

Abstract
AIMS
Pregnant women can be exposed to numerous drugs during the gestational period. For obvious ethical reasons, in vivo studies of fetal exposure to drugs are limited. Information about the transplacental transfer of drugs prior to their administration to pregnant women would be highly useful. In the present study, a novel approach was developed quantitatively predict or to predict the fetal exposure to drugs administered to the mother quantitatively.
Methods
Transplacental parameters estimated from ex vivo human placenta perfusion experiments were implemented in pregnancy–physiologically based pharmacokinetic (p‐PBPK) models in order to predict fetal PK. Thereafter, fetal PK profiles for two antiretroviral drugs, tenofovir (TFV) and emtricitabine (FTC) were simulated. These predictions were then compared to observed cord blood concentrations, to validate these models.
Results
Parameters obtained from the ex vivo experiments enabled a good prediction of observed cord blood concentrations without additional a scaling factor. Moreover, a sensitivity analysis showed that fetal predictions were sensitive to changes in transplacental parameters values obtained ex vivo.
Conclusion
The integration of ex vivo human placental perfusion parameters in a p‐PBPK model should be a promising new approach for predicting human fetal exposure to xenobiotics.
Keywords: emtricitabine, fetus, PBPK, pharmacokinetics, pregnancy, tenofovir
What is Already Known About This Subject
Pregnant women and fetuses are orphan populations with respect to the safety and efficacy of drugs.
The cotyledon perfusion experiment is the gold standard that provides an insight into placental transfer. After maternal intake, fetal exposure is estimated by cord blood sampling.
There is no validated method that is able to predict fetal drug concentrations.
What This Subject Adds
Transplacental transfer parameters ([i.e. diffusion (Dcot), elimination constant (kPE) and placental partition coefficient (Kppl)] were estimated from the cotyledon perfusion model.
A novel approach to predict fetal drug exposure quantitatively, incorporating estimated transplacental transfer parameters in p‐PBPK models, was proposed and validated by comparing predictions to in vivo observations.
Introduction
Drug prescriptions and over‐the‐counter medications are common in pregnancy, and the average pregnant patient in the US and Canada uses more than two drugs during the course of their pregnancy 1. However, pregnant women and fetuses are orphan populations with respect to the safety and efficacy of drugs. Fetal toxicity and efficacy are thought to depend both upon the maternal‐to‐fetal transfer of drugs [pharmacokinetics (PK)] and intrinsic toxicity [pharmacodynamics (PD)].
Assessing drug transport across the human placental barrier is mandatory in order to guarantee drug safety during pregnancy 2. However, for obvious ethical reasons, in vivo fetal risk assessment studies related to maternal drug exposure remain extremely limited. Some studies have evaluated fetal exposure using cord blood plasma samples. Although the cord‐to‐maternal concentration ratio is informative as an index of relative fetal drug exposure, it is highly variable due to the various delays between drug administration and blood sampling 3, 4. Population PK analyses enable fetal PK to be estimated but usually require a large number of exposed patients 5, 6. To ensure drug safety during pregnancy, information about transplacental transfer prior to administration would be highly desirable. As animal studies may not be helpful for predicting human fetal PK because of interspecies differences in the structural and functional features of the placenta, other models have been developed. The ex vivo human placental perfusion model is the gold standard and offers a better insight into the various placental drug transporters, xenobiotic metabolism and tissue binding. Nevertheless, this method cannot directly predict fetal PK profiles.
The present study presents a novel approach predicting drug fetal exposure quantitatively. Transplacental parameter estimated from the ex vivo human placenta perfusion model were implemented in pregnancy–physiologically based PK (p‐PBPK) models in order to predict fetal PK. Physiologically based PK (PBPK) models are based to a large extent on the actual physiology of the organism, whereas conventional PK models use virtual compartments. PBPK models incorporate both physiological parameters that are important for absorption, distribution, metabolism and excretion processes and drug‐specific parameters. These models have already been used to predict PK profiles in specific populations, such as pregnant women 7, 8, 9, 10, 11. In regard to their structure, PBPK models are fully suitable to incorporate ex vivo data. The aim of the present study was to evaluate this new method to simulate the fetal PK of two antiretroviral drugs, tenofovir (TFV) and emtricitabine (FTC). These simulations were compared to observed cord plasma concentrations, to validate our models.
Materials and methods
Figure 1 shows the workflow of the present study. Briefly, we had previously developed PBPK models for FTC and TFV. When the models were able to accurately describe the PK for different routes of administration and dosing regimens in nonpregnant adults, we implemented the physiological changes occurring during pregnancy. PK simulations were then compared to observed concentrations from pregnant women. Thereafter, the feto‐placental compartment was used in the model, with the placenta, amniotic fluid and fetus considered as separate compartments. From the ex vivo experiments on cotyledons, the transplacental transfer parameters [diffusion (Dcot), placental elimination constant (kPE) and placental partition coefficient (Kppl)] were estimated. After scaling Dcot by placental weight, they were implemented in the PBPK models. Finally, simulated fetal and amniotic fluid PK profiles were compared to observed in vivo data.
Figure 1.
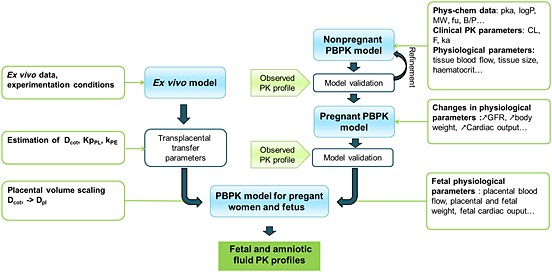
Schematic representation of the workflow of physiologically based pharmacokinetic (PBPK) model development. B/P, blood to plasma ratio; CL, clearance; Dcot, diffusion (cotyledon); Dpl, diffusion (placenta); F, bioavailability; fu, free fraction; GFR, glomerular filtration rate; ka: absorption rate constant; KpPL, placental partition coefficient, kPE, placental elimination; MW, molar mass; Phys‐chem, physicochemical; PK, pharmacokinetic
PBPK modelling in the nonpregnant population
In a previous study, we built up whole‐body PBPK models for pregnant and nonpregnant adults for FTC and TFV by using the Simcyp® software 12. To study transplacental transfer, simplified PBPK models had to be coded for the R software 13. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R‐project.org/) 14. Full PBPK models with first‐order rate absorption were used. To code our models in R language, only the physiological parameters were checked. Drug‐specific parameters used in the previously developed models in Simcyp® remained unchanged (Table 1). For each step of modelling, simulated PK profiles were compared to observed data and PK profiles previously simulated by Simcyp®. The physiological parameters used for these models are summarized in Table 2 15, 16, 17, 18.
Table 1.
Input values
| Tenofovir | Emtricitabine | |
|---|---|---|
| MW (g mol–1) | 287.21 19 | 247.25 20 |
| pKa | 3.7‐6.5 19 | 2.65 20 |
| log P | –2.21 21 | –0.43 20 |
| F | 0.18 22 | 0.93 20 |
| ka (h–1) | 0.56 23 | 0.54 5 |
| fu | 0.993 22 | 0.96 20 |
| B:P ratio | 0.58 24 | 1 20 |
| Total CL (l h–1) | 14.2 25 | 18 26 |
| CLR (l h–1) | 10.6 25 | 13 20, 26 |
B:P ratio, blood‐to‐plasma ratio; CL, clearance; CLR, renal clearance; F, bioavailability; fu, free fraction; ka, first‐order absorption rate; MW, molecular weight; P, partition coefficient; pKa, acid dissociation constant at logarithmic scale.
Table 2.
Mean adult physiological parameters
| Men | Women | Pregnant women (GA = 40) | |
|---|---|---|---|
| Height (cm) | 175 | 165 | 165 |
| Body weight (kg) | 80 | 60 | 74.8 |
| Haematocrit | 0.45* | 0.39* | 0.33*,‡ |
| Glomerular filtration rate (l h–1) | 6.1† | 4.8† | 6.4†,‡ |
| Organ volumes (l) | |||
| Adipose | 20.2* | 20.2* | 27.9*,‡ |
| Bones | 3.6* | 2.1* | 2.1* |
| Blood | 5.8* | 4.2* | 6.1*,‡ |
| Lung | 1.3* | 0.8* | 0.8* |
| Brain | 1.4* | 1.4* | 1.4* |
| Heart | 0.4* | 0.3* | 0.3* |
| Kidney | 0.3* | 0.3* | 0.3* |
| Muscle | 32.1* | 19.6* | 19.6* |
| Skin | 3.8* | 3.2* | 3.2* |
| Liver | 1.5* | 1.2* | 1.2* |
| Spleen | 0.1* | 0.1* | 0.1* |
| Pancreas | 0.1* | 0.1* | 0.1* |
| Gut | 1.3* | 1.1* | 1.1* |
| Blood flows rates (l h –1 ) | |||
| Cardiac output | 359.8 | 296.8 | 381.2 |
| Adipose | 24.2* | 24.3* | 33.5* |
| Bones | 17.5* | 10.4* | 10.4* |
| Brain | 43.0* | 42.5* | 42.5* |
| Heart | 15.7* | 12.6* | 12.6* |
| Kidney | 74.4* | 61.6* | 62.5* |
| Muscle | 57.8* | 35.3* | 35.6* |
| Skin | 27.4* | 23.0* | 23.0* |
| Liver | 90.8*,¶ | 81.3*,¶ | 108.7*,¶,§ |
| Spleen | 7.8* | 7.8* | 7.8a |
| Pancreas | 3.8* | 3.0* | 3.0* |
| Gut | 58.4* | 50.8* | 78.2*, d |
PBPK modelling in pregnant women
Figure 2 shows the PBPK model. The distinct placental and fetal compartments enable ex‐vivo transplacental transfer parameters to be incorporated. Moreover, as amniotic fluid can affect fetal exposure, this compartment was added to the model. As the two drugs are poorly bound to plasma proteins, the free fractions (fu) were assumed to be unchanged during pregnancy. Fetal fu values were assumed to be equal to maternal ones (Table 1). Numerous types of exchange can affect fetal and amniotic fluid PK. To study fetal‐to‐placental exchange, the fetal compartment was split into fetal blood and other fetal tissues (Figure 2). This exchange is driven by the blood flow rate between the placenta and the fetus (QplaF). In late pregnancy, one‐fifth of the fetal cardiac output is distributed to the placenta, with the remainder distributed to the rest of the fetal body (Qrbf) 27. The main exchanges between the amniotic space and the surrounding tissues in late pregnancy come from fetal urine, fetal swallowing (ksw), the intramembranous pathway from the amniotic cavity to the fetal circulation (kINT) and pulmonary excretion (kL) 28, 29, 30, 31. Fetal renal excretion was indexed on fetal glomerular filtration rate (GFRf) 32. The two drugs studied are poorly metabolized, so this elimination pathway was assumed to be negligible for fetus 33. The maternal physiological changes occurring during pregnancy that can affect the distribution and elimination are implemented in this model 12. All of the basal values of the nonpregnant population, except for portal vein blood flow, were modified according to the gestational age (GA) of the pregnant population, as described by Abduljalil et al. (Table 2) 17, 18. All fetal physiological constants are reported in Table 3. A sensitivity analysis was performed on some parameters to evaluate their impact on fetal and amniotic fluid PK.
Figure 2.
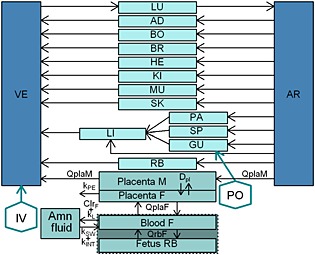
Schematic representation of physiologically based pharmacokinetic (PBPK) models used. AD, adipose; Amn fluid, amniotic fluid; AR, arterial blood; BO, bones; BR, brain; CLrF, fetal urinary excretion; Dpl, diffusion; F, fetus; GU, gut; HE, heart; IV, intravenous; KI, Kidney; kint, intramembranous pathway; kL, oral, nasal, tracheal and pulmonary secretion constant; kPE, placental elimination; kSW, swallowing constant; LI, liver; LU, lung; M, mother; MU, muscle; PA, pancreas; PO, per os; QPlaF, blood flow from the fetus to the placental tissue; QplaM, blood flow from the mother to the placental tissue; QrbF, blood flow to the fetal body; RB, rest of the body; SK, skin; SP, spleen; VE, venous blood
Table 3.
Fetal physiological parameter values
| (GA = 40) | Parameters | Ref. |
|---|---|---|
| Maternal‐to‐placental blood flow, QplaM (l h–1) | 46.5 | Abduljalil et al. 17 |
| Fetal‐to‐placental blood flow, QplaF (l h–1) | 14.3 | Kiserud et al. 27 |
| Fetal cardiac output, Qca (l h–1) | 85.5 | Kiserud et al. 27 |
| Placental weight, Vpla (kg) | 0.65 | Aduljalil et al. 17 |
| Fetal weight, Vfo (kg) | 3.56 | Aduljalil et al. 17 |
| Amniotic fluid volume, Vamf (l) | 0.86 | Aduljalil et al. 17 |
| Fetal blood volume, VbloodF (l) | 0.24 | Smith et al. 34 |
| Fetal haematocrit | 0.5 | Zanardo et al. 35, Chang et al. 36, Eskoka et al. 37 |
| Fetal glomerular filtration rate, GFRf (l h–1) | 0.136 | Arant et al. 32 |
| Swallowing volume, kSW | 0.8 | Underwood et al. 29 |
| Secretion of oral, nasal, tracheal and pulmonary fluids, kL (l/day–1) | 0.126 | Underwood et al. 29 |
| Intramembranous pathway, kINT l day–1 | 0.35 | Underwood et al. 29 |
GA, gestational age.
Ex vivo model
Placenta tissue collections
Thirty‐four placentas from normal pregnancies were obtained from Port Royal Hospital (Paris, France) after uncomplicated vaginal delivery or caesarean section. All mothers were seronegative for HIV infection, were not infected by hepatitis B or C viruses and took no medication other than oxytocin or epidural anaesthesia during labour. All placentas were obtained after a full‐term pregnancy (GA from 37 to 41 weeks and 4 days). Written informed consent was obtained for all participants in the study.
Placental perfusion
Placentas were perfused in a recirculating (closed–closed) circuit, according to a method adapted from those of Schneider et al. 38 and Forestier et al. 39 . Perfusion experiments started within 30 min after delivery. After a visual examination for lack of evident lesions on the chorionic plate, a truncal branch of the chorionic artery and the associated vein were cannulated. The fetal circulation was established at a flow rate of 6 ml min–1 (Qf). After confirmation of the absence of vascular leakage, the perfused area progressively whitened and enabled visualization of the selected cotyledon. The perfusion was subsequently initiated by insertion of two catheters into the intervillous space on the maternal side. The maternal circulation was established at a flow rate of 12 ml min–1 (Qm). The pHs of maternal and fetal solutions, prepared using Earle medium containing 30 g l–1 and 40 g l–1 of human serum albumin, were adjusted to 7.4 ± 0.1 and 7.2 ± 0.1, respectively. The validation of the cotyledon's viability during the experiment was carried out using antipyrine (20 mg l–1). TFV, FTC and antipyrine were perfused into the maternal reservoir. Maternal and fetal reservoir volumes (Vm and Vf, respectively) were 200 ml and 250 ml, respectively. Samples were collected every 10 min during the first half‐hour and then every 30 min until 150 min from both the fetal and maternal sides to determine the concentrations (Cf and Cm, respectively). Samples were then stored at –20°C until analysis.
Sample analysis
TFV and FTC concentrations were determined using ultra performance liquid chromatography coupled with tandem mass spectrometry (UPLC–MS/MS), using the Acquity UPLC/TQD (Applied Biosystems, Foster City, CA, USA) 40. Antipyrine concentrations were determined by high‐performance liquid chromatography with ultraviolet detection at 290 nm 41.
The maternal‐to‐fetal transfer was described by the fetal transfer rate (FTR). It was calculated as follows:
Equation 1:
where Cf and Cm are the drug concentrations in fetal and maternal perfusates, and Vf and Vm are the fetal and maternal perfusate volumes. An FTR of antipyrine >20% was required to validate each experiment. Clearance index (CLI) was calculated as the ratio of the TFV or FTC FTR to that of antipyrine. The FTR and CLI parameters are only useful for comparison purposes between drugs and are not applied in the PBPK model.
Estimation of transplacental transfer parameters
Drug transfer across the placenta was modelled as a cotyledon split into maternal and fetal compartments (Figure 3). The cotyledon volume averaged 58 ml. Maternal cotyledon volume (Vmp) was assumed to be 23 ml 42. Several transplacental transfer models for TFV and FTC were investigated – i.e. simple diffusion, linear transfer, saturable transfer and addition of placental elimination rate or tissue protein binding. Data were analysed with a nonlinear mixed‐effect modelling approach, using NONMEM program version 6.2 (Icon Development Solutions, Ellicott City, MD, USA). Several error models (proportional, additive, mixed) were investigated to describe the residual variability (ε).The objective function value (OFV) was used to test different hypotheses. A model was kept if the OFV was decreased by at least 3.84 (chi‐square test with one degree of freedom). For evaluation of the goodness of fit, the following graphs were performed: observed concentrations vs. predictions, weighted residuals vs. time, and weighted residuals vs. predictions. Diagnostic graphics and distribution statistics were performed using R for Nonmem (RfN) (http://wfn.sourceforge.net) from the R program. Simulated concentrations in the maternal and fetal reservoirs were compared to concentrations from the ex vivo experiment previously described.
Figure 3.
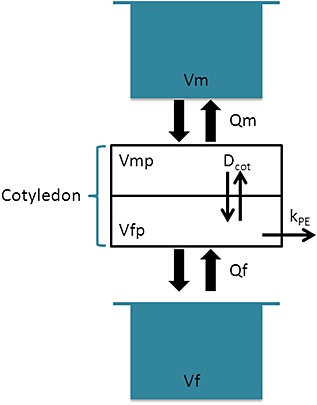
Schematic representation of the ex vivo model used for tenofovir (TFV) and emtricitabine (FTC) (recirculating circuit). Dcot, diffusion parameter; f, fetus; fp, fetal placenta; kPE, placental elimination constant (h–1); m, mother; mp, maternal placenta; Q, flow rate (l h–1); V, volume (l)
As an example, equations 2–5 were used to describe the ex vivo experiment considering passive transfer (Dcot) and placental elimination (kPE).
Equation 2: maternal reservoir
Equation 3: maternal cotyledon
Equation 4: fetal cotyledon
Equation 5: fetal reservoir
where C denotes a concentration (mg l–1), Q a flow rate (l h–1) and V a volume (l). Subscripts m, f and p denote mother, fetus and placenta, respectively. Kppl, is the placental partition coefficient,Dcot is the diffusion parameter (l h–1) and kPE is the placental elimination parameter (h–1). Kppl, Dcot and kPE were estimated.
In vivo fetal and amniotic fluid simulations
The best model describing the transplacental transfer in ex vivo experimentation was implemented in the p‐PBPK model. Cotyledon volume and experimental flow rates were replaced by placental volume and in vivo perfusion rates (Table 3). Transplacental transfer parameters estimated from ex vivo experiments were more integrated. The in vivo diffusion was related to the ex vivo parameters weighed by their respective volumes as shown in Equation 6.
Equation 6: in vivo diffusion parameter
where Dpl and Dcot stand for the in vivo and ex vivo diffusion parameters, and Vpl and Vcot for the placental and cotyledon volumes.
The equations describing the feto‐placental compartments of the p‐PBPK models are shown below.
Equation 7: in vivo maternal placenta
Equation 8: in vivo fetal placenta
Equation 9: fetal blood
Equation 10: fetal body
Equation 11: amniotic fluid
where Q denotes blood flow (l h–1),V tissue volume (l), B/P the blood‐to‐plasma concentration ratio, Dpl a diffusion parameter (l h–1), GFR the glomerular filtration rate (l h–1), Clr the maternal renal clearance (l h–1), fu the free drug fraction, kINT the intramembranous constant (l h–1), kSW the swallowing constant (l h–1) and kL the oral, nasal, tracheal and pulmonary secretion constant (l h–1). The subscripts ab, plaM, plaF, bloodF, rbF and amf denote maternal blood, maternal placenta, fetal placenta, fetal blood, rest of the fetal body (fetal body – fetal blood) and amniotic fluid.
Women in labour had smaller maximal concentrations (Cmax) compared to pregnant women, so changes in absorption were assumed to occur during labour for the two drugs. Therefore, absorption rates were reduced by 50% and the FTC bioavailability was fixed at 0.75.
Model validation
Models were validated by comparing maternal, fetal and amniotic fluid concentration simulations to observed data from women in labour. We simulated the administration of a single dose of 400 mg of FTC or 600 mg of TFV to an average 35‐year‐old patient with a GA of 39 weeks. We compared the simulated maternal and fetal concentrations to the observed concentrations found by Hirt et al. 5, 6. In the Tenofovir/Emtricitabine in Africa and Asia (TEmAA) Agence Nationale de Recherches sur le SIDA et les hépatites virales (ANRS) 12109 study, drugs were given during labour and all women underwent blood sampling for PK analysis at delivery and at 1, 2, 3, 5, 8, 12, and 24 h after drug administration. A cord blood sample was obtained at delivery. We also compared fetal‐to‐maternal and amniotic fluid‐to‐maternal concentration ratios for TFV to data obtained by digitalization 43 from another study during labour 4. Maternal and fetal exposures were compared using the fetal‐to‐maternal area under the curve (AUC) ratios for a single dose and at steady state. The predicted exposures at the 39th week of gestation are reported in Table 5.
Table 5.
AUC ratio obtained after simulation during labour and during pregnancy at steady state
| GA = 39 | FTC | TFV | |
|---|---|---|---|
| Single dose | Plasma AUCf/AUCm | 0.63 | 0.41 |
| Amniotic fluid AUC/plasma AUCm | 1.93 | 1.39 | |
| Steady state | Plasma AUCf/AUCm | 0.64 | 0.45 |
| Amniotic fluid AUC/plasma AUCm | 3.43 | 2.76 |
AUC, area under the curve; f, fetus; FTC, emtricitabine; m, mother; TFV, tenofovir.
Sensitivity analyses on physiological constants such as the placental maternal blood flow parameter (QplaM), kSW, GFRf and on parameters obtained ex vivo such as diffusion, kPE and Kppl were performed. Each time the reference value was multiplied or divided by 1.3, 2 and 5, and simulated FTC fetal and amniotic fluid PK profiles at steady state were obtained.
Results
Ex vivo model
A total of 26 experiments (TFV = 16, FTC = 10) could be validated for the integrity of the placental membrane and adequate conditions of perfusion. The mean (± standard deviation) antipyrine FTR was 53.9% (±5.9 %) for TFV and 42.9 % (± 3.7 %) for FTC. The mean FTR and CLI (30–150 min) were 21.0 ± 5.7 % and 0.39 ± 0.11, respectively, for TFV, and 24.3 ± 6.4 % and 0.53 ± 0.17, respectively, for FTC.
The transplacental transfer for these two drugs was best described by the diffusion model with estimation of Kppl and kPE (Figure 3).
Figure 4 compares observed to simulated concentrations for the ex vivo human perfusion model. Parameter estimates are summarized in Table 4.
Figure 4.
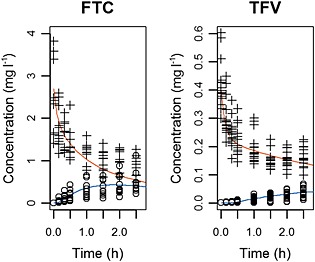
Ex vivo human placental perfusion model for emtricitabine (FTC; left) and tenofovir (TFV; right). Evolution of observed fetal concentration (open circles) and maternal concentration (crosses) compared to fetal (orange) and maternal (blue) simulated profiles in the ex vivo human placental model
Table 4.
Estimated values for transplacental transfer parameters obtained in the ex vivo model, mean (range)
| Dcot(l h–1) | KPE (h–1) | Kppl | |
|---|---|---|---|
| Emtricitabine | 0.104 (0.025–0.395) | 1.49* | 3.94 (0.92–9.33) |
| Tenofovir | 0.013 (0.003–0.020) | 0.443 (0.167–1.2) | 7.15 (3.73–1.45) |
Dcot, diffusion parameter; KPE, placental elimination constant; Kppl, coefficient partition.
No variability was estimated.
In vivo simulations
The parameters estimated ex vivo (Kppl, kPE, Dcot) were applied in the p‐PBPK models. PK profiles during labour were simulated for a single dose and compared to observed data 5, 6. Figure 5 shows that simulated fetal PK profiles using our p‐PBPK model were in agreement with observed cord concentrations. As shown in Figure 5, fetal Cmax values were lower than maternal ones. Table 5 summarizes the simulated fetal‐to‐maternal plasma and amniotic fluid‐to‐maternal plasma AUC ratios. A previous population approach estimated fetal as 10.7 mg h l–1 and 1.64 mg h l–1 at delivery for a single dose of FTC and TFV, respectively, whereas our model predicted 9.23 mg h l–1 and 1.26 mg h l–1, respectively 5, 6.
Figure 5.
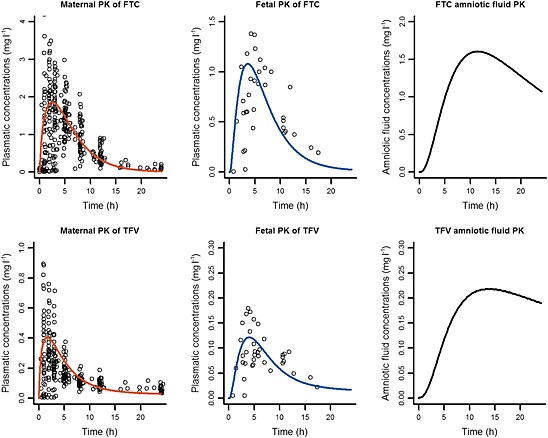
Simulation vs. observation of administration of emtricitabine (FTC) (top) and tenofovir (TFV) (bottom) during labour. Simulation (lines) of mean maternal (left), fetal (middle) and amniotic fluid (right) pharmacokinetic profile for a single dose compared to observations (points) 5, 6. ‘Time’ represents the delay between the last dose and sampling time
PK profiles for a single dose and at steady state obtained after administration of 200 mg of FTC once daily and 300 mg of TFV once daily in 39‐week pregnant women were simulated. Figure 6 shows the variations of fetal‐to‐maternal and amniotic fluid‐to‐maternal concentration ratios over time. For TFV, the fetal‐to‐maternal plasma ratio stayed <1, whereas amniotic fluid accumulation was observed. TFV simulations were close to the observed data obtained by Mirochnick et al. 4.
Figure 6.
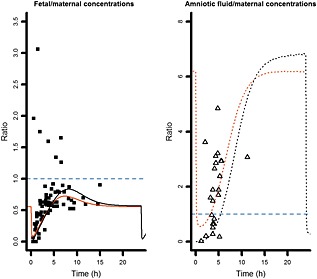
Simulations of fetal‐to‐maternal concentration ratios and amniotic fluid‐to‐maternal concentration ratios for tenofovir, for a single dose and at steady state. Evolution of fetal‐to‐maternal drug concentration ratios (solid lines) and amniotic fluid‐to‐maternal concentration ratios (dashed lines) for a single dose (black bold lines) and at steady state (thin orange lines). Ratios were compared to the available observed data: fetal‐to‐maternal drug concentration ratios (solid squares) and amniotic fluid‐to‐maternal concentration ratios (triangles) obtained by Mirochnick et al. 4
Sensitivity analyses showed that the diffusion and kPE values were strong determinants of simulated fetal and amniotic fluid PK profiles (Figures 7, 8). The modification in Kppl values influenced Tmax but the effect on AUC was weaker. The daily volume of amniotic fluid swallowed by the fetus and the GFRf could be multiplied or divided by 5 without significant modification of the simulated fetal PK but with a significant change in amniotic fluid PK. Finally, the change in maternal‐to‐placental blood flow rate had a negligible effect on both fetal and amniotic fluid PK.
Figure 7.
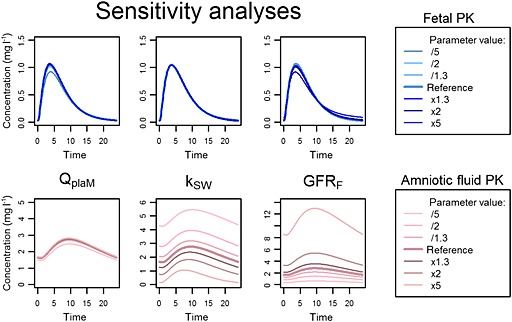
Sensitivity analyses of physiological parameters. Simulation of emtricitabine fetal pharmacokinetic (PK) profile (top) and amniotic fluid PK profile (bottom) after changes in placental perfusion (first column), daily swallowing volume (second column), fetal renal clearance (third column). kSW, swallowing constant; GFRF, fetal glomerular filtration rate; QplaM placental maternal blood flow parameter
Figure 8.
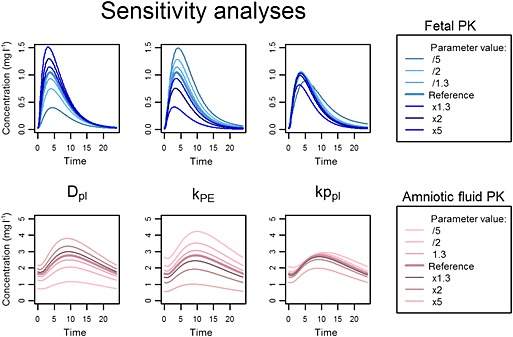
Sensitivity analyses of parameters estimated ex vivo. Simulation of emtricitabine fetal pharmacokinetic (PK) profile (top) and amniotic fluid PK profile (bottom) after changes in placental diffusion parameter Dpl (first column) and placental elimination constant kPE (second column) and placental coefficient partition, Kppl (third column)
Discussion
We developed a novel approach quantitatively predicting the fetal exposure of drugs administered to the mother. The p‐PBPK models which implemented parameters estimated from human placental perfusion ex vivo experiments enabled the prediction of the fetal and amniotic fluid PK of FTC and TFV at full term.
This is the first study to report the ex vivo transplacental transfer of TFV and FTC. Only one other study has implemented parameters obtained from human ex vivo placental perfusion experiments in a mechanistic model 42, 44. However, these authors used animal data to validate their model because no human data were available for a PK profile comparison.
Data from TEmAA ANRS 12109 enable us to validate our models 5, 6 by comparing simulated with actual fetal PK profiles. The predictions were also compared to other published data. Delays between sampling time and last dose were not available individually for these published data, and as concentrations and ratios are highly variable depending on these delays, these data were used only to obtain an estimate of orders of magnitude.
There are few data on FTC placental transfer; the geometric mean of cord blood concentration was found to be 0.26 mg l–1 [90% confidence interval (CI) 0.17‐0.39 mg l–1; n = 11) 45. Moreover, Colbers et al. reported that the cord‐to‐maternal blood ratio 8.5 h (range 0–32 h) after dosing was 1.63 (90% CI 0.46‐1.82; n = 10) 46. Based on model predictions, the mean ratio was 1.6 and mean fetal concentration was 0.19 mg l–1 (delay range: 0–24 h). No amniotic fluid data were available.
TFV placental transfer has been described to be significant. The Pediatric AIDS Clinical Trials Group (PACTG) study observed a median cord blood concentration at delivery of 0.076 mg l–1 (range: 0.000–0.309 mg l–1; n = 10) in the group receiving 600 mg of TFV 47. Moreover the current study found simulated cord‐to‐maternal plasma concentration ratios to be close to those measured by Mirochnick et al. (Figure 6) 4. TFV has been reported to accumulate in the amniotic fluid, with highly variable concentrations 4, 48, 49. Figure 6 shows that the p‐PBPK model is able to predict this accumulation 4.
For TFV, the observed amniotic fluid concentrations indicated two features: accumulation and great variability. This drug is mainly excreted unchanged by the kidney 25, 26. Accumulation in the amniotic fluid could be explained by a greater excretion rate into amniotic fluid (renal excretion, lung excretion) relative to the absorption rate (swallowing, intramembranous pathway).The high variability of the amniotic fluid concentration can be partially explained by the variability of the volume swallowed by the fetus and the renal excretion volume 28, 29, 32. Indeed, as showed by sensitivity analyses, modifications of these two phenomena greatly influence amniotic fluid concentrations (Figure 7).
Values for the fetal swallowing constant and renal excretion are not well documented and are difficult to obtain 29, 32, 50, 51. However, even if the true values are higher or lower than those used in the models, the effect on fetal PK profiles would not be significant (Figure 7). Therefore, uncertainty about these physiological constants was not a major issue. The sensitivity analyses were carried out on one fetal‐to‐amniotic fluid transfer parameter (GFRf) and one amniotic fluid‐to‐fetal transfer parameter (kSW); the impact of analogous parameters (kL, kINT) on fetal and amniotic fluid PK was similar (data not shown). This sensitivity analysis also suggested that amniotic fluid can change considerably without a significant change in fetal PK. Therefore, amniotic fluid may not be an accurate surrogate of fetal concentrations. Moreover, our model shows little effect of amniotic fluid concentrations on fetal PK. Thus, if the aim is only to elucidate the fetal PK profile, this compartment could be ignored. Otherwise, the p‐PBPK models provided a good prediction of the fetal PK profiles for these two renally excreted drugs but it might not be suitable for drugs that are mainly metabolized. The placental elimination pathway has not been elucidated for these drugs. In our models, placental elimination (kPE) influenced fetal (and amniotic fluid) PK but did not significantly affect maternal PK.
Protein binding can lead to misleading results with the placental ex vivo model, so we added albumin to the media to ensure equivalent protein binding. These drugs are poorly plasma bound, so protein binding should not significantly influence the transplacental transfer. However, for drugs with high protein binding, this potential issue should be considered. Two options are available to decrease the risk of obtaining misleading results: (i) the ex vivo free fraction should be the same as the in vivo one or (ii) the transplacental transfer should be corrected for the in vivo free fraction.
The diffusion and placental elimination parameters obtained from the ex vivo experiments enabled good predictions of fetal PK profiles to be obtained. No random scaling factors were needed. Moreover, the sensitivity analyses performed on these parameters showed that any modification had a significant impact on fetal exposure. Thus, the ex vivo parameters, integrated into the PBPK model, seem to be a sensitive and accurate method for predicting fetal exposure. However, as the placental structure changes throughout gestation, this approach reflects the placental barrier only at delivery.
The present approach enables a basic prediction to be made of fetal PK prior to drug administration to the mother. This should be a useful tool for the discovery of drugs targeting the fetus or those that can potentially be used in pregnant women at term. In the future, fetal tissue exposure might also be simulated through a more complex fetal PBPK model.
Conflict of Interest
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years, no other relationships or activities that could appear to have influenced the submitted work.
We acknowledge the French Agence Nationale de Recherche contre le VIH/SIDA et les hépatites virales (ANRS) for sponsoring this work.
De Sousa Mendes, M. , Hirt, D. , Vinot, C. , Valade, E. , Lui, G. , Pressiat, C. , Bouazza, N. , Foissac, F. , Blanche, S. , Lê, M. P. , Peytavin, G. , Treluyer, J.‐M. , Urien, S. , and Benaboud, S. (2016) Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br J Clin Pharmacol, 81: 646–657. doi: 10.1111/bcp.12815.
References
- 1. Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández‐Díaz S. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol 2011; 205: 51.e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. US Food and Drug Administration . Guidance for industry pharmacokinetics in pregnancy – study design, data analysis, and impact on dosing and labeling [online]. Available at http://www.fda.gov/ScienceResearch/SpecialTopics/WomensHealthResearch/ucm133348.htm (last accessed 15 September 2015).
- 3. Chappuy H, Tréluyer J‐M, Jullien V, Dimet J, Rey E, Fouché M, Firtion G, Pons G, Mandelbrot L. Maternal–fetal transfer and amniotic fluid accumulation of nucleoside analogue reverse transcriptase inhibitors in human immunodeficiency virus‐infected pregnant women. Antimicrob Agents Chemother 2004; 48: 4332–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mirochnick M, Taha T, Kreitchmann R, Nielsen‐Saines K, Kumwenda N, Joao E, Pinto J, Santos B, Parsons T, Kearney B, Emel L, Herron C, Richardson P, Hudelson SE, Eshleman SH, George K, Fowler MG, Sato P, Mofenson L; HPTN 057 Protocol Team . Pharmacokinetics and safety of tenofovir in HIV‐infected women during labor and their infants during the first week of life. J Acquir Immune Defic Syndr 2014; 65: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hirt D, Urien S, Rey E, Arrive E, Ekouevi DK, Coffie P, Leang SK, Lalsab S, Avit D, Nerrienet E, McIntyre J, Blanche S, Dabis F, Treluyer J‐M. Population pharmacokinetics of emtricitabine in human immunodeficiency virus type 1‐infected pregnant women and their neonates. Antimicrob Agents Chemother 2008; 53: 1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirt D, Urien S, Ekouévi DK, Rey E, Arrivé E, Blanche S, Amani‐Bosse C, Nerrienet E, Gray G, Kone M, Leang SK, McIntyre J, Dabis F, Tréluyer J‐M; ANRS 12109. Population pharmacokinetics of tenofovir in HIV‐1‐infected pregnant women and their neonates (ANRS 12109). Clin Pharmacol Ther 2009; 85: 182–9. [DOI] [PubMed] [Google Scholar]
- 7. Ke AB, Nallani SC, Zhao P, Rostami‐Hodjegan A, Unadkat JD. A PBPK model to predict disposition of CYP3A‐metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT Pharmacomet Syst Pharmacol 2012; 1: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami‐Hodjegan A. A pregnancy physiologically based pharmacokinetic (p‐PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4: PBPK for pregnancy with time‐varying physiological parameters. Br J Clin Pharmacol 2012; 74: 873–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ke AB, Nallani SC, Zhao P, Rostami‐Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos Biol Fate Chem 2013; 41: 801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Loccisano AE, Longnecker MP, Campbell JL Jr, Andersen ME, Clewell HJ 3rd. Development of PBPK models for PFOA and PFOS for human pregnancy and lactation life stages. J Toxicol Environ Health A. 2013; 76: 25–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luecke RH, Wosilait WD, Young JF. Mathematical representation of organ growth in the human embryo/fetus. Int J Biomed Comput 1995; 39: 337–47. [DOI] [PubMed] [Google Scholar]
- 12. De Sousa Mendes M, Hirt D, Urien S, Valade E, Bouazza N, Foissac F, Blanche S, Treluyer J‐M, Benaboud S. Physiologically‐based pharmacokinetic modeling of renally excreted antiretroviral drugs in pregnant women. Br J Clin Pharmacol 2015; May 22;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R‐project.org (last accessed: 2015 Nov 17).
- 14. Soetaert K, Petzoldt T, Setzer RW. Solving differential equations in R: package deSolve. J Stat Softw 2010; 33 [online]. Available at http://www.jstatsoft.org/v33/i09/ (last accessed 2015 Nov 17).
- 15. Price PS, Conolly RB, Chaisson CF, Gross EA, Young JS, Mathis ET, Tedder DR. Modeling interindividual variation in physiological factors used in PBPK models of humans. Crit Rev Toxicol 2003; 33: 469–503. [PubMed] [Google Scholar]
- 16. Peters AM, Perry L, Hooker CA, Howard B, Neilly MDJ, Seshadri N, Sobnack R, Irwin A, Snelling H, Gruning T, Patel NH, Lawson RS, Shabo G, Williams N, Dave S, Barnfield MC. Extracellular fluid volume and glomerular filtration rate in 1878 healthy potential renal transplant donors: effects of age, gender, obesity and scaling. Nephrol Dial Transplant 2012; 27: 1429–37. [DOI] [PubMed] [Google Scholar]
- 17. Abduljalil K, Furness P, Johnson TN, Rostami‐Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet 2012; 51: 365–96. [DOI] [PubMed] [Google Scholar]
- 18. Clapp JF, Stepanchak W, Tomaselli J, Kortan M, Faneslow S. Portal vein blood flow – effects of pregnancy, gravity, and exercise. Am J Obstet Gynecol 2000; 183: 167–72. [DOI] [PubMed] [Google Scholar]
- 19. Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharmacokinet 2004; 43: 595–612. [DOI] [PubMed] [Google Scholar]
- 20. Gilead Sciences, Inc . Product Information: Emtriva(TM), emtricitabine capsules. 2003.
- 21. Janneh O, Khoo S. Interactions of tenofovir, lamivudine, abacavir and didanosine in primary human cells. Pharmaceutics 2011; 3: 326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilead Sciences, Inc . Product Information: VIREAD(R) oral tablets, tenofovir disoproxil fumarate oral tablets. 2010.
- 23. Benaboud S, Hirt D, Launay O, Pannier E, Firtion G, Rey E, Bouazza N, Foissac F, Chappuy H, Urien S, Treluyer JM. Pregnancy‐related effects on tenofovir pharmacokinetics: a population study with 186 women. Antimicrob Agents Chemother 2011; 56: 857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee WA, He G‐X, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, Cundy KC. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother 2005; 49: 1898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deeks SG, Barditch‐Crovo P, Lietman PS, Hwang F, Cundy KC, Rooney JF, Hellmann NS, Safrin S, Kahn JO. Safety, pharmacokinetics, and antiretroviral activity of intravenous 9‐[2‐(R)‐(phosphonomethoxy)propyl]adenine, a novel anti‐human immunodeficiency virus (HIV) therapy, in HIV‐infected adults. Antimicrob Agents Chemother 1998; 42: 2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gish RG, Leung NWY, Wright TL, Trinh H, Lang W, Kessler HA, Fang L, Wang LH, Delehanty J, Rigney A, Mondou E, Snow A, Rousseau F. Dose range study of pharmacokinetics, safety, and preliminary antiviral activity of emtricitabine in adults with hepatitis B virus infection. Antimicrob Agents Chemother 2002; 46: 1734–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kiserud T, Ebbing C, Kessler J, Rasmussen S. Fetal cardiac output, distribution to the placenta and impact of placental compromise. Ultrasound Obstet Gynecol 2006; 28: 126–36. [DOI] [PubMed] [Google Scholar]
- 28. Heller M, Burd L. Review of ethanol dispersion, distribution, and elimination from the fetal compartment. Birth Defects Res A Clin Mol Teratol 2014; 100: 277–83. [DOI] [PubMed] [Google Scholar]
- 29. Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: not just fetal urine anymore. J Perinatol 2005; 25: 341–8. [DOI] [PubMed] [Google Scholar]
- 30. Beall MH, van den Wijngaard JPHM, van Gemert MJC, Ross MG. Amniotic fluid water dynamics. Placenta 2007; 28: 816–23. [DOI] [PubMed] [Google Scholar]
- 31. Gilbert WM, Newman PS, Eby‐Wilkens E, Brace RA. Technetium Tc 99m rapidly crosses the ovine placenta and intramembranous pathway. Am J Obstet Gynecol 1996; 175: 1557–62. [DOI] [PubMed] [Google Scholar]
- 32. Arant BS Jr Developmental patterns of renal functional maturation compared in the human neonate. J Pediatr 1978; 92: 705–12. [DOI] [PubMed] [Google Scholar]
- 33. Morgan DJ. Drug disposition in mother and foetus. Clin Exp Pharmacol Physiol 1997; 24: 869–73. [DOI] [PubMed] [Google Scholar]
- 34. Smith GCS, Cameron AD. Estimating human fetal blood volume on the basis of gestational age and fetal abdominal circumference. Int J Obstet Gynaecol 2002; 109: 721–2. [DOI] [PubMed] [Google Scholar]
- 35. Zanardo V, Gabrieli C, De Luca F, Trevisanuto D, De Santis M, Scambia G, Straface G. Head‐to‐body delivery by ‘two‐step’ approach: effect on cord blood hematocrit. J Matern‐Fetal Neonatal Med 2013; 26: 1234–8. [DOI] [PubMed] [Google Scholar]
- 36. Chang Y‐H, Yang S‐H, Wang T‐F, Lin T‐Y, Yang K‐L, Chen S‐H. Complete blood count reference values of cord blood in Taiwan and the influence of gender and delivery route on them. Pediatr Neonatol 2011; 52: 155–60. [DOI] [PubMed] [Google Scholar]
- 37. Eskola M, Juutistenaho S, Aranko K, Sainio S, Kekomäki R. Association of cord blood platelet count and volume with hemoglobin in healthy term infants. J Perinatol 2010; 31: 258–62. [DOI] [PubMed] [Google Scholar]
- 38. Schneider H, Panigel M, Dancis J. Transfer across the perfused human placenta of antipyrine, sodium and leucine. Am J Obstet Gynecol 1972; 114: 822–8. [DOI] [PubMed] [Google Scholar]
- 39. Forestier F, de Renty P, Peytavin G, Dohin E, Farinotti R, Mandelbrot L. Maternal–fetal transfer of saquinavir studied in the ex vivo placental perfusion model. Am J Obstet Gynecol 2001; 185: 178–81. [DOI] [PubMed] [Google Scholar]
- 40. Jung BH, Rezk NL, Bridges AS, Corbett AH, Kashuba ADM. Simultaneous determination of 17 antiretroviral drugs in human plasma for quantitative analysis with liquid chromatography –tandem mass spectrometry. Biomed Chromatogr 2007; 21: 1095–104. [DOI] [PubMed] [Google Scholar]
- 41. Vinot C, Gavard L, Treluyer JM, Manceau S, Courbon E, Scherrmann JM, Decleves X, Duro D, Peytavin G, Mandelbrot L, Giraud C. Placental transfer of maraviroc in an ex vivo human cotyledon perfusion model and influence of ABC transporter expression. Antimicrob Agents Chemother 2013; 57: 1415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shintaku K, Arima Y, Dan Y, Takeda T, Kogushi K, Tsujimoto M, Nagata H, Satoh S, Tsukimori K, Nakano H, Hori S, Ohtani H, Sawada Y. Kinetic analysis of the transport of salicylic acid, a nonsteroidal anti‐inflammatory drug, across human placenta. Drug Metab Dispos 2007; 35: 772–8. [DOI] [PubMed] [Google Scholar]
- 43. Huwaldt JA. Plot digitizer [online]. Available at http://plotdigitizer.sourceforge.net (last accessed 2015 Nov 17).
- 44. Shintaku K, Hori S, Satoh H, Tsukimori K, Nakano H, Fujii T, Taketani Y, Ohtani H, Sawada Y. Prediction and evaluation of fetal toxicity induced by NSAIDs using transplacental kinetic parameters obtained from human placental perfusion studies. Br J Clin Pharmacol 2012; 73: 248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stek AM, Best BM, Luo W, Capparelli E, Burchett S, Hu C, Li H, Read JS, Jennings A, Barr E, Smith E, Rossi SS, Mirochnick M. Effect of pregnancy on emtricitabine pharmacokinetics. HIV Med 2012; 13: 226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Colbers APH, Hawkins DA, Gingelmaier A, Kabeya K, Rockstroh JK, Wyen C, Weizsäcker K, Sadiq ST, Ivanovic J, Giaquinto C, Taylor GP, Moltó J, Burger DM, PANNA network . The pharmacokinetics, safety and efficacy of tenofovir and emtricitabine in HIV‐1‐infected pregnant women. AIDS Lond Engl 2013; 27: 739–48. [DOI] [PubMed] [Google Scholar]
- 47. Flynn PM, Mirochnick M, Shapiro DE, Bardeguez A, Rodman J, Robbins B, Huang S, Fiscus SA, van Rompay KKA, Rooney JF, Kearney B, Mofenson LM, Watts DH, Jean‐Philippe P, Heckman B, Thorpe E, Cotter A, Purswani M, PACTG 394 Study Team . Pharmacokinetics and safety of single‐dose tenofovir disoproxil fumarate and emtricitabine in HIV‐1‐infected pregnant women and their infants. Antimicrob Agents Chemother 2011; 55: 5914–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yeh RF, Rezk NL, Kashuba ADM, Dumond JB, Tappouni HL, Tien H‐C, Chen Y‐C, Vourvahis M, Horton AL, Fiscus SA, Patterson KB. Genital tract, cord blood, and amniotic fluid exposures of seven antiretroviral drugs during and after pregnancy in human immunodeficiency virus type 1‐infected women. Antimicrob Agents Chemother 2009; 53: 2367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Else LJ, Taylor S, Back DJ, Khoo SH. Pharmacokinetics of antiretroviral drugs in anatomical sanctuary sites: the fetal compartment (placenta and amniotic fluid). Antivir Ther 2011; 16: 1139–47. [DOI] [PubMed] [Google Scholar]
- 50. Czuba MA, Morgan DJ, Ching MS, Mihaly GW, Hardy KJ, Smallwood RA. Ontogeny of fetal renal organic cation excretion: a study with cimetidine and ranitidine during the latter half of gestation in the pregnant ewe. J Pharmacol Exp Ther 1990; 255: 1177–82. [PubMed] [Google Scholar]
- 51. Beall MH, van den Wijngaard JPHM, van Gemert MJC, Ross MG. Regulation of amniotic fluid volume. Placenta 2007; 28: 824–32. [DOI] [PubMed] [Google Scholar]