

Abstract
Key points
Hypertension is a risk factor for sudden cardiac death caused by ventricular tachycardia and fibrillation.
Whether hypertension in its early stage is associated with an increased risk of ventricular tachyarrhythmias is not known.
Based on experiments performed at the cellular and whole heart levels, we show that, even early in chronic hypertension, the hypertrophied and fibrotic ventricles of spontaneously hypertensive rats aged 5 to 6 months have already developed increased stress‐induced arrhythmogenicity, and this increased susceptibility to ventricular arrhythmias is primarily a result of tissue remodelling rather than cellular electrophysiological changes.
Our findings highlight the need for early hypertension treatment to minimize myocardial fibrosis, ventricular hypertrophy, and arrhythmias.
Abstract
Hypertension is a risk factor for sudden cardiac death caused by ventricular tachycardia and fibrillation (VT/VF). We hypothesized that, in early hypertension, the susceptibility to stress‐induced VT/VF increases. We compared the susceptibility of 5‐ to 6‐month‐old male spontaneously hypertensive rats (SHR) and age/sex‐matched normotensive rats (NR) to VT/VF during challenge with oxidative stress (H2O2; 0.15 mmol l−1). We found that only SHR hearts exhibited left ventricular fibrosis and hypertrophy. H2O2 promoted VT in all 30 SHR but none of the NR hearts. In 33% of SHR cases, focal VT degenerated to VF within 3 s. Simultaneous voltage‐calcium optical mapping of Langendorff‐perfused SHR hearts revealed that H2O2‐induced VT/VF arose spontaneously from focal activations at the base and mid left ventricular epicardium. Microelectrode recording of SHR hearts showed that VT was initiated by early afterdepolarization (EAD)‐mediated triggered activity. However, despite the increased susceptibility of SHR hearts to VT/VF, patch clamped isolated SHR ventricular myocytes developed EADs and triggered activity to the same extent as NR ventricular myocytes, except with larger EAD amplitude. During the early stages of hypertension, when challenged with oxidative stress, SHR hearts showed an increased ventricular arrhythmogenicity that stems primarily from tissue remodelling (hypertrophy, fibrosis) rather than cellular electrophysiological changes. Our findings highlight the need for early hypertension treatment to minimize myocardial fibrosis, ventricular hypertrophy, and arrhythmias.
Key points
Hypertension is a risk factor for sudden cardiac death caused by ventricular tachycardia and fibrillation.
Whether hypertension in its early stage is associated with an increased risk of ventricular tachyarrhythmias is not known.
Based on experiments performed at the cellular and whole heart levels, we show that, even early in chronic hypertension, the hypertrophied and fibrotic ventricles of spontaneously hypertensive rats aged 5 to 6 months have already developed increased stress‐induced arrhythmogenicity, and this increased susceptibility to ventricular arrhythmias is primarily a result of tissue remodelling rather than cellular electrophysiological changes.
Our findings highlight the need for early hypertension treatment to minimize myocardial fibrosis, ventricular hypertrophy, and arrhythmias.
Abbreviations
- AP
action potential
- APD
action potential duration
- APD90
action potential at 90% duration
- CaMKII
calcium/calmodulin‐dependent protein kinase II
- CaT
calcium transient
- CaTD90
calcium transient at 90% duration
- CI
confidence interval
- DBP
diastolic blood pressure
- EAD/DAD
early/delayed after‐depolarization
- HR
heart rate
- ICC
interclass correlation
- ICa,L
L‐type calcium current
- IKs
slow delayed rectifier potassium current
- INa
sodium current
- Ito
transient outward potassium current
- IVS(d,s) interventricular septum thickness (during diastole
during systole)
- LV
left ventricle
- LVEF
left ventricular ejection fraction
- LVFS
left ventricular fractional shortening
- LVH
left ventricular hypertrophy
- LVID(d,s) left ventricular internal diameter (during diastole
during systole)
- MV
mitral valve
- NR
normotensive rats
- PA peak vel
pulmonary artery peak velocity
- (P)CL
(pacing) cycle length
- PW
posterior wall
- P‐ECG
pseudo‐electrocardiogram
- RV
right ventricle
- RWT
relative wall thickness
- SHR
spontaneously hypertensive rats
- SHHF
spontaneously hypertensive heart failure
- SBP
systolic blood pressure
- VT/VF
ventricular tachycardia and fibrillation
Introduction
Hypertension is recognized as an important and treatable contributor to cardiovascular disease, including heart failure, atherosclerosis, stroke, and sudden cardiac death (McLenachan et al. 1987; Wolf‐Maier et al. 2003). With a prevalence of 30% in the USA in 1999–2010, up to 30% of hypertensive adults were unaware of their condition (Guo et al. 2012). Moreover, a 2012 report released by the Centres for Disease Control showed the lack of effective control of blood pressure in more than half of the 67 million Americans with hypertension (Go et al. 2014). Although it is established that chronic uncontrolled hypertension eventually leads to heart failure, which increases the risk of ventricular tachycardia and fibrillation (VT/VF) (Francis, 1986; McLenachan et al. 1987; Wolf‐Maier et al. 2003), whether uncontrolled hypertension prior to the onset of severe hypertrophy and heart failure directly increases the susceptibility of the heart to VT/VF remains undefined.
Tremendous insight into the temporal progression of persistent pressure overload on cardiac structure and function has been achieved with the spontaneously hypertensive rat (SHR), a chronic hypertension model developed over 50 years ago through selective breeding of the Wistar‐Kyoto strain (Okamoto & Aoki, 1963). In SHR, systolic blood pressure (SBP) exceeding 180 mmHg emerges 3–4 months after birth (Okamoto & Aoki, 1963). Although it is recognized that, by the time that SHR reach middle age (∼1 year of age), the left ventricle (LV) has already manifested significant fibrosis and hypertrophy, contractile dysfunction and increased susceptibility to pacing‐induced VT/VF (Pfeffer et al. 1979; Pahor et al. 1991; Zaugg et al. 1997; Kapur et al. 2010; Maron, 2010), the risk of cardiac remodelling during earlier stages of uncontrolled hypertension on VT/VF, spontaneous or pacing‐induced, remains unclear. Accordingly, we studied SHR rats at 5–6 months of age, some 2–3 months after the onset of significant uncontrolled hypertension.
To assess susceptibility to VT/VF, we exposed SHR hearts to oxidative stress using H2O2. There is a growing awareness of the contribution of oxidative stress and plasma H2O2 elevation to the progression of various cardiovascular diseases, including hypertension. For signalling purposes at baseline, cells produce low steady‐state levels of H2O2 (10–20 μmol l−1) (Droge, 2002; Baek et al. 2012), which cause no appreciable oxidative injury (Rhee, 2006). Cytoplasmic peroxidases and catalases maintain steep H2O2 membrane gradients such that intracellular concentrations rarely reach >10–14% of extracellular concentrations (Antunes & Cadenas, 2000; Stone & Yang, 2006). Still, even at baseline, patients with personal or family history of hypertension produce higher plasma H2O2 levels compared to normotensive controls without genetic risk (Lacy et al. 2000). These differences in baseline H2O2 levels are further amplified during oxidative stress. In inflamed or injured tissue during an oxidative burst, cells can produce significant amounts of H2O2 (100 μmol l−1) (Droge, 2002) as rapidly as 2–6 × 10−14 mol h–1 cell–1 (Droge, 2002; Rhee, 2006) or 40 nmol min−1 g−1 (Granger, 1988). For example, within 6 h after cardiac surgery, patients with new onset postoperative atrial fibrillation have significantly more H2O2 elevation compared to patients in sinus rhythm (up to 8‐fold vs. 2‐fold increase, respectively) (Ramlawi et al. 2007). Similarly, upon reperfusion after ischaemia, rat hearts (Droge, 2002) and rat striata (Hyslop et al. 1995) produce substantial fluxes of superoxide and H2O2 (Granger, 1988). Additionally, during oxidative stress in atherosclerosis, arterial binding of oxidized low‐density lipoprotein activates macrophages, stimulating significant H2O2 production.
In the present study, we examined whether SHR hearts at 5–6 months of age were susceptible to VT/VF if exposed to H2O2 concentrations typical of a pathophysiological oxidative burst (Morita et al. 2009; Morita et al. 2011 a). We found that even 2–3 months of uncontrolled hypertension was sufficient to promote mild structural and electrical remodelling that increased susceptibility to H2O2‐induced VT/VF.
Methods
The present study was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of California Institutional Animal Care and Use Committee. Age‐ and sex‐matched male SHR and Wistar‐Kyoto normotensive rats (NR) of 5–6 months of age were euthanized by an i.v. injection of heparin sulfate (1000 U) and sodium pentobarbital (100 mg kg−1). Adequacy of anaesthesia was confirmed by the lack of pedal withdrawal reflex, corneal reflex, and motor response to pain stimuli by scalpel tip.
Echocardiography
The transthoracic echocardiographic technique and protocols used in the present study were analogous to those recommended by the European Association of Echocardiography and the American Society for Echocardiography guideline for use in humans (Lang et al. 2015) and were validated for normotensive and hypertensive rats (de Simone et al. 1990). Echocardiographic examinations were performed as described previously for rats, particularly hypertensive rats (Haas et al. 1995; Ono et al. 2002), by a single technician blinded to the type of animal. Following anaesthesia induction, rats were placed supine on an electrical heating pad at 37°C under mild anaesthesia maintenance with 1.5% isoflurane/98.5% oxygen. Two‐dimensional B‐mode, M‐mode, and pulsed‐wave Doppler images were obtained using the high‐frequency ultrasound scanner Vevo 2100 Micro‐Ultrasound (VisualSonics, Toronto, Canada) equipped with an ultra‐high frequency MS250 linear array transducer (13–24 MHz). Left ventricular internal diameter (LVID), as well as the thickness of the interventricular septum (IVS) and LV posterior wall (PW), were measured at end‐diastole and end‐systole from two‐dimensional or M‐mode tracings in short‐ and long‐axis views using the leading‐edge method, as recommended by the European Association of Echocardiography/American Society for Echocardiography guideline (Lang et al. 2015). Characterization of left ventricular hypertrophy (LVH) was achieved by evaluating the thickness of the IVS and PW during diastole (de Simone et al. 1994). Calculating the relative wall thickness (RWT) based on the formula [(PWd + IVSd)/LVIDd, where d is diastolic] (Haas et al. 1995) permitted further categorization of the LVH as either concentric (RWT >0.45) or eccentric (RWT ≤0.45) (Ganau et al. 1992; Barbieri et al. 2012; Lang et al. 2015). Because no definite qualitative classification scheme exists for rodents, to classify the severity of LVH, we modelled in accordance with the existing classification scheme for human LVH (Ono et al. 2002; Lang et al. 2015) and determined a priori an increase of septal (IVSd) thicknesses over control NR in the range of 10–30% as mild, 40–60% as moderate, and ≥70% as severe LVH. Cardiac chamber volume and function were quantified in accordance with prevalent standards for rats (Schwarz et al. 1998; Brown et al. 2002), which are analogous to those for humans (Lang et al. 2015). Once the LVID measurement was obtained, the LV volume (LV Vol), LV fractional shortening (FS) and LV ejection fraction (EF) were calculated automatically using built‐in formulas and algorithms (Folland et al. 1979) from the VisualSonics cardiac measurement software package for the Vevo 2100 imaging system (VisualSonics). Formulation for the LV Vol was based on a modified ellipsoid model (Teichholz et al. 1976):
where ‘d’ is diastolic and ‘s’ is systolic.
Pulsed‐wave Doppler was used to measure pulmonary arterial peak velocity from the long‐axis view. Mitral flow velocity patterns were obtained in the apical four‐chamber view with colour Doppler mode. Sample volume (1 mm) was placed between the tips of the mitral leaflets and adjusted to the position where velocity was maximal and the flow pattern was laminar. Velocity recordings were made with simultaneous recordings of the electrocardiogram. Mitral flow velocity patterns were analysed to determine peak early diastolic filling velocity (E velocity) and peak late diastolic filling velocity during atrial contraction (A velocity), as well as their ratio (E/A ratio).
Non‐invasive blood pressure measurements
Conscious rats were restrained in a warming chamber at 38°C (model 306; IITC Life Science, Woodland Hills, CA, USA) for 10–20 min before non‐invasive SBP measurements. An integrated sensor‐cuff occluder (B60‐1/4; IITC Life Science) stopped tail pulsation on manual inflation and detected the return of tail pulsations with each deflation cycle (Whitesall et al. 2004). The blood pressure monitor was set for desired sensitivity with maximum tail‐cuff inflation pressure set to at least 40 mmHg above the anticipated SBP. The mean SBP is an average of triplicate measurements made over a 10‐min period.
Electrophysiological studies of isolated perfused Langendorff hearts
Using the Langendorff method, isolated spontaneously‐beating rat hearts were perfused at a constant rate of 5 ml min−1 with standard normal Tyrode's solution at 37°C containing albumin (100 mg l−1) and (in mmol l−1) 125 NaCl, 5.4 KCl, 1.8 NaH2PO4, 24 NaHCO3, 1.8 CaCl2, 0.5 MgCl2 and 5.5. glucose (pH 7.4 adjusted with NaOH). Hearts were also superfused with normal Tyrode's at 37°C and allowed 20 min to equilibrate. Cytochalasin D (1 μmol l−1) was added to the arterial perfusate to suppress contraction, thereby reducing motion artefacts during optical mapping and microelectrode recording.
Hearts were stained with voltage‐ (RH‐237; 2 μmol l−1) and calcium‐sensitive (rhod 2 AM; 2 μmol l−1) fluorescent dyes for dual voltage and calcium optical mapping using a dual CMOS camera system (MiCAM Ultima; BrainVision, Tokyo, Japan). The emitted fluorescence was collected by the dual CMOS camera system using a 690‐nm long‐pass filter for RH‐237 and a 585 ± 20 nm filter for rhod 2 (Numata et al. 2012) The acquisition rate was 1 ms frame–1 and recordings made from 100 × 100 pixels with a spatial resolution of 0.35 × 0.35 mm2 per pixel covering the entire anterior left ventricular epicardial surface. A bipolar electrode was used to record pseudo‐ECG (P‐ECG) continuously during optical mapping and microelectrode recording. To simulate oxidative stress, H2O2 of 0.15 mmol l−1, consistent with reported concentrations during an oxidative burst in vivo (Droge, 2002), was added to the perfusate (Morita et al. 2009). To inhibit calcium/calmodulin‐dependent protein kinase II (CaMKII), KN‐93 (1 μmol l−1) was added to the perfusate; KN‐92 (1 μmol l−1) was used as negative control. If no VF occurred spontaneously after at least 90 min of H2O2 exposure, rapid pacing to induce VT/VF was attempted. Using electrical stimuli of 4‐ms duration and at least 2.5 times the action potential threshold, pacing was also performed to measure action potential duration (APD) and calcium transient (CaT) duration at a constant cycle length (CL).
Patch clamp studies of single myocytes
Single ventricular myocytes were freshly isolated for whole‐cell patch clamp (Bapat et al. 2012; Nguyen et al. 2012). Hearts were perfused using the standard Langendorff retrograde perfusion method at 22 ml min−1, 37°C with Ca2+‐free Tyrode's solution for 4 min to remove blood from the vessels, then with the enzyme solution for 25–27 min and, finally, with 0.05 mmol l−1‐Ca2+ Tyrode's solution to wash out the enzyme solution. The enzyme solution for myocyte isolation was a calcium‐free Tyrode's solution containing 2.0 mg ml−1 collagenase (type II; Worthington Biochemical Corp., Lakewood, NJ, USA) and 0.17 mg ml−1 protease (type XIV; Sigma, St Louis, MO, USA). Hearts were subsequently removed from the perfusion apparatus and gently agitated to dissociate the myocytes. The Ca2+ concentration was gradually increased to 1.8 mmol l−1 over 60 min. Myocytes were stored at room temperature and used within 8 h.
The EGTA‐free pipette solution for whole cell recordings contained (in mmol l−1) 110 K‐aspartate, 30 KCl, 5 NaCl, 10 Hepes, 5 MgATP, 5 creatine phosphate and 0.1 cAMP (pH 7.2 adjusted with KOH). Standard normal Tyrode's solution containing (in mmol l−1) 136 NaCl, 5.4 KCl, 0.33 NaH2PO4, 1.8 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose (pH 7.4 adjusted with NaOH) was used for myocyte isolation and for extracellular perfusion in patch clamp studies. Experiments were performed at 34–36°C (Nguyen et al. 2012). Action potentials were recorded using borosilicate glass electrodes (tip resistance 2.0–2.5 MΩ) and standard whole‐cell patch clamp methods in the current clamp mode. Action potentials were triggered using stimuli of 2‐ms duration and 2.5 times the threshold potential. Corrections were made for liquid junction potentials. Data were acquired by Axopatch 200B patch clamp amplifier, Digidata 1200 acquisition board and Clampex, version 8.0 (Axon Instruments, Inc., Foster City, CA, USA) and filtered at 2 kHz. To induce early afterdepolarizations (EADs) in ventricular myocytes, H2O2 (0.15 mmol l−1) was added to the perfusate. Electrophysiological data were analysed using Clampfit, version 9.2 (Axon Instruments, Inc.) and Origin, version 7.5 (MicroCal Inc., Northampton, MA, USA).
Quantification of tissue fibrosis, ventricular mass and myocyte dimensions
The extent of myocardial tissue fibrosis was assessed by Masson's trichrome staining of 5‐μm transverse sections of formalin‐fixed, paraffin‐embedded hearts. Ventricular mass was determined by weighing formalin‐fixed isolated LVs and right ventricles (RVs). Cellular dimensions of freshly isolated ventricular myocytes were measured using bright‐field microscopy. The isolated ventricular myocytes from both groups were imaged in the bright‐field mode using QIClick Digital CCD Camera (QImaging, Surrey, BC, Canada) installed on a Leica DM5500 B microscope (Leica Microsystems CMS GmbH, Wetzlar, Germany). Single and multiple myocyte views were acquired using 63× and 10× magnification objectives, respectively. Myocyte length, width, and top surface area were measured using Image‐Pro Premier, version 9.0 (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
We examined normal quantile plots and Shapiro–Wilk statistics to confirm that data followed a normal distribution in all cases. We also computed the interclass correlation coefficients (ICC; ratio of inter‐animal variation to total inter‐ and intra‐animal variation) for all measurements to determine whether observations of different myocytes from the same rat were independent (ICC near 0 indicates no ‘clustering’) or non‐independent (ICC near 1 indicates ‘clustering’). Because, overall, our ICCs ranged from 0 to 0.4, we demonstrated non‐independence for almost all cases and had to account for both inter‐ and intra‐animal variation. Therefore, means were compared using a ‘mixed’ analysis of variance model with both the usual random error effect and a random animal effect to take into account the non‐independence of observations of different myocytes from the same rat. The mean, SD, SEM, confidence interval (CI), and P value were based on the mixed model. P < 0.05 was considered statistically significant.
Results
In vivo studies
Non‐invasive SBP measurement confirmed hypertension at 5–6 months of age in the SHR group and normotension in the age‐ and sex‐matched NR group (185 ± 15 vs. 125 ± 10 mmHg, P < 0.01; n = 5 rats per group). Cardiac structure and function was assessed by echocardiography (n = 6 rats per group) (Fig. 1 A and Table 1). SHR hearts had preserved systolic function as indicated by the normal left ventricular ejection fraction (LVEF). However, compared to NR hearts, mild‐to‐moderate concentric LVH was present in SHR hearts as indicated by a RWT of 0.78 (RWT >0.45 denotes concentricity) and global thickening with increased interventricular septum thickness by 37% and LV posterior wall thickness by 20% (Table 1).
Figure 1. Cardiac remodelling in early hypertension of 5‐ to 6‐month‐old male SHR .
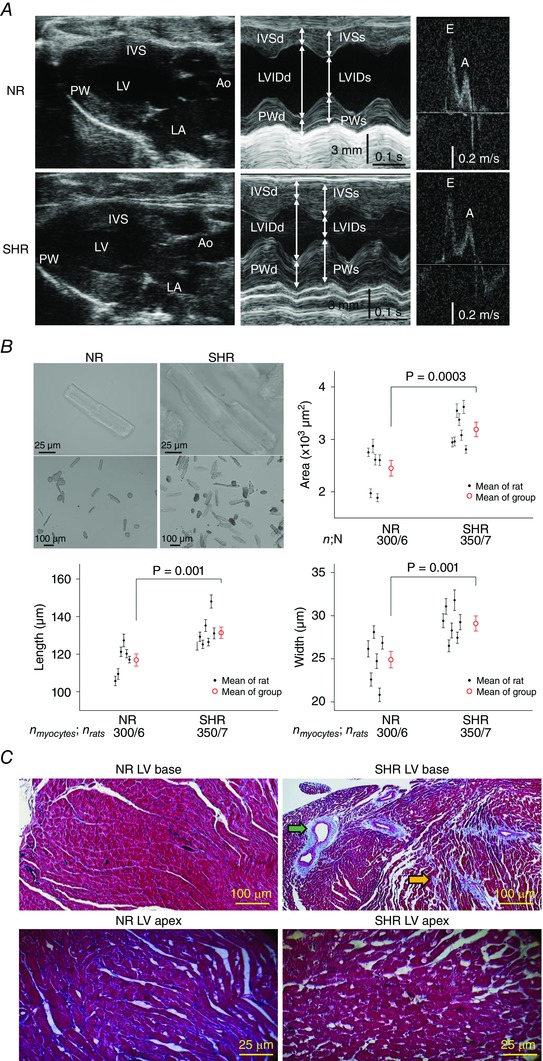
A, although both SHR and NR hearts had preserved LV systolic function, only SHR hearts manifested concentric LVH with global increased wall thicknesses seen on B‐mode (longitudinal, panel 1) and M‐mode (cross‐section, panel 2). Ao, aorta. E and A, peak velocities in early and late filling. B, SHR hypertrophy at the myocyte level is evident on bright‐field micrographs of representative myocytes and is confirmed by direct measurements of myocyte cross‐sectional area, length, and width. The mean ± SEM for each rat is represented by a small black circle with whiskers and, for each group by a large red circle with whiskers. n myocytes/n rats, number of myocytes/number of rats. C, although fibrosis is not apparent in the LV base from NR hearts (upper left) or the LV apex of both NR and SHR hearts (bottom), focal fibrosis (blue trichrome stain) is evident in the LV base from SHR hearts in the perivascular (green arrow) and interstitial (orange arrow) regions.
Table 1.
Transthoracic echocardiographic parameters
| NR (n = 6) | SHR (n = 6) | P value | Mean difference [95% CI] | |
|---|---|---|---|---|
| HR (beats min–1) | 310 ± 21 | 333 ± 48 | 0.3 | 22 [−18,58] |
| IVSd (cm)** | 1.9 ± 0.2 | 2.6 ± 0.2 | 0.001 | 0.7 [0.5,0.9] |
| IVSs (cm)** | 3.0 ± 0.1 | 3.7 ± 0.3 | 0.002 | 0.7 [0.4,0.9] |
| LVIDd (cm) | 6.7 ± 0.4 | 6.5 ± 0.9 | 0.7 | −0.2 [−0.9,0.6] |
| LVIDs (cm) | 3.8 ± 0.3 | 3.8 ± 1.1 | 0.9 | 0.0 [−0.8,0.9] |
| LVPWd (cm)* | 2.0 ± 0.3 | 2.4 ± 0.3 | 0.03 | 0.4 [0.1,0.8] |
| LVPWs (cm)* | 2.9 ± 0.2 | 3.4 ± 0.5 | 0.03 | 0.5 [0.2,0.9] |
| RWT | 0.58 ± 0.07 | 0.78 ± 0.16 | 0.02 | 0.2 [0.07,0.33] |
| LVFS (%) | 44 ± 2 | 43 ± 9 | 0.9 | −1 [−8,6] |
| LVEF (%) | 74 ± 3 | 72 ± 11 | 0.6 | −2 [−11,6] |
| MV E (mm s−1) | 652 ± 76 | 729 ± 128 | 0.2 | 77 [−29,188] |
| MV A (mm s−1) | 460 ± 106 | 477 ± 63 | 0.7 | 17 [−66,113] |
| MV E/A | 1.5 ± 0.4 | 1.5 ± 0.3 | 0.7 | 0.0 [−0.3,0.4] |
| PA peak vel (mm s−1) | 1048 ± 92 | 1080 ± 130 | 0.6 | 32 [−82,146] |
Data are reported as the mean ± SD. *P < 0.05, **P < 0.005; the bootstrap method. LV thickness was measured at the mid LV level. HR, heart rate; IVSd and IVSs, interventricular septal thickness during diastole and systole, respectively; LVIDd and LVIDs, left ventricular internal diameter during diastole and systole, respectively; LVPWd and LVPWs, left ventricular posterior wall thickness during diastole and systole, respectively; LVFS, left ventricular fractional shortening; E, early ventricular filling phase; A, ventricular filling during atrial contraction; MV E, mitral valve E velocity; MV A, mitral valve A velocity; MV E/A, mitral valve E/A ratio; PA peak vel, pulmonary artery peak velocity.
Gross pathology and histology
Despite comparable body weights between and among rats from the two groups (SHR 312 ± 20 vs. NR 300 ± 12 g; not statistically significant), SHR hearts demonstrated increased LV weight compared to NR hearts (607 ± 96 vs. 455 ± 77 mg, P < 0.03; n = 6 hearts per group). By contrast, RVs isolated from the two groups revealed similar weights (SHR 99 ± 16 vs. NR 92 ± 14 mg, P = 0.48, n = 6 hearts per group), suggesting that early hypertension resulted in hypertrophy of primarily the LV but not the RV.
To further assess ventricular hypertrophy, we compared the size of isolated ventricular myocytes from the two groups (Fig. 1 B). SHR myocytes (n = 350 myocytes from seven hearts) were significantly larger than NR myocytes (n = 300 myocytes from six hearts) in both length (131 ± 3 vs. 117 ± 3 μm, P = 0.001) and width (29 ± 0.9 vs. 25 ± 0.9 μm, P = 0.001). The increased size of the SHR ventricular myocyte was consistent with hypertrophy, rather than hyperplasia, as the explanation for the increased mass of the SHR ventricles.
Additionally, histological studies (Fig. 1 C) revealed that SHR hearts had increased fibrosis at the LV base compared to NR hearts (17.8 ± 2 vs. 2.1 ± 0.2%, P < 0.05; n = 5 hearts per group) but not at the mid LV (2.5 ± 0.1% vs. 2.2 ± 0.2%; not statistically significant) or LV apex (1.7 ± 0.1% vs. 1.2 ± 0.2%; not statistically significant). The fibrosis pattern at the LV base of SHR hearts was perivascular and interstitial, as reported previously for pressure overload conditions (Lorell & Carabello, 2000).
Ex vivo electrophysiological studies of intact hearts
Spontaneous VT/VF induced by oxidative stress
To assess susceptibility to VT/VF during oxidative stress with H2O2, we used bipolar electrodes to record P‐ECG concurrently with epicardial microelectrode recordings and/or dual voltage‐calcium optical mapping in isolated Langendorff‐perfused hearts. At baseline, hearts from both SHR and NR groups were in sinus rhythm with similar cycle lengths (SHR CL 395 ± 145 ms vs. NR CL = 430 ± 185 ms; not statistically significant).
During 90 min of H2O2 (0.15 mmol l−1) perfusion, 13% of NR hearts (n = 3 of 24 NR hearts) manifested occasional ventricular premature ventricular complexes and <3‐s runs of non‐sustained VT, although no sustained VT (>30 s) or VF was observed. In three NR hearts, increasing the H2O2 concentration to 2.0 mmol l−1 also failed to induce spontaneous VT/VF. In comparison, in all 30 SHR hearts, within only 10–17 min, H2O2 initiated spontaneous VT that arose abruptly from sinus rhythm. VT was monomorphic in 54% of cases (CL 94 ± 12 ms, 95% CI [85,110]) and polymorphic in 46% of cases (CL 62 ± 8 ms, 95% CI [45,75], P < 0.05). In 33% of cases, spontaneous VT degenerated into VF within 3 s (Table 2).
Table 2.
Incidence of spontaneous and pacing‐induced VT/VF
| Non‐sustained VT (≤30 s) | Sustained VT (>30 s) | VF | ||
|---|---|---|---|---|
| NR (n = 24 hearts) | Spontaneous | 13% (3/24) | 0% | 0% |
| Rapid pacing | 38% (9/24)* | 0% | 0% | |
| SHR (n = 30 hearts) | Spontaneous | 73% (22/30) | 27% (8/30) | 33% (10/30) |
| Rapid pacing | NA | NA | 66% (13/20)* |
Intact hearts were perfused with H2O2 to induce spontaneous VT/VF. *Rapid pacing to induce VF was attempted only on the hearts that had not manifested spontaneous VF. NA, not available.
Figure 2 A illustrates the spontaneous initiation of monomorphic VT that arose during sinus rhythm at a CL of 350 ms and degenerated to VF after eight beats. Optical APs revealed that this monomorphic VT was initiated by late phase 3 EAD‐mediated triggered activity arising from the LV base. The CL of locally recorded triggered activity averaged 86 ± 8 ms, similar to the CL of the VT on the P‐ECG (92 ± 6 ms). By contrast, the CL of the ensuing VF shortened to 48 ± 12 ms. This sequence of sinus rhythm followed by late phase 3 EAD‐mediated triggered VT at the LV base and subsequent degeneration to VF was captured by optical mapping in a total of eight SHR hearts. Figure 2 B shows four isochronal activation maps for selected beats (red *) of optical APs (traces 1–5) propagating from base to apex of the SHR LV during sinus under control condition prior to H2O2 exposure (panel 1), then during the onset of VT (panels 2 and 3) and the subsequent degeneration of VT into VF (panel 4) following H2O2 exposure. The activation map in panel 1 (Fig. 2 B) shows a sinus beat propagating under control condition from left to right (or base to apex) as the arrow indicates. The activation map in panel 2 shows the last sinus beat under H2O2 condition immediately preceding VT onset; panel 2 is similar to panel 1 except for slightly slower conduction and the presence of H2O2‐induced EAD in the corresponding optical AP traces that lengthened APD, thereby lengthening the wavelength (which is defined as the product of APD and conduction velocity and often used as a surrogate estimate of the refractory period). Panel 3 shows two foci during the onset of spontaneous VT originating from the top left and bottom right that collided and fused to propagate from left to right. Panel 4 shows at least two foci originating during the degeneration of VT into VF from the top left and bottom left, and propagating from left to right, consistent with multifocal and probably also mixed focal‐re‐entrant VF, although re‐entry was not evident in the panels illustrated.
Figure 2. Degeneration of EAD‐mediated focal VT into VF in SHR hearts exposed to H2O2 .
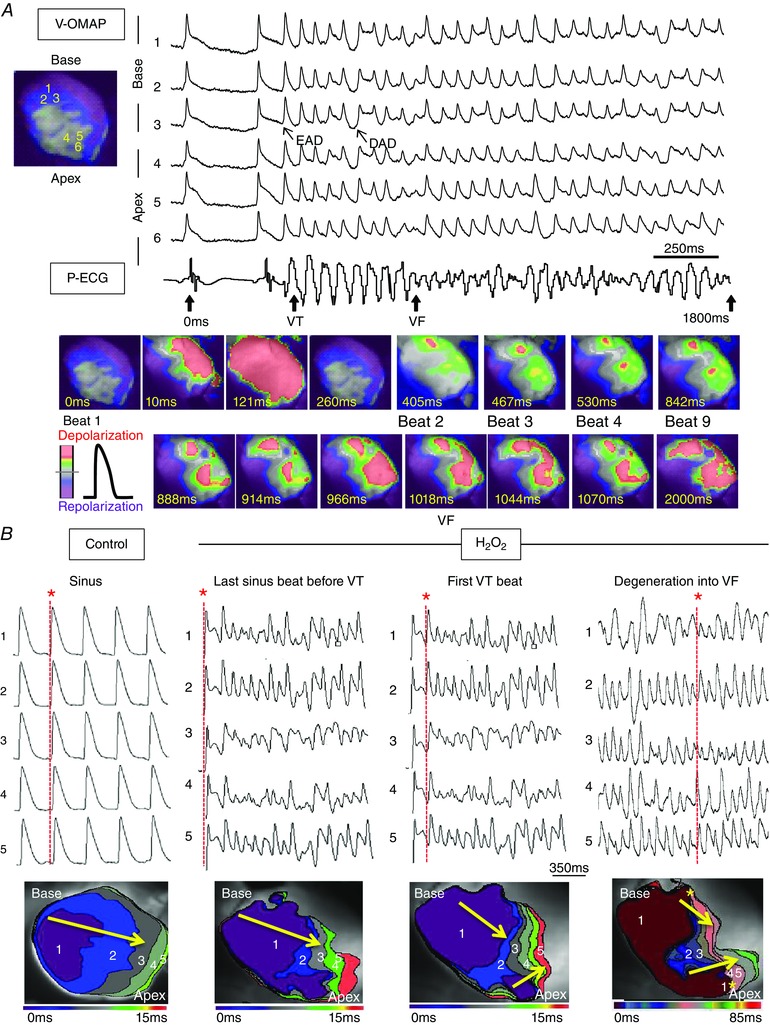
A, voltage epifluorescence optical mapping (V‐OMAP) from the LV base (AP traces 1–3) and apex (AP traces 4–6) of a representative SHR heart exposed to H2O2 with simultaneous P‐ECG recording (bottom trace) captured the onset of a spontaneous focal 8‐beat VT episode arising from prior baseline sinus rhythm. The initial five VT beats were initiated by late phase 3 EADs (APs 1–3) at the base of heart. The subsequent two VT beats (6 and 7) were DAD‐mediated triggered beats, whereas the very last VT beat (8) was another EAD‐mediated triggered beat. After this initial eight rapid triggered beats, the activation pattern degenerated into VF with multifocal irregular wavelets. The snapshots (bottom) illustrate the onset of VT and degeneration of VT into VF. B, isochronal activation maps (lower row) were constructed for selected beats (red * and red dashed lines) from optical APs propagating from base to apex (traces 1–5, upper row) during sinus under control conditions prior to H2O2 exposure (panel 1), then during the onset of VT (panels 2–3) and subsequent degeneration of VT into VF (panel 4) following H2O2 exposure. During VT degeneration into VF, base‐to‐apex epicardial activation was prolonged and multiple foci (yellow *) of conduction slowing and heterogeneous wavelengths emerged and collided, predisposing the LV to wavebreak and re‐entry.
To further explore possible EAD and afterdepolarization (DAD) mechanisms underlying VT/VF as captured by optical mapping, we performed microelectrode recording at the LV base, where most VT foci arose. In eight spontaneous VT episodes from six SHR hearts, VT was typically initiated by EAD‐mediated triggered activity (representative case in Fig. 3). In three such hearts, once VT was initiated, in addition to EADs, DADs also emerged, consistent with findings from optical mapping. However, DADs never initiated VT (Fig. 3).
Figure 3. Microelectrode recording of EAD‐mediated triggered activity degenerating into VF in SHR hearts .
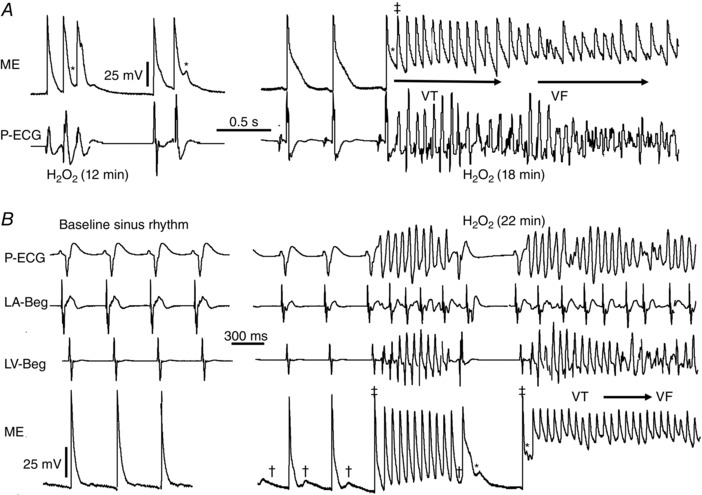
A, microelectrode (ME) with simultaneous P‐ECG recording 12 and 18 min post‐H2O2 exposure illustrates longer lasting triggered activity (‡) that degenerates to VF. B, another representative SHR heart manifests no EADs at stress‐free baseline. After 22 min of H2O2 exposure, DADs (†) emerge, followed by EAD (*)‐mediated triggered activity (‡) causing VT, then VF ensues. LA/LV‐Beg, left atrial/left ventricular bipolar electrogram.
Pacing‐induced VT/VF under oxidative stress
For the 24/24 NR and 20/30 SHR hearts that failed to manifest spontaneous VF after at least 90 min of H2O2 exposure, we investigated their susceptibility to pacing‐induced VF by rapidly pacing from the LV base at pacing cycle lengths (PCLs) of 120–250 ms. In SHR hearts, rapid pacing induced VF in 66% of cases. In NR hearts, rapid pacing induced only non‐sustained VT (≤30 s) in 38% of cases, although degeneration into VF was not observed (Table 2).
H2O2‐induced VF aborted by CaMKII inhibition
We have previously shown, in isolated rabbit ventricular myocytes, that H2O2 induces EADs through activation of CaMKII, which in turn enhances late sodium current (I Na) and L‐type calcium current (I Ca,L) by channel phosphorylation (Xie et al. 2009). Similarly, in intact SHR hearts in the present study, we found that KN‐93 (1 μmol l−1; n = 4 hearts) converted H2O2‐induced VF in three of four fibrillating hearts to sinus rhythm and, in the fourth heart, to a sustained VT that self‐terminated within 20 s. We interpret this finding to indicate that, even when CaMKII had been activated by phosphorylation, phosphatase activity was sufficient to decrease CaMKII activity significantly when rephosphorylation was prevented by KN‐93. By contrast, the inactive isoform KN‐92 (1 μmol l−1) failed to suppress H2O2‐induced VF (n = 3 SHR hearts), implicating CaMKII activation in the maintenance of H2O2‐induced VF.
APD dispersion and calcium handling dynamics
We next compared AP and intracellular CaT characteristics between SHR and NR hearts during rapid pacing at four different PCLs of 300, 350, 200 and 150 ms using simultaneous dual voltage‐calcium fluorescence optical mapping of the LV epicardium (n = 6 hearts per group), both before and after H2O2 perfusion (Table 3). In the absence of EADs and EAD‐mediated triggered activity during rapid pacing, only modest intergroup differences in action potential at 90% duration (APD90) and CaT at 90% duration (CaTD90) existed between SHR and NR hearts both before and after H2O2 exposure. In the intragroup analysis of NR hearts under both baseline and H2O2 conditions, the APD90 and CaTD90 were essentially uniform over the entire LV epicardial surface. However, in the intragroup analysis of SHR hearts at baseline, a slight APD prolongation at the LV base compared to apex was seen during PCL of 300 ms (P = 0.0001). After H2O2 exposure, the APD90 base‐to‐apex gradient was seen for other PCLs tested as well. Also, a new CaTD90 base‐to‐apex gradient developed for PCL of 300 ms. Additionally, exclusively in the SHR (but not NR) ventricles, CaT alternans was detected throughout the LV epicardium from base to apex during pacing at PCL of 150 ms (Fig. 4), as indicated by the increased ratio of minimal‐to‐maximal peak CaT amplitudes (SHR 0.6 ± 0.2 vs. NR 0.05 ± 0.02, P = 0.01, 95% CI [0.49,0.77]). Tall‐short CaT alternans was closely associated with short‐long APD alternans (Fig. 4 C), suggesting a state of increased dispersion of refractoriness, which could potentially predispose the SHR ventricles to wavebreak and initiation of re‐entry.
Table 3.
Left ventricular epicardial APD90 and CaTD90
| Baseline | H₂O₂ | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PCL | LV base | LV apex | LV base | LV apex | |||||
| (ms) | APD90 | CaTD90 | APD90 | CaTD90 | APD90 | CaTD90 | APD90 | CaTD90 | |
| 300 | NR | 112 ± 2 | 195 ± 4 | 115 ± 6 | 190 ± 3 | 113 ± 3 | 195 ± 16 | 116 ± 1 | 185 ± 15 |
| SHR | 128 ± 7***, ††† | 178 ± 21 | 108 ± 4† | 170 ± 8††† | 135 ± 14**, †† | 200 ± 2* | 107 ± 7 | 185 ± 3 | |
| 250 | NR | 98 ± 9 | 167 ± 7 | 96 ± 5 | 164 ± 8 | 100 ± 4 | 143 ± 4 | 99 ± 12 | 136 ± 12 |
| SHR | 114 ± 19 | 142 ± 7††† | 99 ± 18 | 141 ± 9†, † | 125 ± 11*, ††† | 149 ± 4† | 104 ± 19 | 149 ± 20 | |
| 200 | NR | 94 ± 8 | 123 ± 3 | 93 ± 17 | 125 ± 2 | 94 ± 9 | 125 ± 12 | 92 ± 10 | 124 ± 18 |
| SHR | 110 ± 9† | 120 ± 3 | 99 ± 15 | 121 ± 7 | 119 ± 14*, †† | 130 ± 7 | 102 ± 18 | 124 ± 7 | |
| 150 | NR | 88 ± 16 | 112 ± 9 | 87 ± 14 | 114 ± 1 | 91 ± 6 | 115 ± 7 | 88 ± 10 | 114 ± 2 |
| SHR | 100 ± 8 | 116 ± 6 | 88 ± 15 | 115 ± 6 | 115 ± 7*, ††† | 124 ± 6† | 89 ± 17 | 120 ± 11 | |
Simultaneous voltage and calcium imaging (four pairs of recordings at the LV base and four pairs at the LV apex per heart, per PCL) were performed for six NR and six SHR hearts before and after perfusion with H2O2. Data are reported as the mean ± SD. Intragroup comparison: *P < 0.05, **P < 0.005, ***P < 0.0005; intergroup comparison: † P < 0.05, †† P < 0.005, ††† P < 0.0005; the bootstrap method.
Figure 4. Rate‐dependent CaT alternans in SHR hearts .
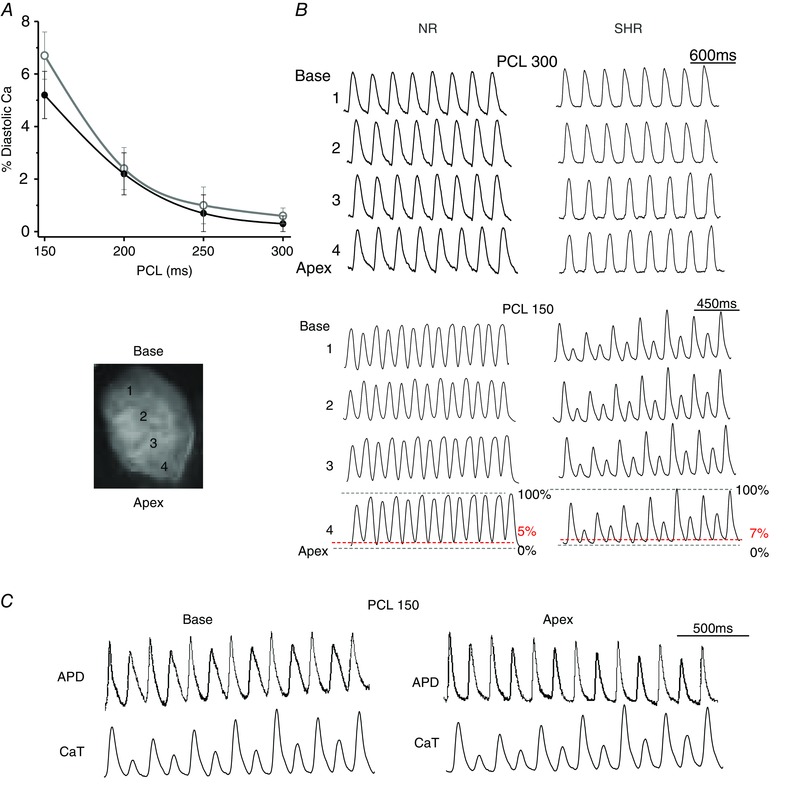
A, calcium epifluorescence optical mapping of SHR and NR hearts exposed to H2O2 (n = 5 hearts per group) during four different PCLs of between 300 and 150 ms reveal similar rate‐dependent diastolic calcium level. During PCL of 150 ms, the diastolic calcium amplitude averages 5–7% of the peak CaT in both groups. B, however, only SHR hearts manifest CaT alternans during PCL of 150 ms (bottom right). C, concurrent CaT alternans and APD alternans during PCL of 150 ms in SHR hearts, but not in NR hearts, may facilitate wavebreak and re‐entry.
By contrast, during spontaneous rhythm typically at a rate (sinus CL ≥350 ms) slower compared to the rapid pacing rate (PCL ≤300 ms), H2O2 often induced the chaotic irregular emergence of a variable number of EADs and/or EAD‐mediated triggered activity per AP, thereby causing a marked increase of APD dispersion. For example, in this representative SHR heart exposed to H2O2 (Fig. 2 A), APD was 102 ± 7 ms and maximal APD dispersion was 15 ms during sinus rhythm. However, during spontaneous VT, APD was irregularly lengthened up to 200 ± 150 ms, causing maximal APD dispersion to increase to 265 ms. During the subsequent degeneration of VT into VF, APD was irregularly lengthened to 192 ± 172 ms, causing maximal APD dispersion to further increase to 400 ms.
Patch clamp studies of isolated single ventricular myocytes
To determine whether the increased susceptibility of SHR hearts to arrhythmias during oxidative stress was also observed at the isolated myocyte level, we performed current clamp studies on LV myocytes isolated from SHR and NR hearts. After 4–6 min of exposure to H2O2 (0.15 mmol l−1), myocytes from both SHR and NR hearts readily developed EADs and repolarization failure during slow pacing (PCL from 6 to 0.5 s). As the PCL was shortened further to 0.25 s, EADs were eventually suppressed in myocytes from both SHR and NR groups to a similar extent (Fig. 5 and Table 4). Compared to NR myocytes (n = 24 myocytes from nine NR hearts), SHR myocytes (n = 22 myocytes from six SHR hearts) displayed slightly larger EAD amplitudes (SHR 39 ± 5 vs. NR 24 ± 6 mV, P = 0.02) but no significant baseline APD90 prolongation (Fig. 5 B). These findings suggest that, although there may be ion channel expression differences between myocytes from the two groups, their susceptibility to H2O2‐induced EAD formation was similar.
Figure 5. Arrhythmia triggers in patch‐clamped ventricular myocytes .
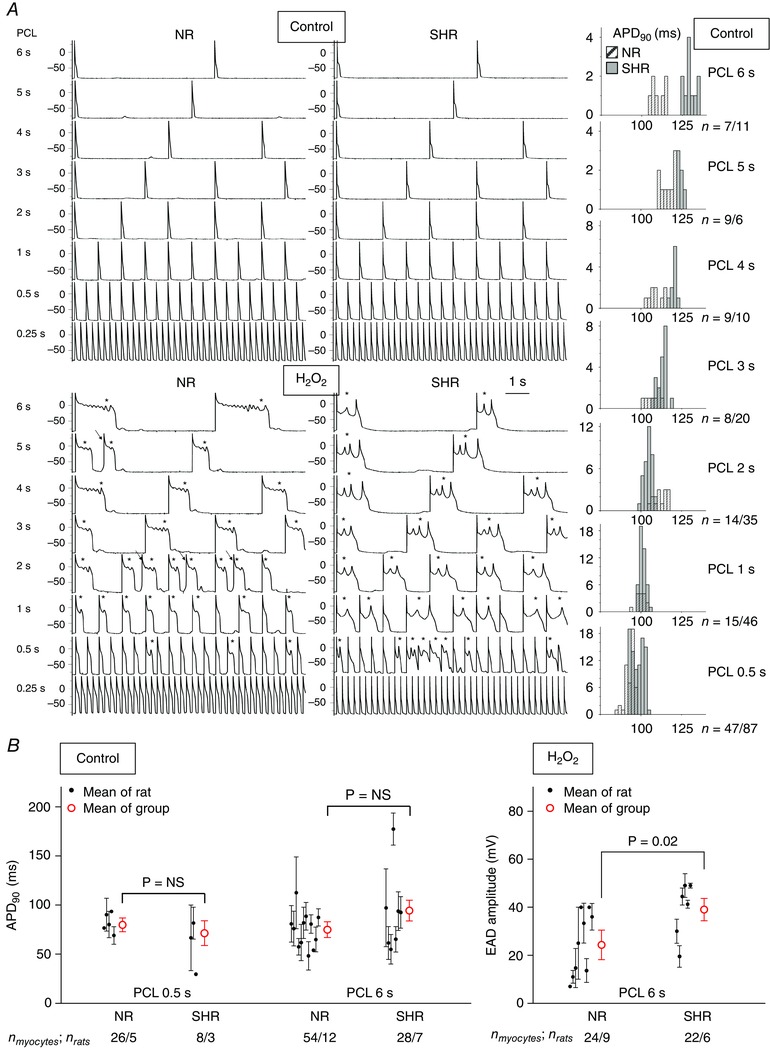
A, although even 2 mmol l−1 H2O2 cannot induce VT/VF in NR intact hearts, 0.15 mmol l−1 H2O2 induces VT/VF arrhythmia triggers, such as EADs (*) and triggered activity (arrows) in this representative NR myocyte just as readily as in the representative SHR myocyte. Compared to the NR myocyte, the SHR myocyte has slightly more prolonged APs during slow pacing (PCL ≥3 s) at baseline (upper panels and histograms) and EADs with lower frequencies and larger amplitudes during H2O2 exposure (bottom). n, number of APs from the representative NR/or SHR myocyte. B, compared to NR myocytes, SHR myocytes do not have excessively prolonged APD90 during either PCL at stress‐free baseline but have slightly larger stress‐induced EAD amplitudes during of PCL 6 s. The mean ± SEM for each rat is represented by a small black circle with whiskers, for each group by a large red circle with whiskers. n myocytes/n rats, number of myocytes/number of rats.
Table 4.
Incidence of EADs and triggered activity induced by H2O2
| Incidence of EADs (%, n) | ||||
|---|---|---|---|---|
| PCL | 6 s | 1 s | 0.5 s | 0.25 s |
| NR | 100 (25) | 86 (14) | 60 (15) | 14 (14) |
| SHR | 100 (25) | 57 (21) | 50 (16) | 6 (18) |
| P value | 1.0 | 0.1 | 0.7 | 0.6 |
| Incidence of triggered activity (%, n) | ||||
| PCL | 6 s | 1 s | 0.5 s | 0.25 s |
| NR | 60 (25) | 50 (14) | 40 (15) | 7 (14) |
| SHR | 76 (25) | 57 (21) | 31 (16) | 11 (18) |
| P value | 0.4 | 0.2 | 0.7 | 1.0 |
Summary of the incidence, or fraction of myocytes, in which EADs or triggered activity were successfully induced by H2O2 in single patch‐clamped ventricular myocytes from NR vs. SHR hearts (n): number of myocytes. The statistical significance was assessed by a two‐tailed Fisher's exact test.
Discussion
A major finding of the present study is the novel demonstration that, long before the development of overt heart failure, hearts from 5‐ to 6‐month‐old SHR have already manifested an increased susceptibility to spontaneous VT/VF mediated by oxidative stress. Moreover, the increased susceptibility to H2O2‐induced VT/VF appears to be primarily attributable to tissue (rather than cellular) electrophysiological differences between SHR and NR ventricles because isolated ventricular myocytes from SHR and NR responded similarly to oxidative stress.
Cardiac remodelling in early hypertension
The inexorable progression to ventricular failure in chronic hypertension entails costly pathophysiological alterations. In the present study, we determined that adverse structural and electrical remodelling changes have already started in early hypertension, long before any clinical signs of heart failure.
Structurally, even when biventricular systolic function was preserved, SHR LV developed interstitial myocardial fibrosis (Fig. 1 C) and, in keeping with systemic hypertensive haemodynamics, LV geometry adopted a concentric hypertrophy pattern (Fig. 1 A) with markedly elevated RWT >0.45, increased LV wall thickness (Table 1) and increased LV mass, which are all reliable prognostic markers associated with elevated peripheral resistance (Ganau et al. 1992). LVH reflected increases in ventricular myocyte length, diameter, and cross‐sectional area (Fig. 1 B), consistent with previously reported remodelling of myocyte shape prior to heart failure development in age‐matched spontaneously hypertensive heart failure (SHHF) rats, which are related to SHR (Onodera et al. 1998). When investigating the temporal development of hypertrophy in 1‐ to 12‐month‐old SHHF rats prior to the development of heart failure at ∼24 months of age, Onodera et al. (1998) found that the increase of LV myocyte length paralleled the continuous rise of systolic wall stress, although the myocyte transverse growth plateaued after reaching its upper limit by 3 months of age, such that the myocyte cross‐sectional area reached a terminal upper value of twice the upper normal range at 3 months of age without further growth thereafter; therefore, the LV was ‘robbed’ of an important adaptive mechanism to normalize wall stress.
In conjunction with structural remodelling, electrical remodelling has also begun early in hypertension long before pump failure. In the present study, when isolated NR and SHR ventricular myocytes were exposed to H2O2, both readily developed EADs during PCL ≥0.5 s (Fig. 5). However, the EADs in NR myocytes had smaller amplitudes and higher frequencies, whereas the EADs in SHR myocytes had larger amplitudes and lower frequencies, with the last EAD before full repolarization having the largest amplitude, typical of a Hopf‐homoclinic bifurcation (Tran et al. 2009; Chang et al. 2012; Qu & Chung, 2012). To the best of our knowledge, these divergent EAD behaviours are reported in the present study for the first time and suggest underlying basic differences in intracellular calcium cycling dynamics, wherein ventricular myocytes from SHR as young as 5–6 months of age have steeper slopes of I Ca,L activation and inactivation kinetics (Qu et al. 2013). Other reported differences in SHR ionic current remodelling include a reduction of transient outward potassium current (I to) current density (Li & Jiang, 2000; Goltz et al. 2007), similar to that occurring in heart failure and a reduction of inward rectifier potassium current density (Brooksby et al. 1993), which may also account for the larger amplitude of EADs. Despite distinct characteristic profiles of ionic currents contributing to EAD genesis, both SHR and NR ventricular myocytes displayed remarkably similar EAD responses to H2O2 induction, which could not per se account for the divergent arrhythmogenic vulnerability of the fibrotic SHR ventricular tissue.
Bradycardia‐dependence of EAD emergence and APD lengthening by H2O2
Arising in the setting of reduced repolarization reserve and often serving as triggers for VT/VF, EADs are membrane voltage oscillations that interrupt and retard repolarization of the cardiac action potential, resulting in the lengthening of APD (Qu & Chung, 2012). In the present study, when SHR ventricular tissue and myocytes were exposed to 150 μmol l−1 H2O2, EADs emerged during CL ≥350 ms (Figs 2 and 5 A, H2O2 panel, PCL ≥0.5 s) but were suppressed during CL ≤300 ms (Fig. 5 A, H2O2 panel, PCL of 0.25 s and Table 3), indicating that the emergence H2O2‐induced EADs is ‘bradycardia’‐related. With complete suppression of EADs and EAD‐mediated triggered activity during rapid pacing (PCL ≤300 ms), there was very little H2O2‐induced lengthening of APD (Table 3, H2O2 panel, PCL of 0.25 s), consistent with our previous observations in isolated rabbit ventricular myocytes exposed to higher doses of H2O2 (Sato et al. 2010; Nguyen et al. 2012). By contrast, during a slower rate (CL ≥350 ms), either arising spontaneously from the tissue (Fig. 2) or by slow pacing of the patch‐clamped myocytes (Fig. 5 A, H2O2 panel, PCL ≥ 0.5 s), the same H2O2 concentration (150 μmol l−1) caused significant irregular lengthening of APD with the presence of variable numbers of EADs and EAD‐mediated triggered activity such that the resultant repolarization failure caused APD to increase up to 2‐fold during VT and up to 5‐fold during VF (Fig. 2). Our findings of the bradycardia‐dependence of APD lengthening reported in the present study are consistent with prior simulated predictions (Qu & Chung, 2012) and experimental observations of significant APD lengthening (recorded by glass microelectrode) in aged rat hearts by H2O2 (100 μmol l−1) only during slow rate (spontaneous CL of 350 ms in Pezhouman et al. 2014; CL of 410 and 600 ms in Morita et al. 2011 b).
The underlying mechanisms for these bradycardia‐dependent effects of H2O2 were demonstrated by our group (Xie et al. 2009) and others (Ward & Giles, 1997; Zhao et al. 2012) to include (1) impairment of INa inactivation; (2) enhancement of the window and pedestal I Ca,L; both (1) and (2) through activation of CaMKII; and (3) enhancement of the conductance, retardation of inactivation, and acceleration of the recovery from the inactivation of the I to. These effects of H2O2 on induction of EADs and triggered activity leading to AP prolongation are eliminated by either increasing the pacing rate or inhibiting CaMKII or I to. However, it is worth noting that this rate‐dependence of EAD emergence and suppression is independent of the stress intervention used to induce EADs (Bapat et al. 2012; Pezhouman et al. 2015) and has been observed across species (Sato et al. 2010; Nguyen et al. 2012; Nguyen et al. 2015), assumed to be related, at least in part, to the delayed activation kinetics of the slow delayed rectifier potassium current (I Ks) from more deeply closed states during bradycardia, as reported previously (Zeng & Rudy, 1995; Qu et al. 2013).
The bradycardia‐dependence of EAD emergence and APD lengthening during oxidative stress may have clinical relevance in at least two respects: (1) anti‐tachycardia pacing (also known as overdrive pacing or entrainment), which has been routinely used by electrophysiologists to terminate refractory ventricular tachycardia in human patients (Llewellyn & Ramsdale, 1987) or mammalian models (Furukawa et al. 1990; Arnar et al. 2005) by taking advantage of a CL that is more rapid than the CL of the VT to minimize the APD lengthening induced by a state of reduced repolarization reserve in the vulnerable cardiac substrate and (2) the differential rate‐related fatal cardiac arrhythmias in LQT2 and LQT3 vs. LQT1 patients: sudden cardiac deaths of LQT2 and LQT3 patients are bradycardia‐related and tend to occur during sleep or at rest because repolarization reserve is reduced disproportionately during bradycardia as a result of delayed activation kinetics of I Ks from deeply closed states of the channel. By contrast, cardiac events of LQT1 patients tend to be tachycardia‐related and are often exercise‐induced because of the drastic reduction of I Ks such that repolarization reserve is reduced during tachycardia when adrenergic stimulation of I Ca,L overwhelms adrenergic stimulation of I Ks (Qu et al. 2013).
The dynamics of ventricular tachycardia
A potentially important risk factor for the increased arrhythmogenicity in SHR hearts is the regional fibrosis at the LV epicardial base (Fig. 1 C), where EAD‐mediated triggered activity most frequently, as revealed by both optical mapping and microelectrode recording (Fig. 2). Fibrosis can increase arrhythmia risk by altering tissue properties (diffusive coupling between myocytes), without otherwise altering the arrhythmogenic properties of individual cells in the tissue (Nguyen et al. 2014). This is because the loading (source‐sink) effects in well‐coupled tissue make it very difficult for a small number of myocytes with an EAD or DAD (the source) to excite adjacent myocytes without an EAD or DAD (the sink). As gap junction coupling between myocytes decreases (either as a result of gap junction remodelling or fibrosis, which equivalently eliminated gap junction coupling by interposing collagen bundles), the sink effect is markedly weakened, allowing EADs or DADs to emerge and trigger focal activity. In addition, fibrosis also slows conduction, allowing those triggers to induce re‐entry. This phenomenon has been readily demonstrated in computer simulations (Xie et al. 2010; Nguyen et al. 2012), in which the cells exhibiting the exact same EAD or DAD regime are unable to generate triggered activity in well‐coupled tissue, whereas, when gap junction coupling is reduced either directly or by simulating fibrosis, triggered activity readily emerges.
Why the SHR LV epicardial base is preferentially susceptible to fibrosis remains unknown, although it may relate to a higher wall tension at the LV base because of its greater radius of curvature compared to the LV apex (Sandler & Dodge, 1963). A previous study in 1‐year‐old SHR rats showed that the extent of fibrosis in the foci of replacement fibrosis in the LV correlated with the occurrence of VF (Pahor et al. 1991). Moreover, chronic treatment with the angiotensin‐converting enzyme inhibitor, enalapril, attenuated myocardial fibrosis and decreased VF vulnerability (Pahor et al. 1991). Besides LV fibrosis, LVH is another feature of structural remodelling that may also contributes to the vulnerability to oxidative‐stress‐induced VT/VF (Wachtell et al. 2007). Even though the underlying mechanisms for such a contribution are not precisely known, a similar correlation between LVH and sudden cardiac death was reported for patients with isolated LVH and sudden cardiac death (Tamarappoo et al. 2012).
Degeneration of ventricular tachycardia into fibrillation
In the present study, we found that, in 5‐ to 6‐month‐old SHR hearts, CaT alternans emerged readily during PCL of 150 ms (Fig. 4 B) in both the LV base and apex exclusively in SHR hearts, coinciding with APD alternans (Fig. 4 C), although, during this very rapid pacing rate, there was only mild APD lengthening at the LV base. This is an important issue because APD alternans reflects a marked increase in dispersion of refractoriness that can predisposes the ventricles to wavebreak and initiation of alternans‐induced re‐entry for VT/VF (Qu et al. 2000). The vulnerable window of re‐entry is much larger for SHR compared to NR ventricles as a result of the presence of fibrosis because the structural barriers to wave propagation that developed during fibrotic remodelling lower the threshold for repolarization alternans (Pastore & Rosenbaum, 2000; Krogh‐Madsen & Christini, 2007). Although the mechanism underlying CaT alternans resides in the disturbance of calcium signalling, APD alternans is a secondary consequence, mediated by calcium‐dependent AP modulation.
Previous simulation studies on different animal models have demonstrated that the degeneration of VT into VF is associated with the breakup of a spiral wave of the action potential in the ventricular tissue with steep APD restitution (Qu et al. 1999; Xie et al. 2014). In the present study, although rapid pacing could induce VT in NR hearts, progression to VF failed. This failure of pacing‐induced VT to degenerate into VF in the NR hearts may stem from the protective shallow APD restitution slope. By contrast, because fibrosis causes random cellular uncoupling that promotes APD dispersion and alternans (Qu et al. 2004), the fibrotic SHR ventricles were more vulnerable to re‐entry as a result of a steeper APD restitution slope. When APD restitution is shallow, such as in NR hearts, wavebreak may be induced through conduction slowing but only in regions with very large pre‐existing electrical heterogeneity and, even then, re‐entry cannot be easily induced because the wavelength (hence refractoriness) of the NR tissue in this region is long and the broken wave does not have sufficient surrounding excitable local tissue to execute a full turn. However, when APD restitution is sufficiently steep to produce alternans, such as in SHR hearts (Fig. 4 B and C), the vulnerable window of re‐entry is much larger because alternans can promote wavebreak through dynamical instability with wavelength oscillation, conduction slowing, and markedly increased dispersion of refractoriness (Qu et al. 2000). In the present study, the fact that multiple foci were perpetuating the spontaneous VF in SHR hearts (Fig. 2 B, panel 4) explains why the ongoing VF could be terminated by KN‐93, which suppressed the foci necessary for the maintenance of wavebreak and VF.
Limitations
Although our method of blood pressure measurement has the advantage of being non‐invasive, the disadvantage is that such a method precludes reliable concomitant measurement of diastolic blood pressure (DBP). Accurate assessment of DBP often requires the invasive surgical implantation of a catheter into a rat artery (e.g. a carotid artery); we did not employ such invasive method in the present study to avoid potential data confounding with the inevitable post‐surgical stress, inflammation, and associated elevated oxidant state possibly altering the SHR and NR hearts differently. However, another group of investigators recently performed direct intravascular recordings using a catheter inserted into the abdominal aorta of 4‐ to 5‐month‐old SHR and found SBP elevation of 181 ± 5 mmHg (comparable to our findings of SBP 185 ± 15 mmHg) associated with DBP elevation of 138 ± 3 mmHg (Jackson et al. 2015). It is also worth noting that pure systolic hypertension in humans predisposes to cardiovascular disease and is a harbinger of increased morbidity and mortality.
Another limitation in our methods is the use of cytochalasin D in optical mapping studies to suppress cardiac contraction and reduce motion artefacts. The well‐studied side effects of cytochalasin D on ion channels and intracellular signalling pathways reportedly develop, especially with concentrations ≥3 μmol l−1, although concentrations as high as 10 μmol l−1 were used in some studies (Hayashi et al. 2003; Nygren et al. 2006). However, we have not noted any arrhythmogenic adverse effects for NR hearts under basal or stress conditions or for SHR hearts under basal conditions with the use of 1 μmol l−1 of cytochalasin D.
As a prototype for oxidative stress, in the present study, we used H2O2 and did not test other reactive oxygen species. However, we have previously shown that, similar to H2O2, angiotensin II (which activates CaMKII and endogenous reactive oxygen species generation via NADPH oxidase) produced EAD‐mediated arrhythmias, including VT/VF, in aged fibrotic rat hearts as well as EADs and triggered activity in isolated adult rat ventricular myocytes (Bapat et al. 2012). Our group used yet a third oxidant‐generating system, the xanthine‐xanthine oxidase system, and discovered that oxidative stress‐mediated afterpotentials were not unique to H2O2 because they could also be induced by xanthine‐xanthine oxidase (Goldhaber, 1996). Additionally, although the KN‐93 experiments demonstrated that CaMKII activation is a necessary component of H2O2‐induced VT/VF, CaMKII activation alone may not be sufficient. Other effects of H2O2, such as the redox effects on proteins or the activation of other signalling pathways (e.g. guanylate cyclase), may also be important and could be tested with guanylate cyclase inhibitors in the absence of KN‐93.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
HSK and TPN conceived, designed, supervised the project, and drafted the manuscript. AAS, AP, HC, AB and NV performed electrophysiological experiments on intact perfused hearts. SI performed echocardiography. CYK performed gross pathological measurements. MCF performed histology studies. TPN performed electrophysiological experiments on isolated myocytes. TPN, AAS, AP, SI, CYK, MG, MCF and HSK analysed data. TPN, AP, SI, CYK, MCF and HSK interpreted the experimental findings. TPN, AAS, AP, SI, HC, CYK, AB, NV, MG, MCF and HSK prepared the figures. TPN, AP, SI and CYK performed statistical analyses with the invaluable assistance of Dr Jeffrey Gornbein from the Statistical/Biomathematical Consulting Clinic of the UCLA Department of Biomathematics. TPN and HSK edited and revised the manuscript with the invaluable assistance of Dr James N. Weiss. Experiments were performed in the Cardiovascular Research Laboratory at UCLA. All authors approved the manuscript final version and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the National Institutes of Health National Centre for Advancing Translational Sciences (UL1TR000124 to TPN); the Lauren B. Leichtman and Arthur E. Levine Cardiovascular Discovery Fund Investigator Award (to TPN); and the American Heart Association NCRP Scientist Development Grant (13SDG 14640095 to TPN).
Translational perspective
We show that, long before pump failure in hypertension, 5‐ to 6‐month‐old hypertensive rat hearts have already manifested increased susceptibility to both spontaneous and pacing‐induced ventricular tachycardia and fibrillation when challenged with oxidative stress. This increased susceptibility to ventricular arrhythmias is attributable to properties primarily at the tissue level (rather than cellular level), such as left ventricular hypertrophy or fibrosis. The implication of our findings, to the extent that they can be extrapolated to humans, is that uncontrolled hypertension prior to overt development of systolic dysfunction and congestive heart failure increases the risk of ventricular tachyarrhythmias. Early treatment of hypertension with the goal towards the optimization of blood pressure control and the regression of left ventricular hypertrophy may be warranted.
Acknowledgements
We thank Dr James N. Weiss for critical reading of the manuscript, Dr Jonathan Hoffman for echocardiography assistance, and Dr Jeffrey Gornbein for statistical support.
References
- Antunes F & Cadenas E (2000). Estimation of H2O2 gradients across biomembranes. FEBS Lett 475, 121–126. [DOI] [PubMed] [Google Scholar]
- Arnar DO, Xing D & Martins JB (2005). Overdrive pacing of early ischemic ventricular tachycardia: evidence for both reentry and triggered activity. Am J Physiol Heart Circ Physiol 288, H1124–H1130. [DOI] [PubMed] [Google Scholar]
- Baek JY, Han SH, Sung SH, Lee HE, Kim YM, Noh YH, Bae SH, Rhee SG & Chang TS (2012). Sulfiredoxin protein is critical for redox balance and survival of cells exposed to low steady‐state levels of H2O2 . J Biol Chem 287, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat A, Nguyen TP, Lee JH, Sovari AA, Fishbein MC, Weiss JN & Karagueuzian HS (2012). Enhanced sensitivity of aged fibrotic hearts to angiotensin II‐ and hypokalemia‐induced early afterdepolarization‐mediated ventricular arrhythmias. Am J Physiol Heart Circ Physiol 302, H2331–H2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri A, Bursi F, Mantovani F, Valenti C, Quaglia M, Berti E, Marino M & Modena MG (2012). Left ventricular hypertrophy reclassification and death: application of the Recommendation of the American Society of Echocardiography/European Association of Echocardiography. Eur Heart J Cardiovasc Imaging 13, 109–117. [DOI] [PubMed] [Google Scholar]
- Brooksby P, Levi AJ & Jones JV (1993). The electrophysiological characteristics of hypertrophied ventricular myocytes from the spontaneously hypertensive rat. J Hypertens 11, 611–622. [DOI] [PubMed] [Google Scholar]
- Brown L, Fenning A, Chan V, Loch D, Wilson K, Anderson B & Burstow D (2002). Echocardiographic assessment of cardiac structure and function in rats. Heart Lung Circ 11, 167–173. [DOI] [PubMed] [Google Scholar]
- Chang MG, Chang CY, de Lange E, Xu L, O'Rourke B, Karagueuzian HS, Tung L, Marban E, Garfinkel A, Weiss JN, Qu Z & Abraham MR (2012). Dynamics of early afterdepolarization‐mediated triggered activity in cardiac monolayers. Biophys J 102, 2706–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Simone G, Devereux RB & Wallerson DC (1994). Echocardiographic assessment of left ventricular hypertrophy in rats using a simplified approach. Am J Hypertens 7, 555–558. [DOI] [PubMed] [Google Scholar]
- de Simone G, Wallerson DC, Volpe M & Devereux RB (1990). Echocardiographic measurement of left ventricular mass and volume in normotensive and hypertensive rats. Necropsy validation. Am J Hypertens 3, 688–696. [DOI] [PubMed] [Google Scholar]
- Droge W (2002). Free radicals in the physiological control of cell function. Physiol Rev 82, 47–95. [DOI] [PubMed] [Google Scholar]
- Folland ED, Parisi AF, Moynihan PF, Jones DR, Feldman CL & Tow DE (1979). Assessment of left ventricular ejection fraction and volumes by real‐time, two‐dimensional echocardiography. A comparison of cineangiographic and radionuclide techniques. Circulation 60, 760–766. [DOI] [PubMed] [Google Scholar]
- Francis GS (1986). Development of arrhythmias in the patient with congestive heart failure: pathophysiology, prevalence and prognosis. Am J Cardiol 57, 3B‐7B. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Kimura S, Catstellanos A, Bassett AL & Myerburg RJ (1990). In vivo induction of ‘focal’ triggered ventricular arrhythmias and responses to overdrive pacing in the canine heart. Circulation 82, 549–559. [DOI] [PubMed] [Google Scholar]
- Ganau A, Devereux RB, Roman MJ, de Simone G, Pickering TG, Saba PS, Vargiu P, Simongini I & Laragh JH (1992). Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J Am Coll Cardiol 19, 1550–1558. [DOI] [PubMed] [Google Scholar]
- Go AS, Bauman M, King SM, Fonarow GC, Lawrence W, Williams KA & Sanchez E (2014). An effective approach to high blood pressure control: a science advisory from the American Heart Association, the American College of Cardiology, and the Centers for Disease Control and Prevention. Hypertension 63, 1230–1238. [DOI] [PubMed] [Google Scholar]
- Goldhaber JI (1996). Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am J Physiol Heart Circ Physiol 271, H823–H833. [DOI] [PubMed] [Google Scholar]
- Goltz D, Schultz JH, Stucke C, Wagner M, Bassalay P, Schwoerer AP, Ehmke H & Volk T (2007). Diminished Kv4.2/3 but not KChIP2 levels reduce the cardiac transient outward K+ current in spontaneously hypertensive rats. Cardiovasc Res 74, 85–95. [DOI] [PubMed] [Google Scholar]
- Granger DN (1988). Role of xanthine oxidase and granulocytes in ischemia‐reperfusion injury. Am J Physiol Heart Circ Physiol 255, H1269–H1275. [DOI] [PubMed] [Google Scholar]
- Guo F, He D, Zhang W & Walton RG (2012). Trends in prevalence, awareness, management, and control of hypertension among United States adults, 1999 to 2010. J Am Coll Cardiol 60, 599–606. [DOI] [PubMed] [Google Scholar]
- Haas GJ, McCune SA, Brown DM & Cody RJ (1995). Echocardiographic characterization of left ventricular adaptation in a genetically determined heart failure rat model. Am Heart J 130, 806–811. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Miyauchi Y, Chou CC, Karagueuzian HS, Chen PS & Lin SF (2003). Effects of cytochalasin D on electrical restitution and the dynamics of ventricular fibrillation in isolated rabbit heart. J Cardiovasc Electrophysiol 14, 1077–1084. [DOI] [PubMed] [Google Scholar]
- Hyslop PA, Zhang Z, Pearson DV & Phebus LA (1995). Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res 671, 181–186. [DOI] [PubMed] [Google Scholar]
- Jackson EK, Mi Z, Tofovic SP & Gillespie DG (2015). Effect of dipeptidyl peptidase 4 inhibition on arterial blood pressure is context dependent. Hypertension 65, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Aistrup GL, Sharma R, Kelly JE, Arora R, Zheng J, Veramasuneni M, Kadish AH, Balke CW & Wasserstrom JA (2010). Early development of intracellular calcium cycling defects in intact hearts of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 299, H1843–H1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh‐Madsen T & Christini DJ (2007). Action potential duration dispersion and alternans in simulated heterogeneous cardiac tissue with a structural barrier. Biophys J 92, 1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy F, Kailasam MT, O'Connor DT, Schmid‐Schonbein GW & Parmer RJ (2000). Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension 36, 878–884. [DOI] [PubMed] [Google Scholar]
- Lang RM, Badano LP, Mor‐Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W & Voigt JU (2015). Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging 16, 233–270. [DOI] [PubMed] [Google Scholar]
- Li X & Jiang W (2000). Electrical remodeling of membrane ionic channels of hypertrophied ventricular myocytes from spontaneously hypertensive rats. Chin Med J (Engl) 113, 584–587. [PubMed] [Google Scholar]
- Llewellyn MJ & Ramsdale DR (1987). Termination of refractory ventricular tachycardia by a combination of intravenous sotalol and overdrive ventricular pacing. Clin Cardiol 10, 416–418. [DOI] [PubMed] [Google Scholar]
- Lorell BH & Carabello BA (2000). Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 102, 470–479. [DOI] [PubMed] [Google Scholar]
- Maron BJ (2010). Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation 121, 445–456. [DOI] [PubMed] [Google Scholar]
- McLenachan JM, Henderson E, Morris KI & Dargie HJ (1987). Ventricular arrhythmias in patients with hypertensive left ventricular hypertrophy. N Engl J Med 317, 787–792. [DOI] [PubMed] [Google Scholar]
- Morita N, Lee JH, Bapat A, Fishbein MC, Mandel WJ, Chen PS, Weiss JN & Karagueuzian HS (2011. a). Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am J Physiol Heart Circ Physiol 301, H180–H191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita N, Lee JH, Xie Y, Sovari A, Qu Z, Weiss JN & Karagueuzian HS (2011. b). Suppression of re‐entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol 57, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, Lin SF, Chen PS, Xie LH, Chen F, Qu Z, Weiss JN & Karagueuzian HS (2009). Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol 297, H1594–H1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TP, Qu Z & Weiss JN (2014). Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol 70, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TP, Singh N, Xie Y, Qu Z & Weiss JN (2015). Repolarization reserve evolves dynamically during the cardiac action potential: effects of transient outward currents on early afterdepolarizations. Circ Arrhythm Electrophysiol 8, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TP, Xie Y, Garfinkel A, Qu Z & Weiss JN (2012). Arrhythmogenic consequences of myofibroblast‐myocyte coupling. Cardiovasc Res 93, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata A, Miyauchi Y, Ono N, Fishbein MC, Mandel WJ, Lin SF, Weiss JN, Chen PS & Karagueuzian HS (2012). Spontaneous atrial fibrillation initiated by tyramine in canine atria with increased sympathetic nerve sprouting. J Cardiovasc Electrophysiol 23, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren A, Baczko I & Giles WR (2006). Measurements of electrophysiological effects of components of acute ischemia in Langendorff‐perfused rat hearts using voltage‐sensitive dye mapping. J Cardiovasc Electrophysiol 17 (Suppl 1), S113‐S123. [DOI] [PubMed] [Google Scholar]
- Okamoto K & Aoki K (1963). Development of a strain of spontaneously hypertensive rats. Jpn Circ J 27, 282–293. [DOI] [PubMed] [Google Scholar]
- Ono K, Masuyama T, Yamamoto K, Doi R, Sakata Y, Nishikawa N, Mano T, Kuzuya T, Takeda H & Hori M (2002). Echo doppler assessment of left ventricular function in rats with hypertensive hypertrophy. J Am Soc Echocardiogr 15, 109–117. [DOI] [PubMed] [Google Scholar]
- Onodera T, Tamura T, Said S, McCune SA & Gerdes AM (1998). Maladaptive remodeling of cardiac myocyte shape begins long before failure in hypertension. Hypertension 32, 753–757. [DOI] [PubMed] [Google Scholar]
- Pahor M, Bernabei R, Sgadari A, Gambassi G, Jr. , Lo Giudice P, Pacifici L, Ramacci MT, Lagrasta C, Olivetti G & Carbonin P (1991). Enalapril prevents cardiac fibrosis and arrhythmias in hypertensive rats. Hypertension 18, 148–157. [DOI] [PubMed] [Google Scholar]
- Pastore JM & Rosenbaum DS (2000). Role of structural barriers in the mechanism of alternans‐induced reentry. Circ Res 87, 1157–1163. [DOI] [PubMed] [Google Scholar]
- Pezhouman A, Madahian S, Stepanyan H, Ghukasyan H, Qu Z, Belardinelli L & Karagueuzian HS (2014). Selective inhibition of late sodium current suppresses ventricular tachycardia and fibrillation in intact rat hearts. Heart Rhythm 11, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezhouman A, Singh N, Song Z, Nivala M, Eskandari A, Cao H, Bapat A, Ko CY, Nguyen T, Qu Z, Karagueuzian HS & Weiss JN (2015). Molecular basis of hypokalemia‐induced ventricular fibrillation. Circulation 132, 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer JM, Pfeffer MA, Fishbein MC & Frohlich ED (1979). Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 237, H461–H468. [DOI] [PubMed] [Google Scholar]
- Qu Z & Chung D (2012). Mechanisms and determinants of ultralong action potential duration and slow rate‐dependence in cardiac myocytes. PLoS ONE 7, e43587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Karagueuzian HS, Garfinkel A & Weiss JN (2004). Effects of Na+ channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart CircPhysiol 286, H1310–H1321. [DOI] [PubMed] [Google Scholar]
- Qu Z, Weiss JN & Garfinkel A (1999). Cardiac electrical restitution properties and stability of reentrant spiral waves: a simulation study. Am J Physiol Heart Circ Physiol 45, H269–H283. [DOI] [PubMed] [Google Scholar]
- Qu Z, Xie LH, Olcese R, Karagueuzian HS, Chen PS, Garfinkel A & Weiss JN (2013). Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res 99, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu ZL, Garfinkel A, Chen PS & Weiss JN (2000). Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation 102, 1664–1670. [DOI] [PubMed] [Google Scholar]
- Ramlawi B, Otu H, Mieno S, Boodhwani M, Sodha NR, Clements RT, Bianchi C & Sellke FW (2007). Oxidative stress and atrial fibrillation after cardiac surgery: a case‐control study. Ann Thorac Surg 84, 1166–1172. [DOI] [PubMed] [Google Scholar]
- Rhee SG (2006). Cell signaling. H2O2, a necessary evil for cell signaling. Science 312, 1882–1883. [DOI] [PubMed] [Google Scholar]
- Sandler H & Dodge HT (1963). Left ventricular tension and stress in man. Circ Res 13, 91–104. [DOI] [PubMed] [Google Scholar]
- Sato D, Xie LH, Nguyen TP, Weiss JN & Qu Z (2010). Irregularly appearing early afterdepolarizations in cardiac myocytes: random fluctuations or dynamical chaos? Biophys J 99, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz ER, Pollick C, Meehan WP & Kloner RA (1998). Evaluation of cardiac structures and function in small experimental animals: transthoracic, transesophageal, and intraventricular echocardiography to assess contractile function in rat heart. Basic Res Cardiol 93, 477–486. [DOI] [PubMed] [Google Scholar]
- Stone JR & Yang S (2006). Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal 8, 243–270. [DOI] [PubMed] [Google Scholar]
- Tamarappoo BK, John BT, Reinier K, Teodorescu C, Uy‐Evanado A, Gunson K, Jui J & Chugh SS (2012). Vulnerable myocardial interstitium in patients with isolated left ventricular hypertrophy and sudden cardiac death: a postmortem histological evaluation. J Am Heart Assoc 1, e001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichholz LE, Kreulen T, Herman MV & Gorlin R (1976). Problems in echocardiographic volume determinations: echocardiographic‐angiographic correlations in the presence of absence of asynergy. Am J Cardiol 37, 7–11. [DOI] [PubMed] [Google Scholar]
- Tran DX, Sato D, Yochelis A, Weiss JN, Garfinkel A & Qu Z (2009). Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys Rev Lett 102, 258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtell K, Okin PM, Olsen MH, Dahlof B, Devereux RB, Ibsen H, Kjeldsen SE, Lindholm LH, Nieminen MS & Thygesen K (2007). Regression of electrocardiographic left ventricular hypertrophy during antihypertensive therapy and reduction in sudden cardiac death: the LIFE Study. Circulation 116, 700–705. [DOI] [PubMed] [Google Scholar]
- Ward CA & Giles WR (1997). Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol 500(Pt 3), 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesall SE, Hoff JB, Vollmer AP & D'Alecy LG (2004). Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail‐cuff methods. Am J Physiol Heart Circ Physiol 286, H2408–H2415. [DOI] [PubMed] [Google Scholar]
- Wolf‐Maier K, Cooper RS, Banegas JR, Giampaoli S, Hense HW, Joffres M, Kastarinen M, Poulter N, Primatesta P, Rodriguez‐Artalejo F, Stegmayr B, Thamm M, Tuomilehto J, Vanuzzo D & Vescio F (2003). Hypertension prevalence and blood pressure levels in 6 European countries, Canada, and the United States. JAMA 289, 2363–2369. [DOI] [PubMed] [Google Scholar]
- Xie LH, Chen F, Karagueuzian HS & Weiss JN (2009). Oxidative stress‐induced afterdepolarizations and calmodulin kinase II signaling. Circ Res 104, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Grandi E, Bers DM & Sato D (2014). How does beta‐adrenergic signalling affect the transitions from ventricular tachycardia to ventricular fibrillation? Europace 16, 452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Sato D, Garfinkel A, Qu Z & Weiss JN (2010). So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 99, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaugg CE, Wu ST, Lee RJ, Wikman‐Coffelt J & Parmley WW (1997). Intracellular Ca2+ handling and vulnerability to ventricular fibrillation in spontaneously hypertensive rats. Hypertension 30, 461–467. [DOI] [PubMed] [Google Scholar]
- Zeng J & Rudy Y (1995). Early afterdeporizations in cardiac myocytes: mechanism and rate dependence. BiophysJ 68, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Xie Y, Wen H, Xiao D, Allen C, Fefelova N, Dun W, Boyden PA, Qu Z & Xie LH (2012). Role of the transient outward potassium current in the genesis of early afterdepolarizations in cardiac cells. Cardiovasc Res 95, 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]