

Abstract
AIMS
The aim of the present study was to investigate whether differences in total and peak drug exposure upon generic substitution are due to differences between formulations or to intrasubject pharmacokinetic variability of the active substance.
Methods
The study was designed as a retrospective reanalysis of existing studies. Nine replicate design bioequivalence studies representing six drug classes – i.e. for alendronate, atorvastatin, cyclosporin, ebastine, exemestane, mycophenolate mofetil, and ropinirole – were retrieved from the Dutch Medicines Regulatory Authority.
Results
In most studies, the intrasubject variability in total and peak drug exposure was comparable for the brand‐name [in the range 0.01–0.24 for area under the concentration–time curve (AUCt) and 0.02–0.29 for peak plasma concentration (Cmax) on a log scale] and generic (0.01–0.23 for AUCt and 0.08–0.33 for Cmax) drugs, and was comparable with the intrasubject variability upon switching between those drugs (0.01–0.23 for AUCt and 0.06–0.33 for Cmax). The variance related to subject‐by‐formulation interaction could be considered negligible (–0.069 to 0.047 for AUCt and –0.091 to 0.02 for Cmax).
Conclusion
In the investigated studies, the variation in total and peak exposure seen when a patient is switched from a brand‐name to a generic drug is comparable with that seen following repeated administration of the brand‐name drug in the patient. Only the intrasubject variability seems to play a crucial and decisive role in the variation in drug exposure seen; no additional formulation‐dependent variation in exposure is observed upon switching. Thus, our data support that, for the medicines that were included in the present investigation, from a clinical pharmacological perspective, the benefit–risk balance of a generic drug is comparable with that of the brand‐name drug.
Keywords: bioequivalence study, generic drugs, intrasubject variability
What is Already Known About This Subject
Differences in drug exposure that are reported upon switching between brand‐name and generic drugs often lead to concerns about the therapeutic equivalence of brand‐name and generic drugs.
As generic drugs are widely used, it is necessary to address the concerns regarding the observed differences in exposure upon switching.
The reported differences in exposure have been postulated to be related to individual‐specific variability in pharmacokinetics but it is not known whether intrasubject variability of the active substance is a key factor in this altered drug exposure or whether this is dependent on differences between the brand‐name and generic formulations.
What This Study Adds
Only the intrasubject variability in the pharmacokinetics of an active substance plays a crucial and decisive role in the variation in total and peak drug exposure seen when switching from a brand‐name to a generic drug; differences in the formulation of the brand‐name and generic drugs do not add to this variation.
The differences in exposure observed after switching from a brand‐name to a generic drug are comparable with those observed upon repeated administration of the brand‐name drug without switching.
Thus, from a clinical pharmacological point of view, the benefit–risk balance of a generic drug is expected to be comparable with that of the brand‐name drug, even directly after the switch.
Introduction
The 80–125% criterion is used in bioequivalence studies worldwide as the standard limit for the 90% confidence intervals (CIs) of the ln‐transformed ratios between generic and brand‐name drugs for the area under the drug concentration–time curve (AUCt) and the maximum plasma concentration (Cmax). Meeting this criterion has been the basic requirement for registration of a generic drug for more than 25 years 1, 2, known as the average bioequivalence approach. Thus, a small difference in average total or peak exposure between a generic drug and a brand‐name drug is permissible and is considered not to have clinical consequences. However, bioequivalence between a generic drug and a brand‐name drug does not exclude the possibility that differences in the exposure occur at an individual level, owing to e.g. intrasubject variability and the variance due to subject‐by‐formulation effects. Thus, in principle, it is possible that there is a detectable difference in plasma drug levels after an individual patient switches from a brand‐name to a generic drug and vice versa. Indeed, a difference in exposure upon switching (either decreased or increased exposure) has been reported 3, 4. This difference is partly responsible for concerns among users and prescribers about the interchangeability of generic drugs. Some reports suggest that brand‐name and generic drugs may not be equally effective or safe because they may be associated with a difference in exposure in individual patients. For example, an increased risk of seizures, frequent visits to the emergency department and a high switching‐back rate have been reported when patients with epilepsy were switched from a brand‐name to a generic drug 4, 5, 6, 7, 8, 9. Yet, a systematic review and meta‐analysis of clinical studies of switching between antiepileptic drugs found no difference in the odds of uncontrolled seizures in patients treated with generic vs. brand‐name drugs 10. A similar clinical equivalence of generic and brand‐name drugs has been reported for drugs for cardiovascular diseases 11.
As traditional bioequivalence studies, based on the average bioequivalence approach, cannot evaluate the differences in drug exposure levels in terms of AUCt and Cmax in individuals, population bioequivalence and individual bioequivalence approaches have been proposed for assessing the total variability in AUCt and Cmax in the population, and intrasubject variability for generic and brand‐name drugs and the subject‐by‐formulation interaction, respectively 12, 13. In 1997, the US Food and Drug Administration (FDA) drafted guidelines on the investigation of bioequivalence based on population and individual bioequivalence approaches 14, 15, aiming to resolve the concerns about prescribability and switchability in individual patients. However, the use of these alternative approaches was not successful owing to the issues of masking effect, power and sample size determination, statistical procedures and study design 16. In the literature, it has been shown that the proposed criteria using a mixed‐scaling aggregate strategy would lead to a relaxation of the 80–125% average bioequivalence standard, particularly for highly variable drugs 17. A positive correlation between the intrasubject variability and the estimation of the variance for the subject‐by‐formulation interaction was identified, which consequently eliminated the possibility of using a constant level (such as 0.0225, as suggested by the FDA) as a basis for demonstrating substantial subject‐by‐formulation interactions 18.
In the literature, the reported individual differences in exposure between brand‐name and generic drugs have been postulated to be related to individual‐specific variability in pharmacokinetics 19, 20, which leads to a variation in drug exposure in individuals. Given that the human body is a dynamic environment when it comes to pharmacokinetics, it is perhaps not surprising that drug absorption and excretion may vary from time to time, resulting in variable drug exposure from dose to dose, either with repeated administration of the same drug or when switching between brand‐name and generic drugs. However, to our knowledge, it has not yet been studied whether intrasubject variability is a key factor in the altered total or peak drug exposure when a patient is switched to a generic drug, or the extent to which the intrasubject variability contributes to this variation.
The aim of the present study was to investigate the reason for the difference in total and peak drug exposure in individuals that is observed when switching between a brand‐name and generic drug, and to clarify the role of intrasubject variability in the pharmacokinetics of this switch. To our knowledge, the role of intrasubject variability in explaining differences in exposure obtained upon switching between brand‐name and generic drugs has not yet been fully clarified. For this purpose, we made use of data obtained from replicate design studies. Typically, these are four‐way crossover studies in which each subject randomly receives, under fasting conditions, the brand‐name drug twice and the generic drug twice. This design makes it possible to investigate the intrasubject variability in pharmacokinetic parameters (i.e. total and peak drug exposure) with the brand‐name drug, the generic drug and the process of switching between the two drugs. It should be noted that in the latter case, the intrasubject variability includes the effect of the difference in formulations (the brand‐name vs. generic) and other unknown factors caused by the study design or trial handling. In addition, the variance related to the subject‐by‐formulation interaction may be critical for demonstrating bioequivalence in individuals. In our investigation, replicate design studies were used to study whether differences in exposure between brand‐name and generic drugs are actually due to intrasubject variability in exposure in an individual and a population. We also investigated whether pharmacokinetic parameters (i.e. AUCt and Cmax) measured after repeated administration of a brand‐name drug (R2 : R1) or a generic drug (T2 : T1) or upon switching (R : T) differ between individuals (i.e. owing to the variance related to the subject‐by‐formulation interaction). By doing so, we aimed to obtain an insight into the potential difference in drug exposure that can be seen when a patient switches between brand‐name and generic drugs.
Methods
We retrieved nine replicate design bioequivalence studies from the Dutch Regulatory Medicines Authority's database in June 2013. The use of these data has been approved for publication by the Dutch Medicine Evaluation Board (MEB). These studies were submitted to the authority in support of the registration of generic drugs, and most of these generic drugs are currently available in the Netherlands and some other European countries. Basic information about these studies and their outcomes is provided in Table 1. These studies investigated seven different active substances – namely, alendronate, atorvastatin, cyclosporin, ebastine, exemestane, mycophenolate mofetil, and ropinirole, which were from six drug classes (i.e. drugs for the treatment of bone diseases, lipid‐modifying agents, immunosuppressants, antihistamines for systemic use, endocrine therapies and anti‐parkinsonian drugs). With the exception of the study on ebastine, which had a three‐way crossover, partial replicate design study (in the sequence: R‐R‐T, R‐T‐R or T‐R‐R), all studies were designed as a four‐way crossover replicate study, in which each subject received two single treatments with the brand‐name drug (R1 and R2) and two treatments with the generic drug (T1 and T2) in the sequence: R‐T‐R‐T or T‐R‐T‐R. No carry‐over effects were identified in any of the studies.
Table 1.
Summary of the investigated replicate design bioequivalence studies (n = 9)
| Active substances | Drug classes (ATC‐5 ) | Formulation | Strength (mg) | Subjects (n) | Remarks | Study results | |||
|---|---|---|---|---|---|---|---|---|---|
| AUCt (T Ae ) | C max (R max ) | ||||||||
| Point estimate | 90% CI | Point estimate | 90% CI | ||||||
| Alendronate | Drugs for treatment of bone diseases (M05BA04) | Immediate release | 10 | 26 | Bioequivalence based on urine data under fasting conditions | 0.93 | 0.81–1.07 | 0.88 | 0.76–1.01 |
| Alendronate | Drugs for treatment of bone diseases (M05BA04) | Immediate release | 70 | 70 | Bioequivalence based on urine data under fasting conditions | 1.01 | 0.89–1.11 | 0.97 | 0.86–1.07 |
| Atorvastatin | Lipid‐modifying agents (C10AA05) | Immediate release | 40 | 63 | Bioequivalence under fasting conditions | 0.94 | 0.90–0.99 | 0.98 | 0.89–1.08 |
| Cyclosporin | Immunosuppressants (L04AD01) | Immediate release | 100 | 137 | Bioequivalence under fed conditions | 1.02 | 1.00–1.05 | 1.02 | 0.97–1.08 |
| Ebastine * | Antihistamines for systemic use (R06AX22) | Immediate release | 20 | 42 | Semi‐replicate design (R‐R‐T); bioequivalence under fasting conditions | 1.08 | 0.89–1.29 | 0.95 | 0.81–1.10 |
| Exemestane | Endocrine therapy (L02BG06) | Immediate release | 25 | 56 | Bioequivalence under fed conditions | 1.04 | 1.01–1.07 | 1.00 | 0.94–1.07 |
| Mycophenolate mofetil | Immunosuppressants (L04AA06) | Immediate release | 250 | 38 | Bioequivalence based on mycophenolic acid under fasting conditions | 0.99 | 0.97–1.02 | 1.01 | 0.92–1.10 |
| Mycophenolate mofetil | Immunosuppressants (L04AA06) | Immediate release | 500 | 42 | Bioequivalence based on mycophenolic acid under fasting conditions | 0.95 | 0.83–1.09 | 1.05 | 0.86–1.28 |
| Ropinirole | Anti‐parkinsonian drugs (N04BC04) | Extended release | 2 | 34 | Bioequivalence under fed conditions | 0.94 | 0.91–0.98 | 1.11 | 1.02–1.20 |
ATC‐5, Anatomical Therapeutic Chemical (ATC) Classification System (5 levels); AUCt, area under the concentration–time curve; CI, confidence interval; Cmax, maximum concentration; TAe, total amount of unchanged active substance excreted in the urine (for alendronate studies); Rmax, maximum rate of drug excretion (for alendronate studies).
This generic formulation was not approved, as acceptance criteria for AUCt were not met.
Total and peak drug exposure were investigated in terms of the pharmacokinetic parameters of AUCt and Cmax, with the exception of alendronate (for which drug urinary excretion was measured), where the total amount of unchanged active substance excreted in the urine (TAe) and the maximum rate of drug excretion (Rmax) were used 1.
The calculations were conducted using SPSS® (IBM, New York, U. S.) version 20 (all data were log transformed), and a Microsoft Excel spreadsheet and R program 3.0.1 were used to prepare graphs. To compare total or peak drug exposure, the ratios of variables in individual subjects between the first and second administration of the brand‐name drug (R2 : R1), the first and second administration of the generic drug (T2 : T1), and the brand‐name drug and the generic drug (T1 : R1 and T2 : R2) were calculated for AUCt (or TAe) and Cmax (or Rmax). As the AUCt and Cmax of the brand‐name drug (R) and the generic drug (T) are both log‐normally distributed, the ratios between the treatments are also log‐normally distributed. The geometric coefficients of variation of the treatment ratios (R2 : R1, T1 : R1, T2 : R2 and T2 : T1) were calculated to estimate the intrasubject variability in exposure for the two drugs, and between the brand‐name and generic drugs at a population level.
The method of moment (MM) recommended by FDA guidelines 12 and in the literature 18, 21 for investigating individual bioequivalence was used to estimate the variance related to the subject‐by‐formulation interaction (S2 D), based on the estimated intrasubject variances of generic (S2 WT) and brand‐name drug (S2 WR) and the variance of the difference between the average individual responses to the two drugs (S2 SD). The raw data of total and peak drug exposure from the studies, and the formula described by Endrenyi et al. 18 for four‐period studies were used here (e.g. ). The upper boundary of the 95% CI for S2 D was also calculated, and was subsequently used to estimate as the worst‐case scenario the probability of having an individual exposure ratio beyond the boundary of the 80–125% acceptance range upon switching from generic to brand‐name drugs, compared with the probability upon repeated administration of the same drug. In addition, difference plots were used to visualize the variation in exposure in individual subjects. For this purpose, the generic : brand‐name ratios (T1 : R1 and T2 : R2) were normalized against either the brand‐name ratio (R2 : R1) or the generic ratio (T2 : T1). Lastly, we investigated whether the variations in exposure for the brand‐name and the generic ratios were correlated.
Results
In general, bioequivalence studies found a marked individual difference in total and peak drug exposure after repeated administration of either the brand‐name or the generic drug (Figure 2 for representative example, and Supplementary Figure S1 for all investigated drugs). To illustrate this, a representative series of plasma concentration–time curves obtained for repeated administration of brand‐name and generic formulations are shown in Figure 1.
Figure 2.
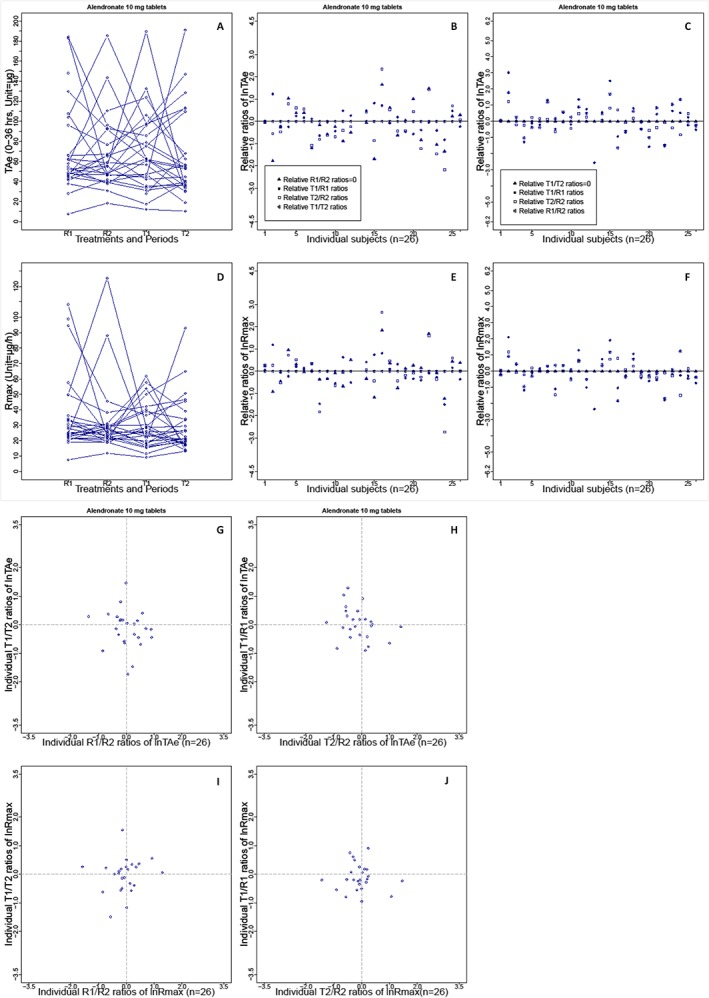
Intrasubject variability in the total amount of unchanged active substance excreted in the urine (TAe) and the maximum rate of drug excretion (Rmax) seen with brand‐name and generic formulations of alendronate (10 mg) in a replicate design bioequivalence study (n = 26). (A) Distribution of TAe in the trial population after administration of the brand‐name or generic formulation of alendronate (R1, R2, T1 or T2); lines indicate the TAe levels per subject. (B) Difference plots for the ln‐transformed treatment ratios for TAe (T1 : R1, T2 : R2 and T1 : T2), corrected by the brand‐name drug ratios (R1 : R2) in individuals. (C) Difference plots for the ratios corrected by the generic drug ratios (T1 : T2). (G) Correlation of the generic ratios (y‐axis) and the brand‐name ratios (x‐axis) for TAe on a logarithmic scale. (H) Correlation of generic : brand‐name ratios (T1 : R1 and T2 : R2) after the first and second drug administration. (D, E, F, I and J) Graphs of Rmax in the same sequence for TAe. Intrasubject variability based on the other eight bioequivalence studies is shown in Supplementary Figures S1–3
Figure 1.

Individual illustrative atorvastatin plasma concentration–time curves for the brand‐name and generic formulations in a single subject in the replicate design bioequivalence study (t = 24 h) on an arithmetic scale (A) and in a semi‐logarithmic plot (B). ‘T1’ and ’T2’ represent the first and the second administration of the generic atorvastatin drug to the subject. The plasma concentration (y‐axis) at every sampling time (x‐axis) is shown; ‘R1’ and ‘R2’ represent the first and the second administration of the brand‐name formulation of atorvastatin. The mean predose level for each formulation is indicated at time = 0
Variability in total and peak exposure at a population level
In the trial population, the distribution of subjects' AUCt and Cmax values for either brand‐name or generic drugs was reasonably comparable (for a representative distribution, see Figure 2, and Supplementary Figure S1 for all investigated drugs), with no period effect or difference in the range of drug exposure. The coefficients of variation (CVs) of individual treatment ratios (R2 : R1, T1 : R1, T2 : R2 and T2 : T1) for both AUCt and Cmax were reasonably comparable for each investigated active substance (see Table 2, with data presented on a log scale) and were not markedly different between brand‐name and generic drugs. The CVs of the AUCt ratio for the generic drugs (T2 : T1) and for the brand‐name drugs (R2 : R1) differed by less than 10%, except for alendronate 10 mg tablets, where the CV was larger for the generic drug (77%) than for the brand‐name drug (57%). The CVs for the Cmax ratio were also similar for the generic and brand‐name drugs, with the exception of atorvastatin and ropinirole, where the CVs were higher for the generic than the brand‐name drug (97% vs. 65% and 42% vs. 21%, respectively). On the other hand, for both mycophenolate studies, the CVs for generic drug were lower than for the brand‐name drug (46% vs. 56% and 46% vs. 70%, respectively).
Table 2.
Summary of individual ratios of AUC0–t and Cmax (ln‐scale) at a population level
| Active substances (Strength ) | N | Ratios (ln‐scale) | AUC 0–t | C max | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Min. | Max. | CV | Mean | Min. | Max. | CV | |||
| Alendronate * (10 mg) | 25 | R2 : R1 | 0.02 | –1.35 | 0.91 | 0.57 | –0.07 | –1.58 | 1.29 | 0.60 |
| 26 | T1 : R1 | –0.02 | –1.67 | 1.29 | 0.72 | –0.17 | –1.71 | 0.91 | 0.61 | |
| 25 | T2 : R2 | –0.12 | –1.26 | 1.41 | 0.63 | –0.08 | –1.43 | 1.46 | 0.60 | |
| 26 | T2 : T1 | –0.11 | –1.73 | 1.46 | 0.77 | –0.01 | –1.49 | 1.55 | 0.66 | |
| Alendronate * (70 mg) | 68 | R2 : R1 | –0.04 | –2.06 | 1.42 | 0.78 | –0.13 | –2.65 | 1.17 | 0.89 |
| 70 | T1 : R1 | –0.04 | –1.78 | 2.77 | 0.92 | –0.12 | –1.60 | 2.18 | 0.92 | |
| 67 | T2 : R2 | 0.03 | –1.81 | 1.41 | 0.72 | 0.04 | –1.73 | 1.57 | 0.83 | |
| 67 | T2 : T1 | 0.01 | –1.78 | 1.33 | 0.75 | 0.01 | –1.84 | 1.36 | 0.84 | |
| Atorvastatin (40 mg) | 54 | R2 : R1 | 0.10 | –0.73 | 0.92 | 0.33 | –0.06 | –1.32 | 1.42 | 0.65 |
| 63 | T1 : R1 | –0.07 | –1.05 | 0.42 | 0.28 | –0.12 | –1.56 | 2.71 | 0.71 | |
| 54 | T2 : R2 | –0.03 | –0.71 | 0.90 | 0.30 | 0.15 | –0.98 | 1.24 | 0.64 | |
| 58 | T2 : T1 | 0.13 | –0.65 | 1.48 | 0.36 | 0.22 | –3.32 | 2.52 | 0.97 | |
| Cyclosporin (100 mg) | 133 | R2 : R1 | 0.01 | –0.72 | 0.86 | 0.28 | –0.02 | –1.44 | 1.57 | 0.64 |
| 137 | T1 : R1 | 0.02 | –0.51 | 0.76 | 0.24 | 0.00 | –1.35 | 1.46 | 0.53 | |
| 133 | T2 : R2 | 0.03 | –0.55 | 0.61 | 0.24 | 0.05 | –1.50 | 1.48 | 0.58 | |
| 134 | T2 : T1 | 0.02 | –0.86 | 0.77 | 0.27 | 0.03 | –1.60 | 1.49 | 0.62 | |
| Ebastine (20 mg) | 42 | R2 : R1 | –0.28 | –1.98 | 1.91 | 0.99 | –0.09 | –1.37 | 1.40 | 0.80 |
| 42 | T1 : R1 | –0.07 | –1.80 | 1.92 | 0.88 | –0.10 | –1.40 | 1.50 | 0.70 | |
| Exemestane (25 mg) | 54 | R2 : R1 | –0.02 | –0.50 | 0.44 | 0.20 | –0.02 | –0.92 | 0.94 | 0.45 |
| 56 | T1 : R1 | 0.03 | –0.37 | 0.45 | 0.18 | –0.04 | –0.76 | 1.31 | 0.48 | |
| 54 | T2 : R2 | 0.05 | –0.32 | 0.37 | 0.15 | 0.05 | –0.99 | 0.69 | 0.38 | |
| 54 | T2 : T1 | 0.00 | –0.42 | 0.38 | 0.20 | 0.07 | –0.74 | 0.79 | 0.42 | |
| Mycophenolate mofetil (250 mg) | 37 | R2 : R1 | 0.00 | –0.36 | 0.24 | 0.15 | 0.05 | –1.03 | 1.68 | 0.56 |
| 38 | T1 : R1 | 0.02 | –0.22 | 0.33 | 0.12 | 0.03 | –1.20 | 1.38 | 0.51 | |
| 37 | T2 : R2 | –0.02 | –0.43 | 0.23 | 0.14 | –0.01 | –0.68 | 1.21 | 0.42 | |
| 37 | T2 : T1 | –0.03 | –0.48 | 0.38 | 0.17 | 0.01 | –0.86 | 0.88 | 0.46 | |
| Mycophenolate mofetil (500 mg) | 41 | R2 : R1 | 0.00 | –1.05 | 0.39 | 0.23 | 0.11 | –1.26 | 1.73 | 0.70 |
| 42 | T1 : R1 | 0.01 | –0.44 | 0.43 | 0.16 | 0.11 | –0.96 | 1.54 | 0.59 | |
| 40 | T2 : R2 | –0.03 | –0.50 | 0.87 | 0.21 | –0.09 | –1.23 | 1.04 | 0.50 | |
| 40 | T2 : T1 | –0.04 | –0.37 | 0.22 | 0.13 | ‐0.06 | –1.12 | 0.75 | 0.46 | |
| Ropinirole (2 mg) | 33 | R2 : R1 | –0.01 | –0.40 | 0.29 | 0.17 | 0.04 | –0.29 | 0.45 | 0.21 |
| 31 | T1 : R1 | –0.06 | –0.41 | 0.31 | 0.17 | 0.09 | –0.53 | 0.70 | 0.36 | |
| 31 | T2 : R2 | –0.05 | –0.43 | 0.40 | 0.16 | 0.15 | –0.59 | 0.87 | 0.41 | |
| 29 | T2 : T1 | 0.01 | –0.33 | 0.32 | 0.18 | 0.14 | –0.70 | 0.89 | 0.42 | |
AUC0–t, area under the drug concentration–time curve from time zero to the last sampling time point; Cmax, peak plasma concentration; N, number of subjects; CV, coefficient of variation.
Alendronate data are given as total of unchanged active substance excreted in the urine (TAe) and the maximum rate of drug excretion (Rmax).
Variability in total and peak exposure at an individual level
To elucidate the difference in drug exposure in individuals, the parameters of variance used for investigation of individual bioequivalence were estimated (listed on a ln‐scale in Table 3). Of note, the variances cannot be estimated for the ebastine study owing to the three‐treatment study design (R‐R‐T). The results showed that the intrasubject variance of generic drugs (on a ln‐scale) was comparable with that of brand‐name drugs, and also with the variance of switching from one to another. Further, variances related to the subject‐by‐formulation interaction for both AUCt and Cmax appeared relatively small (<0.05), and most were negatively estimated by MM, meaning that they were close to zero. The small subject‐by‐formulation interaction‐related variances indicated that the difference in drug exposure upon switching from brand‐name to generic drug was very similar to repeated administration of the same drug for every subject. The calculated upper boundary of 95% CI for the variances of the subject‐by‐formulation interaction was also fairly small for both AUCt and Cmax in all studies (<0.06, except for alendronate 70 mg, <0.16).
Table 3.
Estimations of intrasubject variances, variance due to subject‐by‐formulation interaction, and the probability of exposure ratio being beyond the boundary of the 80–125% range in individuals
| Active substances (Strength ) | Ratios | N | AUC0‐t (ln‐scale) | Cmax (ln‐scale) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Intrasubject variances | Variance of SbyF interaction (the upper boundary of 95% CI * ) | Probability of ratio beyond 80–125% (%) | Mean | Intrasubject variances | Variance of SbyF interaction (the upper boundary of 95% CI * ) | Probability of ratio beyond 80–125% (%) | |||
| Alendronate (10 mg) | R2 : R1 | 25 | 0.023 | 0.140 | –0.069 (0.033) | 67.3 | –0.069 | 0.155 | –0.042 (0.057) | 68.9 |
| T2 : T1 | 26 | –0.106 | 0.233 | 74.4 | –0.014 | 0.181 | 71.1 | |||
| T : R | 25 | –0.039 | 0.118 | 72.8 | –0.093 | 0.126 | 72.8 | |||
| Alendronate (70 mg) | R2 : R1 | 68 | –0.042 | 0.237 | 0.047 (0.158) | 74.6 | –0.135 | 0.291 | 0.021 (0.147) | 77.0 |
| T2 : T1 | 67 | 0.006 | 0.223 | 73.9 | 0.010 | 0.269 | 76.1 | |||
| T : R | 67 | 0.005 | 0.277 | 77.7 | –0.039 | 0.301 | 79.1 | |||
| Atorvastatin (40 mg) | R2 : R1 | 54 | 0.098 | 0.051 | –0.015 (0.007) | 48.7 | –0.057 | 0.176 | –0.091 (0.003) | 70.7 |
| T2 : T1 | 58 | 0.126 | 0.062 | 52.6 | 0.216 | 0.332 | 78.4 | |||
| T : R | 54 | –0.043 | 0.042 | 52.7 | 0.022 | 0.163 | 75.5 | |||
| Cyclosporin (100 mg) | R2 : R1 | 133 | 0.015 | 0.037 | –0.006 (0.003) | 41.5 | –0.019 | 0.170 | –0.026 (0.019) | 70.2 |
| T2 : T1 | 134 | 0.022 | 0.034 | 39.3 | 0.034 | 0.161 | 69.5 | |||
| T : R | 133 | 0.025 | 0.029 | 41.5 | 0.025 | 0.140 | 70.6 | |||
| Exemestane 2(25 mg) | R2 : R1 | 54 | –0.016 | 0.020 | –0.008 (–0.001) | 26.2 | –0.025 | 0.093 | –0.007 (0.032) | 60.6 |
| T2 : T1 | 54 | –0.001 | 0.020 | 26.2 | 0.069 | 0.080 | 57.7 | |||
| T : R | 54 | 0.039 | 0.011 | 26.3 | 0.006 | 0.079 | 62.2 | |||
| Mycophenolate mofetil (250 mg) | R2 : R1 | 37 | 0.001 | 0.011 | –0.003 (0.003) | 12.8 | 0.051 | 0.135 | –0.029 (0.026) | 66.8 |
| T2 : T1 | 37 | –0.032 | 0.015 | 19.2 | 0.008 | 0.095 | 60.8 | |||
| T : R | 37 | –0.007 | 0.009 | 18.3 | 0.012 | 0.086 | 65.9 | |||
| Mycophenolate mofetil (500 mg) | R2 : R1 | 41 | –0.003 | 0.026 | 0.002 (0.013) | 32.9 | 0.112 | 0.199 | –0.038 (0.030) | 72.4 |
| T2 : T1 | 40 | –0.041 | 0.009 | 9.3 | –0.061 | 0.097 | 61.3 | |||
| T : R | 40 | –0.012 | 0.020 | 32.1 | –0.014 | 0.110 | 69.6 | |||
| Ropinirole (2 mg) | R2 : R1 | 33 | –0.006 | 0.014 | 0.000 (0.010) | 18.5 | 0.042 | 0.022 | 0.009 (0.047) | 29.3 |
| T2 : T1 | 29 | 0.007 | 0.016 | 21.9 | 0.138 | 0.080 | 57.7 | |||
| T : R | 28 | –0.067 | 0.016 | 29.3 | 0.144 | 0.060 | 57.8 | |||
CI, confidence interval; SbyF interaction, subject‐by‐formulation interaction.
The upper boundary of the 95% CI for the variance of the SbyF interaction is used in the calculation of the probability of the T : R ratio beyond the boundary of the 80–125% range.
In the worst‐case scenario, in most studies, the probability of a lack of bioequivalence was similar (<10% difference) for an individual subject upon repeated administration of the brand‐name drug or switched from a brand‐name to generic drug. As the variances of the subject‐by‐formulation interaction were very small, the probability of an exposure ratio beyond the boundary of the 80–125% range was predominantly dependent on the intrasubject variability of the drugs. The difference between the formulation of generic and brand‐name drugs seems not to have contributed to the observed differences in drug exposure upon switching in individual subjects. However, for the ropinirole study, the variance in Cmax (0.08) for the generic drug was much larger than that for the brand‐name drug (0.022), which leads to the probability of the ratio beyond the boundary of the 80–125% range upon repeated administration of the generic drug (57%) being higher than that upon repeated administration of the brand‐name drug (29%). In addition, the probability of the ratio beyond the boundary of the 80–125% range upon switching from the generic to brand‐name drug (57%) was higher than that of repeated administration of the brand‐name drug. By contrast, in the mycophenolate (500 mg) study, the probability of the ratio beyond the boundary of the 80–125% range upon repeated administration of the generic drug (9%) for AUCt was much lower than that for repeated administration of the brand‐name drug (32%) because of the difference in variances (0.009 vs. 0.026).
Furthermore, the variations in drug exposure (AUCt and Cmax) between the four treatments (T1, T2, R1 and R2) were monitored. For this purpose, the generic : brand‐name ratios (T1 : R1 and T2 : R2) were corrected by subtracting the reference ratios (R2 : R1) for both AUCt and Cmax for individual subjects in each study. The graphs for the replicate study of alendronate 10 mg tablets are shown as an example in Figure 2, and the results for all studies are shown in Supplementary Figure S2A–F. The individually corrected generic : brand‐name ratios had a comparable distribution for both AUCt and Cmax. Moreover, the corrected individual generic : brand‐name ratios and the generic ratios (T2 : T1) in the study population were distributed symmetrically around the zero line for both AUCt and Cmax. Virtually identical results were obtained after correction for the observed individual ratios with T2 : T1 instead of R2 : R1 (Figure 2 for representative example, and Supplementary Figure S2G–L for all investigated studies). On the basis of these data, drug exposure appeared to vary randomly in individuals in a similar manner for the generic and the brand‐name drugs. This hypothesis is supported by our finding that the variances for the subject‐by‐formulation interaction were negligible.
Correlation between treatment ratios
A comparison of individual AUCt and Cmax ratios for generic drugs (T2 : T1) and brand‐name drugs (R2 : R1) is shown in Figure 2 for alendronate 10 mg (and in Supplementary Figure S3A–F for all investigated drugs). In all graphs, the points were randomly scattered around the zero point, forming a circular area, which shows that the ratios for generic drugs and brand‐name drugs were not correlated. This means that, in the trial population, the distribution of the individual variations in exposure to a generic drug was no different from that of a brand‐name drug. Furthermore, the possible variation in exposure in the individual subjects after repeated administration of a brand‐name drug was comparable with that of repeated administration of the generic drug, regardless of whether data for the first or second drug administration were compared (i.e.,T1 : R1 vs. T2 : R2) as shown in Figure 2H,J (and in Supplementary Figure S3G–L for all investigated studies), and the circular area formed by the individual generic : brand‐name ratios was no different from the area in the graphs for the generic and brand‐name ratios.
Discussion
In the present study, we attempted to explain why an individual's total or peak drug exposure is sometimes different when a brand‐name drug is exchanged for a generic drug. Using data from several replicate design bioequivalence studies, we found that the variances in AUCt and Cmax of individual treatment ratios (T vs. T, T vs. R and R vs. R) were similar for the seven investigated active substances. This outcome at a population level indicates that the intrasubject variability in AUCt and Cmax is comparable upon repeated administration of brand‐name drugs or of generic drugs, and upon the exchange of brand‐name and generic drugs. Therefore, in general, differences in the variability of brand‐name and generic drugs offer no valid explanation for the difference in individual drug exposure that is sometimes seen after switching from a brand‐name drug to a generic drug. Based on the results of the estimation of intrasubject variances using the individual bioequivalence approach, an exception to this conclusion may be ropinirole. In the ropinirole bioequivalence study included in our investigation, the intrasubject variance in the Cmax of the generic drug was double that of the brand‐name drug. However, the opposite situation has been seen in mycophenolate studies, where the intrasubject variance of the generic drug was much lower than that of the brand‐name drug. As the variances due to the subject‐by‐formulation interaction can be considered negligible, the variation in an individual's exposure upon switching appears to be predominantly dependent on the intrasubject variances of the drug in question. Overall, the estimations at an individual level regarding the role of intrasubject variances and subject‐by‐formulation interaction‐related variance in the difference in drug exposure between generic and brand‐name drugs were in line with our findings at a population level. This was further confirmed by the findings in the difference plots, demonstrating that the individual difference in exposure (AUCt and Cmax) between the brand‐name and generic drugs shows a random distribution within a range in a given population. In addition, based on the correlation graphs (Figure 2), the variation in exposure was similar when either the generic or brand‐name drug was given repeatedly or when one drug was switched for another.
As a retrospective analysis, the investigation was limited by the data available. We had access to data held by the Dutch Medicines Regulatory Authority for drugs investigated in replicate design studies as part of registration requirements. For the purpose of registration of generic drugs, it is most likely that applicants do not submit failed studies. Consequently, it is conceivable that only successful bioequivalence studies are available to regulatory authorities. However, in our investigation, the retrieved study for ebastine failed to demonstrate bioequivalence between the generic and the brand‐name drug. In that study, the ratio of R2 : R1 was larger than T1 : R1 for AUCt, meaning that the total exposure appeared to be more different upon repeated administration of the brand‐name drug than following a switch to the generic drug. However, repeated administration of the generic drug (T2 : T1) was not investigated in the present study. Nevertheless, for other investigated generic drugs, unidentified failed studies may exist. The impact of such potentially failed studies on our results cannot be estimated and a potential selection bias cannot be excluded. Most active substances investigated in replicate design studies are known to give rise to a highly variable peak exposure (i.e. intrasubject variability >30% for Cmax) 1, and this is why these studies are necessary for regulatory purposes. The active substances investigated covered various therapeutic areas. Although the generic drugs for epilepsy, for example, and for other reported medicines in the literature 3, 4 were not investigated, there seems to be no a priori reason to assume that a different result would be obtained for these drugs from different therapeutic areas. Similarly, although for the present study only replicate design studies for drugs with a high intrasubject variability were available, we would not expect different results for drugs with a low intrasubject variability. Moreover, for these drugs with low variability, intrasubject variations are likely to explain the major part of the differences in exposure that are obtained following switching between brand‐name and generic drugs. A further limitation is that all studies involved healthy volunteers, and so did not mimic the actual clinical situation in a patient setting. It is known that the degree of drug exposure and pharmacokinetic variability can differ between patients and healthy subjects, for multiple physiological reasons. However, the principal cause of variation in exposure after repeated administration or switching from one (brand‐name or generic) drug to another as identified in the present study – namely, intrasubject variability in exposure – although potentially increased in patients, is expected to be comparable in patients and in healthy subjects. Thus, in this sense, the findings for healthy volunteers can be extrapolated to patient populations. In addition, although single‐dose study conditions do not mimic the actual clinical situation (i.e. often involving multiple dosing to achieve a steady state), a single‐dose study is considered to be more sensitive than a multiple‐dose/steady‐state study for detecting differences in drug exposure between generic and brand‐name drugs 1. Thus, if bioequivalence is demonstrated under single‐dose conditions, the generic drug is also assumed to be bioequivalent with the brand‐name drug under steady‐state conditions. Therefore, the single‐dose replicate design studies used in the present investigation are also considered to be relevant and sensitive for exploring the issue of intrasubject variability in clinical practice.
In the literature, it is suggested that, although bioequivalence has been demonstrated at a population level, exposure following a switch to a generic drug may be different at an individual patient level, presumably because of differences between the brand‐name and generic drug 22. While we found differences in exposure (either increased or decreased) after switching drugs, we do not agree that this difference is primarily due to differences in the formulation of brand‐name and generic drugs. Instead, our results clearly indicate that the difference in exposure after switching to a generic drug is almost exclusively due to the intrasubject variability in the pharmacokinetics of the active substance, independent of the formulation used – the variation in exposure was similar whether repeated administrations of the same (brand‐name or generic) drug were given or one (brand‐name or generic) drug was switched for another, at both a population and individual level. This variation in exposure after repeated administration of the same (either brand‐name or generic) drug has not been acknowledged in the literature. In the current investigation, using the individual bioequivalence approach, the variance related to subject‐by‐formulation interaction can be considered negligible. Even in the worst‐case scenario, the subject‐by‐formulation interaction did not affect the probability of obtaining an equivalent exposure when switching from a brand‐name to generic drug, and thus this probability was comparable with that obtained upon repeated administration of the same drug. This supports one of the reasons to withdraw the FDA guideline that a potential lack of interchangeability due to subject‐by‐formulation interaction in average bioequivalence was not considered to be sufficiently substantiated. Furthermore, our findings are in line with those of a clinical cohort study involving patients with epilepsy, in which there was an increased risk of seizures when prescriptions were refilled, regardless of whether the same brand‐name drug was prescribed or a generic drug was substituted 23. In addition, our findings do not support substantial intersubject differences in the AUCt and Cmax ratios when comparing brand‐name and generic drugs (among T vs. T, T vs. R, and R vs. R), as postulated by Bialer et al. 13, but instead propose that the difference in exposure is based on a random intrasubject variation in exposure upon repeated administration of one drug (either brand‐name or generic).
In conclusion, in the investigated nine studies, the variation in total and peak drug exposure seen in individual patients after switching to a generic drug was generally comparable with the variation in exposure seen with repeated administration of a brand‐name drug. In such cases, only the intrasubject variability in the pharmacokinetics of the active substance seems to play a crucial and decisive role in the variation in drug exposure seen in individuals when switching from a brand‐name drug to a generic drug. No additional effect of the formulation of generic drugs on the variation in drug exposure was identified. Differences in exposure observed after switching from a brand‐name to a generic drug are therefore generally within the range of exposures observed upon repeated administration of the brand‐name drug. Thus, from a clinical pharmacological point of view, our data support that the benefit–risk balance of a generic drug is comparable with that of the brand‐name drug for the medicines that were included in the present investigation. Further confirmatory investigation is warranted, in a larger spectrum of drugs, in order to broaden this conclusion to other drugs.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: YY, ST, CF and MM had support from the Medicine Evaluation Board in the Netherlands; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
We thank Christine Gispen‐de Wied MD, PhD, at the Medicine Evaluation Board, for research support. No compensation was provided for her role in the study.
Contributors
YY performed the study and wrote the manuscript, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. ST was responsible for statistical analyses. CN and DB were involved in the preparation of the study and evaluation of the results. MM was involved in preparation of the study, and was responsible for supervising the project. All authors contributed to the preparation of this manuscript.
Disclaimer
The opinions in this article are only those of the authors. This article is not intended to reflect the opinion of the Medicines Evaluation Board in the Netherlands, or any of the working parties or scientific committees of the European Medicines Agency.
Supporting information
Appendix S1.
Part 1
Distribution of AUCt (TAe) and Cmax (Rmax) in the trial population after administration of the brand‐name or generic drugs (R1, R2, T1 or T2) in nine bioequivalence studies.
Appendix S1.
Part 2
Distribution of AUCt (TAe) and Cmax (Rmax) in the trial population after administration of the brand‐name or generic drugs (R1, R2, T1 or T2) in nine bioequivalence studies.
Appendix S1.
Part 3
Distribution of AUCt (TAe) and Cmax (Rmax) in the trial population after administration of the brand‐name or generic drugs (R1, R2, T1 or T2) in nine bioequivalence studies.
Appendix S2.
Part 1
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the brand‐name drug ratios (R1:R2) in individuals.
Appendix S2.
Part 2
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the brand‐name drug ratios (R1:R2) in individuals.
Appendix S2.
Part 3
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the brand‐name drug ratios (R1:R2) in individuals.
Appendix S2.
Part 4
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the generic drug ratios (T1:T2) in individuals.
Appendix S2.
Part 5
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the generic drug ratios (T1:T2) in individuals.
Appendix SII.
Part 6
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the generic drug ratios (T1:T2) in individuals.
Appendix S3.
Part 1
Correlation of the generic ratios and the brand‐name ratios for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 2
Correlation of the generic ratios and the brand‐name ratios for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 3
Correlation of the generic ratios and the brand‐name ratios for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 4
Correlation of generic:brand‐name ratios (T1:R1 and T2:R2) after the first and second drug administration for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 5
Correlation of generic:brand‐name ratios (T1:R1 and T2:R2) after the first and second drug administration for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 6
Correlation of generic:brand‐name ratios (T1:R1 and T2:R2) after the first and second drug administration for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Yu, Y. , Teerenstra, S. , Neef, C. , Burger, D. , and Maliepaard, M. (2016) A comparison of the intrasubject variation in drug exposure between generic and brand‐name drugs: a retrospective analysis of replicate design trials. Br J Clin Pharmacol, 81: 667–678. doi: 10.1111/bcp.12828.
References
- 1. Committee for Medicinal Products for Human use (CHMP), European Medicine Agency . Guideline on the investigation of bioequivalence, 2010.
- 2. US Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Guidance for industry. Bioavailability and bioequivalence. Studies for orally administered drug products – general considerations, 2003.
- 3. Robertsen I, Asberg A, Ingero AO, Vethe NT, Bremer S, Bergan S, Midtvedt K. Use of generic tacrolimus in elderly renal transplant recipients: precaution is needed. Transplantation 2014; 99: 528–32. [DOI] [PubMed] [Google Scholar]
- 4. Borgheini G. The bioequivalence and therapeutic efficacy of generic versus brand‐name psychoactive drugs. Clin Ther 2003; 25: 1578–92. [DOI] [PubMed] [Google Scholar]
- 5. Crawford P, Hall WW, Chappell B, Collings J, Stewart A. Generic prescribing for epilepsy. Is it safe? Seizure 1996; 5: 1–5. [DOI] [PubMed] [Google Scholar]
- 6. Meredith PA. Potential concerns about generic substitution: bioequivalence versus therapeutic equivalence of different amlodipine salt forms. Curr Med Res Opin 2009; 25: 2179–89. [DOI] [PubMed] [Google Scholar]
- 7. Andermann F, Duh MS, Gosselin A, Paradis PE. Compulsory generic switching of antiepileptic drugs: high switchback rates to branded compounds compared with other drug classes. Epilepsia 2007; 48: 464–9. [DOI] [PubMed] [Google Scholar]
- 8. Zachry WM III, Doan QD, Clewell JD, Smith BJ. Case‐control analysis of ambulance, emergency room, or inpatient hospital events for epilepsy and antiepileptic drug formulation changes. Epilepsia 2009; 50: 493–500. [DOI] [PubMed] [Google Scholar]
- 9. Bautista RE, Gonzales W, Jain D. Factors associated with poor seizure control and increased side effects after switching to generic antiepileptic drugs. Epilepsy Res 2011; 95: 158–67. [DOI] [PubMed] [Google Scholar]
- 10. Kesselheim AS, Stedman MR, Bubrick EJ, Gagne JJ, Misono AS, Lee JL, Brookhart MA, Avorn J, Shrank WH. Seizure outcomes following the use of generic versus brand‐name antiepileptic drugs: a systematic review and meta‐analysis. Drugs 2010; 70: 605–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kesselheim AS, Misono AS, Lee JL, Stedman MR, Brookhart MA, Choudhry NK, Shrank WH. Clinical equivalence of generic and brand‐name drugs used in cardiovascular disease: a systematic review and meta‐analysis. JAMA 2008; 300: 2514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. US Department of Health and Human Services, FDA, Center for Drug Evaluation and Research (CDER) . Guidance for industry: statistical approaches to establishing bioequivalence, 2001.
- 13. Bialer M. Generic products of antiepileptic drugs (AEDs): is it an issue? Epilepsia 2007; 48: 1825–32. [DOI] [PubMed] [Google Scholar]
- 14. US Department of Health and Human Services, FDA, Center for Drug Evaluation and Research (CDER) . Guidance for industry: average, population, and individual approaches to establishing bioequivalence, 1999.
- 15. US Department of Health and Human Services FDA, Center for Drug Evaluation and Research (CDER) . In vivo bioequivalence studies based on population and individual bioequivalence approaches, 1997.
- 16. Chow S‐C. Individual bioequivalence – a review of FDA draft guidance. Drug Inf J 1999; 33: 10. [Google Scholar]
- 17. Hsuan FC. Some statistical considerations on the FDA draft guidance for individual bioequivalence. Stat Med 2000; 19: 2879–84. [DOI] [PubMed] [Google Scholar]
- 18. Endrenyi L, Taback N, Tothfalusi L. Properties of the estimated variance component for subject‐by‐formulation interaction in studies of individual bioequivalence. Stat Med 2000; 19: 2867–78. [DOI] [PubMed] [Google Scholar]
- 19. Bialer M, Midha KK. Generic products of antiepileptic drugs: a perspective on bioequivalence and interchangeability. Epilepsia 2010; 51: 941–50. [DOI] [PubMed] [Google Scholar]
- 20. Hauck WW, Hyslop T, Chen ML, Patnaik R, Williams RL. Subject‐by‐formulation interaction in bioequivalence: conceptual and statistical issues. FDA Population/Individual Bioequivalence Working Group. Food and Drug Administration. Pharm Res 2000; 17: 375–80. [DOI] [PubMed] [Google Scholar]
- 21. Endrenyi L. A simple approach for the evaluation of individual bioequivalence. Drug Inf J 1995; 29: 847–55. [Google Scholar]
- 22. van Gelder T. Within‐patient variability in immunosuppressive drug exposure as a predictor for poor outcome after transplantation. Kidney Int 2014; 85: 1267–8. [DOI] [PubMed] [Google Scholar]
- 23. Gagne JJ, Avorn J, Shrank WH, Schneeweiss S. Refilling and switching of antiepileptic drugs and seizure‐related events. Clin Pharmacol Ther 2010; 88: 347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Part 1
Distribution of AUCt (TAe) and Cmax (Rmax) in the trial population after administration of the brand‐name or generic drugs (R1, R2, T1 or T2) in nine bioequivalence studies.
Appendix S1.
Part 2
Distribution of AUCt (TAe) and Cmax (Rmax) in the trial population after administration of the brand‐name or generic drugs (R1, R2, T1 or T2) in nine bioequivalence studies.
Appendix S1.
Part 3
Distribution of AUCt (TAe) and Cmax (Rmax) in the trial population after administration of the brand‐name or generic drugs (R1, R2, T1 or T2) in nine bioequivalence studies.
Appendix S2.
Part 1
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the brand‐name drug ratios (R1:R2) in individuals.
Appendix S2.
Part 2
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the brand‐name drug ratios (R1:R2) in individuals.
Appendix S2.
Part 3
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the brand‐name drug ratios (R1:R2) in individuals.
Appendix S2.
Part 4
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the generic drug ratios (T1:T2) in individuals.
Appendix S2.
Part 5
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the generic drug ratios (T1:T2) in individuals.
Appendix SII.
Part 6
Difference plots for the ln‐transformed treatment ratios for AUCt (TAe) and Cmax (Rmax) (T1:R1, T2:R2 and T1:T2) corrected by the generic drug ratios (T1:T2) in individuals.
Appendix S3.
Part 1
Correlation of the generic ratios and the brand‐name ratios for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 2
Correlation of the generic ratios and the brand‐name ratios for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 3
Correlation of the generic ratios and the brand‐name ratios for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 4
Correlation of generic:brand‐name ratios (T1:R1 and T2:R2) after the first and second drug administration for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 5
Correlation of generic:brand‐name ratios (T1:R1 and T2:R2) after the first and second drug administration for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Appendix S3.
Part 6
Correlation of generic:brand‐name ratios (T1:R1 and T2:R2) after the first and second drug administration for AUCt (TAe) and Cmax (Rmax) on a logarithmic scale.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item