

Abstract
Over the past 10 years, thousands of first‐into‐human (FIH) clinical trials have been performed in Europe, with few severe adverse events (SAEs). Each has received detailed prior safety review at both the local clinical research facility and at national drug regulatory authority level. The recent fatal SAE in the BIA‐102474‐101 clinical trial shows the limitations of this process. Although criticized for not sequentially dosing subjects both within and between cohorts – as recommended by the European Medicines Agency for high‐risk compounds after the TeGenero clinical trial disaster in 2006 – BIA‐102474‐101 was not considered to be high risk. Indeed, compounds with similar mechanisms of action had previously been taken through phase I and II trials without incident, and higher doses had been safely given for longer durations to nonhuman primates. If the available data are comprehensive and accurate, and further investigation does not reveal unreported warning signs, this study has serious implications for ongoing and future review of FIH clinical trials. All preclinical study documents and clinical data collected during the BIA‐102474‐101 trial should be made available urgently so that lessons can be learnt. In the meantime, reviewers and clinical researchers should always ask for information on drug and target interactions and full reports of preclinical toxicity studies, and plan sequential dosing with longer delays between patients and cohorts, particularly if late SAEs might be anticipated. The use of individual patient pharmacokinetic and dynamic data should guide sequential dosing. A process for systematic risk assessment, like that currently used in the Netherlands, should be applied routinely to all trials with novel compounds.
Introduction
Drug development requires first‐into‐human (FIH) studies to gain initial information on tolerability, pharmacokinetics/dynamics and basic elements of drug safety 1. All such study protocols are closely scrutinized by investigators, local review committees and the relevant national drug regulatory authority (DRA) as well as by ethics committees. The TeGenero trial, performed in the UK in 2006 2, resulted in major changes in design 3, first in the UK, following the Duff committee report in 2006 4, and then with revised EU guidelines in 2007 5.
The risk assessment of FIH studies in Europe includes a review of the applicability of the preclinical toxicity studies to humans 5. Studies that include nonhuman primates (NHPs) might be considered more applicable than those carried out in rodents and dogs, and therefore reassuring if no adverse events are noted. The risk assessment also includes consideration of whether compounds of similar structure and/or mechanism have previously been administered to humans 6. A lack of adverse events in such clinical studies would generally be reassuring for reviewers. However, the disastrous events of the BIA‐102474‐101 clinical trial bring these assumptions into question.
The clinical trial
The trial aimed to take the Bial‐Portela compound BIA‐102474‐101 (Figure 1), a fatty acid amide hydroxylase (FAAH) inhibitor, into humans for the first time. The design was a standard combination of eight single ascending dose (SAD) cohorts, food studies (pharmacokinetics with or without food) and four multiple ascending dose (MAD) cohorts receiving the compound for 10 days 7. After receiving regulatory approval from the French DRA [Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM)] and ethics approval, the first patient was dosed on 9 July 2015 8 (see timeline, Figure 2). Forty‐eight patients received single doses between 0.25 mg and 100 mg (16 received placebo) without any severe adverse events (SAEs) being reported to ANSM. The first MAD cohort was dosed with 2.5 mg in early October 2015, just before the final SAD cohort received the highest dose. A food cohort was completed between the first and third MAD cohorts. The first four MAD cohorts completed doses of 2.5 mg up to 20 mg daily for 10 days without reported incident 8.
Figure 1.
Molecular structure of the fatty acid amide hydroxylase inhibitor BIA‐102474‐101. From Wikipedia, under the Creative Commons Attribution–ShareAlike licence
Figure 2.
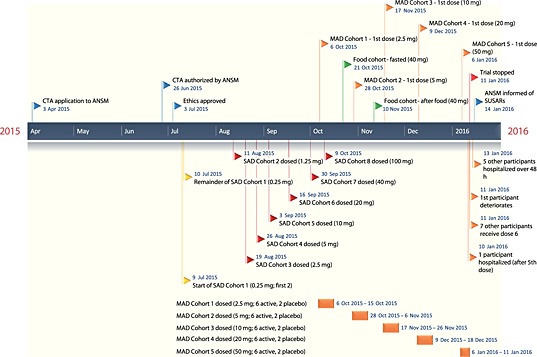
Timeline of the BIA‐102474‐101 clinical trial. ANSM, Agence Nationale de Sécurité du Médicament et des Produits de Santé; CTA, clinical trial application; MAD, multiple ascending dose; SAD, single ascending dose; SUSAR, suspected unexpected serious adverse reaction
The fifth MAD cohort (the first extra cohort, added because the maximum tolerated dose was not reached in the first four MAD cohorts), receiving 50 mg daily for 10 days, was first dosed on 6 January 2016 8. One participant became ill after the fifth dose on 10 January and was admitted to Rennes University Hospital. Despite fulfilling the criteria for an SAE, the study was not stopped or SAE reported, and the remaining seven participants in the cohort received the sixth dose the next day. Deterioration in the first participant's condition later that morning resulted in the trial being stopped. Four of the five participants receiving active therapy became ill over the following few days and each was hospitalized. The first subject to become ill died on 17 January. Brain magnetic resonance imaging (MRI) of the five symptomatic participants showed a consistent picture of deep‐brain haemorrhage and necrosis 9. Subsequent MRI imaging of participants who received lower SAD and MAD doses has shown no evidence of subclinical brain injury 10.
Follow‐up
At the time of writing this editorial, the amount of information available for public scrutiny was low, despite a call for the immediate release of all data by us 11 and others 12. The protocol was made public by ANSM shortly after the incident 7. Bial has thus far declined to make the Investigator's Brochure (IB) and Investigational Medicinal Product Dossier (IMPD) publically available. No preclinical toxicity study reports are available.
A preliminary report was produced by the French Inspection Générale des Affaires Sociales (IGAS) in early February, which criticized the trial on three points – continuing the study after admission of the first participant into hospital, not informing ANSM within 24 hours of the participant being admitted to hospital and not reconsenting the other participants after the SAE 13. No other major criticisms were made of the trial design in that report.
On 16 February, the ANSM's Temporary Specialist Scientific Committee on Inhibitors of FAAH published its preliminary conclusions 9. It noted that it considered preclinical studies to have been adequate for testing in humans, the pharmaceutical quality to be acceptable and the appearance of SAEs after five or six daily doses of 50 mg to demonstrate a threshold effect. The preferred hypotheses were that the molecule had effects additional to FAAH inhibition or that the effects were due to a metabolite 9.
Sequential dosing
After the first reports of the SAEs in the trial, there was much discussion in the press and scientific literature. The UK's Royal Society of Statistics (RSS), which produced its own report on FIH trial design after the TeGenero trial 14, expressed concerns with the trial design 12. A news article in Nature 15 reported particular concerns among statisticians about the time intervals between dosing in the SAD and MAD cohorts. Of note, this discussion took place before it became clear that the SAE had occurred in the fifth MAD cohort. Since the TeGenero study, the first dose of a new high‐risk compound (Box 1) has had to be given to just one participant (with one receiving placebo), with a delay before dosing the others in the cohort until the effects on this participant have been assessed 4, 5, 14 – termed sequential dosing. The need for such dosing is based upon careful risk assessment.
Box 1. MHRA guidance on identification of high‐risk trials
Examples of trials where expert advice may be needed include first‐in‐human (FIH) trials with novel compounds where:
The mode of action involves a target that is connected to multiple signalling pathways (target with pleiotropic effects), e.g. leading to various physiological effects or targets that are ubiquitously expressed.
The compound acts (directly or indirectly) via a cascade system, where there may be an amplification effect which might not be sufficiently controlled by a physiological feedback mechanism.
The compound acts (directly or indirectly) via the immune system, with a target or mechanism of action which is novel or currently not well characterized.
There is novelty in the structure of the active substance; e.g. a new type of engineered structural format, such as those with enhanced receptor interaction as compared with the parent compound.
The level of expression and biological function of the target receptor may differ between healthy individuals and patients with the relevant disease.
There is insufficient available knowledge of the structure, tissue distribution, cell specificity, disease specificity, regulation, level of expression and biological function of the human target, including downstream effects.
The compound acts via a possible or likely species‐specific mechanism or where animal data are unlikely to be predictive of activity in humans.
From: https://www.gov.uk/guidance/clinical‐trials‐for‐medicines‐apply‐for‐authorisation‐in‐the‐uk
The Duff report states that:
‘The intervals of observation between administration to the first, second and subsequent subjects should be determined by the kind of adverse reaction that might be anticipated based on the nature of the agent, its target, and the intended recipient'.
The EU FIH guidelines for high‐risk compounds are more specific about sequential dosing in later cohorts, stating:
‘Further dose administration should be sequential within each cohort to mitigate the risk. Any non‐sequential dose administration within each cohort should be justified. There must be an adequate period of observation between the administration of the medicinal product to the first, second and subsequent subjects in a cohort to observe and interpret reactions and adverse events. The duration of the interval of observation should be justified and will depend on the properties of the product and the data available, including non‐clinical PK and PD. Experience and identified risk factors from trials with comparable medicinal products should also be considered'.
The UK approach from 2006 has included a clear distinction between high‐ and low‐risk compounds – based largely on mode of action, nature of the target and relevance of the animal species and models (Box 1) – with high‐risk compounds being referred to a specialist Medicines and Healthcare products Regulatory Agency (MHRA) Expert Advisory Group 16. The European Medicines Agency (EMA) guidelines do not recommend such a dichotomous approach to risk assessment, all compounds requiring some element of explicit risk assessment. A structured systematic process for risk assessment was proposed after the TeGenero trial 6 and is now part of the standard protocol format used by the Netherlands Competent Authority for Clinical Trials (www.ccmo.nl).
Sequential dosing in the BIA‐102474‐101 protocol
According to the protocol, sequential dosing was carried out for the first dose on 9 July 2015 but the delay was for just 24 h 7. Thereafter, all cohorts were dosed together – dosing was not sequential within cohorts. The delay between SAD and MAD cohorts was between 7 and 14 days, except for the second SAD cohort and the fifth MAD cohort, which started 31 and 18 days, respectively, after the final doses of the prior cohort 8. Except for the second cohort, the delay between cohorts did not allow the previous cohort's pharmacokinetics to be considered before starting another 7, something recommended in the RSS report 14.
Unfortunately, and contrary to the EMA guidance, the protocol provides minimal discussion of the risk assessment for sequential dosing 7. There is no discussion of whether sequential dosing should be done for each cohort or why a 24‐hour interval was selected for SAD cohort 1. Of note, it states that if there were drug safety concerns for the MAD cohorts:
‘…the subjects dosing will be staggered (a maximum of 4 subjects dosed on the same day and 24 hours of follow‐up necessary before doing the remaining subjects)… ’
rather than sequential dosing of all subjects for each MAD cohort.
Only rapid‐onset adverse effects appear to have been considered as dosing was planned to take place with just 10 min between subjects dosed on the same day 7.
Risk assessment of BIA‐102474‐101
The drug was not apparently considered as high risk by ANSM or the ethics committee and it is therefore not clear that sequential dosing throughout the SAD and MAD protocols would ever have been recommended.
The protocol reports that no SAEs were noted in any preclinical toxicity study and that multiple‐dose studies were performed in NHPs using much higher doses and for a longer duration than those received by the fifth MAD cohort (50 mg, representing doses of 0.67 mg kg–1 in 75 kg adults). No‐observed‐adverse‐effect‐levels (NOAEL) of 100 mg kg–1 day–1 and 75 mg kg–1 day–1 were reported after 4‐week and 3‐month oral repeat administration studies, respectively, in NHPs. Somewhat lower, but still reassuring, NOAELs of 50 mg kg–1 day–and 20 mg kg–1 day–1 were noted in similar dog studies. However, no indication was given in the protocol of whether higher doses were given to NHPs or dogs with resulting toxicity.
The highest area under the plasma drug concentration–time curve reported from any preclinical study was in the 4‐week 100 mg day–1 NHP study. It is unclear whether the pharmacokinetic data from the prior MAD cohorts 1–4 indicated differential elimination of the compounds in humans compared with NHPs as they have not been made available. The preclinical pharmacokinetic information available at the beginning of the study does not suggest the need for a long‐duration pause between patients at the first dose or any other dose level or duration.
BIA‐102474‐101 is also not first in mechanistic class, although post‐hoc analysis has shown that it is different to the other FAAH inhibitors that have previously been taken through phase I and II clinical trials without reported problems. Although no comparative enzyme and receptor binding assays were reported in the protocol, an in silico study performed by Sean Ekins of Collaborative Chemistry 17 after the problems were reported found that BIA‐102474‐101 was predicted to bind to other targets as well as FAAH. It is possible that the toxicity seen was due to off‐target effects seen suddenly at high cumulative dose, as considered by the ANSM Expert Committee 9.
The protocol reports that FAAH inhibition was to be measured in the clinical study 7 but no results have been made available. Such measurements of effect are important for FIH trials as they may show maximum effect before the onset of toxicity. It is surprising that the trial aimed to establish a maximum tolerated dose ‐ indeed, the SAEs occurred in the first additional cohort performed to seek the maximum tolerated dose. Dosing could have better used pharmacological FAAH inhibition assays to guide dosing, instead of the absence of side effects at each dose 1, 18. It is possible that the SAEs occurred at doses well past maximum FAAH inhibition.
Conclusions
The occurrence of SAEs in most participants of a cohort, without any previous signal from preclinical or prior SAD and MAD cohorts, is not known to have occurred before. The total dose received by these participants, from 250 mg to 300 mg, is only 25–33% greater than the total dose received by the prior MAD cohort (200 mg), which had no reported signs of toxicity. The consistent picture across the cohort of six suggests a dose effect and not an idiosyncratic reaction.
Public release of the results from the preclinical toxicity studies and the clinical trial, as well as the IB and IMPD, is required urgently so that lessons can be learnt from this trial. Bial should now provide a timetable for the release of this information.
While we wait for these data to become available and new guidelines to be produced, what can reviewers do differently while considering protocols? We should ask for more information on drug and target interactions by the compound, more information on clinical studies with similar compounds via greater inter‐company sharing of data, and copies of the full reports of preclinical toxicity studies. We can insist on longer delays between patients and between cohorts, and use of individual patient pharmacokinetic and ‐dynamic data to guide sequential dosing within and between cohorts, slowing down the trials – but are we able to identify the compounds for which this is appropriate?
Regulation should be adapted urgently to ensure immediate and comprehensive release of data into the public domain after such an incident, to allow lessons to be learnt. Invoking commercial considerations, when volunteers have suffered such devastating outcomes, is both immoral and irrelevant, as the drugs causing SAEs will never be developed and there may be important lessons of immediate relevance to research programmes looking at similar drugs. We should also require information on previous reviews of the protocol by Clinical Research Facilities (CRFs) or Contract Research Organisations (CROs) that decide not to take on such trials – valuable information may be present in these reviews that has never been made public.
The BIA trial has shown again that drug development will always have risks. It is not possible to identify all ‘high‐risk’ compounds that need elevated risk assessment; therefore, all compounds need to undergo graded risk assessment. A process for systematic risk assessment currently used in the Netherlands could be applied routinely to all trials with novel compounds. Careful monitoring of pharmacological and clinical effects during dose escalation may not prevent all SAEs but it currently offers the best guarantee that dangerous effects of novel compounds are picked up early. Lessons from such disasters can best be learned by full and immediate transparency of documents, including, at least, the protocol (including a full risk assessment), IB, IMPD and all preclinical study reports.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: ME had no support from any organization for the submitted work. AFC is the Editor‐in‐Chief, British Journal of Clinical Pharmacology. DJW is the Non‐Executive Director, Medicines and Healthcare products Regulatory Agency (MHRA), and President, British Pharmacological Society (BPS). The views expressed in this article are our personal views and do not necessarily represent those of the MHRA or BPS.
Eddleston, M. , Cohen, A. F. , and Webb, D. J. (2016) Implications of the BIA‐102474‐101 study for review of first‐into‐human clinical trials. Br J Clin Pharmacol, 81: 582–586. doi: 10.1111/bcp.12920.
References
- 1. Cohen A Should we tolerate tolerability as an objective in early drug development? Br J Clin Pharmacol 2007; 64: 249–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello‐Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355: 1018–28. [DOI] [PubMed] [Google Scholar]
- 3. Milton MN, Horvath CJ. The EMEA guideline on first‐in‐human clinical trials and its impact on pharmaceutical development. Toxicol Pathol 2009; 37: 363–71. [DOI] [PubMed] [Google Scholar]
- 4. Expert Scientific Group on Phase One Clinical Trials . Final Report, 2006. [online]. Available at http://webarchive.nationalarchives.gov.uk/+/dh.gov.uk/en/publicationsandstatistics/publications/publicationspolicyandguidance/dh_063117 (last accessed 21 February 2016).
- 5. Committee for Medical Products for Human Use (CHMP) . Guideline on strategies to identify and mitigate risks for first‐in‐human clinical trials with investigational medicinal products. EMEA/CHMP/SWP/28367/07, 2007. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf (last accessed 21 February 2016).
- 6. Kenter MJ, Cohen AF. Establishing risk of human experimentation with drugs: lessons from TGN1412. Lancet 2006; 368: 1387–91. [DOI] [PubMed] [Google Scholar]
- 7. BIAL – Portela & Ca SA . Clinical Study Protocol N° BIA‐102474‐101. Version 1.2, 2015. [online]. Available at http://ansm.sante.fr/S‐informer/Actualite/Essai‐clinique‐BIA‐102474‐101‐du‐laboratoire‐ BIAL ‐Publication‐du‐protocole‐clinique‐Point‐d‐Information (last accessed 21 February 2016)
- 8. L’Agence Nationale de Sécurité du Médicament et des Produits de Santé [ANSM] . Chronology of the evaluation and performance of the clinical trial sponsored by BIAL laboratories and conducted by BIOTRIAL in Rennes, 2016. [online]. Available at http://ansm.sante.fr/content/download/85117/1074133/version/2/file/Chrono+point+dinfo+27.01.16_EN_1.pdf (last accessed 21 February 2016).
- 9. L’Agence Nationale de Sécurité du Médicament et des Produits de Santé [ANSM] . Essai clinique de Rennes: relevé de conclusions du CSST ‘Inhibiteurs de la FAAH’, 2016. [online]. Available at http://www.ansm.sante.fr/S‐informer/Actualite/Essai‐clinique‐de‐Rennes‐Releve‐de‐conclusions‐du‐CSST‐Inhibiteurs‐de‐la‐FAAH (last accessed 21 February 2016).
- 10. Ministère des Affaires Sociales et de la Santé . Intervention de Marisol Touraine – Conférence de presse – Accident d’essai clinique – point d’étape, 2016. 04/02/2016 [online]. Available at http://social‐sante.gouv.fr/actualites/presse/discours/article/intervention‐de‐marisol‐touraine‐conference‐de‐presse‐accident‐d‐essai‐clinique (last accessed: 21 February 2016).
- 11. Alexander S, Cohen A, Pirmohamed M, Webb D. Improve early access to data from catastrophic clinical trials: a statement on behalf of the British Pharmacological Society, 2016. 22/01/2016 [online]. Available at https://www.bps.ac.uk/news‐events/news/society‐news/articles/improve‐early‐access‐to‐data‐from‐catastrophic‐cli (last accessed 24 February 2016).
- 12. Royal Statistical Society . RSS statement on publication of the study protocol BIA‐102474‐101 for the French ‘first‐in‐man’ trial in healthy volunteers, 2016. 22/01/2016 [online]. Available at http://www.rss.org.uk/Images/PDF/about/press‐releases/2016‐01‐22‐rss‐statment‐BIA‐102474‐101‐french‐first‐trial‐in‐healthy‐volunteers.pdf (last accessed: 21 February 2016).
- 13. d'Autume C, Duhamel G. Note d'étape: enquête sur des incidents graves survenus dans le cadre de la réalisation d'un essai clinique, 2016. [online]. Available at http://social‐sante.gouv.fr/IMG/pdf/fevrier_2016_‐_note_etape_‐_accident_essai_clinique.pdf (last accessed: 21 February 2016).
- 14. Senn S, Amin D, Bailey RA, Bird SM, Bogacka B, Colman P, Garrett A, Grieve A, Lachmann P. Statistical issues in first‐in‐man studies. J R Statist Soc A 2007; 170: 517–79. [Google Scholar]
- 15. Callaway E, Butler D. Researchers question design of fatal French clinical trial. Nature 2016; 529: 263–4. [DOI] [PubMed] [Google Scholar]
- 16. MHRA . Clinical trials for medicines: apply for authorisation in the UK, 2016. [online[.Available at https://www.gov.uk/guidance/clinical‐trials‐for‐medicines‐apply‐for‐authorisation‐in‐the‐uk (last accessed: 21 February 2016).
- 17. Ekins S. Differences between similar FAAH inhibitors and their in silico target predictions, 2016. 21/01/2016 [online]. Available at http://www.collabchem.com/2016/01/21/differences‐between‐similar‐faah‐inhibitors‐and‐their‐in‐silico‐target‐predictions/ (last accessed: 24 February 2016).
- 18. Cohen AF. Developing drug prototypes: pharmacology replaces safety and tolerability? Nat Rev Drug Discov 2010; 9: 856–65. [DOI] [PubMed] [Google Scholar]