

Abstract
Key points
Endothelin‐1 (ET‐1) is upregulated upon intermittent hypoxia and has lipolytic effects.
Intermittent hypoxia induces adipose tissue lipolysis that may be mediated by ET‐1.
In the present study, we show that ET‐1 is involved in adipose tissue remodelling induced by intermittent hypoxia and that phosphorylation of hormone‐sensitive lipase could be one mechanism mediating this effect.
We also show that ET‐1 upregulates its own and its type A endothelin receptor expression, possibly leading to an autoactivatory loop.
These results help us better understand the mechanisms of dyslipidaemia in disorders associated with intermittent hypoxia, such as obstructive sleep apnoea.
Abstract
Obstructive sleep apnoea syndrome is characterized by repetitive episodes of upper airway collapse during sleep resulting in chronic intermittent hypoxia (IH). Obstructive sleep apnoea syndrome, through IH, promotes cardiovascular and metabolic disorders. Endothelin‐1 (ET‐1) secretion is upregulated by IH, and is able to modulate adipocyte metabolism. Therefore, the present study aimed to characterize the role of ET‐1 in the metabolic consequences of IH on adipose tissue in vivo and in vitro. Wistar rats were submitted to 14 days of IH‐cycles (30 s of 21% FiO2 and 30 s of 5% FiO2; 8 h day−1) or normoxia (air–air cycles) and were treated or not with bosentan, a dual type A and B endothelin receptor (ETA‐R and ETB‐R) antagonist. Bosentan treatment decreased plasma free fatty acid and triglyceride levels, and inhibited IH‐induced lipolysis in adipose tissue. Moreover, IH induced a 2‐fold increase in ET‐1 transcription and ETA‐R expression in adipose tissue that was reversed by bosentan. In 3T3‐L1 adipocytes, ET‐1 upregulated its own and its ETA‐R transcription and this effect was abolished by bosentan. Moreover, ET‐1 induced glycerol release and inhibited insulin‐induced glucose uptake. Bosentan and BQ123 inhibited these effects. Bosentan also reversed the ET‐1‐induced phosphorylation of hormone‐sensitive lipase (HSL) on Ser660. Finally, ET‐1‐induced lipolysis and HSL phosphorylation were also observed under hypoxia. Altogether, these data suggest that ET‐1 is involved in IH‐induced lipolysis in Wistar rats, and that upregulation of ET‐1 production and ETA‐R expression by ET‐1 itself under IH could amplify its effects. Moreover, ET‐1‐induced lipolysis could be mediated through ETA‐R and activation of HSL by Ser660 phosphorylation.
Key points
Endothelin‐1 (ET‐1) is upregulated upon intermittent hypoxia and has lipolytic effects.
Intermittent hypoxia induces adipose tissue lipolysis that may be mediated by ET‐1.
In the present study, we show that ET‐1 is involved in adipose tissue remodelling induced by intermittent hypoxia and that phosphorylation of hormone‐sensitive lipase could be one mechanism mediating this effect.
We also show that ET‐1 upregulates its own and its type A endothelin receptor expression, possibly leading to an autoactivatory loop.
These results help us better understand the mechanisms of dyslipidaemia in disorders associated with intermittent hypoxia, such as obstructive sleep apnoea.
Abbreviations
- ATGL
adipose triglyceride lipase
- DMEM
Dulbecco's modified Eagle's medium
- ET‐1
endothelin‐1
- ETA‐R
type A endothelin receptor
- ETB‐R
type B endothelin receptor
- FBS
fetal bovine serum
- FFA
free fatty acid
- HIF‐1
hypoxia‐inducible factor 1
- HSL
hormone‐sensitive lipase
- IH
intermittent hypoxia
- OSA
obstructive sleep apnoea
- PKA
protein kinase A
- PS
penicillin–streptomycin
- qPCR
quantitative PCR
Introduction
Obstructive sleep apnoea (OSA) syndrome is a highly prevalent disease affecting 5–20% of the population and representing a growing health concern as a result of its association with cardiovascular mortality and morbidity (Levy et al. 2012). It is characterized by periodic upper airway collapse during sleep, leading to sleep fragmentation, respiratory efforts and chronic intermittent hypoxia, which appears to be the most deleterious consequence promoting cardiovascular disease through oxidative stress and an inflammatory cascade (Garvey et al. 2009; Levy et al. 2009).
Intermittent hypoxia (IH) is a hallmark of OSA and is associated with metabolic dysfunction (Drager et al. 2010). Experimental data suggest that IH leads to dyslipidaemia (Li et al. 2005; Trzepizur et al. 2013) through up‐regulation of liver lipid biosynthesis (Savransky et al. 2008), increased lipolysis accompanied by free fatty acid (FFA) release and fat remodelling (Jun et al. 2010; Poulain et al. 2014) and decreased lipoprotein clearance as a result of inhibition of lipoprotein lipase (Drager et al. 2013). IH has also been shown to induce IL‐6 release by epididymal fat and to decrease adiponectin secretion (Magalang et al. 2009; Poulain et al. 2014).
Finally, IH induces insulin resistance as a result of its pleiotropic effects through the sympathetic nervous system and the response of different tissues such as the muscle, liver and adipose tissue (Drager et al. 2010). Indeed, FFAs released by lipolysis of adipose tissue upon IH reduce insulin‐mediated glucose uptake in muscles (Delarue & Magnan, 2007).
Endothelin‐1 (ET‐1) is a potent vasoconstrictor peptide mainly produced by vascular endothelial cells. It is upregulated upon hypoxia by hypoxia‐inducible factor‐1 (HIF‐1) transcription factor (Yamashita et al. 2001). Moreover, we have shown that ET‐1 plays a major role in the development of IH‐induced cardiovascular alterations such as hypertension and increased myocardial infarction (Belaidi et al. 2009). Beside its cardiovascular consequences, endothelin also has a strong impact on adipose tissue, in particular on adipocyte glucose and lipid metabolism. Indeed, ET‐1 induces lipolysis in 3T3‐L1 adipocytes (Juan et al. 2005), rat adipocytes (Juan et al. 2006) and primary human adipocytes (Eriksson et al. 2009), an effect mediated by type A endothelin receptors (ETA‐R). Finally, ET‐1 decreases insulin sensitivity, possibly through inhibition of Glut4 translocation (Ishibashi et al. 2001), resulting in decreased fatty acid and glucose uptake (Chien et al. 2011). The experimental data therefore outline various similarities between the effects of IH and of ET‐1 on the adipose tissue, suggesting that ET‐1 might be one of the mediators of the metabolic consequences of IH.
In adipocytes, lipolysis is regulated by several key enzymes, including adipose triglyceride lipase (ATGL; also known as PNPLA2) and hormone‐sensitive lipase (HSL; also known as LIPE)(Fruhbeck et al. 2014). ATGL, HSL and monoglyceride lipase sequentially hydrolyse triglycerides into glycerol and fatty acids. The activity of both ATGL and HSL is submitted to transcriptional and post‐transcriptional regulation. HSL regulation requires serine phosphorylations with opposite effects: phosphorylation of Ser563 and Ser659‐660 by protein kinase A (PKA) increases its activity, whereas that of Ser565 by cyclic AMP‐dependent protein kinase inhibits its activity by preventing Ser563 phosphorylation (Lampidonis et al. 2011).
The present study aimed to determine whether activation of the endothelin system by IH could contribute to its metabolic and lipolytic effects on adipose tissue. Using a combination of in vivo and in vitro approaches, we demonstrate that endothelin and its ETA‐Rs participate in the lipolytic effects of intermittent hypoxia and that regulation of HSL activity could be one of the mechanisms involved.
Methods
Animals and ethical approval
Thirty‐two male Wistar rats (8 weeks old, weight 300–350 g; Janvier Labs, Le Genest‐Saint‐Isle, France) were used in the present study. The animals were kept under control conditions (21 ± 1°C, 12:12 h light/dark cycle) and fed ad libitum with standard chow for 1 week. The animals were then divided into four experimental groups (n = 8 per group) exposed to intermittent hypoxia or normoxia and treated or not with bosentan (100 mg kg−1 day−1 admixed in chow). All experiments were conducted in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (Council of Europe, European Treaties ETS 123, Strasbourg, 18 March 1986) and were approved by an Institutional Animal Care and Use Committee (Cometh, University Grenoble Alpes).
IH
IH was performed as described previously (Arnaud et al. 2011). Animals were exposed in their cages during their daytime sleeping period to 8 h of consecutive 1 min cycles (alternating 30 s of 21% and 30 s of 5% FiO2, 60 cycles h–1) for 14 consecutive days. FiO2 was measured with a gas analyser (ML206; ADInstruments, Oxford, UK) throughout the experiment. Control normoxic rats were exposed to similar air–air cycles to reproduce the noise and air turbulences of the IH stimulus. Ambient air temperature was maintained at 20–22°C.
Blood and tissue collection
At the end of IH exposure, animals were fasted overnight and anaesthetized with sodium pentobarbital (50 mg kg−1, i.p.). Blood was collected in heparinized and EDTA tubes and centrifuged at 1700 g for 10 min at 4°C. Plasma was frozen and stored at −80°C until analysis. Right and left epididymal fat pads were removed, weighted and either stored at −80°C for quantitative PCR (qPCR) analysis or fixed in 95% ethanol and paraffin‐embedded for histological studies.
Plasma analysis
Heparinized plasma glucose and triglyceride concentrations were assayed on a modular analyser (Roche Diagnostics, Mannheim, Germany). Circulating FFA levels were measured in ETDA plasma by an enzymatic assay using the NEFA FS DiaSys® kit (Diasys Diagnostic Systems, Holzheim, Germany).
Immunohistochemistry
Paraffin‐embedded epididymal fat was sectioned in 5 μm slices and stained with anti‐ETA antibody (dilution 1:100; Alomone Labs, Jerusalem, Israel) and secondary anti‐rabbit antibody (dilution 1:1000). Slices were viewed and photographed with a Nikon Eclipse 80i® microscope (Nikon, Tokyo, Japan). Quantifications were made using ImageJ (NIH, Bethesda, MD, USA) and NIS (Nikon) software.
Cell culture
Murine 3T3‐L1 preadipocytes were cultured (37°C, 5% CO2) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin (PS) and 0.1% amphotericin B. Two days after confluence, the medium was replaced with differentiation medium, containing DMEM/HAM F‐12 Glutamax (Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with FBS, PS, amphotericin B and insulin (10 μg ml−1), dexamethasone (250 nm) and 3‐isobutyle‐1‐methylxanthine (0.1 mm). After 72 h, differentiation medium was replaced with adipocyte medium: DMEM/HAM F‐12 Glutamax supplemented with FBS, PS, amphotericin B and insulin (5 μg ml−1). Seven days after the end of differentiation, adipocytes were deprived with DMEM without glucose for 24 h. Cells were then pre‐treated with or without BQ123 (a specific ETA‐R, antagonist, 10 μm), BQ788 [a specific endothelin receptor B (ETB), antagonist, 10 μm] or bosentan (mixed ETA and ETB antagonist, 10 μm) for 1 h, followed by insulin (5 nm) or ET‐1 (10 nm) for 4–24 h. For hypoxia experiments, differentiated cells were cultured with 3% O2 and 5% CO2 in a trigaz incubator (MCO‐18 M; Sanyo, Moriguchi, Japan) for 4 or 24 h. At the end of the experiments, supernatants and cell pellets were stored at −80°C until further use.
Glycerol release and glucose uptake
3T3‐L1 glycerol release was measured in the culture medium by colourimetric assay for triglyceride on a modular analyser (Roche Diagnostics) as described previously (Nagele, 1985). The glucose concentration in the culture medium was assayed using the hexokinase method on the same analyser (Schmidt, 1961).
Real‐time qPCR
Total RNA extraction was performed using Trizol followed by RNeasy Mini Kit (Qiagen, Venlo, The Netherlands) in accordance with the manufacturer's instructions. Total RNA was treated with RNase‐free DNase I (Qiagen). cDNA was reverse transcribed from 1 μg of total RNA with the SuperScriptIII First‐Strand Synthesis System (Life Technologies, Saint Aubin, France). RNase H treatment was added as recommended by the manufacturer.
Real‐time PCR was conducted using the QuantiTect SYBR Green RT‐PCR kit (Qiagen) and a Mx3005P qPCR system (Stratagene, La Jolla, CA, USA).
Primers were chosen to include intron spanning and were synthesized by Life Technologies. Primer sequences and experimental qPCR conditions are reported in the Supporting information (Table S1).
Gene expression was quantified using the comparative threshold cycle (C t) method (Giulietti et al. 2001). The amount of target gene, normalized to three endogenous reference genes (ACTB, CYCA and HPRT1) was expressed relative to that of control cells.
Western blot analysis
3T3‐L1 adipocytes and epididymal adipose tissue were lysed in RIPA lysis buffer supplemented with protease and phosphatases inhibitors. Protein concentration was measured using the bicinchoninic acid method (Pierce BCA Protein Assay Kit®; Thermo Fisher Scientific). Proteins were separated by 10% SDS‐PAGE and transferred to nitrocellulose membrane. Membrane were incubated overnight at 4°C with anti‐ET‐1, anti‐ETA, anti‐ETB, anti‐pHSL(Ser565), anti‐pHSL(Ser660) and anti‐ATGL antibodies (dilution 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, membranes were incubated for 1 h with secondary anti‐rabbit HRP‐linked antibody. Results were normalized with anti‐actin antibody (dilution 1:1000; Sigma‐Aldrich, St Louis, MO, USA). Signal density was analysed with ImageJ software.
Statistical analysis
Results are expressed as the mean ± SEM and were analysed using Prism (GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered statistically significant. Comparison of data from the four groups of rats was performed by two‐way ANOVA followed by Tukey's post hoc analysis. Comparison of data from cell culture experiments was performed by one‐ or two‐way ANOVA depending on the number of factors, or by non‐parametric ANOVA on ranks when a normal distribution of the values was not achieved.
Results
In vivo, ET‐1 is involved in IH‐induced metabolic perturbations and lipolysis
The body weights of Wistar rats submitted to 14 days of normoxia or IH with or without oral bosentan treatment are reported in Table 1. Rats submitted to IH did not gain weight compared to normoxic animals and this difference was significantly attenuated by bosentan treatment. Plasma FFAs were not modified by IH exposure but were significantly reduced by bosentan treatment in both normoxic and IH groups (Fig. 1 A). Similarly, plasma triglycerides were not affected by IH exposure but were significantly reduced by bosentan treatment in the IH group (Fig. 1 B). Finally, a significant interaction between IH exposure and bosentan treatment was found for glycaemia (P = 0.033) (Fig. 1 C).
Table 1.
Body weights before and after 14 days of exposure to normoxia or intermittent hypoxia with or without bosentan treatment
| Group | Day 0 | Day 14 | Weight change |
|---|---|---|---|
| Normoxia | 353.3 ± 4.6 | 383.9 ± 6.8 | 30.6 ± 5.5 |
| Intermittent hypoxia | 353.8 ± 6.7 | 346.7 ± 7.1 | −7.1 ± 3.3* |
| Normoxia + bosentan | 363.8 ± 4.4 | 402.5 ± 6.0 | 38.7 ± 4.0 |
| Intermittent hypoxia + bosentan | 350.4 ± 3.9 | 359.7 ± 8.1 | 9.3 ± 6.2*† |
*P < 0.05 vs. corresponding normoxia group. † P < 0.05 vs. corresponding untreated group; two‐way ANOVA.
Weights (g) are expressed as the mean ± SEM.
Figure 1. Plasma metabolic parameters in rats .
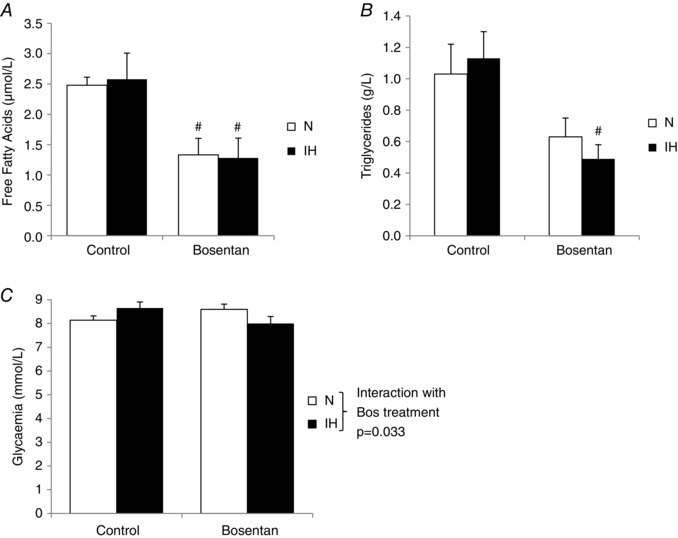
Plasma levels of (A) FFAs (μmol L–1), (B) triglycerides (g L–1) and (C) glucose (mmol L–1) in rats submitted to 14 days of normoxia (N) or intermittent hypoxia (IH) with or without oral bosentan treatment (100 mg kg–1 day–1) (n = 8 rats per group). # P < 0.05 vs. control, two‐way ANOVA. There was a significant interaction between IH exposure and bosentan treatment for glycaemia (P = 0.033).
IH induced a significant loss of epididymal adipose tissue mass in untreated animals, which was significantly reversed by bosentan treatment (Fig. 2 A). This was associated with an IH‐induced decrease in mean adipose cell volume in untreated but not in bosentan‐treated rats, as shown in haematoxylin‐stained slices of epididymal fat (Fig. 2 B and C).
Figure 2. ET‐1 contributes to intermittent hypoxia‐induced adipose tissue lipolysis .
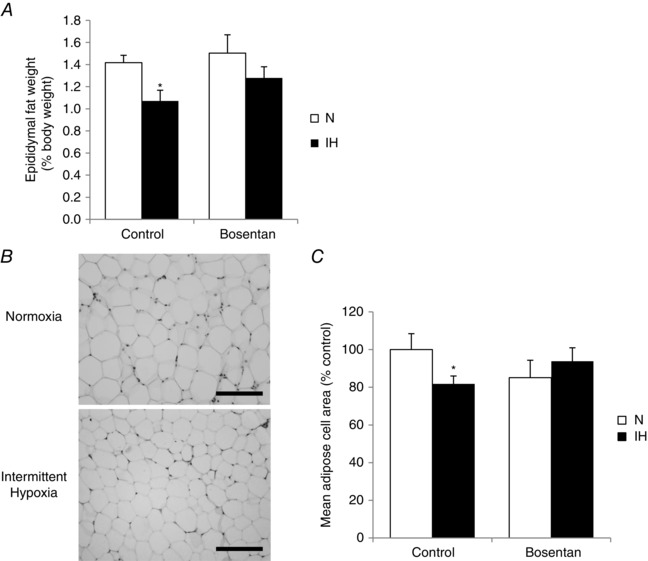
A, ratio (%) of epididymal fat to total body weight after 14 days of exposure to normoxia (N) or intermittent hypoxia (IH) with or without oral bosentan treatment (100 mg kg–1 day–1). B, hematoxylin staining of representative slices of adipose tissue from untreated N and IH rats. Scale bar = 40 μm. C, quantification of mean adipose cell area on at least three epididymal fat sections per rat (n = 8 rats per group) *P < 0.05 vs. N; two‐way ANOVA.
ET‐1 and ETA‐R expression in adipose tissue in response to IH exposure
We observed that ET‐1 mRNA levels were increased almost two‐fold in the epididymal adipose tissue of IH rats compared to normoxic rats (Fig. 3 A). The ET‐1 protein level in epididymal fat extracts was not significantly modified by IH but was significantly decreased by bosentan (Fig. 3 B).
Figure 3. Intermittent hypoxia regulates adipose tissue ET‐1 and ETA‐R expression .
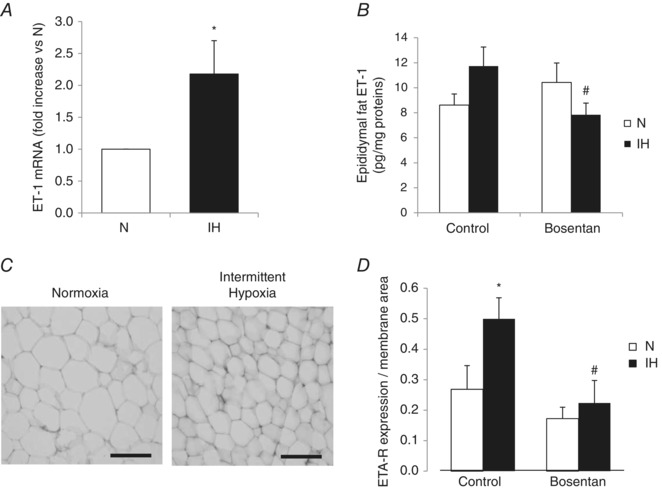
A, qPCR quantification of ET‐1 mRNA in epididymal fat of rats exposed to 14 days of normoxia (N) or intermittent hypoxia (IH). *P < 0.05 vs. N; Mann–Whitney rank sum test. B, ET‐1 protein level in epididymal fat extracts of rats exposed to 14 days of N or IH with or without oral bosentan treatment (100 mg kg–1 day–1). # P < 0.05 vs. control; two‐way ANOVA. C, immunohistochemistry of epididymal fat stained with anti‐ETA‐R antibody. Scale bar = 30 μm. D, quantification of ETA‐R expression normalized to the area of cell membrane in the field, from at least three epididymal fat sections per rat (n = 8 rats per group). *P < 0.05 vs. N and # P < 0.05 vs. control; two‐way ANOVA.
Adipose tissue ETA‐R and ETB‐R were not induced by intermittent hypoxia at the transcriptional level (data not shown). However, we observed that IH induced a marked increase in protein ETA‐R expression (normalized to total cell area because of a decreased cell size after IH exposure) in immunostained epididymal fat sections, with this effect being abolished by bosentan treatment (Fig. 3 D).
ET‐1 regulates its own expression in 3T3‐L1 cells
In differentiated 3T3‐L1 adipocytes, ET‐1 mRNA levels were induced 12‐fold when cells were incubated for 24 h with recombinant ET‐1 (10 nm) and this effect was abolished by co‐incubation with a selective ETA‐R antagonist (BQ123), a selective ETB‐R antagonist (BQ788) or a non‐selective ETA‐R/ETB‐R antagonist (bosentan) (Fig. 4 A). Similarly, although ETB‐R expression was low (data not shown), ETA‐R gene expression was increased 1.4‐fold in ET‐1 stimulated adipocytes and this effect was also abolished by all three antagonists (Fig. 4 B). At the protein level, we observed a 20% increase in ETA‐R content in 3T3‐L1 cells incubated with ET‐1, which was abolished by bosentan co‐treatment (Fig. 4 C).
Figure 4. ET‐1 regulates its expression and that of ETA‐R in 3T3‐L1 adipocytes .
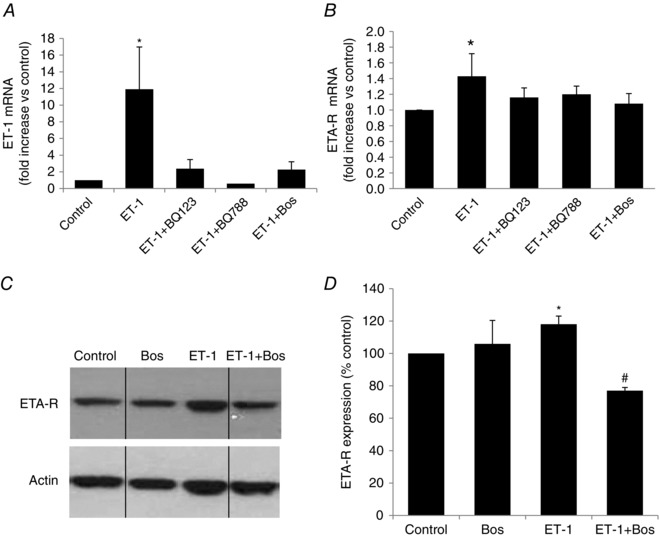
qPCR quantification of ET‐1 (A) and ETA‐R (B) mRNA in mature 3T3‐L1 adipocytes exposed to ET‐1 (10 nm) with or without BQ123, BQ788 or bosentan (Bos) (10 μm each) for 24 h. *P < 0.05; one‐way ANOVA on ranks. Western blot (C) and quantification (D) of ETA‐R protein expression in mature 3T3‐L1 adipocytes submitted to ET‐1 (10 nm) with or without bosentan (10 μm) for 24 h. Quantification was performed on at least three independent experiments. *P < 0.05 vs. control and # P < 0.05 vs. ET‐1; two‐way ANOVA. Vertical dividing lines indicate the components parts of a single image, where intercalating bands were suppressed.
ET‐1 induces lipolysis in 3T3‐L1 cells
Because bosentan treatment abolished the decrease in adipocyte size induced by IH exposure, we wanted to further investigate the metabolic effects of ET‐1 in vitro. We measured supernatant glycerol release and glucose concentration in culture medium as markers of lipolysis and glucose uptake, respectively. 3T3‐L1 cells were cultured under hypoxia (3% O2) for 4 or 24 h. The addition of ET‐1 (10 nm) to the culture medium resulted in an increased glycerol release at both time points that was blocked by bosentan (Fig. 5A). We further evaluated the effects of ET‐1 in normoxic conditions. As shown in Fig. 5 B, the addition of ET‐1 (10 nm) for 24 h induced a 50% increase in glycerol release that was totally abolished by ETA‐R antagonist (BQ123) or mixed antagonist (bosentan) and partially blocked by ETB‐R antagonist (BQ788). As expected, insulin (5 nm) inhibited glycerol release and the different endothelin receptor antagonists did not modify this effect. Co‐incubation of adipocytes with insulin and ET‐1 attenuated the endothelin‐induced glycerol release (Fig. 5 B). Insulin also significantly increased glucose uptake in 3T3‐L1 cells (+72%) (Fig. 5 C). ET‐1 per se had no significant effect on glucose uptake but was able to reverse insulin‐induced glucose uptake and this effect was inhibited by BQ123 and bosentan but not by BQ788 (Fig. 5 C).
Figure 5. ET‐1 induces lipolysis and reduces glucose uptake in 3T3‐L1 adipocytes .
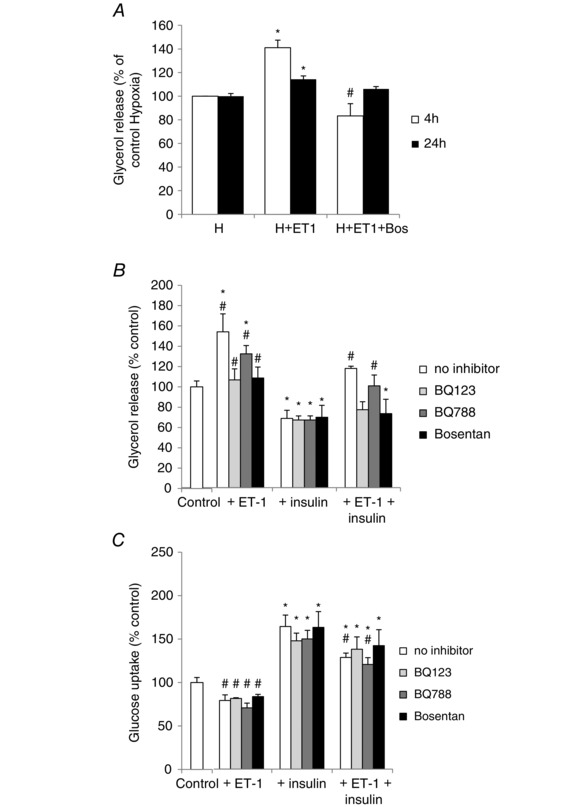
A, lipolysis assessed by glycerol release (% of control hypoxic cells) in culture media of 3T3‐L1 adipocytes cultivated under hypoxia (3% O2) and treated or not with ET‐1 (10 nm) and bosentan (Bos) (10 μm) for 4 or 24 h. *P < 0.05 vs. control untreated cells. # P < 0.05 vs. the corresponding ET‐1‐treated cells; two‐way ANOVA (n = 4 independent experiments). Lipolysis (B), assessed by glycerol release (% of control untreated cells) in culture media and glucose uptake (C) (% of control untreated cells) in 3T3‐L1 adipocytes and treated or not for 24 h with ET‐1 (10 nm), insulin (5 nm), BQ123, BQ788 or bosentan (10 μm each) under normoxia. *P < 0.05 vs. control untreated cells. # P < 0.05 vs. the corresponding insulin‐treated cells; one‐way ANOVA on ranks (n = 5 independent experiments).
ET‐1 regulates the phosphorylation of HSL
To better understand the mechanisms underlying the endothelin‐induced lipolysis, we investigated its regulation of HSL and ATGL in 3T3‐L1 adipocytes. We observed that total protein expression of HSL (Fig. 6 A) and ATGL (Fig. 6 B and C) did not change in the presence of ET‐1 for 5 or 10 h. However, the activating phosphorylation of HSL on Ser660 was significantly increased by exposure to ET‐1 for 5 and 10 h and this was abolished by bosentan (Fig. 6 C and D). The inhibitory phosphorylation of HSL on Ser565 was not modified by ET‐1 (Fig. 6 E). Finally, HSL phosphorylation on Ser660 upon ET‐1 treatment was also observed when the adipocytes were cultivated under a hypoxic environment with 3% oxygen (Fig. 7).
Figure 6. ET‐1 regulates HSL phosphorylation but not HSL or ATGL expression .
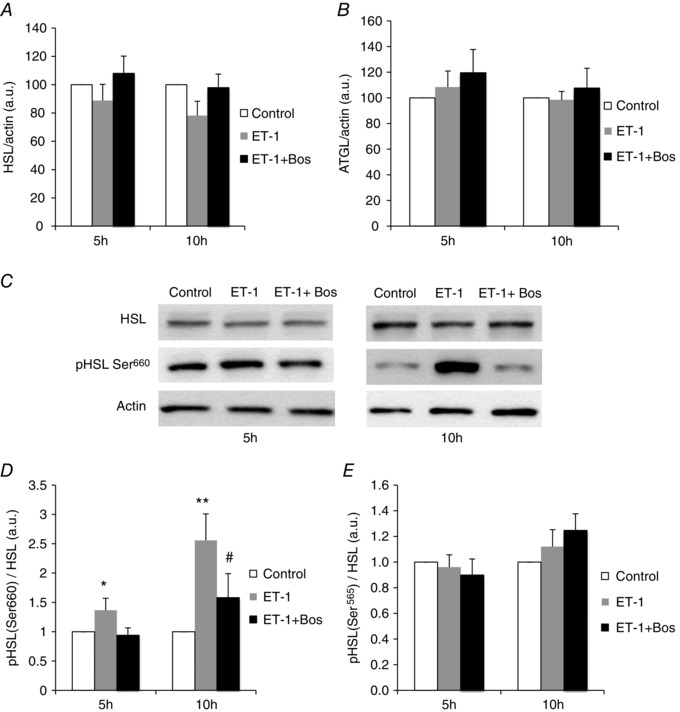
Western blot quantification (normalized to actin) of HSL (A) and ATGL (B) expression in 3T3‐L1 adipocytes treated or not for 5 or 10 h with ET‐1 (10 nm) and bosentan (Bos) (10 μm). C, western blots of HSL and phosphorylated (pHSL) on Ser660. Quantification of pHSL (normalized to total HSL) on (D) activatory Ser660 and (E) inhibitory Ser565. Western blots are representative of five independent experiments performed in duplicate except for (B) and (E) (n = 3 experiments performed in duplicate). *P < 0.05 and **P < 0.01 vs. untreated cells. # P < 0.05 vs. ET1‐treated cells; two‐way ANOVA.
Figure 7. Endothelin‐1 induces HSL phosphorylation in 3T3‐L1 cultivated in hypoxia .
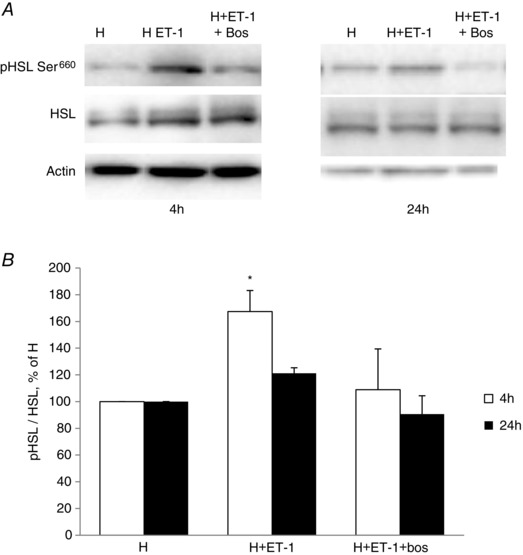
A, HSL phosphorylation on Ser660 in 3T3‐L1 adipocytes cultivated under hypoxia (H) (3% O2) and treated or not with ET‐1 (10 nm) and bosentan (Bos) (10 μm) for 4 or 24 h. B, quantification of the western blots. Western blots are representative of three independent experiments performed in triplicate. *P < 0.05 vs. control H; two‐way ANOVA. P = 0.054 at 4 h for H + ET1 + Bos vs. H + ET1.
Discussion
By combining in vivo and in vitro approaches, the present study demonstrates that activation of the endothelin system in adipocytes is involved in the structural and functional adipose tissue remodelling induced by intermittent hypoxia exposure. We showed that endothelin promotes adipocyte lipolysis and also inhibits glucose uptake in vitro. We have also identified modulation of HSL activity as a potential mechanism behind the effects of endothelin on adipocyte metabolism (Fig. 8).
Figure 8. Proposed mechanisms explaining the dual action of endothelin‐1 on glucose and lipid metabolism in adipocytes .
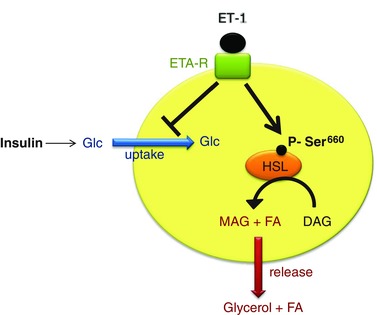
ET‐1 binding to its ETA receptor inhibits insulin‐induced glucose uptake and activates HSL phosphorylation on Ser660, thereby inducing diacylglycerol (DAG) lipolysis into monoacylglycerol (MAG) + fatty acid (FA). MAG conversion by monoglyceride lipase (not represented) results in glycerol and fatty acid release. Glc, glucose.
The decrease in adipose tissue mass and in adipocyte size induced by IH exposure is in agreement with recent data reported by our group (Poulain et al. 2014) and is indicative of increased lipolysis. We observed an epididymal fat loss of ∼1 g but it is probable that other fat compartments, such as subcutaneous fat, are also affected. Loss of body fat could be one of the mechanisms behind the lack of weight gain seen in IH‐exposed rats compared to control rats. In agreement, a recent meta‐analysis in OSA patients reported that continuous positive airway pressure treatment significantly increases the body mass index and body weight (Drager et al. 2015). Also relevant to the present study, there is clinical evidence for circulating ET‐1 levels becoming elevated in OSA patients and being normalized by continuous positive airway pressure treatment (Jordan et al. 2005; Gjorup et al. 2007). In addition, triglyceride levels were improved in patients treated with atrasantan, an ET‐1 receptor antagonist (Reriani et al. 2010). Similarly, in favour of a role of endothelin in the metabolic response to IH, we observed that bosentan treatment partially reversed fat loss and improved weight gain. Induction of the gene for ET‐1 is under the control of the HIF‐1 transcription factor in sustained hypoxia (Stow et al. 2011) and we have shown that the gene for ET‐1 was upregulated by HIF‐1 under intermittent hypoxia (Belaidi et al. 2009). In accordance, we observed that ET‐1 transcription was increased in epididymal fat of rats exposed to IH. Nevertheless, other lipolytic factors, upregulated or not by HIF‐1, might be induced by intermittent hypoxia. Indeed, functional changes potentially leading to lipolysis were reported in adipocytes exposed to hypoxia, including a loss of insulin sensitivity, leptin upregulation and adiponectin downregulation (Trayhurn, 2014). Leptin, in particular, is associated with respiratory drive control (Campo et al. 2007) and has been shown to be upregulated in rodents exposed to CIH (Fu et al. 2015); moreover, leptin is involved in lipolysis in white adipocytes (Fruhbeck et al. 2001). Therefore, leptin may participate in the lipolytic remodelling of adipose tissue induced by IH exposure.
In accordance with the lipolytic effects of endothelin, we observed an ET‐1‐induced lipolysis in vitro in 3T3‐L1 adipocytes along with inhibition of insulin‐induced glucose uptake, and these findings were maintained in cells cultured under hypoxic conditions, supporting our hypothesis that endothelin may be one of the main mediators of the IH‐induced reduction in adipocyte size. We also observed that 4 h, but not 24 h, of moderate hypoxia alone (3% O2) induced lipolysis (124.7% vs. 100% in hypoxia vs. normoxia, respectively, P = 0.028). Thus, at 4 h of exposure, the effects of hypoxia and ET‐1 were additive, leading to a global 75% increase in lipolysis compared to control normoxia. At 24 h, it appears that, in these cells and under moderate hypoxia (3% O2), the effect of ET‐1 is predominant over that of hypoxia alone. Lipolytic effects of hypoxia on 3T3‐L1 adipocytes have also been reported in several other studies (Hashimoto et al. 2013; Regazzetti et al. 2009; Yin et al. 2009).
The effects of endothelin on lipolysis and glucose uptake in 3T3‐L1 cells appear to be related to ETA, and not ETB, receptors, because they were blocked by BQ123 and bosentan but not by BQ788. It was also reported that ET‐1‐mediated lipolysis (Juan et al. 2005) and adiponectin secretion (Juan et al. 2007) were blocked by selective ETA‐R but not by ETB‐R antagonists. Similarly, a study on human adipocytes reported that ET‐1 increases lipolysis via the activation of ETA‐R (Eriksson et al. 2009). Therefore, endothelin appears to have anti‐insulin effects on lipolysis and glucose uptake. In agreement, another study showed that ET‐1 affects insulin signalling and counteracts insulin‐induced inhibition of lipolysis by decreasing the expression of insulin receptor, IRS‐1 and PDE‐3B, although, by contrast with our results, in this study ETB‐R through protein kinase C and calmodulin pathways, rather than ETA‐Rs, appear to mediate ET‐1 signalling (van Harmelen et al. 2008).
Although endothelin‐induced lipolysis in vitro has already been reported, very little is known about the mechanisms involved. We therefore investigated whether ET‐1 could modify the expression and/or phosphorylation of the two main lipases involved in adipose tissue lipolysis, namely HSL and ATGL. Although the expression of both lipases was not modified, ET‐1 induced the phosphorylation of HSL on Ser660 without affecting Ser565 phosphorylation. Because Ser660 phosphorylation is known to be activatory (Anthonsen et al. 1998), the ET‐1‐induced lipolysis could thus be explained by increased HSL activity, which promotes the second step of lipolysis. The classical pathway of Ser660HSL phosphorylation and lipolysis activation in adipocytes is cAMP‐dependent PKA activation. Although ET‐1 signalling is not coupled to cAMP production, it has the ability to activate PKA in a cAMP‐independent fashion (Dulin et al. 2001). Moreover, ET‐1 receptors are coupled to various signalling pathways, including protein kinase C, extracellular signal regulated kinase 1/2 and p38 mitogen‐activated protein kinase (Sugden, 2003), which can also modulate HSL and ATGL activity (Chaves et al. 2011) and be involved in its lipolytic effects.
In addition to the lipolytic effect of ET‐1, it could be hypothesized that the adipose tissue remodelling induced by IH could also be a result of the inhibition of FFA intake and triglyceride lipogenesis. Indeed, ET‐1 decreases adipogenesis during 3T3‐L1 adipocyte differentiation (Bhattacharya & Ullrich, 2006) and increases the expression of resistin, an adipogenesis‐inhibiting hormone (Tang et al. 2014). Although bosentan treatment decreased plasma triglycerides and FFAs in our rats, we did not observe any significant effect of intermittent hypoxia on these parameters. Most studies investigating the effects of IH on plasma lipid levels have been performed in mice and have shown that FFAs and/or triglycerides increase in response to short‐term or long‐term IH exposure (Li et al. 2005; Jun et al. 2010; Poulain et al. 2014). A possible explanation for our results could be that no lipoprotein lipase inhibitor was added to our samples. However, Perry et al. (2007) have shown that the duration of exposure was an important factor in determining the metabolic response to IH in rats because short‐term exposure did not affect plasma lipids, whereas a 3‐week exposure significantly increased triglyceride levels. Therefore, the lack of an effect observed in the present study could be the result of a species difference and/or the duration of IH exposure.
A novel finding of the present study is that intermittent hypoxia stimulates ET‐1 and ETA receptor expression in adipose tissue in vivo. Although ETA‐Rs were expressed by adipocytes, the origin of ET‐1 mRNA in adipose tissue remains unclear. Indeed, endothelial cells are well known for synthesizing endothelin in response to hypoxia (Yamashita et al. 2001). Although ET‐1 production by primary adipocyte has not been demonstrated, we observed a production of ET‐1 mRNA in differentiated 3T3‐L1 adipocytes suggesting, for the first time, that fat‐like cells could also contribute to adipose tissue ET‐1 secretion.
Another important finding is our observation that ET‐1 per se stimulates its own expression, as well as its ETA‐R expression, in cultured adipocytes. This could potentially induce a positive feedback loop contributing to enhanced endothelin system activation in response to IH. Our results, showing that bosentan treatment abolishes the adipose tissue increase in ET‐1 and ET‐A receptors, are in accordance with this hypothesis. However, the potential mechanisms involved remain to be determined. As already noted, the ET‐1 gene is induced by the HIF‐1 transcription factor and ET‐1 is known to activate HIF‐1 (Wilson et al. 2006; Spinella et al. 2007), possibly through its ability to promote NADPH expression and the generation of superoxide anions (Duerrschmidt et al. 2000; Li et al. 2003), which are powerful activators of HIF‐1 (Semenza, 2009). Promoting HIF‐1 activity could thereby provide a possible mechanism by which ET‐1 might induce a positive retroactive effect on its own transcription.
To conclude, the present study shows that blockade of the effects of endothelin by bosentan is able to reverse the intermittent hypoxia‐induced lipolysis in vivo. This could be explained by the ability of bosentan to inhibit the expression of both ET‐1 and ETA‐Rs. We confirmed these results in an adipocyte model in vitro, by showing that endothelin increases lipolysis in an ETA‐R‐dependent manner. Finally, we demonstrated that endothelin induces the phosphorylation of HSL on its activatory Ser660, providing a possible mechanism to explain its lipolytic effect in adipocytes.
Additional information
Competing interests
The authors declare that they have no competing interests.
Funding
This study was supported by funding from Université Grenoble Alpes, CHU de Grenoble, INSERM and from the Fonds de Dotation Agir Pour les Maladies Chroniques (APMC). Bosentan was graciously provided by Actelion Pharmaceuticals (Allschwil, Switzerland).
Author contributions
ABM, MH, DM, FHP and PF conceived, designed and performed the experiments, and analysed the data. DGR designed the study. ABM, DM, MH, PF, JLP and DGR were involved in manuscript preparation. All authors revised the manuscript for intellectual and form improvements. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Supporting information
Table S1. Primer sequences and qPCR experimental conditions
Acknowledgements
The authors wish to thank Sandrine Cachot and Aurélie Dariz for their technical assistance, as well as Dr Sabine Steffens for kindly providing 3T3‐L1 cells.
References
- Anthonsen MW, Ronnstrand L, Wernstedt C, Degerman E & Holm C (1998). Identification of novel phosphorylation sites in hormone‐sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem 273, 215–221. [DOI] [PubMed] [Google Scholar]
- Arnaud C, Poulain L, Levy P & Dematteis M (2011). Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein‐E knock out mice. Atherosclerosis 219, 425–431. [DOI] [PubMed] [Google Scholar]
- Belaidi E, Joyeux‐Faure M, Ribuot C, Launois SH, Levy P & Godin‐Ribuot D (2009). Major role for hypoxia inducible factor‐1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. J Am Coll Cardiol 53, 1309–1317. [DOI] [PubMed] [Google Scholar]
- Bhattacharya I & Ullrich A (2006). Endothelin‐1 inhibits adipogenesis: role of phosphorylation of Akt and ERK1/2. FEBS Lett 580, 5765–5771. [DOI] [PubMed] [Google Scholar]
- Campo A, Fruhbeck G, Zulueta JJ, Iriarte J, Seijo LM, Alcaide AB, Galdiz JB & Salvador J (2007). Hyperleptinaemia, respiratory drive and hypercapnic response in obese patients. Eur Respir J 30, 223–231. [DOI] [PubMed] [Google Scholar]
- Chaves VE, Frasson D & Kawashita NH (2011). Several agents and pathways regulate lipolysis in adipocytes. Biochimie 93, 1631–1640. [DOI] [PubMed] [Google Scholar]
- Chien Y, Lai YH, Kwok CF & Ho LT (2011). Endothelin‐1 suppresses long‐chain fatty acid uptake and glucose uptake via distinct mechanisms in 3T3‐L1 adipocytes. Obesity (Silver Spring) 19, 6–12. [DOI] [PubMed] [Google Scholar]
- Delarue J & Magnan C (2007). Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care 10, 142–148. [DOI] [PubMed] [Google Scholar]
- Drager LF, Brunoni AR, Jenner R, Lorenzi‐Filho G, Bensenor IM & Lotufo PA (2015). Effects of CPAP on body weight in patients with obstructive sleep apnoea: a meta‐analysis of randomised trials. Thorax 70, 258–264. [DOI] [PubMed] [Google Scholar]
- Drager LF, Jun JC & Polotsky VY (2010). Metabolic consequences of intermittent hypoxia: relevance to obstructive sleep apnea. Best Pract Res Clin Endocrinol Metab 24, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drager LF, Yao Q, Hernandez KL, Shin MK, Bevans‐Fonti S, Gay J, Sussan TE, Jun JC, Myers AC, Olivecrona G, Schwartz AR, Halberg N, Scherer PE, Semenza GL, Powell DR & Polotsky VY (2013). Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin‐like 4. Am J Respir Crit Care Med 188, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ & Morawietz H (2000). Endothelin‐1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun 269, 713–717. [DOI] [PubMed] [Google Scholar]
- Dulin NO, Niu J, Browning DD, Ye RD & Voyno‐Yasenetskaya T (2001). Cyclic AMP‐independent activation of protein kinase A by vasoactive peptides. J Biol Chem 276, 20827–20830. [DOI] [PubMed] [Google Scholar]
- Eriksson AK, van Harmelen V, Stenson BM, Astrom G, Wahlen K, Laurencikiene J & Ryden M (2009). Endothelin‐1 stimulates human adipocyte lipolysis through the ET A receptor. Int J Obes (Lond) 33, 67–74. [DOI] [PubMed] [Google Scholar]
- Fruhbeck G, Gomez‐Ambrosi J & Salvador J (2001). Leptin‐induced lipolysis opposes the tonic inhibition of endogenous adenosine in white adipocytes. Faseb J 15, 333–340. [DOI] [PubMed] [Google Scholar]
- Fruhbeck G, Mendez‐Gimenez L, Fernandez‐Formoso JA, Fernandez S & Rodriguez A (2014). Regulation of adipocyte lipolysis. Nutr Res Rev 27, 63–93. [DOI] [PubMed] [Google Scholar]
- Fu C, Jiang L, Zhu F, Liu Z, Li W, Jiang H, Ye H, Kushida CA & Li S (2015). Chronic intermittent hypoxia leads to insulin resistance and impaired glucose tolerance through dysregulation of adipokines in non‐obese rats. Sleep Breath 19, 1467–1473. [DOI] [PubMed] [Google Scholar]
- Garvey JF, Taylor CT & McNicholas WT (2009). Cardiovascular disease in obstructive sleep apnoea syndrome: the role of intermittent hypoxia and inflammation. Eur Respir J 33, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R & Mathieu C (2001). An overview of real‐time quantitative PCR: applications to quantify cytokine gene expression. Methods 25, 386–401. [DOI] [PubMed] [Google Scholar]
- Gjorup PH, Sadauskiene L, Wessels J, Nyvad O, Strunge B & Pedersen EB (2007). Abnormally increased endothelin‐1 in plasma during the night in obstructive sleep apnea: relation to blood pressure and severity of disease. Am J Hypertens 20, 44–52. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Yokokawa T, Endo Y, Iwanaka N, Higashida K & Taguchi S (2013). Modest hypoxia significantly reduces triglyceride content and lipid droplet size in 3T3‐L1 adipocytes. Biochem Biophys Res Commun 440, 43–49. [DOI] [PubMed] [Google Scholar]
- Ishibashi KI, Imamura T, Sharma PM, Huang J, Ugi S & Olefsky JM (2001). Chronic endothelin‐1 treatment leads to heterologous desensitization of insulin signaling in 3T3‐L1 adipocytes. J Clin Invest 107, 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan W, Reinbacher A, Cohrs S, Grunewald RW, Mayer G, Ruther E & Rodenbeck A (2005). Obstructive sleep apnea: plasma endothelin‐1 precursor but not endothelin‐1 levels are elevated and decline with nasal continuous positive airway pressure. Peptides 26, 1654–1660. [DOI] [PubMed] [Google Scholar]
- Juan CC, Chang CL, Lai YH & Ho LT (2005). Endothelin‐1 induces lipolysis in 3T3‐L1 adipocytes. Am J Physiol Endocrinol Metab 288, E1146–E1152. [DOI] [PubMed] [Google Scholar]
- Juan CC, Chang LW, Huang SW, Chang CL, Lee CY, Chien Y, Hsu YP, Ho PH, Chen YC & Ho LT (2006). Effect of endothelin‐1 on lipolysis in rat adipocytes. Obesity (Silver Spring) 14, 398–404. [DOI] [PubMed] [Google Scholar]
- Juan CC, Chuang TY, Chang CL, Huang SW & Ho LT (2007). Endothelin‐1 regulates adiponectin gene expression and secretion in 3T3‐L1 adipocytes via distinct signaling pathways. Endocrinology 148, 1835–1842. [DOI] [PubMed] [Google Scholar]
- Jun J, Reinke C, Bedja D, Berkowitz D, Bevans‐Fonti S, Li J, Barouch LA, Gabrielson K & Polotsky VY (2010). Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E‐deficient mice. Atherosclerosis 209, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampidonis AD, Rogdakis E, Voutsinas GE & Stravopodis DJ (2011). The resurgence of Hormone‐Sensitive Lipase (HSL) in mammalian lipolysis. Gene 477, 1–11. [DOI] [PubMed] [Google Scholar]
- Levy P, Pepin JL, Arnaud C, Baguet JP, Dematteis M & Mach F (2009). Obstructive sleep apnea and atherosclerosis. Prog Cardiovasc Dis 51, 400–410. [DOI] [PubMed] [Google Scholar]
- Levy P, Tamisier R, Arnaud C, Monneret D, Baguet JP, Stanke‐Labesque F, Dematteis M, Godin‐Ribuot D, Ribuot C & Pepin JL (2012). Sleep deprivation, sleep apnea and cardiovascular diseases. Front Biosci (Elite Ed) 4, 2007–2021. [DOI] [PubMed] [Google Scholar]
- Li J, Thorne LN, Punjabi NM, Sun CK, Schwartz AR, Smith PL, Marino RL, Rodriguez A, Hubbard WC, O'Donnell CP & Polotsky VY (2005). Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res 97, 698–706. [DOI] [PubMed] [Google Scholar]
- Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ & Chen AF (2003). Endothelin‐1 increases vascular superoxide via endothelin(A)‐NADPH oxidase pathway in low‐renin hypertension. Circulation 107, 1053–1058. [DOI] [PubMed] [Google Scholar]
- Magalang UJ, Cruff JP, Rajappan R, Hunter MG, Patel T, Marsh CB, Raman SV & Parinandi NL (2009). Intermittent hypoxia suppresses adiponectin secretion by adipocytes. Exp Clin Endocrinol Diabetes 117, 129–134. [DOI] [PubMed] [Google Scholar]
- Nagele U, Wahlefeld, AW ., Ziegernhorn, J (1985). Lipids: Fatty acids and derivatives. Triglycerides. Colorimetric method In Method in Enzymatic Analysis, 3rd edn. ed. Bergmeyer HU, pp. 12–18. VHC Publishers, Deerfield Beach, FL. [Google Scholar]
- Perry JC, D'Almeida V, Souza FG, Schoorlemmer GH, Colombari E & Tufik S (2007). Consequences of subchronic and chronic exposure to intermittent hypoxia and sleep deprivation on cardiovascular risk factors in rats. Respir Physiol Neurobiol 156, 250–258. [DOI] [PubMed] [Google Scholar]
- Poulain L, Thomas A, Rieusset J, Casteilla L, Levy P, Arnaud C & Dematteis M (2014). Visceral white fat remodeling contributes to intermittent hypoxia‐induced atherogenesis. Eur Respir J 43, 513–522. [DOI] [PubMed] [Google Scholar]
- Regazzetti C, Peraldi P, Gremeaux T, Najem‐Lendom R, Ben‐Sahra I, Cormont M, Bost F, Le Marchand‐Brustel Y, Tanti JF & Giorgetti‐Peraldi S (2009). Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes 58, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reriani M, Raichlin E, Prasad A, Mathew V, Pumper GM, Nelson RE, Lennon R, Rihal C, Lerman LO & Lerman A (2010). Long‐term administration of endothelin receptor antagonist improves coronary endothelial function in patients with early atherosclerosis. Circulation 122, 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savransky V, Jun J, Li J, Nanayakkara A, Fonti S, Moser AB, Steele KE, Schweitzer MA, Patil SP, Bhanot S, Schwartz AR & Polotsky VY (2008). Dyslipidemia and atherosclerosis induced by chronic intermittent hypoxia are attenuated by deficiency of stearoyl coenzyme A desaturase. Circ Res 103, 1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FH (1961). [Enzymatic determination of glucose and fructose simultaneously]. Klin Wochenschr 39, 1244–1247. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2009). Regulation of oxygen homeostasis by hypoxia‐inducible factor 1. Physiology (Bethesda) 24, 97–106. [DOI] [PubMed] [Google Scholar]
- Spinella F, Rosano L, Di Castro V, Decandia S, Nicotra MR, Natali PG & Bagnato A (2007). Endothelin‐1 and endothelin‐3 promote invasive behavior via hypoxia‐inducible factor‐1alpha in human melanoma cells. Cancer Res 67, 1725–1734. [DOI] [PubMed] [Google Scholar]
- Stow LR, Jacobs ME, Wingo CS & Cain BD (2011). Endothelin‐1 gene regulation. Faseb J 25, 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden PH (2003). An overview of endothelin signaling in the cardiac myocyte. J Mol Cell Cardiol 35, 871–886. [DOI] [PubMed] [Google Scholar]
- Tang YC, Liu CW, Chang HH, Juan CC, Kuo YC, Kao CC, Huang YM & Kao YH (2014). Endothelin‐1 stimulates resistin gene expression. Endocrinology 155, 854–864. [DOI] [PubMed] [Google Scholar]
- Trayhurn P (2014). Hypoxia and adipocyte physiology: implications for adipose tissue dysfunction in obesity. Annu Rev Nutr 34, 207–236. [DOI] [PubMed] [Google Scholar]
- Trzepizur W, Le Vaillant M, Meslier N, Pigeanne T, Masson P, Humeau MP, Bizieux‐Thaminy A, Goupil F, Chollet S, Ducluzeau PH & Gagnadoux F (2013). Independent association between nocturnal intermittent hypoxemia and metabolic dyslipidemia. Chest 143, 1584–1589. [DOI] [PubMed] [Google Scholar]
- van Harmelen V, Eriksson A, Astrom G, Wahlen K, Naslund E, Karpe F, Frayn K, Olsson T, Andersson J, Ryden M & Arner P (2008). Vascular peptide endothelin‐1 links fat accumulation with alterations of visceral adipocyte lipolysis. Diabetes 57, 378–386. [DOI] [PubMed] [Google Scholar]
- Wilson JL, Burchell J & Grimshaw MJ (2006). Endothelins induce CCR7 expression by breast tumor cells via endothelin receptor A and hypoxia‐inducible factor‐1. Cancer Res 66, 11802–11807. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Discher DJ, Hu J, Bishopric NH & Webster KA (2001). Molecular regulation of the endothelin‐1 gene by hypoxia. Contributions of hypoxia‐inducible factor‐1, activator protein‐1, GATA‐2, AND p300/CBP. J Biol Chem 276, 12645–12653. [DOI] [PubMed] [Google Scholar]
- Yin J, Gao Z, He Q, Zhou D, Guo Z & Ye J (2009). Role of hypoxia in obesity‐induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab 296, E333–E342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences and qPCR experimental conditions