

Abstract
We discuss recent evidence which suggests that the principal central respiratory chemoreceptors are located within the retrotrapezoid nucleus (RTN) and that RTN neurons are directly sensitive to [H+]. RTN neurons are glutamatergic. In vitro, their activation by [H+] requires expression of a proton‐activated G protein‐coupled receptor (GPR4) and a proton‐modulated potassium channel (TASK‐2) whose transcripts are undetectable in astrocytes and the rest of the lower brainstem respiratory network. The pH response of RTN neurons is modulated by surrounding astrocytes but genetic deletion of RTN neurons or deletion of both GPR4 and TASK‐2 virtually eliminates the central respiratory chemoreflex. Thus, although this reflex is regulated by innumerable brain pathways, it seems to operate predominantly by modulating the discharge rate of RTN neurons, and the activation of RTN neurons by hypercapnia may ultimately derive from their intrinsic pH sensitivity. RTN neurons increase lung ventilation by stimulating multiple aspects of breathing simultaneously. They stimulate breathing about equally during quiet wake and non‐rapid eye movement (REM) sleep, and to a lesser degree during REM sleep. The activity of RTN neurons is regulated by inhibitory feedback and by excitatory inputs, notably from the carotid bodies. The latter input operates during normo‐ or hypercapnia but fails to activate RTN neurons under hypocapnic conditions. RTN inhibition probably limits the degree of hyperventilation produced by hypocapnic hypoxia. RTN neurons are also activated by inputs from serotonergic neurons and hypothalamic neurons. The absence of RTN neurons probably underlies the sleep apnoea and lack of chemoreflex that characterize congenital central hypoventilation syndrome.
Abbreviations
- CCHS
congenital central hypoventilation syndrome
- cVRG
caudal ventral respiratory group (VRC segment that contains abdominal premotor neurons)
- DIA
depolarization‐induced intracellular alkalization
- GPCR
G protein‐coupled receptor
- NTS
nucleus of the solitary tract
partial pressure of CO2 in arterial blood
- preBötC
preBötzinger complex (VRC segment that contains the rhythmogenic kernel)
- REM sleep
rapid eye movement sleep
- RPG
respiratory pattern generator (network located in the ventrolateral medulla, the dorsal pons and the nucleus of the solitary tract, responsible for generating the various respiratory outflows)
- RTN
retrotrapezoid nucleus
- rVRG
rostral ventral respiratory group (VRC segment that contains phrenic premotor neurons)
- TRH
thyrotropin‐releasing hormone
- VRC
ventral respiratory column (portion of the RPG located in the ventrolateral medulla)
Introduction
The homeostatic regulation of arterial () relies on the respiratory chemoreflexes and several other neural mechanisms (for reviews see Forster et al. 2012; Nattie, 2012; Prabhakar, 2013; Guyenet, 2014). The central respiratory chemoreflex refers to the breathing stimulation elicited by increases in brain . The peripheral chemoreflex is the stimulation of breathing elicited by activation of the carotid and aortic bodies, which respond to hypoxia in a pH‐dependent manner and to select circulating factors (Kumar & Prabhakar, 2012).
Despite dedicated investigations for well over a century (Haldane & Priestley, 1905), the central respiratory chemoreflex has revealed its secrets grudgingly. We know at present that this reflex is modulated by countless brain pathways and transmitters (Nattie, 2011). However, far less is known regarding the nature and location of the proton/CO2 sensor(s) that trigger(s) this reflex. There is still disagreement as to which molecule or ion is detected (molecular CO2, protons, hydroxyl radicals, bicarbonate), further disagreement as to which types of brain cells (neurons or glia) express the cognate receptors and yet more uncertainty regarding how many brain regions contain CO2‐sensitive cells (neurons or others) capable of stimulating breathing (the definition of a central respiratory chemoreceptor). Until the early 1980s, central respiratory chemoreceptors were thought to be neurons that detect [H+], directly or indirectly, and reside at the ventral surface of the medulla oblongata (Loeschcke, 1982; Eldridge et al. 1984). From the mid‐1980s to the present time, the dominant view has been that respiratory chemoreception is an emergent property of the brain caused by widespread effects of CO2/H+ on elements within the respiratory pattern generator (RPG) and many of its inputs (Nattie, 2012; Teran et al. 2014). While the chemoreflex is undeniably regulated by a multitude of CNS neurons, recent work suggests that, as proposed by earlier investigators, central respiratory chemoreceptors encode [H+] and reside predominantly at the ventral surface of the medulla oblongata, more specifically within the retrotrapezoid nucleus (RTN) (Goridis & Brunet, 2010; Guyenet, 2014; Ruffault et al. 2015) (Fig. 1 A). The purpose of this review is to scrutinize the evidence that supports this claim.
Figure 1. Location and phenotype of RTN neurons .
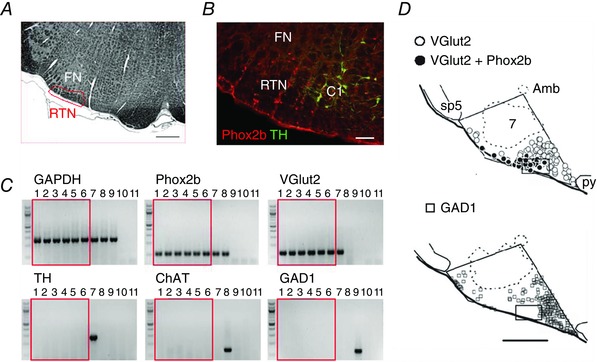
A, transverse section through the caudal end of the facial motor nucleus (FN, Sprague–Dawley adult rat; bregma −11.6 mm; myelin stain; scale 500 μm). Box showing approximate location of retrotrapezoid nucleus. B, RTN identified as Phox2b‐immunoreactive (Phox2b‐ir), non‐catecholaminergic (TH‐negative) neurons in a transverse section of medulla oblongata (bregma −11.6 mm; adult Sprague–Dawley rat; scale bar: 100 μm). The C1 (adrenergic/glutamatergic) neurons also express Phox2b (reproduced from Guyenet, 2008). C, single‐cell RT‐PCR data showing presence of Phox2b and VGlut2 transcripts in enhanced green fluorescent protein (eGFP)‐expressing neurons dissociated from the RTN of a Phox2b‐eGFP mouse (lanes 1–6), including three pH‐sensitive neurons examined after recording (lanes 4–6); the RTN neurons did not express tyrosine hydroxylase (TH), choline acetyl‐transferase (ChAT), or glutamate decarboxylase 1 (GAD1). Control experiments verified detection of the following: TH (with Phox2b and VGlut2) in a C1 neuron (lane 7); ChAT (and Phox2b) in a facial motoneuron (lane 8); and GAD1 in a striatal medium spiny cell (lane 9). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) expression was seen in all cells, and negative controls for each PCR reaction included bath solution (lane 10) and water (lane 11) substituted for cell contents (reproduced from Wang et al. 2013 b). D, plots of glutamatergic (VGlut2‐mRNA+) and GABAergic (GAD1‐mRNA+) neurons in a representative transverse section through the rat's medulla oblongata (bregma level −11.4 mm; calibration 500 μm). Box shows predominance of glutamatergic neurons in RTN. All Phox2b‐ir neurons express VGlut2 (filled circles); none contain GAD1. The Phox2b+ neurons, most of which are non‐catecholaminergic at this coronal level, are closely surrounded by Phox2b‐negative glutamatergic neurons and Phox2b‐negative GABAergic neurons (reproduced from Stornetta et al. 2006). Amb, nucleus ambiguus, compact part; sp5, spinal trigeminal tract; 7, facial motor nucleus; py, pyramidal tract.
The main issues discussed in this review include how RTN is defined anatomically and genetically, which RTN neurons sense CO2, how they encode and whether these neurons also respond to CNS hypoxia. We also examine what type of synaptic inputs RTN neurons receive, which aspects of breathing (inspiratory frequency and amplitude, airway patency, active expiration) they regulate and the state dependence of their contribution to breathing. Finally, we briefly address how the properties of RTN neurons change during development and whether the respiratory signs of the congenital central hypoventilation syndrome, a polynucleotide repeat disease that affects the development of the brainstem, are caused by the absence of RTN neurons.
The retrotrapezoid nucleus (RTN): what's in a name?
Ideally, a brain nucleus should be defined as a group of neurons that have the same developmental lineage and identical or at least closely related gene expression profiles, synaptic inputs and axonal projections. However, there are no metrics to help decide how ‘similar’ neurons should be to qualify as a single nucleus. Single‐neuron ‘transcriptomics’ may soon clarify this basic neuroanatomical question. Until then, one should simply recall that nuclei, especially in the reticular core of the brain, may have to be redefined or further subdivided as knowledge of the properties of their constituent neurons accrues.
The term RTN originally described a region of the reticular formation located roughly below the facial motor nucleus (Fig. 1 A) which contains neurons with axonal projections to more caudal regions of the ventrolateral medulla (Smith et al. 1989). It is also used to describe a subset of neurons defined by their genetic lineage and located in the same region of the brain (Ruffault et al. 2015). Finally, we use the same name to describe a subset of non‐catecholaminergic neurons located in this region that express both paired‐like homeobox 2b (Phox2b) and vesicular glutamate transporter 2 (VGlut2) (Fig. 1 A, B and C). We refer to the above three definitions as ‘anatomical’, ‘genetic’ and ‘histological’.
Histological definition of RTN
In rodents, Stornetta et al. (2006) defined RTN as a collection of neurons that express both VGlut2 and Phox2b, are devoid of catecholaminergic biosynthetic enzymes, and reside below the facial motor nucleus (Fig. 1 C and D). Most of these neurons also express TASK‐2 and GPR4 (Gestreau et al. 2010; Wang et al. 2013 a; Kumar et al. 2015). Rats have ∼2000 such neurons (Takakura et al. 2008); in mice there are ∼800, which can be easily identified using a Phox2b‐eGFP transgenic mouse strain (Lazarenko et al. 2009). Markers associated with glycinergic, GABAergic, cholinergic and serotonergic transmission are absent from the RTN neurons as defined above (Fig. 1 C) (Mulkey et al. 2004). Caudally, RTN neurons are intermingled with substantial numbers of ‘presympathetic’ barosensitive neurons (mostly catecholaminergic) (Stornetta et al. 2006) (Fig. 1 B) and rostrally they are intermixed with the A5 group of noradrenergic neurons (Burke et al. 2015 b). As shown in Fig. 1 D, RTN neurons are surrounded by GABAergic neurons and other types of glutamatergic neurons.
The following findings suggest that RTN neurons are a fairly homogeneous population. Virtually all the Phox2b+ neurons identified below the facial motor nucleus in a Phox2b‐eGFP transgenic mouse are acid activated, in slices (95%) or after complete isolation (80%) (Lazarenko et al. 2009; Wang et al. 2013 b). In rats, histologically defined RTN neurons innervate only four regions of the brain, all of which regulate breathing (Abbott et al. 2009; Bochorishvili et al. 2012) (Fig. 2 B). RTN neurons are also uniformly activated by serotonin, thyrotropin‐releasing hormone (TRH), substance P and orexin (Mulkey et al. 2007; Lazarenko et al. 2011).
Figure 2. Inputs and outputs of RTN neurons .
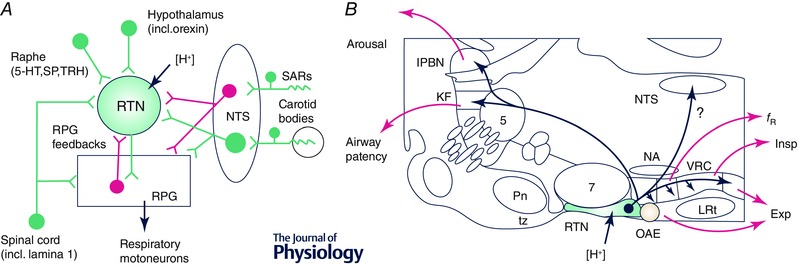
A, main connections of RTN neurons (green, excitatory; magenta inhibitory). The RPG includes the ventral respiratory column (VRC) and the dorsolateral pons. The monosynaptic nature of the RTN inputs shown in this figure has not yet been confirmed by ultrastructural evidence. B, parasagittal section through the rat's brain (∼1.8 mm lateral to the midline) illustrating the location of RTN neurons, their axonal projections and the putative function of each projection. Abbreviations: 5‐HT, serotonin; Exp, active (abdominal) expiration; f R, breathing frequency; Insp, inspiration; KF, Kölliker–Fuse nucleus; lPBN, lateral parabrachial nuclei; LRt, lateral reticular nucleus; NA, nucleus ambiguus; NTS, nucleus of the solitary tract; OAE, oscillator for active expiration; Pn, pontine nuclei; RPG, respiratory pattern generator; SARs, stretch‐activated receptors; SP, substance P; TRH, thyrotropin‐releasing hormone; tz, trapezoid body; VRC, ventral respiratory column (incl. its four subdivisions, from rostral to caudal: Bötzinger, preBötzinger complex, rVRG and cVRG).
Some evidence for heterogeneity of the neuronal population defined here as RTN also exists. For example, a subgroup (∼45%) of RTN neurons express galanin in rats (Stornetta et al. 2009). Although all RTN neurons examined so far have some type of respiratory modulation (Fig. 3 C), this modulation comes in several patterns suggesting that RTN neurons receive inputs from several types of respiratory neurons (Guyenet et al. 2005). It has also been suggested that a subtype of RTN neuron might contribute selectively to active (abdominal) expiration (Abdala et al. 2009 b; Molkov et al. 2010). However, evidence that these late‐expiratory neurons possess the RTN phenotype (genetic and/or histological) is yet to be produced. Finally, as discussed later, the relative level of TASK‐2 and GPR4 transcripts varies between RTN neurons (Wang et al. 2013 a; Kumar et al. 2015).
Figure 3. RTN neurons encode in vivo .
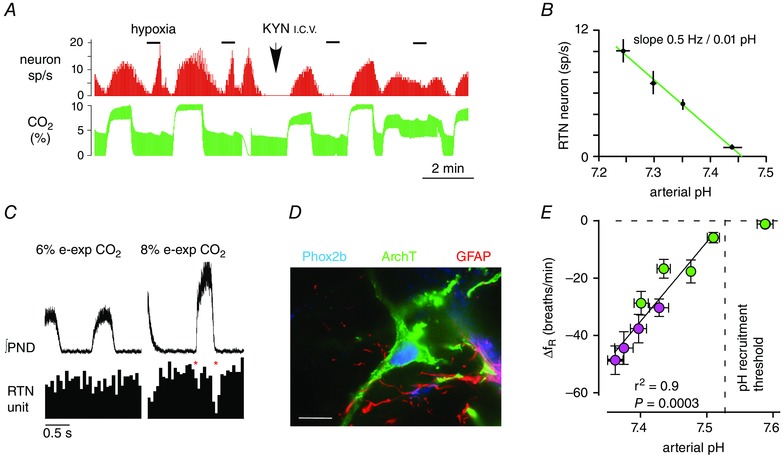
A, single RTN neuron recorded in a halothane‐anaesthetized vagotomized rat. The neuron is activated by elevated CO2 (hypercapnia) and by hypoxia (sp/s, spikes s−1). The effect of hypoxia is selectively eliminated by administration of kynurenic acid (KYN, i.c.v.) to block glutamate transmission in the brainstem (reproduced from Mulkey et al. 2004). B, relationship between the discharge rate of RTN neurons (n = 12) and arterial pH in halothane‐anaesthetized rats in which glutamatergic transmission was impaired (by KYN, i.c.v.). C, respiratory modulation of an RTN neuron in a vagotomized halothane‐anaesthetized rat. The respiratory modulation was enhanced by hypercapnia (right panel). This particular neuron exhibits two periods of reduced firing probability per breathing cycle, one at the onset of inspiration, the other during post‐inspiration (asterisks). Top trace, averaged rectified phrenic nerve discharge (∫PND; 100 sweeps triggered on PND upstroke). Bottom trace: event‐triggered histogram of neuron action potentials (50 ms bins) (redrawn from Guyenet et al. 2005). D, example of one RTN neuron transduced with archaerhodopsin from Halorubrum strain TP009, version 3.0 (ArchT3.0) (astrocytes, identified by the presence of glial fibrillary acid protein, GFAP, do not express the opsin). E, the reduction of breathing frequency caused by bilateral opto‐inhibition of RTN neurons plotted as a function of arterial pH. Bilateral inhibition of archaerhodopsin (ArchT3.0)‐transduced RTN neurons decreases breathing frequency in proportion to plasma pH in conscious rats. Plasma pH was manipulated by lowering or increasing (green symbols) or by combining various degrees of respiratory alkalosis on a background of metabolic acidosis by administering acetazolamide (pink symbols). Adapted from Basting et al. (2015).
Genetic definition of RTN
The name RTN has also been used in mice to define a group of neurons whose precursors originate from rhombomeres 3 and or 5 (Egr‐2‐dependent lineage) and whose differentiation uniquely depends on the co‐expression of transcription factors Phox2b and Atoh‐1 (Dubreuil et al. 2008; Goridis & Brunet, 2010; Ramanantsoa et al. 2011; Ruffault et al. 2015). Genetically defined RTN neurons (Atoh1+/Phox2b+) express VGlut2, reside predominantly but not exclusively under the facial motor nucleus, are acid sensitive, non‐catecholaminergic, express NK1 receptors, mediate the bulk of the chemoreflex in neonates and their number, in mice, has been estimated at around 600–800 in the late embryonic period, a figure that is very close to that of the VGlut2+/Phox2b+ non‐catecholaminergic neurons, defined as RTN in a Phox2b‐eGFP mouse (Lazarenko et al. 2009; Thoby‐Brisson et al. 2009; Ramanantsoa et al. 2011; Ruffault et al. 2015). Finally, both genetically and histologically defined RTN neurons activate breathing by releasing glutamate (Holloway et al. 2015; Ruffault et al. 2015).
Summary and uncertainties
The RTN was first defined anatomically as a segment of the reticular formation located under the facial motor nucleus, then histologically (non‐catecholaminergic VGlut2+/Phox2b+ neurons located within the same region) and finally genetically (one of only two clusters of lower brainstem neurons whose development requires co‐expression of Atoh‐1 and Phox2b; Ruffault et al. 2015). The first definition refers to an area of the reticular formation that contains numerous cell types, particularly towards its caudal, medial and lateral borders (Fig. 1 D). The histological definition is much more restrictive, but this population of neurons is not totally homogeneous; individual cells contain variable levels of several transcripts (TASK‐2, GPR4 and galanin) and the neurons display differences in synaptic inputs (respiratory modulation). There appears to be substantial overlap between genetically and histologically defined RTN neurons in mice but no direct evidence that these two populations are strictly identical. Finally, the existence of several functional subclasses of genetically or histologically defined RTN neurons is a distinct possibility.
RTN neurons drive the RPG by releasing glutamate
Lesioning or inhibiting the RTN region reduces the phrenic nerve discharge at all levels of in anaesthetized cats or rats whereas activation of this brain region, e.g. with glutamate, increases breathing (St John et al. 1989; Nattie et al. 1991; Nattie & Li, 1994). These early results demonstrated that the RTN region (anatomical definition) contains neurons whose activation drives the RPG. The following evidence indicates that the non‐catecholaminergic, Phox2b+ neurons in the RTN (the putative central respiratory chemoreceptors) drive breathing by releasing glutamate.
First, RTN neurons contain VGlut2 mRNA (Fig. 1 C), a diagnostic marker of glutamatergic transmission, and their terminals contain VGlut2 immunoreactivity (Mulkey et al. 2004; Stornetta et al. 2006; Dubreuil et al. 2008; Bochorishvili et al. 2012; Ruffault et al. 2015). Second, optogenetic activation of these neurons increases breathing in both anaesthetized and conscious rodents (rats and mice) (Abbott et al. 2009, 2011). In these experiments, RTN neurons were transduced to express channelrhodopsin‐2(H134R) (ChR2) using a lentiviral vector featuring the PRSx8 promoter (Hwang et al. 2001). PRSx8 is activated by Phox2b and drives transgene expression in both RTN and neighbouring C1 catecholaminergic neurons when the lentiviral vector is accurately delivered below the facial motor nucleus (Abbott et al. 2009, 2011). Destroying C1 catecholaminergic neurons selectively (in rats) has little effect on the breathing stimulation elicited by optogenetically activating the Phox2b+/VGlut2+ neurons located under the facial motonucleus (Abbott et al. 2009; Burke et al. 2015 b). Third, in resting conscious rats, optogenetic inhibition of the same RTN neurons using the archaerhodopsin variant ArchT3.0 (Chow et al. 2010) inhibits breathing in direct proportion to arterial pH below a pH threshold of 7.53 (Fig. 3 E) (Basting et al. 2015). Complete apnoea can be produced by bilateral opto‐inhibition of RTN neurons in rats in which the carotid bodies are functionally inactivated by inhalation of pure O2 (Burke et al. 2015 a). Fourth, after VGlut2 is deleted from RTN neurons, optogenetic activation of these neurons no longer stimulates breathing, including in adult conscious mice (Holloway et al. 2015; Ruffault et al. 2015). Finally, RTN terminals make asymmetric synapses consistent with glutamatergic transmission (Bochorishvili et al. 2012).
In summary, stimulation of RTN neurons activates breathing; inhibition has the opposite effect. RTN neurons synthesize VGlut2 and, in the absence of this vesicular transporter, their activation no longer stimulates breathing. This evidence demonstrates that RTN neurons stimulate breathing primarily by releasing glutamate.
The activity of RTN neurons tracks , not ventilation
Neurons activated by hypercapnia have been recorded in the RTN region for at least 25 years (Connelly et al. 1990; Nattie et al. 1993; Bodineau et al. 2000; Mulkey et al. 2004). However, virtually all brainstem respiratory‐related neurons – this population includes many neurons that regulate the autonomic nervous system – increase their firing rate with hypercapnia regardless of their role in the breathing network. Therefore, this property is weak evidence that such neurons serve as chemoreceptors. The fact that the discharge of RTN neurons tracks rather than lung ventilation, especially in the conscious state, is much more persuasive evidence.
A substantial fraction of RTN neurons express c‐Fos, a surrogate of neuronal activity (Sagar et al. 1988), in rats and mice exposed to hypercapnia (e.g. Sato et al. 1992; Kumar et al. 2015; Wakai et al. 2015). Preventing CO2‐induced hyperventilation with morphine does not detectably reduce Fos expression in this region (Sato et al. 1992). Similarly, in anaesthetized rats, i.v. morphine eliminates the phrenic outflow but has little effect on CO2‐stimulated discharge rate measured directly in single RTN neurons (Mulkey et al. 2004). Thus, the activity of RTN neurons is driven by but not by the RPG. This interpretation is also supported by the fact that inhibiting the ventral respiratory column (VRC) with muscimol or by intracerebroventricular (i.c.v.) administration of kynurenic acid silences the RPG of anaesthetized rats but reduces the sensitivity of RTN neurons to hypercapnia only minimally (Fig. 3 A and B) (Mulkey et al. 2004; Takakura et al. 2006). A discrepant finding should be mentioned: microinjection of small amounts of NMDA receptor antagonists into RTN (anatomical definition), even unilaterally, reduces the phrenic nerve discharge substantially in anaesthetized rats (Nattie & Li, 1995). This result suggests that the activity of RTN neurons could be tonically driven by excitatory synaptic inputs from elsewhere. However, two additional interpretations should be considered. First, the anatomically defined RTN region is very heterogeneous (Fig. 1 D) and may contain neurons other than the chemoreceptors which also provide an important excitatory drive to the RPG. Alternatively, RTN neurons could be re‐exciting each other via NMDA receptors. Such a mechanism could perhaps explain their higher CO2 sensitivity in vivo than in vitro. This issue is unresolved.
The effect of hypocapnic hypoxia on the activity of RTN neurons in conscious rodents also illustrates that these neurons track rather than the level of lung ventilation (Basting et al. 2015; Wakai et al. 2015). Wakai et al. (2015) showed that, in rats, hypercapnia causes Fos expression in RTN neurons whereas hypoxia (10% inspired O2 fraction ()) does not, unless CO2 is added. More detailed optogenetic evidence leads to the same conclusion (Basting et al. 2015): at rest, in conscious rats, RTN neurons are silent above pH 7.53 (respiratory alkalosis) and increasingly activated as the plasma is acidified (Basting et al. 2015). Their firing rate, judged by how much breathing inhibition results from silencing these neurons optogenetically, is tightly correlated with plasma pH, not with ventilation (Basting et al. 2015) (Fig. 3 D and E).
In summary, under anaesthesia, RTN neuron activation by CO2 occurs regardless of whether the RPG is active or silenced with drugs; in conscious rats, the discharge of these neurons tracks arterial (and pH) rather than the intensity of lung ventilation. Such behaviour is very unusual for respiratory neurons. It is consistent with two non‐mutually exclusive possibilities: RTN neurons encode local (i.e. directly or via astrocytes) or RTN neurons are activated by CO2 via synaptic inputs from pH‐responsive neurons.
Whether and how RTN neurons encode pH/ is examined next. The following three mechanisms are under consideration at this time: (a) CO2 activates RTN neurons directly via proton receptors; (b) astrocytes are the ‘real’ chemoreceptors which detect CO2 and/or protons, and activate RTN neurons by releasing ATP or some other gliotransmitter; and (c) the CO2 sensitivity of RTN neurons is mediated by excitatory synaptic inputs from a widely distributed system of chemosensory neurons.
RTN neurons are intrinsically activated by protons
The earliest supportive evidence is that acidification of the ventral medullary surface or, more specifically, of the RTN region, increases resting breathing (Mitchell et al. 1963; Loeschcke, 1982; Li et al. 1999). The breathing stimulation elicited by these manoeuvres was relatively modest (<30%) but perhaps these methods do not acidify more than a tiny fraction of the RTN neurons. In passing, breathing is also mildly enhanced by acidifying many other brain regions with microdialysis; such observations, and the fact that activating or inhibiting these regions biases (up or down) the respiratory chemoreflex, are the key pieces of evidence supporting the distributed chemosensory theory (see Nattie, 2012 for an exhaustive review of these results and Guyenet, 2014 for alternative interpretations of this evidence).
The next critical piece of evidence was that RTN neurons detect local changes in or pH in slices. Most of the histologically defined RTN neurons (>95%) are activated by CO2 in neonatal rodent slices (Fig. 4 B and C) (Mulkey et al. 2004; Lazarenko et al. 2009). The CO2 response is mediated by changes in [H+] and it is observed in the presence of blockers of glutamatergic, GABAergic and glycinergic ionotropic receptors as well as purinergic receptors. Therefore, it is not determined by synaptic inputs that operate via these classical transmitters (Mulkey et al. 2004; Lazarenko et al. 2009). Importantly, the CO2 response observed in slices persists essentially unchanged in 80% of these cells after acute isolation (Wang et al. 2013 b). This result demonstrates that the pH sensitivity of RTN neurons is at least in part an intrinsic property. Two objections can be levied against this evidence: neurons located elsewhere in the brain often respond to acidification in slices or after isolation, and the pH response of RTN neurons in slices or after isolation (0–3 Hz when extracellular pH is changed from 7.5. to 7.0) is weaker than their response in vivo (0–10 Hz when arterial pH varies between 7.5 and 7.25). The use of immature neurons and the low temperature at which the in vitro recordings were made (typically room temperature) probably account for some of the discrepancy. Indeed, RTN neurons were found to respond with a greater dynamic range when studied at 35°C in rat neonatal slices (0–8 Hz, pH 7.5–6.9) (Guyenet et al. 2005). However, the notion that the intrinsic pH sensitivity of RTN neurons is indeed critical to the operation of the respiratory chemoreflex is also supported by other, entirely different observations: first, two proteins that are expressed by RTN neurons and undetectable in glial cells are required for RTN neurons to encode changes in pH in vitro (TASK‐2 and GPR4, Fig. 4 A, D, E and F); and, second, in the absence of these two putative proton sensors the central chemoreflex of conscious mice is reduced by ∼90% (Reyes et al. 1998; Ludwig et al. 2003; Gestreau et al. 2010; Wang et al. 2013 a; Kumar et al. 2015; and see updated data by N. N. Kumar and D. A. Bayliss illustrated in Fig. 4 G).
Figure 4. RTN neurons directly encode [H+] .
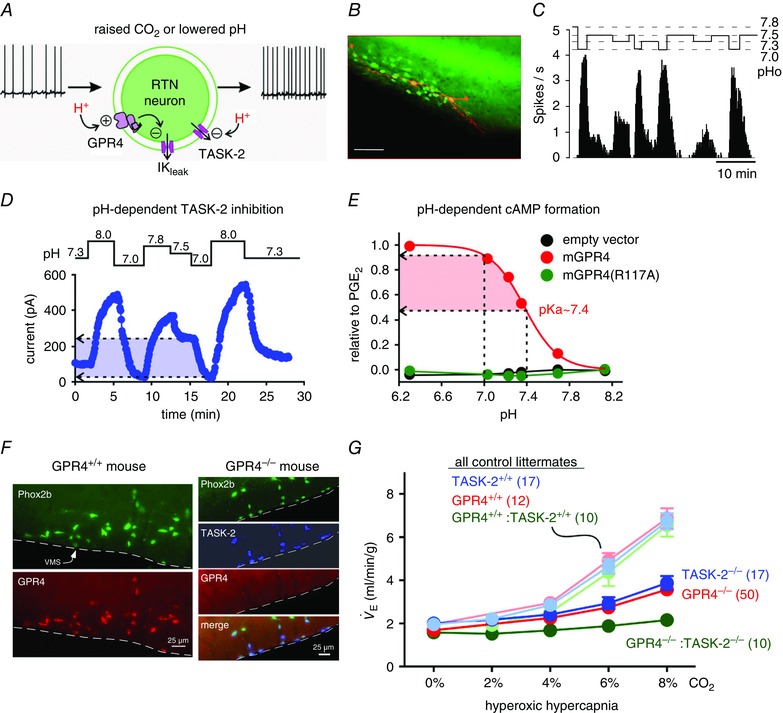
A, molecular basis of the intrinsic proton sensitivity of RTN neurons (reproduced from Kumar et al. 2015). B, RTN neurons identified in a Phox2b‐eGFP transgenic mouse (green cells). Two RTN neurons were filled with biotinamide after intracellular recording (orange; scale bar: 50 μm). C, right, example of pH‐dependent firing rate of one RTN neuron recorded in a Phox2b‐eGFP mouse brain slice (reproduced from Lazarenko et al. 2009). D, current elicited in a HEK293 cell transiently transfected with TASK‐2. Note the large inward shift in holding current elicited by changing extracellular pH from neutral (7.5) to acid (7.0). E, representative pH‐dependent accumulation of cAMP in HEK293 cells transiently transfected with mGPR4 (red curve), by comparison with cell transfected with empty vector (black) or mGPR4(R117A), a signalling‐deficient receptor mutant (green). The response is normalized to cAMP accumulation elicited in these cells by a saturating concentration of PGE2, which activates an endogenous receptor. F, left two panels show colocalization of Phox2b (top) and GPR4 transcripts (bottom) in RTN neurons in a wild‐type mouse. The four panels on the right show an experiment performed in a GPR4 knockout mouse. From top to bottom: Phox2b mRNA, TASK‐2 mRNA, absence of GPR4 transcripts and merged image showing colocalization of Phox2b and TASK‐2 transcripts. VMS, ventral medullary surface. G, central respiratory chemoreflex (, minute ventilation; breathing stimulation elicited by hyperoxic hypercapnia) is reduced by 65% in TASK‐2 and in GPR4 knockout mice relative to control littermates (n provided in parentheses). GPR4−/−:TASK‐2−/− mice deleted for both genes show 90% reduction (with sample size increased to n = 10 double knockout mice, from n = 4 in the original publication, Kumar et al. 2015).
In RTN neurons, acidification inhibits a resting potassium conductance (Mulkey et al. 2004; Onimaru et al. 2012). In about half these cells, this appears to be entirely mediated by the two‐pore domain potassium (channel) (K2P) channel TASK‐2 (Reyes et al. 1998; Mulkey et al. 2004; Gestreau et al. 2010; Wang et al. 2013 a). In TASK‐2−/− mice, the action potential discharge of this particular subset of RTN neurons is normal at pH 7.0 (∼3 Hz, in vitro at room temperature) but it is no longer inhibited by alkalization above this level. In the CNS, TASK‐2 seems to be exclusively expressed by neurons (Gestreau et al. 2010). Accordingly, there is no reason to assume that a glial defect could account for the CO2 insensitivity of RTN neurons in TASK‐2−/− mice. Likewise, GPR4, a proton‐activated G protein‐coupled receptor (GPCR), is expressed at comparatively high levels selectively by RTN neurons but appears absent from surrounding astrocytes (Ludwig et al. 2003; Kumar et al. 2015). Figure 4 F illustrates the remarkable co‐localization of GPR4 and Phox2b transcripts in neurons of the RTN and the fact that GPR4 transcripts are no longer detectable in GPR4−/− mice. Both TASK‐2 and GPR4 show pH‐sensitive activity in a physiologically relevant range. For example, Fig. 4 D illustrates the strong pH sensitivity of whole cell currents recorded from HEK 293 cells expressing recombinant TASK‐2, with significant current inhibition apparent between pH 7.5 and pH 7.0 (Y. Shi and D. A. Bayliss, unpublished observations; Kumar et al. 2015). Likewise, Fig. 4 E illustrates the effect of pH on cAMP production by HEK 293 cells transfected with GPR4 (M.‐G. Ludwig, unpublished observations). The accumulation of cAMP occurs with a pK a ∼7.4 (extracellular pH at which cAMP accumulation is half‐maximal), and the magnitude of the response at pH 7.0 is nearly equal to that observed after activation of endogenous PGE2 receptors in those same cells. By in situ hybridization, neither GPR4 nor TASK‐2 transcripts are detected in TH+ neurons, 95% of GPR4+ neurons located below the facial motor nucleus contain TASK‐2 transcripts and 87% of TASK‐2+ neurons within this region contain GPR4 (Kumar et al. 2015; N. N. Kumar, unpublished observations). When a more sensitive detection method is used (single‐cell PCR), GPR4 and TASK‐2 transcripts can be identified in virtually every RTN neuron (Wang et al. 2013 a; Kumar et al. 2015). A conservative interpretation of the slight discrepancy between single‐cell PCR (scPCR) and in situ hybridization (ISH) is that GPR4 and TASK‐2 are expressed by most chemosensitive RTN neurons, but in varying amounts. The central respiratory chemoreflex (ventilatory response to hyperoxic hypercapnia) is reduced by ∼65% in GPR4−/− mice (Fig. 4 G); this reflex can be restored to normal by viral‐mediated re‐expression of mouse GPR4 selectively into RTN neurons but it remains depressed when a mutated version of GPR4 that cannot interact with G‐proteins (mGPR4(R117A) in Fig. 4 E) is re‐introduced (Kumar et al. 2015). In mice, deletion of GPR4 or TASK‐2 preserves the histological integrity of RTN neurons (numbers and appearance; N. N. Kumar, unpublished observations) and produces a greater reduction of the respiratory chemoreflex than knocking out each gene individually. An update of these recently published results (Kumar et al. 2015), with a larger cohort of double knockout mice (10 instead of 4), demonstrates that the central respiratory chemoreflex (ventilatory response to hyperoxic hypercapnia) is reduced by an average of 90% in GPR4−/−:TASK‐2−/− mice vs. 65% in either TASK‐2 or GPR4 single knockout mice (Fig. 4 G).
In summary, both the central chemoreflex and the response of RTN neurons to acid require at least two putative proton detectors, GPR4 and TASK‐2, that are co‐expressed by these neurons but are undetectable, at least by hydridization histochemistry, in glial and other surrounding cells (endothelium, vascular smooth muscle cells). GPR4 and TASK‐2 have a very restricted neuronal distribution. Although both TASK‐2 and GPR4 are expressed elsewhere in the brain, neither is present in the ventrolateral medulla caudal to RTN, i.e. within the VRC, and co‐expression of these molecules outside RTN has not yet been observed (Gestreau et al. 2010; Kumar et al. 2015). Collectively, this evidence suggests that the central respiratory chemoreflex operates predominantly by modulating the discharge rate of RTN neurons and that the activation of RTN neurons by hypercapnia derives from the intrinsic pH sensitivity of these neurons. Finally, GPR4 and TASK‐2 may represent two independent cellular mechanisms that converge to influence the pH sensitivity of individual RTN neurons and the integrative respiratory chemoreflex.
The evidence has the following limitations. While TASK‐2 conductance increases with alkalization through a broad range that encompasses physiological pH levels (Fig. 4 D) (Reyes et al. 1998), there is no definitive evidence that the intrinsic pH sensitivity of TASK‐2 channels can fully account for their inhibition by extracellular acidification in RTN neurons. Like other K2P channels, TASK‐2 channels may be G‐protein modulated (Sepulveda et al. 2015) and could perhaps act as downstream effectors of other unidentified proton‐activated receptor(s). This hypothetical proton receptor may not be GPR4, since GPR4 fails to couple to TASK‐2 in HEK 293 cells (Kumar et al. 2015), and because, in TASK‐2‐deleted mice, the residual (and presumably GPR4‐mediated) pH‐dependent regulation of RTN neurons occurs via inhibition of a distinct K+ conductance. Nonetheless, it remains possible that GPR4 signalling to TASK‐2 might require some specific mechanism present in RTN neurons but not in HEK 293 cells, and that an alternative K+ channel substitutes for TASK‐2 following its genetic deletion. Finally, although GPR4 is proton activated, this receptor could conceivably have other, still unknown agonists, including a heretofore unidentified substance released by acid‐sensitive astrocytes. In this respect, whereas viral replacement experiments showed that downstream function of GPR4 in RTN neurons was necessary for the phenotypic rescue in the GPR4 knockout mice, they did not demonstrate directly that the pH sensitivity of GPR4 was required (Kumar et al. 2015).
Astrocytes, the RTN and central respiratory chemoreception
Acidification depolarizes a subset of ventral medullary astrocytes (Wenker et al. 2010), increases their intracellular calcium concentration and releases ATP from these cells (Gourine et al. 2010), possibly via exocytosis (Kasymov et al. 2013). Ultimately, ATP depolarizes nearby respiratory neurons, RTN included, via P2(X and/or Y) receptors (Gourine et al. 2010). This sequence of events summarizes the proposed contribution of astrocytes to central respiratory chemoreception according to Gourine and colleagues and is henceforth called the glial theory of central respiratory chemosensitivity (glial theory for short, Fig. 5 B). A variant of this theory posits that ATP exits astrocytes through connexin channels that open when carbamylated by molecular CO2 (Fig. 5 B) (Huckstepp et al. 2010; Meigh et al. 2013). The glial theory is supported by several additional pieces of evidence. First, breathing is increased by activating RTN astrocytes in anaesthetized rats, optogenetically or with the metabolic poison fluorocitrate (Erlichman et al. 1998; Gourine et al. 2010). Second, the response of RTN neurons to high levels of CO2 in organotypic brain slices is reduced by MRS2179, a selective antagonist of ATP receptors (Gourine et al. 2010). Finally, astrocytes are less sensitive to CO2 in Mecp2 −/− mice, a model of Rett syndrome that also features a reduced hypercapnic chemoreflex, among other deficits (Garg et al. 2015; Turovsky et al. 2015). Because acid‐responsive astrocytes are present throughout the ventral medullary surface, ATP can activate most ‘respiratory’ neurons and selected ATP receptor antagonists attenuate the effect of CO2 on preBötzinger complex (preBötC) inspiratory neurons in vivo, the glial theory of chemoreception further postulates that CO2/H+‐sensitive astrocytes activate respiratory neurons throughout the ventral respiratory column (Thomas & Spyer, 2000; Gourine & Kasparov, 2011). In this conception, RTN is viewed as one of many sites where CO2‐sensitive astrocytes activate neurons to increase breathing. This interpretation is generally in line with the distributed chemosensitivity theory championed by Nattie and colleagues (Nattie, 2012), with a few notable discrepancies. First, high pH sensitivity is a distinctive feature of astrocytes residing near the ventral medullary surface (Gourine et al. 2010); however, the respiratory chemoreceptors postulated by Nattie and many other investigators are located in the raphe, the hypothalamus, the nucleus of the solitary tract (NTS) and the locus coeruleus. Second, the pH sensitivity of serotonergic neurons, another prominent group of putative central respiratory chemoreceptors, is also attributed to their intrinsic pH sensitivity rather than to astrocytes because it persists after cell isolation (Hodges & Richerson, 2010). Further, neither the glial theory nor the distributed chemosensitivity theory seem to account for the following results: genetic lesions of RTN neurons eliminate the chemoreflex at birth while resting breathing persists (Ramanantsoa et al. 2011; Ruffault et al. 2015) and knocking out GPR4 and TASK‐2, two molecules that are undetectable in the VRC outside RTN, eliminates 90% of the central respiratory chemoreflex in adult mice (Fig. 4 G; Kumar et al. 2015). These discrepancies need to be resolved.
Figure 5. Possible role of astrocytes in the RTN .
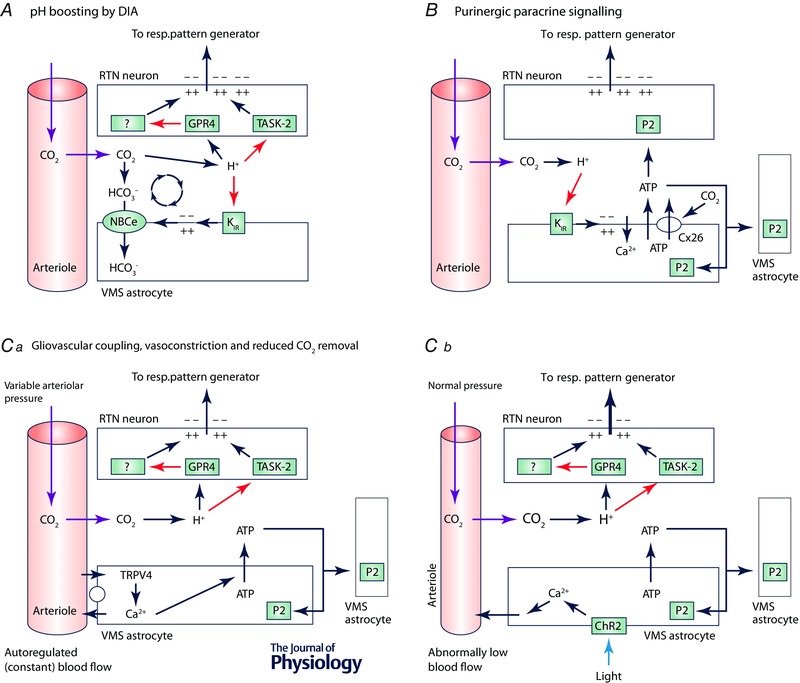
A, proton‐depolarized astrocytes may enhance extracellular acidification via depolarization‐induced alkalization (DIA), thereby increasing the apparent sensitivity of RTN neurons to changes in . KIR, inwardly rectifying potassium channels; NBCe, sodium–bicarbonate exchanger (electrogenic). B, glial theory of chemosensitivity. Ventral medullary surface (VMS) astrocytes are depolarized by acid causing exocytosis of ATP and activation of RTN neurons via P2Y receptors (Gourine et al. 2010). An alternative view is that CO2 activates connexin26 directly, causing ATP release (Meigh et al. 2013). C, astrocyte‐induced vasoconstriction. Ca, TRPV4‐mediated brain blood flow autoregulation by astrocytes. This mechanism is Ca2+‐ and ATP‐dependent (reproduced from Kim et al. 2015). Cb, hypothesis based on the mechanism described in Ca: photoactivation of ChR2 expressed by astrocytes causes a rise in astrocytic intracellular [Ca2+] and arteriolar constriction that propagates to other vessels via ATP release and recruitment of the astrocytic syncytium. Vasoconstriction slows CO2 removal, causing to rise and activation of RTN neurons via [H+].
This disconnect could be quantitative. RTN neurons could indeed be responsible for the bulk of the reflex (90%) (Kumar et al. 2015) and the remainder could be distributed elsewhere. For technical reasons, the importance of RTN to the respiratory chemoreflex may have been greatly underestimated by proponents of the glial or the distributed chemosensitivity theories. For example, the percentage of the reflex that can be attenuated by applying ATP receptor antagonists to the ventral medullary surface has been used to gauge the contribution of RTN to the chemoreflex (Gourine et al. 2005 a). This calculated contribution (∼25%) assumes: (a) that these drugs are selective; (b) that they block the receptors entirely; (c) that ATP mediates entirely the effect of CO2; and (d) that these drugs do not reduce the reflex simply by causing vasodilatation (CO2 wash‐out). The contribution of RTN to the chemoreflex has also been estimated at around 23%, based primarily on the breathing stimulation caused by focal acidification of RTN (Li & Nattie, 2002). However, as noted by the authors, the proportion of RTN neurons impacted by a dialysis probe cannot be determined and the 23% figure could be grossly underestimated.
The apparent disconnect may also reflect our incomplete understanding of the exact role of astrocytes in the lower brainstem. One possibility is that astrocyte activation enhances the acidification of the extracellular space relative to arteriolar pH, thereby sharpening detection by RTN neurons (Fig. 5 A). This hypothesis is appealing given that presumed proton detectors (TASK‐2 and GPR4), expressed by RTN neurons but undetectable in glia, are required for the ventilatory response to CO2 (Gestreau et al. 2010; Kumar et al. 2015). Astrocytes could boost extracellular pH via the mechanism called depolarization‐induced alkalization (DIA). DIA refers to the alkalization of the glial cytoplasm caused by activation of an electrogenic sodium–bicarbonate exchanger (Fig. 5 A) (Erlichman et al. 2004). The inward flux of bicarbonate responsible for DIA, pari passu, acidifies the extracellular space. Astrocyte activation by CO2 (Wenker et al. 2010) or with ChR2 (Gourine et al. 2010) or consequent to astrocyte poisoning with fluorocitrate (Erlichman et al. 2004) could therefore activate or boost the chemoreflex by enhancing extracellular acidification. This mechanism could account for the role of astrocytes in chemoreception and explain why the effectiveness of P2 receptor antagonists on the CO2 response of RTN neurons is relatively modest in slices (25% reduction) (Wenker et al. 2012) and has not even been universally observed (Mulkey et al. 2004; Onimaru et al. 2012): the main function of ATP released by astrocytes could be to recruit more glial cells via P2 receptors to further acidify the extracellular space (via the DIA mechanism). Also, ChR2 is permeable to protons and acidifies the cytoplasm of glial cells; this acidification may cause the release of lactate and glutamate, which can depolarize surrounding neurons (Beppu et al. 2014; Tang et al. 2014). Finally, astrocytes could alter the activity of RTN neurons by regulating local blood flow (Fig. 5 Ca and b). Astrocytes are indeed capable of dynamic and, importantly, bidirectional regulation of local blood flow (Gordon et al. 2011). They also contribute to pressure flow regulation via a mechanism that involves TRPV4 channel activation and a rise in astrocytic calcium; this mechanism is also attenuated by ATP receptor blockade (Kim et al. 2015). Since ChR2 is calcium permeant (Nagel et al. 2003), optogenetic stimulation of astrocytes with this opsin could very well produce vasoconstriction (Fig. 5 Cb). If so, the consequence would be local CO2 retention and acidification, RTN neuron activation and increased breathing. Finally, whilst deleting methyl‐CpG binding protein 2 (MeCP2) selectively from astrocytes in mice attenuates the respiratory chemoreflex (Garg et al. 2015), only the tidal volume (V T) component was reduced. Such an effect could occur for numerous reasons unrelated to CO2 detection per se (e.g. reduced motor or premotor neuron performance, etc.). The frequency component of the reflex, which is the largest contributor to the changes in ventilation in mice and is largely mediated by RTN in this species (Holloway et al. 2015; Kumar et al. 2015) is unaffected by deleting MeCP2 from astrocytes, or from neurons for that matter (Garg et al. 2015). The possibility that astrocytes mediate the effect of pH on the neurons that regulate the tidal volume but not on those that regulate breathing frequency cannot be excluded a priori but seems rather unlikely.
In summary, the notion that CO2‐mediated astrocyte activation throughout the ventral respiratory column underlies the central respiratory chemoreflex fails to explain why this reflex depends to such a large degree on the integrity of RTN neurons (Ramanantsoa et al. 2011) or on the selective expression by these neurons of two putative proton receptors, GPR4 and TASK‐2, which are undetectable in astrocytes and absent from the rest of the ventrolateral medulla (Gestreau et al. 2010; Kumar et al. 2015). The distributed theory of central chemoreception faces much the same issue. The fact that astrocytes have the potential to modulate the CO2 response of RTN neurons is non‐controversial but this evidence can be interpreted in several ways. CO2‐sensitive astrocytes could increase pH detection by RTN neurons by enhancing extracellular acidification or they could do so by releasing gliotransmitters. Alternatively, astrocytes could modulate local blood flow and influence RTN neuron discharge by regulating local tissue . Such mechanisms are undoubtedly elicited when astrocytes are subject to severe depolarization (ChR2, fluorocitrate) but the extent to which these mechanisms contribute to pH detection under physiological conditions remains to be defined.
RTN and the central hypoxic ventilatory response
A thorough discussion of the potential contribution of central O2 sensors to breathing is beyond the scope of this review. After a few general considerations, we will limit our remarks to the known effects of hypoxia on RTN neurons and discuss whether there is any evidence that activation of these neurons could be contributing to a central hypoxic ventilatory reflex. This issue is of some theoretical importance given that lower brainstem astrocytes seem to be activated by both acidification and by hypoxia (Gourine, 2005; Gourine et al. 2005 b; Gourine & Kasparov, 2011; Marina et al. 2015) and, as reviewed above, RTN neurons can be activated by surrounding astrocytes, presumably of the acid‐sensitive variety.
Brainstem oxygen sensors have been proposed to underlie the Cushing response, a sympathoactivation and rise in blood pressure elicited by cerebral ischaemia and, possibly, to contribute to blood pressure regulation under ordinary circumstances (Reis et al. 1989, 1997; Sun & Reis, 1994, 1996; Paton et al. 2009; Teppema & Dahan, 2010). It is also possible that oxygen sensors located in the brainstem could mitigate the general depressant effect of hypoxia on neural circuits and help maintain or even increase breathing under hypoxia (Curran et al. 2000; Teppema & Dahan, 2010; Angelova et al. 2015). The notion that brainstem O2 sensors activate breathing originally derives from the observation that the hypoxic ventilatory reflex (breathing stimulation caused by hypoxia) is only transiently abolished following excision of the carotid bodies and then recovers substantially in a matter of weeks (Martin‐Body et al. 1986; Forster, 2003; Mouradian et al. 2012; Angelova et al. 2015). The recovery is species dependent: virtually complete in rats, partial in many species, recovery has not been observed in humans (Swanson et al. 1978; Timmers et al. 2003; Teppema & Dahan, 2010). One interpretation of this species‐ and time‐dependent recovery is that certain mammals, rats especially, have a central hypoxic ventilatory reflex that slowly appears following carotid body denervation. Another interpretation, which is not mutually exclusive, is that this reflex returns because new connections are established, in rats but not in man, by subsidiary peripheral chemoreceptors (ectopic carotid body glomus cells a.k.a. mini‐glomeruli, aortic bodies or undefined and species‐specific hypoxia‐sensitive afferents) (Chang et al. 2015). A contribution of the aortic bodies to the recovery of the hypoxic ventilatory reflex is supported by denervation experiments in goats and ponies but not in rats (for review see Hodges & Forster, 2012). Deletion of Olf78, a lactate receptor‐encoding gene that is expressed by the carotid body but undetectable in the CNS, eliminates the hypoxic ventilatory reflex of adult mice without altering their hypercapnic response (Chang et al. 2015). This evidence does not support the existence of central hypoxia‐responsive respiratory chemoreceptors.
RTN neurons are silenced in unanaesthetized intact rats exposed to moderate hypoxia for up to 45 min (12% , i.e. present at 5000 m elevation, causing of ∼43 mmHg, Fig. 3 E) (Basting et al. 2015). Furthermore, also unlike hypercapnia, exposure to 10% for half an hour in freely breathing urethane‐anaesthetized rats fails to activate Fos in these neurons (Wakai et al. 2015). Thus, sustained hypocapnic hypoxia clearly does not activate RTN neurons in spontaneous breathing anaesthetized or unanaesthetized rats with intact carotid bodies. This lack of activation could be because the level of hypoxia was insufficient to activate astrocytes or because the hypothetical astrocyte‐mediated hypoxic stimulation of RTN neurons could not overcome the alkalosis‐related inhibition of these neurons. In anaesthetized cats with the brain maintained approximately isocapnic by controlling end‐expiratory CO2, hypoxia produces some acidification of the ventral medullary surface (Xu et al. 1992). It is therefore theoretically possible that, on a background of normo‐ or hypercapnia, CNS hypoxia might activate RTN by further acidifying the environment of these neurons. However, other experiments also performed under approximately isocapnic conditions (ventilated rats anaesthetized with chloralose–urethane or halothane) provided no evidence of a central stimulatory effect of hypoxia on RTN neurons. Specifically, hypoxic stimulation of RTN neurons ( 12%) was eliminated by carotid body denervation or by blocking glutamate transmission in the lower brainstem (Fig. 3 A) (Mulkey et al. 2004; Takakura et al. 2006). These hypoxia trials were brief. Conceivably, more severe and more sustained periods of hypoxia might be able to acidify the RTN region sufficiently to activate the chemosensory neurons.
However, other experimental data support the opposite view, namely that hypoxia might inhibit RTN neurons (Gestreau et al. 2010). These authors proposed that radical oxygen species (ROS) generated by hypoxia open TASK‐2 channels in RTN neurons thereby triggering the respiratory frequency decline that develops during sustained severe hypoxia ( 8–10%). The theory is compatible with our observations that RTN neurons are inhibited by hypocapnic hypoxia in conscious rats (Basting et al. 2015) although we favour a simpler explanation, namely that hypoxia inhibits RTN neurons primarily via respiratory alkalosis (Fig. 3 B and E).
In conclusion, CNS hypoxia could well be a physiological regulator of breathing in unanaesthetized mammals but the evidence is still equivocal and is likely to remain so until the underlying biochemical mechanisms are identified. At this time, there is no evidence that RTN neurons are directly activated by CNS hypoxia. Some degree of breathing stimulation may be elicited by a direct effect of hypoxia on the preBötC (Solomon et al. 2000; Angelova et al. 2015) or elsewhere (Neubauer & Sunderram, 2004). Some breathing stimulation may also result from the direct or paracrine effect of hypoxia on lower brainstem catecholaminergic neurons (Sun, 1995; Roux et al. 2000; Teppema & Dahan, 2010; Marina et al. 2013; Burke et al. 2014). These hypoxia‐responsive structures may simply mitigate the depressant effect of severe brainstem hypoxia on the respiratory network rather than being capable of stimulating breathing beyond the level present under normoxia.
How does RTN regulate breathing?
The relative contribution of frequency and tidal volume to the central chemoreflex is species specific. These details are beyond the scope of this review and we confine our present analysis to rodents. In quietly resting unanaesthetized rats and mice, hypercapnia has a powerful stimulatory effect on the breathing rate (Li & Nattie, 2002). This effect is observed under hyperoxic conditions and after carotid body denervation (e.g. Kumar et al. 2015 for mice) and shows that, in intact unanaesthetized rodents, central chemoreceptor stimulation clearly regulates breathing frequency. Consistent with the presumed role of RTN in central chemoreception, selective optogenetic activation of these neurons likewise produces robust increases in breathing frequency in rats (Burke et al. 2015 b) and mice (Holloway et al. 2015). However, certain anaesthetics such as urethane greatly attenuate the effects of hypercapnia on the breathing rate of freely breathing rats (Wakai et al. 2015) and RTN stimulation seems similarly ineffective at increasing the breathing rate (f R) in such preparations (Huckstepp et al. 2015). The inability to increase f R under these particular conditions may be because the animals are already hypercapnic at rest or it could be secondary to some untoward effect of the anaesthetic on the respiratory rhythm generator.
In terms of circuitry, how RTN neurons regulate breathing remains the most difficult question to answer given the extraordinary complexity of the RPG (Molkov et al. 2014). Only two observations are firmly established at this time. First, as a group, RTN neurons have a very restricted projection pattern (Fig. 2 B). The targeted regions include the four subdivisions of the ventral respiratory column (Bötzinger region, preBötC, rostral and caudal ventral respiratory group) and the Kölliker–Fuse nucleus (Bochorishvili et al. 2012). These regions contain the core components of the respiratory pattern generator (Alheid & McCrimmon, 2008; Smith et al. 2013; Molkov et al. 2014). RTN also targets the caudal NTS, which receives carotid body input and numerous cardiopulmonary afferents (Kubin et al. 2006). Finally, RTN also innervates the lateral parabrachial complex, which projects rostrally; the latter connection could contribute to CO2‐induced arousal (Kaur et al. 2013). Second, consistent with this projection pattern, RTN neurons alter multiple breathing parameters in ways that cooperate to enhance alveolar ventilation (increased rate, increased inspiratory amplitude, reduced post‐inspiratory expiratory flow and enhanced active expiration) (Marina et al. 2010; Abbott et al. 2011; Basting et al. 2015; Burke et al. 2015 a).
An additional complexity stems from the fact that breathing frequency control by RTN varies in different preparations. Specifically, breathing frequency is exquisitely responsive to optogenetic stimulation or inhibition of RTN in conscious unstressed rats (resting f R of ∼70 beats min−1) (Abbott et al. 2011; Basting et al. 2015) but poorly responsive to RTN inhibition in an arterially perfused preparation (Marina et al. 2010) and, as mentioned above, unresponsive to what appeared to have been RTN stimulation in urethane‐anaesthetized spontaneously breathing rats (Huckstepp et al. 2015). Even in unanaesthetized unrestrained rats, the control of breathing frequency by RTN is selectively eliminated during REM sleep (Fig. 6) (Burke et al. 2015 a). These observations suggest that the control of breathing by RTN is subject to countless biases and gating mechanisms that can be triggered by factors as diverse as anaesthesia, the type of preparation (especially the arterially perfused rodent) and, in conscious animals, their state of vigilance, emotional state and behaviour. When the brainstem respiratory pattern is in autorhythmic mode (anaesthetized and normocapnic rats, unanaesthetized rodents that are quietly resting or in non‐REM sleep), the respiratory rate is presumably generated internally by the preBötC (Feldman & Kam, 2015) and immediately adjacent portions of the VRC (Smith et al. 2013). However, during voluntary breathing or REM sleep, the timing of each inspiration is probably imposed on this circuitry by external inputs (Richter & Smith, 2014). Under such conditions, the regulation of the respiratory frequency by chemoreceptors may be gated out so as not to interfere with the precise timing of the respiratory movements required for the selected behaviour.
Figure 6. State‐dependent control of breathing frequency by RTN .
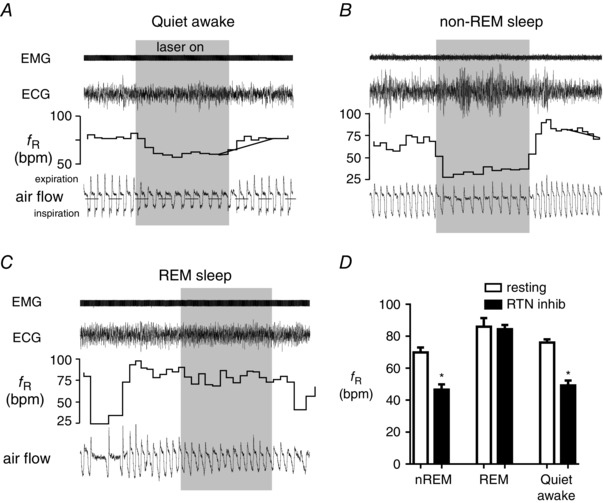
A–C, breathing frequency (bpm, beats min−1) is reduced by bilateral optogenetic inhibition of RTN (ArchT3.0 method) during quiet resting (A) and non‐REM sleep (B) but not during REM sleep (C). Excerpts are from the same rat. D, summary diagram. Data (mean ± SEM of 7 rats; asterisk indicates P < 0.01 from resting, i.e. no laser light). Reproduced from Burke et al. (2015 a).
Prenatally, RTN neurons are electrically coupled and fire synchronously, forming what has been called the embryonic parafacial oscillator (Thoby‐Brisson et al. 2009; Ruffault et al. 2015). This ‘inspiratory’ oscillator is acid‐activated and drives the preBötzinger oscillator at a frequency greater than its intrinsic beating frequency in vitro. The assumption is that the coupling between these two oscillators occurs via ‘direct’ glutamatergic projections from RTN to the preBötC, which then sends a predominantly inhibitory synchronizing feedback to the RTN (Thoby‐Brisson et al. 2009). Monosynaptic excitatory inputs from RTN to the preBötC have indeed been documented with ultrastructural methods in adult rats (Bochorishvili et al. 2012) but the type of preBötC neurons targeted by RTN (‘rhythmogenic’ neuron, interneuron) is undefined. In addition, the breathing rate can be modulated by the other compartments of the ventral respiratory column and by the dorsolateral pons (Smith et al. 2013). All these areas also receive input from RTN and could also contribute to the regulation of the breathing rhythm by brain .
The control of inspiratory amplitude by RTN is less dependent on the state of vigilance of the animal or the type of anaesthetic (Burke et al. 2015 a). This control is plausibly mediated by excitatory projections from RTN to inspiratory premotor neurons located in the rostral ventral respiratory group (rVRG) but inspiratory amplitude could also be regulated via RTN projections to the dorsolateral pons, especially the Kölliker–Fuse nucleus (Bochorishvili et al. 2012; Damasceno et al. 2014). The regulation of early expiratory airflow by RTN could also operate via projections to the Kölliker–Fuse nucleus or a subdivision of the lateral parabrachial complex (Dutschmann & Dick, 2012; Yokota et al. 2015).
The control of active abdominal expiration by RTN neurons (Abdala et al. 2009 a; Marina et al. 2010; Abbott et al. 2011) is presumably mediated by projections from these neurons to the nearby ‘oscillator for active expiration’ (Janczewski & Feldman, 2006; Pagliardini et al. 2011; Huckstepp et al. 2015) and to expiratory premotor neurons located in the caudal ventral respiratory group (cVRG) (Bochorishvili et al. 2012) (Fig. 2 B). An alternative possibility is that, at very high levels of CO2, a subset of bona fide RTN neurons (Phox2b+/VGlut2+ and acid sensitive) develops a late‐expiratory discharge pattern and drives abdominal premotor neurons (Abdala et al. 2009 a; Molkov et al. 2010). A few such late‐E neurons have been recorded close to the ventral medullary surface in an arterially perfused rat preparation but their phenotype has not been identified. These late‐E neurons could be downstream of RTN and belong to the oscillator for active expiration postulated by Feldman and colleagues (Fig. 2 B) (Pagliardini et al. 2011; Huckstepp et al. 2015). They could, however, be a subset of facial motoneurons.
In summary, RTN controls multiple aspects of breathing via its connections to the four segments of the VRC, the NTS and the dorsolateral pons. The control of breathing frequency and active expiration by RTN is most labile and state dependent. The neurons contacted by RTN need to be identified.
Inhibitory inputs to RTN
At the electron microscope level the dendrites of RTN neurons are covered with conventional excitatory and inhibitory synapses (Lazarenko et al. 2009). These synaptic inputs are potentially just as important to breathing and CO2 homeostasis as the intrinsic pH sensitivity of these neurons, probably contributing to the ability of so many brain regions to bias the central respiratory chemoreflex, either positively or negatively (Nattie, 2011). Single‐unit recordings in vivo and tract‐tracing evidence have provided some limited information regarding the origin and potential role of these inputs.
Under anaesthesia, RTN neurons are unaffected by arterial baroreceptors (Mulkey et al. 2004) but they receive numerous respiratory phasic inhibitory inputs which originate from the RPG and from pulmonary stretch receptors (Guyenet et al. 2005; Moreira et al. 2007; Takakura et al. 2007) (Fig. 2 A). In vagotomized rats the inhibitory inputs from the RPG occur during early inspiration, post‐inspiration and late expiration (Guyenet et al. 2005). They disappear when the preBötC/rVRG is inhibited whereas the CO2‐dependent activity of RTN neurons persists (Takakura et al. 2006). Thus, the respiratory modulation of RTN neurons, unlike their CO2 sensitivity, originates somewhere within the respiratory pattern generator. The strength of these inhibitory inputs increases with the degree of activation of the RPG and they seem to impose an upper limit to the discharge rate of RTN neurons as increases (Guyenet et al. 2005). This phenomenon may underlie the curvilinear and saturable relationship between the phrenic outflow and or brain extracellular fluid (ECF) [H+] which is characteristic of vagotomized anaesthetized preparations (Eldridge et al. 1984; Guyenet et al. 2005). In conscious animals, the function of this inhibitory feedback could be to inhibit RTN neurons when the RPG is already highly active for reasons other than hypercapnia. Slowly adapting pulmonary receptors (SARs) also inhibit RTN neurons (Moreira et al. 2007), probably via GABAergic ‘pump cells’ located in the ventrolateral portion of the NTS (Takakura et al. 2007) (Fig. 2 A). The function of this input is probably to reduce the excitatory drive to the RPG when the lungs are overinflated.
Activation of RTN by the carotid bodies
In normocapnic anaesthetized rats RTN neurons are strongly activated by systemic hypoxia (Fig. 3 A) or by intravenously administered cyanide (Mulkey et al. 2004; Takakura et al. 2006). These effects are eliminated by carotid body denervation and thus synaptically mediated. RTN activation by hypoxia is also eliminated by blocking glutamatergic transmission in the brainstem, even as the effect of hypercapnia on these neurons is unaffected (Fig. 3 A and B) (Mulkey et al. 2004; Guyenet et al. 2005). The pathway from the carotid bodies to RTN may have a single relay in the NTS (Fig. 2 A) (Sapru, 1996; Takakura et al. 2006). The evidence summarized so far suggests that the carotid body input to RTN contributes to the activation of these neurons during normo‐ or hypercapnia and could contribute to synergistic interactions between the carotid bodies and central chemoreceptors (Blain et al. 2010).
However, in conscious rats subjected to hypocapnic hypoxia (: 12%, no CO2 added), the excitatory input from the carotid bodies is evidently insufficient to activate RTN neurons because optogenetic inhibition of RTN has no effect on breathing (Fig. 3 E) (Basting et al. 2015). Breathing inhibition returns if hypoxia is more moderate (15% ) or if either CO2 or acetazolamide is administered (Basting et al. 2015). Thus, in conscious rodents exposed to increasingly severe hypoxia, RTN inhibition by alkalosis progressively overrides the excitatory input from the carotid bodies (Basting et al. 2015). Since hypocapnic hypoxia stimulates breathing, the inevitable conclusion is that RTN is not an obligatory relay of the peripheral chemoreflex in conscious rats and peripheral chemoreceptors can activate breathing even when central chemoreceptors are inactivated by CNS alkalosis. This conclusion is consistent with previous observations made in the arterially perfused rat preparation which demonstrated that sustained peripheral chemoreceptor activation is sufficient to drive breathing, even in the presumed absence of central chemoreceptor stimulation (Fiamma et al. 2013).
Modulation of RTN by serotonin and other sleep‐related modulators
RTN neurons are activated by serotonin; in vitro and in vivo the effect of this transmitter is pH independent and blocked by ketanserin, a broad spectrum serotonin receptor antagonist (Mulkey et al. 2007). A more detailed analysis suggests that serotonin activates RTN neurons by modulating hyperpolarization‐activated cyclic‐nucleotide‐gated (HCN) channels and KCNQ channels via 5‐HT7 and 5‐HT2 receptors, respectively (Hawryluk et al. 2012; Hawkins et al. 2015). The serotonergic innervation of RTN may originate from a subset of serotonergic neurons that are themselves mildly acid activated (Brust et al. 2014).
The potential implications of these findings are as follows. Inhibition of medullary raphe serotonergic neurons and natural sleep, during which the serotonergic neurons are much less active, are associated with an attenuation of the chemoreflex (Jacobs & Azmitia, 1992; Brust et al. 2014; Burke et al. 2015 a). A reduction of serotonin release within RTN should hyperpolarize these neurons, causing breathing reduction, and could raise the chemoreflex threshold by increasing the level necessary for these neurons to discharge. This effect could be even more prominent if the release of serotonin is itself CO2 dependent (Brust et al. 2014). However, given the profusion of serotonin inputs to other segments of the respiratory network, the well‐described chemoreflex facilitation elicited by serotonergic neurons (Hodges & Richerson, 2010) is most likely a widely distributed effect that also occurs downstream from RTN, for example in the VRC and at the motoneuronal level where serotonin enhances excitability and responses to glutamate (Rasmussen & Aghajanian, 1990; Ptak et al. 2009). Notably, various raphe‐derived neuropeptides (substance P, TRH) can also activate RTN neurons, cells within the RPG and respiratory motoneurons (e.g. Rekling et al. 2000; Mulkey et al. 2007; Doi & Ramirez, 2008; Ptak et al. 2009).
Much the same can be said about orexin. Orexin activates RTN neurons (Lazarenko et al. 2011) and the activity of orexinergic neurons is also state dependent (Lee et al. 2005). Like serotonin, orexin probably facilitates the chemoreflex during the waking state in part by activating RTN chemoreceptors (Li & Nattie, 2010). Serotonin, orexin or noradrenaline are presumably not released during REM sleep because the cognate neurons are silent (Foote et al. 1980; Lee et al. 2005); REM sleep is the state when the chemoreflex is the weakest and the respiratory drive originating from RTN also the most reduced (Burke et al. 2015 a).
In short, state‐dependent regulation of breathing and the chemoreflex are probably mediated to some extent via changes in the release of serotonin, substance P, TRH, orexin, and possibly noradrenaline, at the level of RTN. The known effects of serotonin and orexin on RTN neurons and those of pH seem purely additive. This characteristic suggests that the transmitters examined so far do not interfere with the chemosensitivity of RTN neurons per se, i.e. they do not modulate the cellular mechanism by which the neurons detect acidification. However, the effects of these modulators should enhance breathing at any given level of and may facilitate the chemoreflex by enhancing the excitability of the respiratory neurons downstream of RTN.
RTN also receives excitatory inputs that could be especially important in the context of exercise. This issue is considered next.
Central respiratory chemoreceptors: potential contribution to the hyperpnoea of exercise
Textbooks and reviews assert that the carotid bodies and central respiratory chemoreceptors do not contribute to the hyperpnoea of low to moderate exercise because neither arterial pH nor arterial rise under such conditions, and thus the central respiratory chemoreceptors will not be activated (Forster et al. 2012). The reasoning rests on the increasingly dubious assumption that central respiratory chemoreceptors are cells whose activity is regulated solely by CO2 or pH.
RTN receives excitatory input from the hypothalamus (Fig. 2 A) (Fortuna et al. 2009). These inputs originate in part from the orexinergic neurons (Dias et al. 2009; Lazarenko et al. 2011). In conscious rats, orexinergic neurons discharge during active waking, when postural muscle tone is high in association with movement; their discharge rate decreases during quiet waking in the absence of movement, and they virtually cease firing during sleep, when postural muscle tone is low or absent (Lee et al. 2005). Given these characteristics, orexinergic neurons are likely to contribute to the so‐called waking drive to breathe or, perhaps, to the central command of exercise hyperpnoea (Eldridge et al. 1981). Supporting the latter possibility, moderate dynamic exercise in rats causes Fos expression in about 20% of putative chemoreceptors (Phox2b+/TH− RTN neurons) (Barna et al. 2014). Additional evidence will be required to rule out stress as the cause of the Fos expression and to ascertain that the small minority of Phox2b+ RTN neurons that were activated by exercise are indeed CO2 sensitive.
RTN activation by exercise could also be mediated by inputs from exercising muscles. The RTN region receives input from lamina I of the spinal cord, which relays cutaneous nociceptive inputs but also muscle metabotropic (type III and IV) primary afferents activated by contracting muscles (Fig. 2 A) (Craig, 1995; Kaufman, 2012). RTN neurons are activated by sciatic nerve stimulation (R. Kanbar & P. G. Guyenet, unpublished observations). Finally, neonatal Phox2b‐positive ‘parafacial respiratory generator (pfRG)’ neurons receive input from the lumbar locomotor pattern generator and contribute to the synchronization of lumbar locomotor and phrenic outflows (Le Gal et al. 2014). These particular Phox2b‐positive cells are a subset of RTN neurons that display a peculiar bursting pattern (pre‐/postinspiratory) in early neonatal brainstem preparations in vitro (Onimaru et al. 2014). This bursting pattern may reflect the persistence, during a brief postnatal period, of their prenatal group pacemaker properties (Thoby‐Brisson et al. 2009; Ruffault et al. 2015).
A subset of serotonergic neurons may also function as central respiratory chemoreceptors (Brust et al. 2014). Given that in awake cats, serotonergic neuron activity correlates best with tonic and repetitive motor activity (Jacobs et al. 2002), a case could also be made for the involvement of serotonergic chemoreceptors in the hyperpnoea of exercise.
In summary, RTN and other central respiratory chemoreceptors may contribute to the hyperpnoea of exercise, not because of a rise in brain or arterial , but because these neurons could receive descending excitatory inputs related to central command and ascending inputs from the spinal cord related to exercising muscles. In addition, RTN activation by exercise could also result from an increase in carotid body afferent input (Prabhakar & Peng, 2004). The available evidence supports all these notions to some extent but is still insufficient to arrive at a firm conclusion.
Is the congenital central hypoventilation syndrome caused by the absence of the RTN neurons?
The following evidence supports the hypothesis that congenital central hypoventilation syndrome (CCHS) results from the loss of the RTN neurons. CCHS is caused by Phox2b mutations (Amiel et al. 2003). The cardinal respiratory signs of CCHS are the absence of a hypercapnic ventilatory reflex and sleep‐related central apnoeas (Weese‐Mayer et al. 2010). RTN neurons mediate a large portion of this reflex in rodents (Kumar et al. 2015; Ruffault et al. 2015). RTN neurons fail to develop in transgenic mice that express a mutated form of Phox2b that cause a severe form of CCHS (Phox2b27/ala) (Dubreuil et al. 2009). These mice die of respiratory failure at birth as would infants with the same mutation were it not for artificial ventilation (Dubreuil et al. 2009). RTN has been tentatively identified in man (Rudzinski & Kapur, 2010).
The following evidence suggests that the loss of RTN is necessary but perhaps not sufficient to explain the respiratory deficits of CCHS. Mice engineered to express the 27Ala Phox2b mutation more selectively (i.e. exclusively by neurons of r3/r5 lineage or even more restrictively by RTN neurons) also have a greatly reduced hypercapnic ventilatory reflex but they survive (Ramanantsoa et al. 2011; Ruffault et al. 2015). An interpretative limitation of these conditional models is that the mutated gene may not have been expressed by all RTN neurons because of incomplete recombination of the floxed allele by Cre recombinase. Finally, CCHS patients have the most severe apnoeas during non‐REM sleep, are less severely affected during REM sleep and breathe normally while awake (Fleming et al. 1980; Huang et al. 2008). RTN is clearly more important to sustain breathing during non‐REM than REM sleep in normal rats (Fig. 5) (Burke et al. 2015 a) but there is no evidence yet that mice with reduced numbers of RTN neurons have especially severe hypoventilation during non‐REM sleep. CCHS patients also experience non‐respiratory (e.g. cardiovascular) deficits that denote the existence of more widespread brainstem defects (Weese‐Mayer et al. 2010). These additional deficits, which may include a reduced carotid body reflex and defects in brainstem catecholaminergic neurons, could further reduce breathing during non‐REM sleep.
In conclusion, the respiratory signs of CCHS could well be caused in large part by the loss of RTN. The reason why RTN neurons are especially vulnerable to Phox2b mutations remains mysterious.
Summary
RTN neurons can be identified histologically in mice and rats as Phox2b+/VGlut2+ non‐catecholaminergic neurons located under the facial motor nucleus or they can be defined by intersectional genetics (transcription factors Phox2b, Atoh‐1 and Egr‐2). The two definitions identify largely similar, possibly identical neuronal populations.
Histologically defined RTN neurons innervate exclusively the pontomedullary regions that harbour the RPG and/or process cardiopulmonary reflexes (NTS). RTN neurons increase breathing by releasing glutamate. Breathing stimulation occurs via at least four mechanisms: by increasing breathing rate and inspiratory amplitude, by triggering active expiration and by slowing airflow during early expiration. In unanaesthetized rodents RTN neurons control breathing frequency when the RPG is in autorhythmic mode (anaesthesia, quiet waking, non‐REM sleep) but not when this network is presumed to be driven externally (voluntary control, REM sleep). RTN neurons regulate inspiratory amplitude even during REM sleep but trigger active expiration only during quiet waking. Breathing rate, inspiratory muscles, active expiration and airway patency could conceivably be regulated by different subsets of RTN neurons. The subnuclear organization of the RTN is unknown.
In order to detect local changes in RTN neurons must express a proton‐modulated potassium channel, TASK‐2, and a proton‐activated GPCR, GPR4. TASK‐2 and GPR4 are expressed at relatively high levels by RTN neurons, but are not detectable in astrocytes or elsewhere in the VRC. The central respiratory chemoreflex of conscious mice is reduced by 65% in single TASK‐2−/− or GPR4−/− mice and by 90% in mice deleted for both genes suggesting that TASK‐2 and GPR4 represent two partially independent pH‐detection mechanisms. Genetic lesions of RTN neurons eliminate the chemoreflex in neonatal mice and cause large deficits in adults. Thus, in mice, the hypercapnic ventilatory reflex relies to a very large extent on the ability of ∼800 neurons (the RTN) to detect [H+] and activate the RPG.
Phox2b is widely expressed in the lower brainstem but RTN neuron development in mice is particularly vulnerable to a mutation that causes a serious form of CCHS (Phox2b27ala). These mice have a greatly reduced central respiratory chemoreflex, a hallmark of the human disease. RTN may also degenerate in CCHS but definitive histological evidence from human samples has not been produced. Based on current knowledge of the function of RTN in rodents, the congenital absence of RTN neurons in humans could account for the loss of the hypercapnic ventilatory reflex and could contribute to the hypoventilation or apnoea during non‐REM sleep; the cardiovascular, gastrointestinal and other deficits associated with CCHS are almost certainly unrelated to RTN.
Astrocyte activation/depolarization with fluorocitrate, ChR2, or with CO2, activates RTN neurons and stimulates breathing. Three mechanisms may be implicated. Astrocytic DIA may raise local proton concentrations to act on the proton receptors expressed by RTN neurons and thereby increase the apparent pH sensitivity of respiratory chemoreflex. This hypothesis could reconcile two seemingly contradictory findings: the existence of acid‐depolarized astrocytes throughout the ventral medulla and the very large contribution of RTN neurons to the chemoreflex; other respiratory neurons may simply lack the necessary receptors (TASK‐2, GPR4) to experience this hypothetical astrocytic ‘pH boost’. Serotonergic neurons, some of which also express GPR4 may be a special case. Other ways in which astrocytes could modify the chemoreflex include the release of gliotransmitters or blood flow reduction and local CO2 accumulation. More work is clearly required to understand the extent to which astrocytes contribute to central respiratory chemoreception and which mechanism is at play in intact conscious mammals.
Both hypercapnia and hypoxia activate astrocytes in vitro and both stimuli acidify the ventral medullary surface in anaesthetized mammals. One would therefore have anticipated that hypoxia, like hypercapnia should be capable of activating RTN neurons. However, no such effect has been found so far. Hypoxia in vivo has no effect on the discharge of RTN neurons in rats with acutely denervated carotid bodies. In conscious rats RTN neurons are actually inhibited by up to 40 min of hypoxia (12% ), presumably because of the overriding inhibitory effect of alkalosis on these neurons. Conceivably, under normo‐ or hypercapnic conditions, severe hypoxia could further activate RTN neurons. However, TASK‐2 activation by radical oxygen species may also contribute to their inhibition during severe hypoxia. The postulated central hypoxic ventilatory reflex of carotid body‐denervated rats may rely on neurons other than RTN, for example brainstem catecholaminergic neurons or the region of the preBötC. Alternatively, breathing stimulation following carotid body excision could be caused by unidentified accessory peripheral chemoreceptors located in different peripheral sites in different mammals.
Conclusions
RTN neurons are proton‐activated glutamatergic neurons; they encode [H+] directly and, probably in cooperation with local astrocytes, they increase lung ventilation by stimulating multiple components of breathing simultaneously. These characteristics are those expected of central respiratory chemoreceptors. The activity of RTN neurons is regulated by multiple inhibitory feedbacks and excitatory inputs, notably from the carotid bodies. RTN neurons drive breathing about equally during quiet waking and non‐REM sleep and to a lesser degree during REM sleep. They are probably essential for the homeostatic regulation of at rest in the waking state and during sleep. RTN neurons are silenced by hypocapnic hypoxia, which accounts for the limited degree of hyperventilation that can be achieved in rodents (mice especially) under even severe hypoxia. RTN neurons are also activated by many types of inputs, including from serotonergic neurons. There is a hint of evidence that RTN neurons might be activated by exercise. Should this be confirmed, the dogma that central respiratory chemoreceptors do not participate in exercise hyperpnoea might have to be revised.
Additional information
Competing interests
None declared.
Funding
This research was funded by the following grants from the National Institutes of Health: HL074011, HL28785 (P.G.G.), HL108609 (D.A.B.).
Acknowledgements
The authors thank Dr Klaus Seuwen (Novartis, Basel, Switzerland) for helpful comments and experimental support related to GPR4 function.
Biography
Douglas A. Bayliss received his PhD in Physiology from the University of North Carolina, USA. After a postdoctoral stint in Physiology & Biophysics at the University of Washington, he joined the Department of Pharmacology at the University of Virginia, where he is now Professor and Chair. His studies involve ion channel function in health and disease with particular emphasis on central apnoea and neuropathic pain. Patrice G. Guyenet was trained at Ecole Normale Supérieure (Paris), College de France (PhD) and Yale University (Postdoc). His principal mentors were Drs Jacques Glowinski and George K. Aghajanian. He is currently Professor of Pharmacology at the University of Virginia. His laboratory studies the neural networks that regulate breathing and blood pressure.

References
- Abbott SB, Stornetta RL, Coates MB & Guyenet PG (2011). Phox2b‐expressing neurons of the parafacial region regulate breathing rate, inspiration, and expiration in conscious rats. J Neurosci 31, 16410–16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, Stornetta RL, Fortuna MG, Depuy SD, West GH, Harris TE & Guyenet PG (2009). Photostimulation of retrotrapezoid nucleus phox2b‐expressing neurons in vivo produces long‐lasting activation of breathing in rats. J Neurosci 29, 5806–5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Rybak IA, Smith JC & Paton JF (2009. a). Abdominal expiratory activity in the rat brainstem–spinal cord in situ: patterns, origins, and implications for respiratory rhythm generation. J Physiol 587, 3539–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Rybak IA, Smith JC, Zoccal DB, Machado BH, St‐John WM & Paton JF (2009. b). Multiple pontomedullary mechanisms of respiratory rhythmogenesis. Respir Physiol Neurobiol 168, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alheid GF & McCrimmon DR (2008). The chemical neuroanatomy of breathing. Respir Physiol Neurobiol 164, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel J, Laudier B, Attié‐Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, Vekemans M, Munnich A, Gaultier C & Lyonnet S (2003). Polyalanine expansion and frameshift mutations of the paired‐like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 33, 459–461. [DOI] [PubMed] [Google Scholar]
- Angelova PR, Kasymov V, Christie I, Sheikhbahaei S, Turovsky E, Marina N, Korsak A, Zwicker J, Teschemacher AG, Ackland GL, Funk GD, Kasparov S, Abramov AY & Gourine AV (2015). Functional oxygen sensitivity of astrocytes. J Neurosci 35, 10460–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna BF, Takakura AC & Moreira TS (2014). Acute exercise‐induced activation of Phox2b‐expressing neurons of the retrotrapezoid nucleus in rats may involve the hypothalamus. Neuroscience 258, 355–363. [DOI] [PubMed] [Google Scholar]
- Basting TM, Burke PG, Kanbar R, Viar KE, Stornetta DS, Stornetta RL & Guyenet PG (2015). Hypoxia silences retrotrapezoid nucleus respiratory chemoreceptors via alkalosis. J Neurosci 35, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beppu K, Sasaki T, Tanaka KF, Yamanaka A, Fukazawa Y, Shigemoto R & Matsui K (2014). Optogenetic countering of glial acidosis suppresses glial glutamate release and ischemic brain damage. Neuron 81, 314–320. [DOI] [PubMed] [Google Scholar]
- Blain GM, Smith CA, Henderson KS & Dempsey JA (2010). Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO2 . J Physiol 588, 2455–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochorishvili G, Stornetta RL, Coates MB & Guyenet PG (2012). Pre‐Bötzinger complex receives glutamatergic innervation from galaninergic and other retrotrapezoid nucleus neurons. J Comp Neurol 520, 1047–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodineau L, Frugiere A, Marlot D & Wallois F (2000). Effect of hypoxia on the activity of respiratory and non‐respiratory modulated retrotrapezoid neurons of the cat. Auton Neurosci 86, 70–77. [DOI] [PubMed] [Google Scholar]
- Brust RD, Corcoran AE, Richerson GB, Nattie E & Dymecki SM (2014). Functional and developmental identification of a molecular subtype of brain serotonergic neuron specialized to regulate breathing dynamics. Cell Rep 9, 2152–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke PG, Abbott SB, Coates MB, Viar KE, Stornetta RL & Guyenet PG (2014). Optogenetic stimulation of adrenergic C1 neurons causes sleep state‐dependent cardiorespiratory stimulation and arousal with sighs in rats. Am J Respir Crit Care Med 190, 1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke PG, Kanbar R, Basting TM, Hodges WM, Viar KE, Stornetta RL & Guyenet PG (2015. a). State‐dependent control of breathing by the retrotrapezoid nucleus. J Physiol 593, 2909–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke PG, Kanbar R, Viar KE, Stornetta RL & Guyenet PG (2015. b). Selective optogenetic stimulation of the retrotrapezoid nucleus in sleeping rats activates breathing without changing blood pressure or causing arousal or sighs. J Appl Physiol (1985) 118, 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AJ, Ortega FE, Riegler J, Madison DV & Krasnow MA (2015). Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 527, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE & Boyden ES (2010). High‐performance genetically targetable optical neural silencing by light‐driven proton pumps. Nature 463, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CA, Ellenberger HH & Feldman JL (1990). Respiratory activity in retrotrapezoid nucleus in cat. Am J Physiol 258, L33–L44. [DOI] [PubMed] [Google Scholar]
- Craig AD (1995). Distribution of brainstem projections from spinal lamina I neurons in the cat and the monkey. J Comp Neurol 361, 225–248. [DOI] [PubMed] [Google Scholar]
- Curran AK, Rodman JR, Eastwood PR, Henderson KS, Dempsey JA & Smith CA (2000). Ventilatory responses to specific CNS hypoxia in sleeping dogs. J Appl Physiol (1985) 88, 1840–1852. [DOI] [PubMed] [Google Scholar]
- Damasceno RS, Takakura AC & Moreira TS (2014). Regulation of the chemosensory control of breathing by Kölliker‐Fuse neurons. Am J Physiol Regul Integr Comp Physiol 307, R57–R67. [DOI] [PubMed] [Google Scholar]
- Dias MB, Li A & Nattie EE (2009). Antagonism of orexin receptor 1 in the retrotrapezoid nucleus inhibits the ventilatory response to hypercapnia predominantly in wakefulness. J Physiol 587, 2059–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A & Ramirez JM (2008). Neuromodulation and the orchestration of the respiratory rhythm. Respir Physiol Neurobiol 164, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil V, Ramanantsoa N, Trochet D, Vaubourg V, Amiel J, Gallego J, Brunet JF & Goridis C (2008). A human mutation in Phox2b causes lack of CO2 chemosensitivity, fatal central apnea and specific loss of parafacial neurons. Proc Natl Acad Sci USA 105, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil V, Thoby‐Brisson M, Rallu M, Persson K, Pattyn A, Birchmeier C, Brunet JF, Fortin G & Goridis C (2009). Defective respiratory rhythmogenesis and loss of central chemosensitivity in Phox2b mutants targeting retrotrapezoid nucleus neurons. J Neurosci 29, 14836–14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutschmann M & Dick TE (2012). Pontine mechanisms of respiratory control. Compr Physiol 2, 2443–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge FL, Kiley JP & Millhorn DE (1984). Respiratory effects of carbon dioxide‐induced changes of medullary extracellular fluid pH in cats. J Physiol 355, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge FL, Millhorn DE & Waldrop TG (1981). Exercise hyperpnea and locomotion: parallel activation from the hypothalamus. Science 211, 844–846. [DOI] [PubMed] [Google Scholar]
- Erlichman JS, Cook A, Schwab MC, Budd TW & Leiter JC (2004). Heterogeneous patterns of pH regulation in glial cells in the dorsal and ventral medulla. Am J Physiol Regul Integr Comp Physiol 286, R289–R302. [DOI] [PubMed] [Google Scholar]
- Erlichman JS, Li A & Nattie EE (1998). Ventilatory effects of glial dysfunction in a rat brain stem chemoreceptor region. J Appl Physiol (1985) 85, 1599–1604. [DOI] [PubMed] [Google Scholar]
- Feldman JL & Kam K (2015). Facing the challenge of mammalian neural microcircuits: taking a few breaths may help. J Physiol 593, 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiamma MN, O'Connor ET, Roy A, Zuna I & Wilson RJ (2013). The essential role of peripheral respiratory chemoreceptor inputs in maintaining breathing revealed when CO2 stimulation of central chemoreceptors is diminished. J Physiol 591, 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming PJ, Cade D, Bryan MH & Bryan AC (1980). Congenital central hypoventilation and sleep state. Pediatrics 66, 425–428. [PubMed] [Google Scholar]
- Foote SL, Aston‐Jones G & Bloom FE (1980). Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc Natl Acad Sci USA 77, 3033–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster HV (2003). Plasticity in the control of breathing following sensory denervation. J Appl Physiol (1985) 94, 784–794. [DOI] [PubMed] [Google Scholar]
- Forster HV, Haouzi P & Dempsey JA (2012). Control of breathing during exercise. Compr Physiol 2, 743–777. [DOI] [PubMed] [Google Scholar]
- Fortuna MG, Stornetta RL, West GH & Guyenet PG (2009). Activation of the retrotrapezoid nucleus by posterior hypothalamic stimulation. J Physiol 587, 5121–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Knopp SJ & Bissonnette JM (2015). Conditional depletion of methyl‐CpG‐binding protein 2 in astrocytes depresses the hypercapnic ventilatory response in mice. J Appl Physiol (1985) 119, 670–676. [DOI] [PubMed] [Google Scholar]
- Gestreau C, Heitzmann D, Thomas J, Dubreuil V, Bandulik S, Reichold M, Bendahhou S, Pierson P, Sterner C, Peyronnet‐Roux J, Benfriha C, Tegtmeier I, Ehnes H, Georgieff M, Lesage F, Brunet JF, Goridis C, Warth R & Barhanin J (2010). Task2 potassium channels set central respiratory CO2 and O2 sensitivity. Proc Natl Acad Sci USA 107, 2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Howarth C & MacVicar BA (2011). Bidirectional control of arteriole diameter by astrocytes. Exp Physiol 96, 393–399. [DOI] [PubMed] [Google Scholar]
- Goridis C & Brunet JF (2010). Central chemoreception: lessons from mouse and human genetics. Respir Physiol Neurobiol 173, 312–321. [DOI] [PubMed] [Google Scholar]
- Gourine AV (2005). On the peripheral and central chemoreception and control of breathing: an emerging role of ATP. J Physiol 568, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV & Kasparov S (2011). Astrocytes as brain interoceptors. Exp Physiol 96, 411–416. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K & Kasparov S (2010). Astrocytes control breathing through pH‐dependent release of ATP. Science 329, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N & Spyer KM (2005. a). ATP is a mediator of chemosensory transduction in the central nervous system. Nature 436, 108–111. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N & Spyer KM (2005. b). Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci 25, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG (2008). The 2008 Carl Ludwig Lecture: Retrotrapezoid nucleus, CO2 homeostasis, and breathing automaticity. J Appl Physiol (1985) 105, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG (2014). Regulation of breathing and autonomic outflows by chemoreceptors. Compr Physiol 4, 1511–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Mulkey DK, Stornetta RL & Bayliss DA (2005). Regulation of ventral surface chemoreceptors by the central respiratory pattern generator. J Neurosci 25, 8938–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JS & Priestley JG (1905). The regulation of the lung‐ventilation. J Physiol 32, 225–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins VE, Hawryluk JM, Takakura AC, Tzingounis AV, Moreira TS & Mulkey DK (2015). HCN channels contribute to serotonergic modulation of ventral surface chemosensitive neurons and respiratory activity. J Neurophysiol 113, 1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk JM, Moreira TS, Takakura AC, Wenker IC, Tzingounis AV & Mulkey DK (2012). KCNQ channels determine serotonergic modulation of ventral surface chemoreceptors and respiratory drive. J Neurosci 32, 16943–16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR & Forster HV (2012). Respiratory neuroplasticity following carotid body denervation: Central and peripheral adaptations. Neural Regen Res 7, 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR & Richerson GB (2010). Medullary serotonin neurons and their roles in central respiratory chemoreception. Respir Physiol Neurobiol 173, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway BB, Viar KE, Stornetta RL & Guyenet PG (2015). The retrotrapezoid nucleus stimulates breathing by releasing glutamate in adult conscious mice. Eur J Neurosci 42, 2271–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Colrain IM, Panitch HB, Tapia IE, Schwartz MS, Samuel J, Pepe M, Bandla P, Bradford R, Mosse YP, Maris JM & Marcus CL (2008). Effect of sleep stage on breathing in children with central hypoventilation. J Appl Physiol (1985) 105, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Cardoza KP, Henderson LE & Feldman JL (2015). Role of parafacial nuclei in control of breathing in adult rats. J Neurosci 35, 1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Eason R, Sachdev A & Dale N (2010). CO2‐dependent opening of connexin 26 and related β connexins. J Physiol 588, 3921–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DY, Carlezon WA Jr, Isacson O & Kim KS (2001). A high‐efficiency synthetic promoter that drives transgene expression selectively in noradrenergic neurons. Hum Gene Ther 12, 1731–1740. [DOI] [PubMed] [Google Scholar]
- Jacobs BL & Azmitia EC (1992). Structure and function of the brain serotonin system. Physiol Rev 72, 165–229. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Martín‐Cora FJ & Fornal CA (2002). Activity of medullary serotonergic neurons in freely moving animals. Brain Res Rev 40, 45–52. [DOI] [PubMed] [Google Scholar]
- Janczewski WA & Feldman JL (2006). Distinct rhythm generators for inspiration and expiration in the juvenile rat. J Physiol 570, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasymov V, Larina O, Castaldo C, Marina N, Patrushev M, Kasparov S & Gourine AV (2013). Differential sensitivity of brainstem versus cortical astrocytes to changes in pH reveals functional regional specialization of astroglia. J Neurosci 33, 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MP (2012). The exercise pressor reflex in animals. Exp Physiol 97, 51–58. [DOI] [PubMed] [Google Scholar]
- Kaur S, Pedersen NP, Yokota S, Hur EE, Fuller PM, Lazarus M, Chamberlin NL & Saper CB (2013). Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J Neurosci 33, 7627–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA & Filosa JA (2015). Astrocyte contributions to flow/pressure‐evoked parenchymal arteriole vasoconstriction. J Neurosci 35, 8245–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L, Alheid GF, Zuperku EJ & McCrimmon DR (2006). Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol (1985) 101, 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar NN, Velic A, Soliz J, Shi Y, Li K, Wang S, Weaver JL, Sen J, Abbott SB, Lazarenko RM, Ludwig MG, Perez‐Reyes E, Mohebbi N, Bettoni C, Gassmann M, Suply T, Seuwen K, Guyenet PG, Wagner CA & Bayliss DA (2015). Regulation of breathing by CO2 requires the proton‐activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 348, 1255–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P & Prabhakar NR (2012). Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol 2, 141–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarenko RM, Milner TA, Depuy SD, Stornetta RL, West GH, Kievits JA, Bayliss DA & Guyenet PG (2009). Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b‐EGFP transgenic mice. J Comp Neurol 517, 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarenko RM, Stornetta RL, Bayliss DA & Guyenet PG (2011). Orexin A activates retrotrapezoid neurons in mice. Respir Physiol Neurobiol 175, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gal JP, Juvin L, Cardoit L, Thoby‐Brisson M & Morin D (2014). Remote control of respiratory neural network by spinal locomotor generators. PLoS One 9, e89670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK & Jones BE (2005). Discharge of identified orexin/hypocretin neurons across the sleep‐waking cycle. J Neurosci 25, 6716–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A & Nattie E (2002). CO2 dialysis in one chemoreceptor site, the RTN: stimulus intensity and sensitivity in the awake rat. Respir Physiol Neurobiol 133, 11–22. [DOI] [PubMed] [Google Scholar]
- Li A & Nattie E (2010). Antagonism of rat orexin receptors by almorexant attenuates central chemoreception in wakefulness in the active period of the diurnal cycle. J Physiol 588, 2935–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Randall M & Nattie EE (1999). CO2 microdialysis in retrotrapezoid nucleus of the rat increases breathing in wakefulness but not in sleep. J Appl Physiol (1985) 87, 910–919. [DOI] [PubMed] [Google Scholar]
- Loeschcke HH (1982). Central chemosensitivity and the reaction theory. J Physiol 332, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig MG, Vanek M, Guerini D, Gasser JA, Jones CE, Junker U, Hofstetter H, Wolf RM & Seuwen K (2003). Proton‐sensing G‐protein‐coupled receptors. Nature 425, 93–98. [DOI] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Trapp S, Li A, Nattie EE, Hewinson J, Smith JC, Paton JF & Gourine AV (2010). Essential role of Phox2b‐expressing ventrolateral brainstem neurons in the chemosensory control of inspiration and expiration. J Neurosci 30, 12466–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Ang R, Machhada A, Kasymov V, Karagiannis A, Hosford PS, Mosienko V, Teschemacher AG, Vihko P, Paton JF, Kasparov S & Gourine AV (2015). Brainstem hypoxia contributes to the development of hypertension in the spontaneously hypertensive rat. Hypertension 65, 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Tang F, Figueiredo M, Mastitskaya S, Kasimov V, Mohamed‐Ali V, Roloff E, Teschemacher AG, Gourine AV & Kasparov S (2013). Purinergic signalling in the rostral ventro‐lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res Cardiol 108, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Body RL, Robson GJ & Sinclair JD (1986). Restoration of hypoxic respiratory responses in the awake rat after carotid body denervation by sinus nerve section. J Physiol 380, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meigh L, Greenhalgh SA, Rodgers TL, Cann MJ, Roper DI & Dale N (2013). CO2 directly modulates connexin 26 by formation of carbamate bridges between subunits. Elife 22, e01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RA, Loeschcke HH, Massion WH & Severinghaus JW (1963). Respiratory responses mediated through superficial chemosensitive areas on the medulla. J Appl Physiol 18, 523–533. [DOI] [PubMed] [Google Scholar]
- Molkov YI, Abdala AP, Bacak BJ, Smith JC, Paton JF & Rybak IA (2010). Late‐expiratory activity: emergence and interactions with the respiratory CpG. J Neurophysiol 104, 2713–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkov YI, Shevtsova NA, Park C, Ben‐Tal A, Smith JC, Rubin JE & Rybak IA (2014). A closed‐loop model of the respiratory system: focus on hypercapnia and active expiration. PLoS One 9, e109894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira TS, Takakura AC, Colombari E, West GH & Guyenet PG (2007). Inhibitory input from slowly adapting lung stretch receptors to retrotrapezoid nucleus chemoreceptors. J Physiol 580, 285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouradian GC, Forster HV & Hodges MR (2012). Acute and chronic effects of carotid body denervation on ventilation and chemoreflexes in three rat strains. J Physiol 590, 3335–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Rosin DL, West G, Takakura AC, Moreira TS, Bayliss DA & Guyenet PG (2007). Serotonergic neurons activate chemosensitive retrotrapezoid nucleus neurons by a pH‐independent mechanism. J Neurosci 27, 14128–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA & Guyenet PG (2004). Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci 7, 1360–1369. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P & Bamberg E (2003). Channelrhodopsin‐2, a directly light‐gated cation‐selective membrane channel. Proc Natl Acad Sci USA 100, 13940–13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E (2011). Julius H. Comroe, Jr., Distinguished Lecture: Central chemoreception: then … and now. J Appl Physiol (1985) 110, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E & Li A (2012). Central chemoreceptors: locations and functions. Compr Physiol 2, 221–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie EE, Fung ML, Li A & St John WM (1993). Responses of respiratory modulated and tonic units in the retrotrapezoid nucleus to CO2 . Respir Physiol 94, 35–50. [DOI] [PubMed] [Google Scholar]
- Nattie EE & Li A (1994). Retrotrapezoid nucleus glutamate injections: long‐term stimulation of phrenic activity. J Appl Physiol (1985) 76, 760–772. [DOI] [PubMed] [Google Scholar]
- Nattie EE & Li A (1995). Rat retrotrapezoid nucleus iono‐ and metabotropic glutamate receptors and the control of breathing. J Appl Physiol (1985) 78, 153–163. [DOI] [PubMed] [Google Scholar]
- Nattie EE, Li AH & St John WM (1991). Lesions in retrotrapezoid nucleus decrease ventilatory output in anesthetized or decerebrate cats. J Appl Physiol (1985) 71, 1364–1375. [DOI] [PubMed] [Google Scholar]
- Neubauer JA & Sunderram J (2004). Oxygen‐sensing neurons in the central nervous system. J Appl Physiol (1985) 96, 367–374. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K & Kawakami K (2012). Postsynaptic mechanisms of CO2 responses in parafacial respiratory neurons of newborn rats. J Physiol 590, 1615–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K, Mariho T & Kawakami K (2014). Cytoarchitecture and CO2 sensitivity of Phox2b‐positive parafacial neurons in the newborn rat medulla. Prog Brain Res 209, 57–71. [DOI] [PubMed] [Google Scholar]
- Pagliardini S, Janczewski WA, Tan W, Dickson CT, Deisseroth K & Feldman JL (2011). Active expiration induced by excitation of ventral medulla in adult anesthetized rats. J Neurosci 31, 2895–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF, Dickinson CJ & Mitchell G (2009). Harvey Cushing and the regulation of blood pressure in giraffe, rat and man: introducing ‘Cushing's mechanism’. Exp Physiol 94, 11–17. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR (2013). Sensing hypoxia: physiology, genetics and epigenetics. J Physiol 591, 2245–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR & Peng YJ (2004). Peripheral chemoreceptors in health and disease. J Appl Physiol (1985) 96, 359–366. [DOI] [PubMed] [Google Scholar]
- Ptak K, Yamanishi T, Aungst J, Milescu LS, Zhang R, Richerson GB & Smith JC (2009). Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J Neurosci 29, 3720–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanantsoa N, Hirsch MR, Thoby‐Brisson M, Dubreuil V, Bouvier J, Ruffault PL, Matrot B, Fortin G, Brunet JF, Gallego J & Goridis C (2011). Breathing without CO2 chemosensitivity in conditional Phox2b mutants. J Neurosci 31, 12880–12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K & Aghajanian GK (1990). Serotonin excitation of facial motoneurons: receptor subtype characterization. Synapse 5, 324–332. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Golanov EV, Galea E & Feinstein DL (1997). Central neurogenic neuroprotection: Central neural systems that protect the brain from hypoxia and ischemia. Ann N Y Acad Sci 835, 168–186. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Ruggiero DA & Morrison SF (1989). The C1 area of the rostral ventrolateral medulla oblongata. A critical brainstem region for control of resting and reflex integration of arterial pressure. Am J Hypertens 2, 363S–374S. [PubMed] [Google Scholar]
- Rekling JC, Funk GD, Bayliss DA, Dong XW & Feldman JL (2000). Synaptic control of motoneuronal excitability. Physiol Rev 80, 767–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N & Lazdunski M (1998). Cloning and expression of a novel pH‐sensitive two pore domain K+ channel from human kidney. J Biol Chem 273, 30863–30869. [DOI] [PubMed] [Google Scholar]
- Richter DW & Smith JC (2014). Respiratory rhythm generation in vivo. Physiology (Bethesda) 29, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux JC, Pequignot JM, Dumas S, Pascual O, Ghilini G, Pequignot J, Mallet J & Denavit‐Saubié M (2000). O2‐sensing after carotid chemodenervation: hypoxic ventilatory responsiveness and upregulation of tyrosine hydroxylase mRNA in brainstem catecholaminergic cells. Eur J Neurosci 12, 3181–3190. [DOI] [PubMed] [Google Scholar]
- Rudzinski E & Kapur RP (2010). PHOX2B immunolocalization of the candidate human retrotrapezoid nucleus. Pediatr Dev Pathol 13, 291–299. [DOI] [PubMed] [Google Scholar]
- Ruffault PL, D'Autréaux F, Hayes JA, Nomaksteinsky M, Autran S, Fujiyama T, Hoshino M, Hägglund M, Kiehn O, Brunet JF, Fortin G & Goridis C (2015). The retrotrapezoid nucleus neurons expressing Phox2b and Atoh1 are essential for the respiratory response to CO2 . Elife 4, DOI: 10.7554/eLife.07051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar SM, Sharp FR & Curran T (1988). Expression of c‐fos protein in brain: Metabolic mapping at the cellular level. Science 240, 1328–1330. [DOI] [PubMed] [Google Scholar]
- Sapru HN (1996). Carotid chemoreflex. Neural pathways and transmitters. Adv Exp Med Biol 410, 357–364. [PubMed] [Google Scholar]
- Sato M, Severinghaus JW & Basbaum AI (1992). Medullary CO2 chemoreceptor neuron identification by c‐fos immunocytochemistry. J Appl Physiol (1985) 73, 96–100. [DOI] [PubMed] [Google Scholar]
- Sepulveda FV, Pablo Cid L, Teulon J & Niemeyer MI (2015). Molecular aspects of structure, gating, and physiology of pH‐sensitive background K2P and Kir K+‐transport channels. Physiol Rev 95, 179–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Borgmann A, Rybak IA & Paton JF (2013). Brainstem respiratory networks: building blocks and microcircuits. Trends Neurosci 36, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Morrison DE, Ellenberger HH, Otto MR & Feldman JL (1989). Brainstem projections to the major respiratory neuron populations in the medulla of the cat. J Comp Neurol 281, 69–96. [DOI] [PubMed] [Google Scholar]
- Solomon IC, Edelman NH & Neubauer JA (2000). Pre‐Bötzinger complex functions as a central hypoxia chemosensor for respiration in vivo. J Neurophysiol 83, 2854–2868. [DOI] [PubMed] [Google Scholar]
- St John WM, Hwang Q, Nattie EE & Zhou D (1989). Functions of the retrofacial nucleus in chemosensitivity and ventilatory neurogenesis. Respir Physiol 76, 159–171. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA & Guyenet PG (2006). Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci 26, 10305–10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Spirovski D, Moreira TS, Takakura AC, West GH, Gwilt JM, Pilowsky PM & Guyenet PG (2009). Galanin is a selective marker of the retrotrapezoid nucleus in rats. J Comp Neurol 512, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK (1995). Central neural organization and control of sympathetic nervous system in mammals. Prog Neurobiol 47, 157–233. [DOI] [PubMed] [Google Scholar]
- Sun MK & Reis DJ (1994). Central neural mechanisms mediating excitation of sympathetic neurons by hypoxia. Prog Neurobiol 44, 197–219. [DOI] [PubMed] [Google Scholar]
- Sun MK & Reis DJ (1996). Medullary vasomotor activity and hypoxic sympathoexcitation in pentobarbital‐anesthetized rats. Am J Physiol Regul Integr Comp Physiol 270, R348–R355. [DOI] [PubMed] [Google Scholar]
- Swanson GD, Whipp BJ, Kaufman RD, Aqleh KA, Winter B & Bellville JW (1978). Effect of hypercapnia on hypoxic ventilatory drive in carotid body‐resected man. J Appl Physiol Respir Environ Exerc Physiol 45, 871–877. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL & Guyenet PG (2006). Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2‐sensitive neurons in rats. J Physiol 572, 503–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Stornetta RL, West GH, Gwilt JM & Guyenet PG (2008). Selective lesion of retrotrapezoid Phox2b‐expressing neurons raises the apnoeic threshold in rats. J Physiol 586, 2975–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, West GH, Gwilt JM, Colombari E, Stornetta RL & Guyenet PG (2007). GABAergic pump cells of solitary tract nucleus innervate retrotrapezoid nucleus chemoreceptors. J Neurophysiol 98, 374–381. [DOI] [PubMed] [Google Scholar]
- Tang F, Lane S, Korsak A, Paton JF, Gourine AV, Kasparov S & Teschemacher AG (2014). Lactate‐mediated glia‐neuronal signalling in the mammalian brain. Nat Commun 5, 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema LJ & Dahan A (2010). The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev 90, 675–754. [DOI] [PubMed] [Google Scholar]
- Teran FA, Massey CA & Richerson GB (2014). Serotonin neurons and central respiratory chemoreception: where are we now? Prog Brain Res 209, 207–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoby‐Brisson M, Karlen M, Wu N, Charnay P, Champagnat J & Fortin G (2009). Genetic identification of an embryonic parafacial oscillator coupling to the preBötzinger complex. Nat Neurosci 12, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Thomas T & Spyer KM (2000). ATP as a mediator of mammalian central CO2 chemoreception. J Physiol 523, 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers HJ, Wieling W, Karemaker JM & Lenders JW (2003). Denervation of carotid baro‐ and chemoreceptors in humans. J Physiol 553, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turovsky E, Karagiannis A, Abdala AP & Gourine AV (2015). Impaired CO2 sensitivity of astrocytes in a mouse model of Rett syndrome. J Physiol 593, 3159–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakai J, Takamura D, Morinaga R, Nakamuta N & Yamamoto Y (2015). Differences in respiratory changes and Fos expression in the ventrolateral medulla of rats exposed to hypoxia, hypercapnia, and hypercapnic hypoxia. Respir Physiol Neurobiol 215, 64–72. [DOI] [PubMed] [Google Scholar]
- Wang S, Benamer N, Zanella S, Kumar NN, Shi Y, Bevengut M, Penton D, Guyenet PG, Lesage F, Gestreau C, Barhanin J & Bayliss DA (2013. a). TASK‐2 channels contribute to pH sensitivity of retrotrapezoid nucleus chemoreceptor neurons. J Neurosci 33, 16033–16044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Shi Y, Shu S, Guyenet PG & Bayliss DA (2013. b). Phox2b‐expressing retrotrapezoid neurons are intrinsically responsive to H+ and CO2 . J Neurosci 33, 7756–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weese‐Mayer DE, Berry‐Kravis EM, Ceccherini I, Keens TG, Loghmanee DA & Trang H (2010). An official ATS clinical policy statement: Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med 181, 626–644. [DOI] [PubMed] [Google Scholar]
- Wenker IC, Kreneisz O, Nishiyama A & Mulkey DK (2010). Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1‐Kir5.1‐like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol 104, 3042–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Sobrinho CR, Takakura AC, Moreira TS & Mulkey DK (2012). Regulation of ventral surface CO2/H+‐sensitive neurons by purinergic signalling. J Physiol 590, 2137–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Sato M, Spellman MJ Jr, Mitchell RA & Severinghaus JW (1992). Topography of cat medullary ventral surface hypoxic acidification. J Appl Physiol (1985) 73, 2631–2637. [DOI] [PubMed] [Google Scholar]
- Yokota S, Kaur S, VanderHorst VG, Saper CB & Chamberlin NL (2015). Respiratory‐related outputs of glutamatergic, hypercapnia‐responsive parabrachial neurons in mice. J Comp Neurol 523, 907–920. [DOI] [PMC free article] [PubMed] [Google Scholar]