, but in our opinion, the presence of a variant in more than five persons makes it unlikely to be pathogenic.
Conclusions
In this analysis, we demonstrate that MYH7 is the predominant gene associated with LVNC. MYH7 variants comprised 24 of the total 60 variants. None of these variants were found in the ESP, but two of the variants were found in the ExAC database. Family members showed a widespread clinical spectrum from asymptomatic to symptomatic forms of LVNC and other cardiomyopathies.
We also identified nine ESP‐positive and 18 ExAC‐positive variants of 60 previously reported LVNC‐associated variants, suggesting that these variants are not necessarily the monogenic cause of LVNC. Thus, additional data from functional studies and genetic screening in family members is still important in order to help clinician to estimate whether a variant is truly disease causing or not.
Conflict of Interest
Javad Jabbari is employed at LEO Pharma A/S. There are no financial interests to report.
Supporting information
Table S1. LVNC‐associated genes and variants.
Acknowledgments
The authors thank the NHLBIGO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL‐102923), the WHI Sequencing Project (HL‐102924), the Broad GO Sequencing Project (HL‐102925), the Seattle GO Sequencing Project (HL‐102926), and the Heart GO Sequencing Project (HL‐103010). We would also the Human Gene Mutation Database for collecting data over the years. The work was supported by The Danish Heart Foundation, The Danish National Research Foundation Center for Cardiac Arrhythmia (DARC), The John and Birth Meyer Foundation, and the Research Foundation at the Heart Center, Rigshospitalet, Copenhagen, Denmark.
References
- Agmon, Y. , Connolly H. M., Olson L. J., Khandheria B. K., and Seward J. B.. 1999. Noncompaction of the ventricular myocardium. J. Am. Soc. Echocardiogr. 12:859–863. [DOI] [PubMed] [Google Scholar]
- Andreasen, C. , Refsgaard L., Nielsen J. B., Sajadieh A., Winkel B. G., Tfelt‐Hansen J., et al. 2013. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can. J. Cardiol. 29:1104–1109. [DOI] [PubMed] [Google Scholar]
- Aras, D. , Tufekcioglu O., Ergun K., Ozeke O., Yildiz A., Topaloglu S., et al. 2006. Clinical features of isolated ventricular noncompaction in adults long‐term clinical course, echocardiographic properties, and predictors of left ventricular failure. J. Card. Fail. 12:726–733. [DOI] [PubMed] [Google Scholar]
- Arndt, A.‐K. , Schafer S., Drenckhahn J.‐D., Sabeh M. K., Plovie E. R., Caliebe A., et al. 2013. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am. J. Hum. Genet. 93:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger, A. R. , Miller M. A., Donthireddi U. R., Najovits A. J., and Goldman M. E.. 2008. New classification scheme of left ventricular noncompaction and correlation with ventricular performance. Am. J. Cardiol. 102:92–96. [DOI] [PubMed] [Google Scholar]
- Bhatia, N. L. , Tajik A. J., Wilansky S., Steidley D. E., and Mookadam F.. 2011. Isolated noncompaction of the left ventricular myocardium in adults: a systematic overview. J. Card. Fail. 17:771–778. [DOI] [PubMed] [Google Scholar]
- Bleyl, S. B. , Mumford B. R., Brown‐Harrison M. C., Pagotto L. T., Carey J. C., Pysher T. J., et al. 1997a. Xq28‐linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am. J. Med. Genet. 72:257–265. [PubMed] [Google Scholar]
- Bleyl, S. B. , Mumford B. R., Thompson V., Carey J. C., Pysher T. J., Chin T. K., et al. 1997b. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am. J. Hum. Genet. 61:868–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde, B. S. , Binner P., Waldmüller S., Höhne W., Blankenfeldt W., Hassfeld S., et al. 2007. Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta‐myosin heavy chain gene. PLoS ONE 2:e1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, T. K. , Perloff J. K., Williams R. G., Jue K., and Mohrmann R.. 1990. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 82:507–513. [DOI] [PubMed] [Google Scholar]
- Dusek, J. , Ostádal B., and Duskova M.. 1975. Postnatal persistence of spongy myocardium with embryonic blood supply. Arch. Pathol. 99:312–317. [PubMed] [Google Scholar]
- Esposito, T. , Sampaolo S., Limongelli G., Varone A., Formicola D., Diodato D., et al. 2013. Digenic mutational inheritance of the integrin alpha 7 and the myosin heavy chain 7B genes causes congenital myopathy with left ventricular non‐compact cardiomyopathy. Orphanet. J. Rare Dis. 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Aggregation Consortium (ExAC) . Cambridge, MA. Available at http://exac.broadinstitute.org (Accessed September 2015).
- Exome Variant Server . NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available at http://evs.gs.washington.edu/EVS/ (Accepted 20 December 2013).
- Finsterer, J. 2009. Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr. Cardiol. 30:659–681. [DOI] [PubMed] [Google Scholar]
- Giudicessi, J. R. , Kapplinger J. D., Tester D. J., Alders M., Salisbury B. A., Wilde A. A. M., et al. 2012. Phylogenetic and physicochemical analyses enhance the classification of rare nonsynonymous single nucleotide variants in type 1 and 2 long‐QT syndrome. Circ. Cardiovasc. Genet. 5:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham, R. 1974. Amino acid difference formula to help explain protein evolution. Science 185:862–864. [DOI] [PubMed] [Google Scholar]
- HGMD (Human Gene Mutation Database for Human Genetics Research) BIOBASE Biological Databases. Available at http://www.biobase-international.com/product/hgmd (Accepted 20 December 2013).
- Hoedemaekers, Y. M. , Caliskan K., Majoor‐Krakauer D., van de Laar I., Michels M., Witsenburg M., et al. 2007. Cardiac beta‐myosin heavy chain defects in two families with non‐compaction cardiomyopathy: linking non‐compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur. Heart J. 28:2732–2737. [DOI] [PubMed] [Google Scholar]
- Hoedemaekers, Y. M. , Caliskan K., Michels M., Frohn‐Mulder I., van der Smagt J. J., Phefferkorn J. E., et al. 2010. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ. Cardiovasc. Genet. 3:232–239. [DOI] [PubMed] [Google Scholar]
- Ichida, F. , Hamamichi Y., Miyawaki T., Ono Y., Kamiya T., Akagi T., et al. 1999. Clinical features of isolated noncompaction of the ventricular myocardium: long‐term clinical course, hemodynamic properties, and genetic background. J. Am. Coll. Cardiol. 34:233–240. [DOI] [PubMed] [Google Scholar]
- Ichida, F. , Tsubata S., Bowles K. R., Haneda N., Uese K., Miyawaki T., et al. 2001. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103:1256–1263. [DOI] [PubMed] [Google Scholar]
- Jabbari, J. , Jabbari R., Nielsen M. W., Holst A. G., Nielsen J. B., Haunsø S., et al. 2013. New exome data question the pathogenicity of genetic variants previously associated with catecholaminergic polymorphic ventricular tachycardia. Circ. Cardiovasc. Genet. 6:481–489. [DOI] [PubMed] [Google Scholar]
- Jenni, R. , Oechslin E., Schneider J., Attenhofer Jost C., and Kaufmann P. A.. 2001. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: a step towards classification as a distinct cardiomyopathy. Heart Br. Card. Soc. 86:666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda, T. , Naruse C., Kawashima A., Fujino N., Oshima T., Namura M., et al. 2008. A novel beta‐myosin heavy chain gene mutation, p.Met531Arg, identified in isolated left ventricular non‐compaction in humans, results in left ventricular hypertrophy that progresses to dilation in a mouse model. Clin. Sci. Lond. Engl. 1979 114:431–440. [DOI] [PubMed] [Google Scholar]
- Klaassen, S. , Probst S., Oechslin E., Gerull B., Krings G., Schuler P., et al. 2008. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 117:2893–2901. [DOI] [PubMed] [Google Scholar]
- Monserrat, L. , Hermida‐Prieto M., Fernandez X., Rodríguez I., Dumont C., Cazón L., et al. 2007. Mutation in the alpha‐cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non‐compaction, and septal defects. Eur. Heart J. 28:1953–1961. [DOI] [PubMed] [Google Scholar]
- Moric‐Janiszewska, E. , and Markiewicz‐Łoskot G.. 2008. Genetic heterogeneity of left‐ventricular noncompaction cardiomyopathy. Clin. Cardiol. 31:201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oechslin, E. N. , Attenhofer Jost C. H., Rojas J. R., Kaufmann P. A., and Jenni R.. 2000. Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J. Am. Coll. Cardiol. 36:493–500. [DOI] [PubMed] [Google Scholar]
- Paterick, T. E. , Gerber T. C., Pradhan S. R., Lindor N. M., and Tajik A. J.. 2010. Left ventricular noncompaction cardiomyopathy: what do we know? Rev. Cardiovasc. Med. 11:92–99. [DOI] [PubMed] [Google Scholar]
- Pignatelli, R. H. , McMahon C. J., Dreyer W. J., Denfield S. W., Price J., Belmont J. W., et al. 2003. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation 108:2672–2678. [DOI] [PubMed] [Google Scholar]
- Postma, A. V. , van Engelen K., van de Meerakker J., Rahman T., Probst S., Baars M. J. H., et al. 2011. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ. Cardiovasc. Genet. 4:43–50. [DOI] [PubMed] [Google Scholar]
- Probst, S. , Oechslin E., Schuler P., Greutmann M., Boyé P., Knirsch W., et al. 2011. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ. Cardiovasc. Genet. 4:367–374. [DOI] [PubMed] [Google Scholar]
- Risgaard, B. , Jabbari R., Refsgaard L., Holst A. G., Haunsø S., Sadjadieh A., et al. 2013. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin. Genet. 84:489–495. [DOI] [PubMed] [Google Scholar]
- Sarma, R. J. , Chana A., and Elkayam U.. 2010. Left ventricular noncompaction. Prog. Cardiovasc. Dis. 52:264–273. [DOI] [PubMed] [Google Scholar]
- Sasse‐Klaassen, S. , Gerull B., Oechslin E., Jenni R., and Thierfelder L.. 2003. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am. J. Med. Genet. A 119A:162–167. [DOI] [PubMed] [Google Scholar]
- Sen‐Chowdhry, S. , and McKenna W. J.. 2008. Left ventricular noncompaction and cardiomyopathy: cause, contributor, or epiphenomenon? Curr. Opin. Cardiol. 23:171–175. [DOI] [PubMed] [Google Scholar]
- SIFT . Tool to predict nonsynonmous/missense variants. Available at http://sift.bii.a-star.edu.sg/ (Accepted 6 June 2014).
- Stanton, C. , Bruce C., Connolly H., Brady P., Syed I., Hodge D., et al. 2009. Isolated left ventricular noncompaction syndrome. Am. J. Cardiol. 104:1135–1138. [DOI] [PubMed] [Google Scholar]
- Stöllberger, C. , and Finsterer J.. 2005. Cardiologic and neurologic findings in left ventricular hypertrabeculation/non‐compaction related to wall thickness, size and systolic function. Eur. J. Heart Fail. 7:95–97. [DOI] [PubMed] [Google Scholar]
- Xing, Y. , Ichida F., Matsuoka T., Isobe T., Ikemoto Y., Higaki T., et al. 2006. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol. Genet. Metab. 88:71–77. [DOI] [PubMed] [Google Scholar]
- Zambrano, E. , Marshalko S. J., Jaffe C. C., and Hui P.. 2002. Isolated noncompaction of the ventricular myocardium: clinical and molecular aspects of a rare cardiomyopathy. Lab. Invest. 82:117–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. LVNC‐associated genes and variants.