

Abstract
Key points
Raised arterial blood CO2 (hypercapnia) is a feature of many lung diseases.
CO2 has been shown to act as a cell signalling molecule in human cells, notably by influencing the levels of cell signalling second messengers: cAMP and Ca2+.
Hypercapnia reduced cAMP‐stimulated cystic fibrosis transmembrane conductance regulator‐dependent anion and fluid transport in Calu‐3 cells and primary human airway epithelia but did not affect cAMP‐regulated HCO3 − transport via pendrin or Na+/HCO3 − cotransporters.
These results further support the role of CO2 as a cell signalling molecule and suggests CO2‐induced reductions in airway anion and fluid transport may impair innate defence mechanisms of the lungs.
Abstract
Hypercapnia is clinically defined as an arterial blood partial pressure of CO2 of above 40 mmHg and is a feature of chronic lung disease. In previous studies we have demonstrated that hypercapnia modulates agonist‐stimulated cAMP levels through effects on transmembrane adenylyl cyclase activity. In the airways, cAMP is known to regulate cystic fibrosis transmembrane conductance regulator (CFTR)‐mediated anion and fluid secretion, which contributes to airway surface liquid homeostasis. The aim of the current work was to investigate if hypercapnia could modulate cAMP‐regulated ion and fluid transport in human airway epithelial cells. We found that acute exposure to hypercapnia significantly reduced forskolin‐stimulated elevations in intracellular cAMP as well as both adenosine‐ and forskolin‐stimulated increases in CFTR‐dependent transepithelial short‐circuit current, in polarised cultures of Calu‐3 human airway cells. This CO2‐induced reduction in anion secretion was not due to a decrease in HCO3 − transport given that neither a change in CFTR‐dependent HCO3 − efflux nor Na+/HCO3 − cotransporter‐dependent HCO3 − influx were CO2‐sensitive. Hypercapnia also reduced the volume of forskolin‐stimulated fluid secretion over 24 h, yet had no effect on the HCO3 − content of the secreted fluid. Our data reveal that hypercapnia reduces CFTR‐dependent, electrogenic Cl− and fluid secretion, but not CFTR‐dependent HCO3 − secretion, which highlights a differential sensitivity of Cl− and HCO3 − transporters to raised CO2 in Calu‐3 cells. Hypercapnia also reduced forskolin‐stimulated CFTR‐dependent anion secretion in primary human airway epithelia. Based on current models of airways biology, a reduction in fluid secretion, associated with hypercapnia, would be predicted to have important consequences for airways hydration and the innate defence mechanisms of the lungs.
Key points
Raised arterial blood CO2 (hypercapnia) is a feature of many lung diseases.
CO2 has been shown to act as a cell signalling molecule in human cells, notably by influencing the levels of cell signalling second messengers: cAMP and Ca2+.
Hypercapnia reduced cAMP‐stimulated cystic fibrosis transmembrane conductance regulator‐dependent anion and fluid transport in Calu‐3 cells and primary human airway epithelia but did not affect cAMP‐regulated HCO3 − transport via pendrin or Na+/HCO3 − cotransporters.
These results further support the role of CO2 as a cell signalling molecule and suggests CO2‐induced reductions in airway anion and fluid transport may impair innate defence mechanisms of the lungs.
Abbreviations
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- Isc
short circuit current
- NBC
Na+/HCO3 − cotransporter
- NHE
Na+/H+ exchanger
- pHi
intracellular pH
- pHe
extracellular pH
- PKA
protein kinase A
- sAC
soluble adenylyl cyclase
- tmAC
transmembrane adenylyl cyclase
- Vte
transepithelial voltage
Introduction
Carbon dioxide constitutes 0.04% by volume of the Earth's atmosphere (van der Laan‐Luijkx et al. 2013) and has major roles in plant, prokaryote and animal biology (Cummins et al. 2014). In plants, CO2 is used to synthesise sugars during photosynthesis whilst in animals, although CO2 is a waste product of cellular respiration, it also has an important roles in maintaining plasma pH via its buffering effect on HCO3 − (Marques et al. 2003) as well as stimulation of peripheral and central chemoreceptors to regulate ventilation (Somers et al. 1989; Guyenet et al. 2010). Elevated CO2 in arterial blood (hypercapnia) is associated with lung disease in humans (Lourenco & Miranda, 1968; Prin et al. 2002), yet the effects of hypercapnia in human physiology are not fully understood. In mammals, recent studies have provided strong evidence that CO2 can act as a bona fide cell signalling molecule, and that changes in CO2 alter the activity of a variety of membrane transporters, including connexin 26 (Huckstepp et al. 2010 a,b; Meigh et al. 2013), the epithelial Na+/HCO3 − cotransporter (NBC) (Adijanto et al. 2009), inwardly rectifying K+ channels (Huckstepp & Dale, 2011) and the Na+/K+‐ATPase (Briva et al. 2007; Vadasz et al. 2008). The action of CO2 on membrane transporters has been shown to involve different mechanisms. For instance, CO2‐dependent downregulation of Na+/K+‐ATPase activity specifically involves the endocytosis of the α subunit of the Na+/K+‐ATPase, demonstrating that CO2 can alter surface expression of ion transporters (Briva et al. 2007). Alternatively, CO2 directly modulates connexin 26 via carbamylation, a post‐translational modification whereby a covalent bond forms between the carbon in CO2 and a primary amine group of the target protein (Meigh et al. 2013). In addition, CO2 also has reported effects on key cell second messengers involved in membrane transporter regulation, specifically cAMP and Ca2+ (Cann et al. 2003; Cann, 2004). cAMP is synthesised from ATP, a reaction catalysed by adenylyl cyclase, of which there exists both membrane‐bound transmembrane adenylyl cyclase (tmAC) and the soluble adenylyl cyclase (sAC) in mammals (Buck et al. 1999). Our laboratory has previously shown that the activity of a recombinant, catalytically active mammalian tmAC, expressed in HEK 293T cells, was significantly higher in cells exposed to 5% CO2 compared to those exposed to 0.03% CO2, demonstrating that tmAC is sensitive to changes in CO2 (Townsend et al. 2009). This study also showed that tmAC was sensitive to CO2 but not HCO3 − in vivo and in vitro, supporting previous findings that first proposed tmAC activity was only sensitive to CO2 and not inorganic carbon per se (Hammer et al. 2006). More recently, we have shown that incubating OK cells (a model of human proximal tubule cells) in 10% CO2 caused a significant reduction in both forskolin and parathyroid hormone‐stimulated increases in intracellular cAMP ([cAMP]i) compared to levels measured under normocapnic conditions of 5% CO2 (Cook et al. 2012). The decrease in cAMP correlated with an enhanced activity of the Na+/H+ exchanger (NHE) 3, a transporter known to be negatively regulated by cAMP/protein kinase A (PKA), thus providing evidence that hypercapnia was able to modulate cAMP‐regulated transporters in human epithelial cells. This work further showed that the effect of raised CO2 on cAMP was dependent on an IP3‐dependent release of Ca2+ which, in turn, led to an inhibition in tmAC activity, thereby demonstrating that CO2 affected Ca2+ as well as cAMP signalling. These data supported earlier studies that demonstrated CO2 modulated Ca2+ signalling in other mammalian and human cells (Nishio et al. 2001; Bouyer et al. 2003; Briva et al. 2011).
In the airways, cAMP plays a major role in regulating the volume and composition of the airway surface liquid (ASL). In the upper airways, ASL secretion occurs predominantly from serous cells of the submucosal glands (SMGs). Studies on intact SMG secretions as well as SMG‐derived secretory cell lines, such as Calu‐3, have found that elevations in intracellular cAMP stimulate cystic fibrosis transmembrane conductance regulator (CFTR)‐dependent Cl−, HCO3 − and fluid transport (Lee et al. 1998; Devor et al. 1999; Joo et al. 2002; Krouse et al. 2004; Ballard et al. 2006; Ianowski et al. 2007; Lee & Foskett, 2010; Garnett et al. 2011; Huang et al. 2012; Shan et al. 2012). Efficient anion secretion in the airways is paramount to maintain ASL hydration and pH, as well as efficient mucus secretion and expansion (Garcia et al. 2009; Chen et al. 2010; Gustafsson et al. 2012; Ridley et al. 2014). Loss of functional expression of CFTR at the apical membrane of HCO3 −‐secreting epithelia underlies the hereditary disease cystic fibrosis (CF) and airways dehydration and impaired ASL alkalinisation have been reported in CF airways (Coakley et al. 2003; Song et al. 2006; Boucher, 2007) consistent with a key role for CFTR in mediating airway HCO3 − secretion. Furthermore, it has been shown that the acidic ASL found in CF pigs compromises the ability to kill airway pathogens (Pezzulo et al. 2012) and provides a plausible explanation as to why CF patients are susceptible to airway bacterial colonisation.
Given the previously reported findings from our laboratory that hypercapnia modulated cAMP signalling in renal epithelial cells (Cook et al. 2012), we hypothesised that hypercapnia would also affect airway epithelial cell function. Our results show that hypercapnia reduced cAMP levels in Calu‐3 cells and this correlated with a drop in cAMP‐dependent anion secretion. The reduction in anion secretion appeared primarily due to a reduction in Cl− transport, given that both CFTR‐dependent HCO3 − efflux via pendrin, and NBC‐dependent HCO3 − import were unaffected by hypercapnia. Furthermore, hypercapnia also reduced the volume of cAMP‐stimulated fluid secretion without affecting the HCO3 − content of the fluid, implying that Cl− secretion and HCO3 − secretion have differential sensitivities to hypercapnia. Hypercapnia also reduced cAMP‐stimulated anion secretion in primary human bronchial epithelial layers, indicating this effect of CO2 would be predicted to occur in vivo. Our results therefore demonstrate that CO2 acts as a signalling molecule in human airway epithelia to downregulate anion and fluid secretion.
Methods
Calu‐3 cell culture
The human serous cell line, Calu‐3 (Shen et al. 1994), was grown in Eagle's minimum essential medium supplemented with 10% (v/v) fetal calf serum, 1% (v/v) non‐essential amino acids, 2 mm l‐glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were incubated at 37°C in humidified air containing 5% (v/v) CO2 and were used between passage 20 and 50. Unless otherwise stated, 250,000 cells were seeded onto either 12 mm Costar Transwells or 12 mm Snapwells, 0.4 μm pore size, polyester membrane inserts, and grown under submerged conditions with 500 μl growth media applied to the apical compartment of membrane inserts. Transepithelial electrical resistance (TEER) was routinely measured using an epithelial voltohmmeter (WPI, Hitchin, UK) and cells generally reached a confluent monolayer, with a TEER of above 600 Ω cm−2 after 6 days of growth on Transwell inserts. Experiments were performed 9–13 days after seeding.
Primary human bronchial epithelial cell culture
Ethical approval was granted for this work from Newcastle and North Tyneside 2 [Min Ref: 2001/179]. Differentiated primary bronchial epithelial cells were derived from bronchial brushings taken from lung transplant recipients during surveillance bronchoscopy as previously described (Forrest et al. 2005). These were grown in a CO2 incubator (37°C; 5% CO2) to 90% confluence using bronchial epithelial growth medium with supplements (BEGM, Lonza, Tewkesbury, UK) in T25 flasks pre‐coated with 32 μg ml−1 collagen. Cells were passaged using a standard trypsin/EDTA technique and cryopreserved for future use. After reconstitution, cells were again expanded to near confluence in T25 flasks, before being seeded onto collagen‐coated 12 mm Costar Snapwells at a density of 100,000 cells per membrane in 0.5 ml BEGM, with 2 ml of this medium applied to the basal chamber. Confluence was reached after 72 h, at which point the cell culture was taken to the air–liquid interface (ALI). Here, the medium above the cells was removed completely, and the cells were subsequently fed only from the basal chamber with an ALI medium as described by Fulcher et al. (2005). Ciliogenesis was first observed at 14 days at the ALI, and short‐circuit current measurements were performed 30–35 days after growth at the ALI.
Short‐circuit current measurements
Cells were grown on Snapwell inserts and mounted into a Ussing chamber in which each chamber was connected to a calomel voltage sensing electrode and an AgCl2 current sensing electrode by 3 m KCl salt bridges containing 3% (w/v) agar. Cells were bathed in 7.5 ml Krebs solution and continually gassed with either 5% (v/v) CO2/95% (v/v) O2 for control conditions or 10% (v/v) CO2/90% (v/v) O2 to induce hypercapnia. To measure the short circuit current (I sc), cells were clamped at 0 mV using a DVC‐1000 Voltage/Current Clamp (WPI) and a Powerlab 1200 feedback amplifier (AD Instruments, Oxford, UK) injected the appropriate current to clamp transepithelial voltage (V te) to 0 mV, which was recorded as the I sc using Scope 3 software (AD Instruments). To monitor transepithelial resistance (R te), a 2 s, 10 mV pulse was applied every 30 s.
Intracellular pH measurements
Calu‐3 cells were grown on Transwell inserts and loaded with the pH‐sensitive, fluorescent dye BCECF‐AM (10 μm) for 1 h in a NaHEPES buffered solution at 37°C. Cells were mounted on to the stage of a Nikon fluor inverted microscope and perfused with a modified Krebs solution gassed with either 5% (v/v) CO2/95% (v/v) O2 or 10% (v/v) CO2/90% (v/v) O2. Solutions were perfused across the apical and basolateral membranes at 37°C at a speed of 3 ml min−1 (apical) and 6 ml min−1 (basolateral). Intracellular pH (pHi) was measured using a Life Sciences Microfluorimeter System in which cells were alternately excited at 490 and 440 nm wavelengths every 1.024 s with emitted light collected at 510 nm. The ratio of 490 to 440 nm emission was recorded using PhoCal 1.6 b software and calibrated to pHi using the high K+/nigericin technique (Hegyi et al. 2003) in which cells were exposed to high K+ solutions containing 10 μm nigericin, set to a desired pH, ranging from 6.6 to 8.4. Total buffering capacity (βtot) was calculated by addition of the intrinsic buffering capacity (βi) to the buffering capacity of the CO2–HCO3 − buffer system (βHCO3 −) in which βi was calculated using the NH4 + technique as described by Roos and Boron (1981). For analysis of pHi measurements, ΔpHi was determined by calculating the mean pHi over 60 s resulting from treatment. The rate of pHi change (ΔpHi/Δt) was determined by performing a linear regression over a period of at least 30 s which was converted to a transmembrane HCO3 − flux [–J(B)] by multiplying ΔpHi/Δt by βtot.
Radiolabelled cAMP assay
Calu‐3 cells were cultured in Corning 12‐well plates at an initial seeding density of 3 × 105 cells per well and used at approximately 80% confluency. Cells were loaded with 2 μCi ml−1 [3H]‐adenine and incubated for 2 h at 37°C in humidified air containing 5% (v/v) CO2. Cells were then washed twice with PBS and incubated for a further 30 min at 37°C in humidified air containing 5% (v/v) CO2/95% (v/v) O2 (normocapnic controls) or 10% (v/v) CO2/90% (v/v) O2 (hypercapnia). Incubation was performed in growth medium containing 1 mm 3‐isobutyl‐1‐methylxanthine (IBMX) that had been pregassed with the appropriate CO2 concentration and titrated to pH 7.4 using 1 m NaOH. Forskolin (5 μm) was then added to the cells for 10 min before the assay was ended by removal of media and lysis of cells by adding 5% (w/v) trichloroacetic acid containing 1 mm ATP and 1 mm cAMP for 1 h at 4°C. cAMP levels in lysates were measured by the twin column chromatography procedure described by Johnson et al. (1994).
Cell surface biotinylation
Calu‐3 cells were grown on Transwell inserts and washed three times with PBS. Cells were then incubated at 37°C in humidified air containing 5% (v/v) CO2 (control) or 10% (v/v) CO2 (hypercapnia) in pregassed high Cl− Krebs solution for 20 min. The solution was removed and cells were incubated for 30 min at 4°C in PBS++ (PBS containing 0.1 mm Ca2+ and 1 mm Mg2+; pH 8.0) with 0.5 mg ml−1 EZ‐Link Sulfo‐NHS‐Biotin (Thermo Scientific, Waltham, MA, USA) added to the apical membrane. Biotinylation was stopped by removal of the apical solution and addition of ice cold PBS++. Cells were then lysed using RIPA buffer containing 150 mm NaCl, 20 mm Tris, 1% Triton‐X‐100, 0.1% SDS and 0.08% sodium deoxycholate (pH8.0) with one protease inhibitor cocktail tablet (Roche Applied Sciences, Penzberg, Germany) added to 50 ml RIPA buffer. The lysate was collected and centrifuged for 15 min at 16,200 × g at 4°C and the protein concentration of the supernatant was assessed using the BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA). One hundred micrograms of protein was taken to be used for analysis of whole cell protein expression. Streptavidin agarose beads (Novagen, Billerica, MA, USA) that had been equilibrated with PBS++ and RIPA buffer were added to the remaining protein at 1 μl beads/20 μg protein and incubated overnight at 4°C with continuous inversion of samples to ensure thorough mixing. These samples were then centrifuged and washed five times with RIPA buffer and heated to 65°C for 5 min. Protein expression was then detected by Western blot.
Western blot
SDS‐PAGE using 7% gels was performed on all samples at 120 V for 2 h. Samples were then transferred to a nitrocellulose membrane at 400 mA for 90 min at 4°C. The membrane was blocked for 1 h in blocking buffer consisting of Tris‐buffered saline (TBS) + 0.1% Tween 20 (TTBS) containing 5% dried skimmed milk powder (Compliments) before primary mouse anti‐CFTR monoclonal antibody 23C5 (1:200 dilution in TBS) and mouse anti‐α tubulin antibody (1:1000 dilution in TBS) were added overnight at 4°C. The membrane was then washed using TTBS before a goat anti‐mouse antibody labelled with HRP was added at 1:5000 dilution in TBS for 1 h. Any unbound secondary antibody was then washed off with TTBS. To detect any HRP activity, equal volumes of the enhanced chemiluminescent substrates Enhanced Luminol Reagent and the Oxidizing Reagent (Thermo Scientific) were added to the blot for 10 min before the blot was exposed to Kodak Scientific Imaging film for 30 s. The film was developed and the band intensity was analysed using ImageJ software.
Fluid secretion assays
Calu‐3 cells were grown on Transwell inserts and washed three times with PBS to remove any mucus that may have accumulated over time. Extra care was taken when removing the PBS to ensure no residual fluid remained in the Transwell at the end of the washes. Solutions were then added to the cells (1 ml basolaterally, 200 μl apically) and cells were incubated at 37°C in humidified air containing 5% (v/v) CO2 (control) or 10% (v/v) CO2 (hypercapnia) for 24 h (Garnett et al. 2011). The apical fluid was then removed and its volume was measured. First, 180 μl was removed and then the rest of the fluid was removed 1 μl at a time to ensure high accuracy. Samples were collected in an Eppendorf tube and after a full equilibration in either 5 or 10 % CO2, pH was assessed using a MiniTrode lab pH electrode (Hamilton, Reno, NV, USA). This enabled the HCO3 − concentration of the secreted fluid to be calculated using the Henderson–Hasselbalch equation, where; where pKa = 6.1 (the negative log of the carbonic acid dissociation constant).
Periodic acid‐Schiffs (PAS) assay
Given that it has been reported that Calu‐3 cells secrete mucins, notably MUC5AC (Kreda et al. 2007, 2010), the PAS assay was used to detect the glycoprotein content of the secreted fluid as an indicator of secreted mucin. To generate a standard curve, pig mucin (a gift from Prof. Jeff Pearson, Newcastle University) was diluted to 100, 50, 20, 10, 5, 2 and 1 μg ml−1 and 100 μl of standards was added to a 96‐well plate in duplicate. Then, 100 μl of sample was made to 1 ml by addition of deionised water and 100 μl was added to wells in duplicate. One hundred microlitres of a periodic acid/acetic acid mix (made from 10 μl periodic acid added to 7% acetic acid) was added to all standards and samples and the plate were incubated for 60 min at 37°C. In total, 100 μl of 1.6% sodium metabisulphate solution in Schiff's reagent was added to all standards and samples. The plate was then incubated at room temperature for 30 min before absorbance was read at 550 nm using an ELx808 Absorbance Microplate Reader (BioTek, Winooski, VT, USA). Absorbance was then converted to mucin concentration using the standard curve.
Solutions and reagents
All reagents were purchased from Sigma Aldrich (Poole, UK) apart from forskolin and ouabain (R & D Systems, Abingdon, UK), BCECF‐AM (Invitrogen, Paisley, UK) and GlyH‐101 and CFTRinh 172 (Calbiochem, Watford, UK). All gas cylinders were purchased from BOC (Guildford, UK) and consisted of the following mixtures: 5% CO2/95% O2 and 10% CO2/90% O2. NaHEPES solution consisted of (in mm) 130 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 NaHEPES and 10 d‐glucose, pH 7.4 at 37°C. High Cl− Krebs solution consisted of (in mm) 25 NaHCO3, 115 NaCl, 5 KCl, 1 CaCl2 1 MgCl2 and 10 d‐glucose (pH 7.4). For high Cl−, Na+‐free solutions, NaHCO3 was replaced with choline bicarbonate and NaCl was replaced with N‐methyl‐d‐glucamine (NMDG)‐Cl. Zero Cl− Krebs solution consisted of (in mm) 25 NaHCO3, 115 Na‐gluconate, 2.5 K2SO4, 1 Ca‐gluconate, 1 Mg‐gluconate and 10 d‐glucose. pHi calibration solutions consisted of (in mm) 5 NaCl, 130 KCl, 1 CaCl2, 1 MgCl2, 10 d‐glucose, 10 HEPES (for solutions set at pH 7.6 or below) or 10 Tris (for solutions set at pH 7.8 or above) as well as 10 μm nigericin. Solutions were set to the desired pH by using 1 m HCl or 1 m NaOH. Solutions used to determine intracellular buffering capacity consisted of (in mm) 4.5 KCl, 1 MgCl2, 2 CaCl2, 5 BaCl, 10 Hepes, 10 d‐glucose as well as varying concentrations of NH4Cl/NMDG‐Cl, ranging from 0 NH4Cl/145 NMDG‐Cl to 30 NH4Cl/115 NMDG‐Cl. All solutions were titrated to pH 7.4 at 37°C using 1 m CsOH.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 4 software. Results are expressed as mean ± SEM of n observations. Student's t test, one‐way ANOVA (with Tukey's multiple comparison post‐test) or two‐way ANOVA (with Bonferroni post‐test) were carried out where applicable to determine statistical significance between measurements. A P value of < 0.05 was considered statistically significant.
Results
Acute hypercapnia attenuates forskolin‐stimulated cAMP levels in Calu‐3 cells independent of changes in pHi
We first assessed the effect of hypercapnia on the pHi of Calu‐3 cells as it is well known that raising CO2 generally induces cytosolic acidification. Cells were first perfused with Krebs solution gassed with 5% (v/v) CO2 to maintain cells in a normocapnic environment. Perfusing cells with 10% (v/v) CO2 caused pHi to decrease by 0.18 ± 0.01 pH units (n = 60). This intracellular acidosis recovered after ∼20 min even upon continuous exposure of cells to 10% (v/v) CO2 (Fig. 1 A). We therefore chose 20 min as an appropriate time to study the effects of acute hypercapnia as cells would have recovered their pHi. Exposure of cells to 10% (v/v) CO2 for 20 min did not alter the integrity of the epithelial monolayer as assessed by recording TEER. In normocapnia, TEER was 671 ± 42Ω cm−2 (n = 3) and 600 ± 42Ω cm−2 in monolayers of Calu‐3 cells exposed to acute hypercapnia (P > 0.05 vs. normocapnia; n = 3). For all experiments, [HCO3 −] in the Krebs solution was maintained at 25 mm in both normocapnia and hypercapnia. This was necessary to ensure that any effects of hypercapnia on cAMP signalling were due to CO2‐dependent effects on tmAC as opposed to effects of HCO3 − on sAC – an enzyme shown to be sensitive to HCO3 − (Chen et al. 2000). In addition, given the scope of our work was to investigate the effect of raised CO2 on bicarbonate secretion, changing [HCO3 −] in hypercapnia would be predicted to compromise these studies.
Figure 1. Acute hypercapnia attenuates forskolin‐stimulated cAMP levels in Calu‐3 cells independent of changes in intracellular pH .
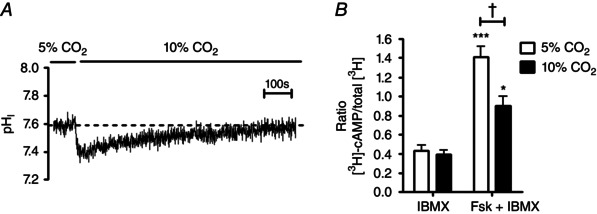
A, the effect of hypercapnia (10% CO2) on the pHi of Calu‐3 cells; cells recovered pHi from CO2‐induced acidosis after ∼20 min. B, the effect of acute hypercapnia on intracellular cAMP in which cells were incubated for 20 min in either 5% CO2 (v/v) in air or 10% CO2 (v/v) in air before being stimulated with either IBMX (1 mm) or forskolin (5 μm) + IBMX (1 mm) for a further 10 min. Intracellular cAMP levels were determined by measuring the amount of [3H]‐cAMP in each sample. ***Significant effect of forskolin (P < 0.001; *P < 0.05); †significant effect of hypercapnia (P < 0.05). Data represent mean ± SEM; n = 6 for each.
As we have previously shown that cAMP signalling is sensitive to changes in CO2 (Townsend et al. 2009; Cook et al. 2012), [cAMP]i was measured in conditions of normocapnia and after 20 min exposure to hypercapnia, with the incubation media buffered to pH 7.4 in each condition to control for differences in extracellular pH (pHe). In the presence of the non‐specific phosphodiesterase (PDE) inhibitor IBMX, there was no effect of hypercapnia on [cAMP]i (Fig. 1 B). Stimulation of cells with the cAMP elevating agonist forskolin (added after 20 min of exposure to 5 or 10% CO2 to allow for pHi recovery) produced a 3.3 ± 0.5‐fold increase in [cAMP]i in normocapnia (P < 0.001; n = 6; Fig. 1 B) but this was significantly reduced to a 2.3 ± 0.4‐fold increase in [cAMP]i in cells exposed to acute hypercapnia (P < 0.05 vs. normocapnia; n = 6; Fig. 1 B). When the cAMP levels produced in IBMX‐stimulated cells were subtracted from the cAMP levels measured in the presence of forskolin + IBMX, acute hypercapnia induced a 48 ± 4% reduction in [cAMP]i. These results demonstrate that cAMP signalling in Calu‐3 cells is responsive to elevated CO2, through a mechanism that is independent of changes in pHe and not due to the CO2‐induced intracellular acidosis.
Forskolin‐stimulated transepithelial anion secretion is reduced in conditions of acute hypercapnia in Calu‐3 cells
To assess whether the CO2‐induced reductions in forskolin‐stimulated [cAMP]i modulated cAMP‐regulated transepithelial ion transport, I sc measurements were made in monolayers of Calu‐3 cells. I sc is the current required to clamp the transepithelial voltage difference (V te) to 0 mV. In Calu‐3 monolayers, the magnitude of V te is mainly accounted for by transepithelial anion secretion (Lee et al. 1998; Devor et al. 1999; Cobb et al. 2003; Cuthbert et al. 2003; Shan et al. 2012) and therefore changes in I sc reflect changes in anion secretion. Figure 2 A shows a representative recording of I sc in normocapnic conditions. To maximise electrogenic Cl− secretion, a basolateral to apical Cl− gradient was applied across the monolayer by reducing apical [Cl−] to 40 mm by substitution of 84 mm NaCl with equimolar Na‐gluconate. In normocapnia, prior to reducing the apical Cl− concentration, Calu‐3 cells displayed a basal I sc of 5.2 ± 0.4 μA and further investigations showed that this basal I sc was insensitive to both the basolateral Na+/K+/2Cl− (NKCC1) inhibitor bumetanide (25 μm) and the NHE inhibitor Ethyl‐isopropyl amiloride (EIPA) (3 μm) (Masereel et al. 2003), whereas application of the CFTR blocker CFTRinh‐172 (20 μm) reduced basal I sc by 48.5 ± 4.2% (P < 0.01; n = 3), indicating that the majority of basal I sc was mediated by CFTR. Interestingly, in cells exposed to 20 min hypercapnia (Fig. 2 B), basal I sc was reduced to 1.3 ± 1.3 μA (P < 0.01 vs. normocapnia; n = 8; Fig. 2 C), implying that acute hypercapnia inhibited CFTR‐dependent anion secretion under resting conditions. After establishing a basolateral to apical Cl− gradient, addition of forskolin stimulated an increase in I sc which peaked after approximately 90 s to a maximal level and then decreased slightly until a new steady state was reached. The forskolin‐stimulated increase in I sc was blocked by a combination of apical CFTRinh‐172 (20 μm) and basolateral bumetanide (25 μm), and both the magnitude and the rate of I sc increase were significantly reduced by 61.8 ± 16.0 and 73.4 ± 6.8%, respectively, by the PKA inhibitor H‐89 (P < 0.05 vs. control; n = 3). These results demonstrated that CFTR‐dependent anion secretion mediated the forskolin‐stimulated increase in I sc, consistent with previous studies (Welsh & Smith, 2001; Kreda et al. 2007; Shan et al. 2012). The maximal forskolin‐stimulated increase in I sc (ΔI sc) was 19.3 ± 2.0 μA cm−2 (n = 10) in normocapnia compared to 14.1 ± 1.1 μA cm−2 in acute hypercapnia (P = 0.053 vs. normocapnia; n = 8; Fig. 2 D). The rate of forskolin‐stimulated increase in I sc in normocapnia was 10.4 ± 1.3 μA cm−2 min−1 (n = 10), which was reduced to 5.7 ± 0.6 μA cm−2 min−1 (P < 0.01 vs. normocapnia; n = 8; Fig. 2 E) in cells exposed to acute hypercapnia. These results, combined with those in Fig. 1, imply that attenuation of forskolin‐stimulated cAMP levels by acute hypercapnia was sufficient to significantly reduce the rate of cAMP‐regulated anion secretion in Calu‐3 cells. In addition, the forskolin‐stimulated I sc that was sensitive to CFTRinh‐172 was also measured. In normocapnia, this was 3.3 ± 0.7 μA cm−2 (n = 10) and although it was lower in hypercapnia (1.6 ± 0.2 μA cm−2; n = 8), this was not statistically significant, although a clear trend existed (P = 0.058 vs. normocapnia; Fig. 2 F). Taken together with the data displayed in Fig. 2 C and E, these findings suggest CFTR activity is lower in hypercapnia in both basal and forskolin‐stimulated conditions.
Figure 2. Forskolin‐stimulated transepithelial anion secretion is reduced in conditions of acute hypercapnia in Calu‐3 cells .
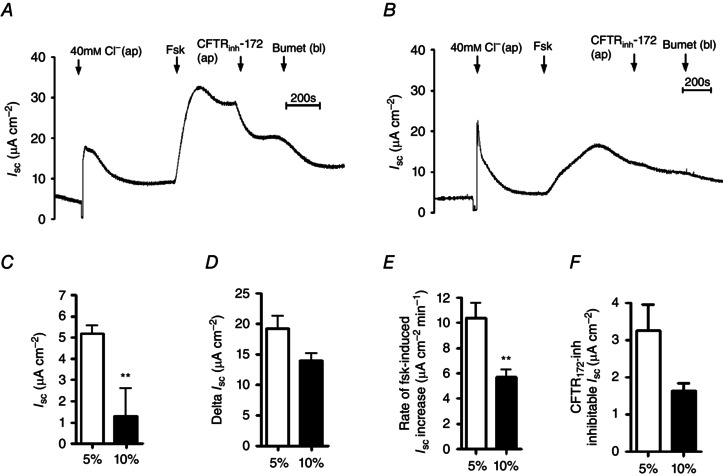
Calu‐3 cells were grown on permeable Snapwell supports and I sc was measured using an Ussing chamber. A, a representative I sc recording of a control experiment in which cells were exposed to 5% (v/v) CO2/95% (v/v) O2; B, a representative recording in which cells were pre‐exposed to 10% (v/v) CO2/90% (v/v) O2 for 20 min prior to being studied. Apical [Cl−] was reduced to 40 mm and cells were stimulated with forskolin (Fsk; 5 μm) before addition of apical CFTRinh‐172 (20 μm) and basolateral bumetanide (Bumet; 25 μm) as indicated. C–F, basal I sc (C), maximal forskolin‐stimulated increase in I sc (D), rate of increase in forskolin‐stimulated I sc (E) and amount of forskolin‐stimulated current that was inhibitied by CFTRinh‐172 (F). **Significant effect of hypercapnia (P < 0.01). Data represent mean ± SEM; n = 10 for normocapnia and n = 8 for hypercapnia.
Acute hypercapnia reduces adenosine but not IBMX‐stimulated transepithelial anion secretion in Calu‐3 cells
Having shown that hypercapnia reduced forskolin‐stimulated I sc in Calu‐3 cells, it was important to investigate whether hypercapnia also elicited similar effects when a more physiological agonist was used to increase [cAMP]i in Calu‐3 cells. For this, cells were stimulated with adenosine (Cobb et al. 2003) and the resulting I sc was measured. In normocapnia, adenosine stimulated a maximal I sc increase of 23.9 ± 3.5 μA cm−2 (n = 5), which was significantly reduced to 6.4 ± 1.4 μA cm−2 in cells exposed to acute hypercapnia (P < 0.05 vs. normocapnia; n = 3; Fig. 3 A). The rate of the adenosine‐stimulated increase in I sc was 13.4 ± 8.4 μA cm−2 min−1 (n = 5) in normocapnia which was reduced to 2.3 ± 0.8 μA cm−2 min−1 in acute hypercapnia (P = 0.06 vs. normocapnia; n = 3; Fig 3 B). Therefore, these data demonstrated that hypercapnia reduced adenosine‐stimulated, CFTR‐dependent anion secretion in Calu‐3 cells, which mimicked what was observed with forskolin. Interestingly, when [cAMP]i levels were increased by stimulation of cells with IBMX, there was no effect of acute hypercapnia on either the IBMX‐stimulated ΔI sc (normocapnia = 3.1 ± 0.9 μA cm−2; hypercapnia = 3.1 ± 1.3 μA cm−2; P > 0.05 vs. normocapnia; n = 3–4; Fig. 3 C) or the rate of IBMX‐stimulated increase in I sc (normocapnia = 1.0 ± 0.31 μA cm−2 min−1; hypercapnia = 1.2 ± 0.8 μA cm−2 min−1; P > 0.05 vs. normocapnia; n = 3–4; Fig. 3 D). Therefore, these data support the observations in Fig. 1 B, which demonstrated that IBMX‐stimulated increases in [cAMP]i was insensitive to CO2, and suggest hypercapnia‐induced changes in [cAMP]i were not due to modulation of IBMX‐sensitive PDE activity.
Figure 3. Acute hypercapnia reduces adenosine but not IBMX‐stimulated transepithelial anion secretion in Calu‐3 cells .
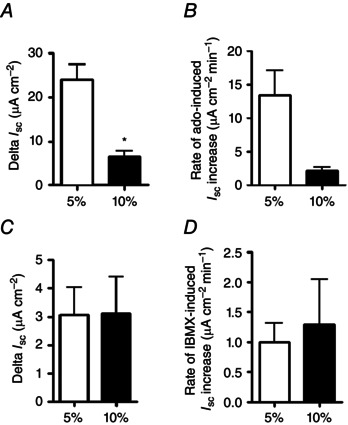
Calu‐3 cells were grown on permeable Snapwell supports and I sc was measured using an Ussing chamber. For control experiments, cells were gassed with 5% (v/v) CO2/95% (v/v) O2 whilst hypercapnia was induced by pre‐exposing cells to 10% (v/v) CO2/90% (v/v) O2 for 20 min prior to being studied. Apical [Cl−] was reduced to 40 mm and cells were stimulated with either adenosine (10 μm) or IBMX (1 mm) before addition of apical CFTRinh‐172 (20 μm) and basolateral bumetanide (25 μm). A, the maximal adenosine‐stimulated increase in I sc; B, the rate of increase in adenosine‐stimulated I sc. *Significant effect of hypercapnia (P < 0.05). Data represent mean ± SEM; n = 5 for normocapnia and n = 3 for hypercapnia. C, the maximal IBMX‐stimulated increase in I sc; D, the rate of increase in IBMX‐stimulated I sc. Data represent mean ± SEM; n = 3 for normocapnia and n = 4 for hypercapnia.
The effect of hypercapnia on cAMP‐dependent transepithelial anion secretion is independent of CO2‐induced intracellular acidosis
Although I sc measurements performed in hypercapnia were made after 20 min of exposure to 10% CO2, during which time pHi had recovered from intracellular acidosis (see Fig. 1 A), it was possible the intracellular acidosis may have induced long‐term modifications to transporters involved in cAMP‐regulated anion secretion. Therefore, cells were acid loaded using 40 mm sodium acetate, which caused an intracellular acidification of 0.17 ± 0.02 (n = 6) that recovered within 20 min (Fig. 4 A and B) and was thus highly similar to the effect of 10% CO2. Thus, the effect of forskolin on I sc was measured in cells exposed to 40 mm sodium acetate or 80 mm mannitol (to compensate for the increased osmolarity of the sodium acetate‐containing solutions). Representative experiments are shown in Fig. 4 C and D. There was no effect of 40 mm sodium acetate on either the magnitude or the rate of forskolin‐stimulated increases in I sc (Fig. 4 E and F), therefore demonstrating that the CO2‐induced intracellular acidosis does not contribute to the effects of hypercapnia on cAMP‐stimulated anion transport in Calu‐3 cells.
Figure 4. The effect of hypercapnia on cAMP‐dependent transepithelial anion secretion is independent of CO2‐induced intracellular acidosis .
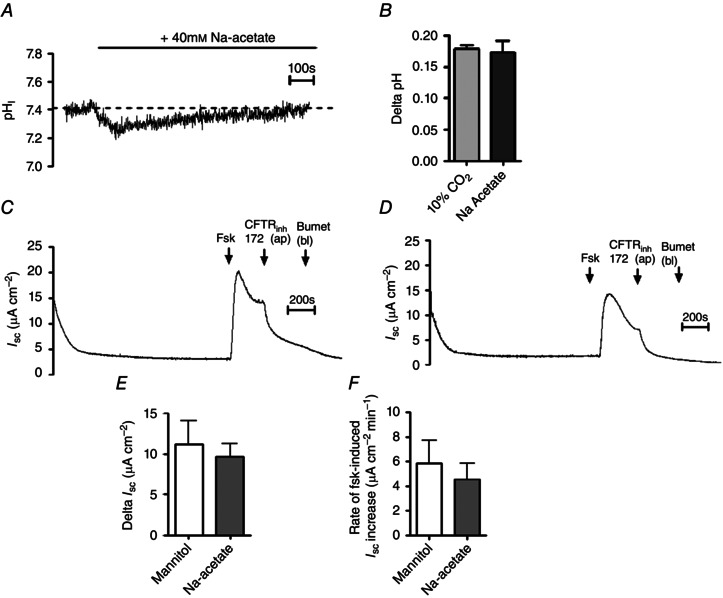
A, a representative experiment in which Calu‐3 cells were gassed with 5% (v/v) CO2/95% (v/v) O2 and exposed to 40 mm sodium acetate and pHi was measured using fluorescence microscopy. B, summary of the magnitude of the intracellular acidosis resulting from either 10% CO2 or sodium acetate. Data represent mean ± SEM; n = 60 for 10% CO2 and n = 6 for sodium acetate. C and D, representative I sc measurements in which cells were exposed to 80 mm mannitol or 40 mm sodium acetate, respectively, for 20 min prior to addition of forskolin (Fsk; 5 μm), apical CFTRinh‐172 (20 μm) and basolateral bumetanide (Bumet; 25 μm) as indicated. E and F, summary of the effect of sodium acetate on the magnitude and the rate, respectively, of the forskolin‐stimulated increase in I sc. Data represent mean ± SEM, n = 5 for each.
Surface expression of CFTR is unaffected by hypercapnia
Our results from the I sc measurements indicated that CO2‐induced reductions in [cAMP]i were sufficient to reduce cAMP‐stimulated, CFTR‐dependent anion secretion in Calu‐3 cells. To investigate if this observation was due to the effect of CO2 on cAMP and not on cell surface levels of CFTR, the amount of CFTR present at the apical membrane was assessed by cell surface biotinylation. Figure 5 shows that after normalising CFTR levels to α‐tubulin, there was no significant effect of CO2 on either total cell CFTR expression (P > 0.05; n = 5, Fig. 5 A) or cell surface CFTR expression (P > 0.05; n = 4, Fig. 5 B), which therefore suggest that mechanisms which control CFTR expression at the plasma membrane are insensitive to hypercapnia.
Figure 5. Cell surface expression of CFTR is unaffected by acute hypercapnia .
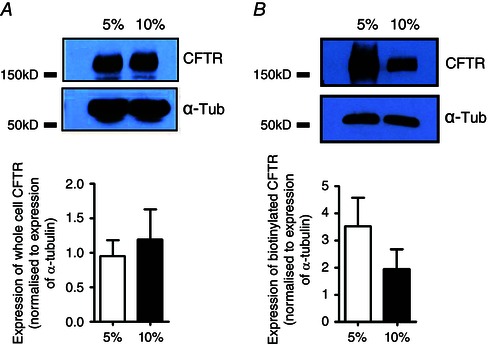
Calu‐3 cells were grown on permeable transwell supports and membrane expression of CFTR was assessed using a biotinylation assay. A, an example blot of whole cell CFTR expression under 5% CO2 and 10% CO2 and the relative expression of whole cell CFTR when normalised to expression of whole cell α‐tubulin. Data represent mean ± SEM; n = 5. B, an example blot of biotinylated CFTR expression, used as a marker of surface expression, under 5% CO2 and 10% CO2 and the relative expression of biotinlayed CFTR when normalised to expression of biotinylated α‐tubulin. Data represent mean ± SEM; n = 4.
CFTR‐regulated, pendrin‐dependent apical HCO3 − secretion is unaffected by hypercapnia
Having identified that hypercapnia reduces cAMP‐stimulated anion secretion in Calu‐3 cells, it was interesting to assess whether CO2 was modulating Cl− or HCO3 − secretion or indeed both. pHi measurements were performed to indirectly measure HCO3 − transport across the cells. At the apical membrane, we have previously shown that Calu‐3 cells express the Cl−/HCO3 − anion exchanger pendrin, which mediates the majority of HCO3 − efflux from the cell (Garnett et al. 2011). Pendrin activity was also shown to be regulated by CFTR. To measure CFTR‐dependent pendrin activity, cells were stimulated with forskolin and pendrin activity was assessed by Cl− removal and re‐addition (Fig. 6 A) (Garnett et al. 2011). In normocapnia, removal of apical Cl− caused pHi to increase by 0.61 ± 0.08 units (n = 6), due to reversal of pendrin‐mediated Cl−/HCO3 − exchange, whilst in hypercapnia this increase in pHi was 0.64 ± 0.10 (P > 0.05 vs. normocapnia; n = 6, Fig. 6 B). Furthermore, reintroduction of apical Cl− caused pHi to re‐acidify at a rate of 0.49 ± 0.08 pH units min−1 in normocapnia and 0.45 ± 0.06 pH units min−1 in hypercapnia (P > 0.05; n = 6; Fig. 6 C) which equated to an HCO3 − efflux of 104 ± 21 mm HCO3 − min−1 and 127 ± 38 mm HCO3 − min−1, respectively (P > 0.05; n = 6; Fig. 6 D). Note that in forskolin‐stimulated conditions, the basolateral anion exchanger, AE2, was almost completely inhibited, both in normocapnia (96.9 ± 1.9% inhibition; n = 4) and in hypercapnia (93.8 ± 4.3% inhibition; n = 4), consistent with previous findings (Garnett et al. 2011). Thus, AE2‐dependent HCO3 − transport can be eliminated from having any effect on these measurements. Therefore, these data show that apical CFTR‐dependent anion exchange activity was unaffected by acute hypercapnia and suggested that HCO3 − transport across the apical membrane was insensitive to changes in CO2.
Figure 6. CFTR‐regulated, pendrin‐dependent apical HCO3− efflux is unaffected by hypercapnia .
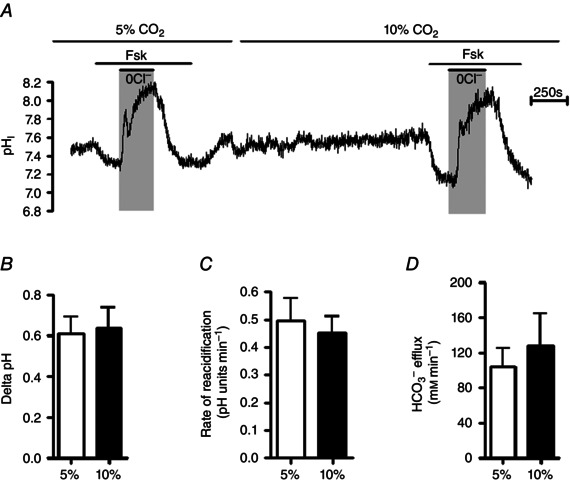
A, a representative pHi experiment in which the effect of acute hypercapnia on 5 μm forskolin‐stimulated, CFTR‐regulated apical HCO3 − transport was assessed by removal and subsequent re‐addition of apical Cl−. B, ΔpH in response to removal of Cl−. C and D, rate of re‐acidification (C) and HCO3 − flux resulting from re‐addition of apical Cl− (D). Data represent mean ± SEM; n = 6 for each.
Acute hypercapnia does not alter cAMP‐stimulated NBC activity in Calu‐3 cells
To investigate HCO3 − transport across the basolateral membrane, we measured the activity of NBC transporters, which have been shown to mediate basolateral membrane HCO3 − import in Calu‐3 cells (Lee et al. 1998; Devor et al. 1999; Shan et al. 2012). NBC activity was monitored by measuring changes in pHi following the removal of basolateral Na+ (to inhibit NBC) and the re‐addition of basolateral Na+ (to re‐activate NBC), as described by Yang et al. (2009), in the presence of EIPA to inhibit NHE activity. However, it was first necessary to determine whether NBC activity in Calu‐3 cells was cAMP‐dependent. Figure 7 A and B shows that both forskolin and adenosine stimulated a 2.3 ± 0.4‐fold (n = 3; P < 0.05) and 2.5 ± 0.5‐fold (n = 3; P < 0.05) increase, respectively, in NBC activity, under normocapnic conditions, indicating that NBC activity in Calu‐3 cells is increased by cAMP. The effect of acute hypercapnia on cAMP‐regulated NBC activity was next assessed. Here, NBC activity was measured in normocapnic conditions (Fig. 7 A) or after cells had been exposed to 20 min of hypercapnia (Fig. 7 C). As summarised in Fig. 7 D, forskolin stimulated an NBC‐dependent HCO3 − influx of 12.5 ± 1.8 mm min−1 (n = 7) under normocapnia whilst in hypercapnia, forskolin‐stimulated NBC‐dependent HCO3 − influx was 11.3 ± 1.7 mm min−1 (n = 7; P > 0.05 vs. normocapnia). These findings suggest that, like pendrin, acute hypercapnia does not affect cAMP‐stimulated NBC activity and thus imply that CO2‐induced effects on cAMP‐regulated anion transport were not due to changes in HCO3 − secretion per se and suggested only Cl− secretion was sensitive to elevated CO2.
Figure 7. Hypercapnia does not alter cAMP‐stimulated NBC activity in Calu‐3 cells .
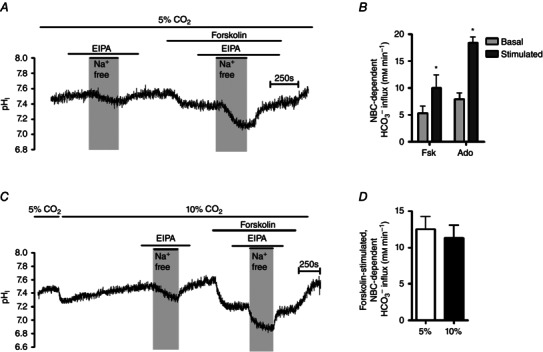
A, a representative pHi experiment in which NBC activity was assessed under basal and forskolin‐stimulated conditions in 5% CO2. EIPA (3 μm) was present to inhibit the NHE. B, the effect of the cAMP agonists forskolin (5 μm) and adenosine (10 μm) on NBC‐dependent HCO3 − influx. *Significant effect of agonist stimulation (P < 0.05). Data represent mean ± SEM; n = 3 for each. C, a representative pHi experiments in which forskolin‐stimulated NBC activity was assessed in conditions of acute hypercapnia. EIPA (3 μm) was present to inhibit the NHE. E, the effect of hypercapnia on forskolin‐stimulated NBC activity. Data represent mean ± SEM; n = 7 for each.
Hypercapnia reduces the volume of forskolin‐stimulated fluid secretion in Calu‐3 cells but has no effect on pH
We have previously shown that stimulation of Calu‐3 cells with forskolin for 24 h increased the secretion of a HCO3 −‐rich fluid. Furthermore, based on pharmacological and genetic knockdown experiments, we suggested that cAMP‐stimulated liquid secretion was primarily regulated by CFTR, while HCO3 − secretion was not directly via CFTR but through Cl−/HCO3 − via pendrin (Garnett et al. 2011, 2013). Given that it appears separate transporters were responsible for Cl− and HCO3 − secretion in Calu‐3 cells, it was of interest to assess if hypercapnia impacted upon forskolin‐stimulated ion and fluid secretion. Calu‐3 cells were stimulated with forskolin in either 5% CO2 (v/v) in air or 10% CO2 (v/v) in air for 24 h and the amount and pH of the secreted fluid were analysed. Note that TEER was not significantly different between normocapnic controls (682 ± 28 Ω cm−2; n = 6) and cells incubated for 24 h in hypercapnia (681 ± 6 Ω cm−2; P > 0.05 vs. control; n = 6), suggesting that chronic hypercapnia did not alter tight junction properties of Calu‐3 cells. In normocapnic conditions, unstimulated cells secreted 12 ± 4 μl fluid over 24 h (n = 3), which was significantly enhanced 3.9 ± 0.2‐fold to 49 ± 3 μl by forskolin stimulation (P < 0.01 vs. unstimulated cells; n = 3; Fig. 8 A). In hypercapnic conditions, unstimulated cells secreted 12 ± 1 μl fluid over 24 h which was almost identical to that seen in normocapnia (P > 0.05; n = 3). However, although forskolin increased fluid secretion to 32 ± 1 μl over 24 h (P < 0.01; n = 3; Fig. 8 A), this 2.7 ± 0.1‐fold increase in the volume of forskolin‐stimulated fluid secretion was significantly lower than that observed in normocapnia (P < 0.05 vs. normocapnia; n = 3; Fig. 8 A). This suggested chronic hypercapnia impaired cAMP‐regulated CFTR‐dependent Cl− secretion in airway epithelia to reduce the osmotic driving force for fluid secretion. The pH of the secreted fluid was also measured. In normocapnia, the pH of secreted fluid increased from 7.52 ± 0.01 to 7.82 ± 0.06 (P < 0.01; n = 3) indicative of a greater [HCO3 −] in forskolin‐stimulated fluid secretion. This pH increase of 0.31 ± 0.01 was not different from the pH increase of 0.30 ± 0.01 observed in hypercapnia (7.21 ± 0.04 to 7.51 ± 0.02; P < 0.01 vs. unstimulated controls; P > 0.05 vs. normocapnia; n = 3; Fig. 8 B) with the lower pH values observed due to acidosis induced by elevated CO2. Using the Henderson–Hasselbalch equation to calculate [HCO3 −] revealed that the forskolin‐stimulated fluid contained 61.6 ± 9.5 mm HCO3 − in normocapnia, which was not significantly different from the 58.2 ± 2.4 mm HCO3 − in the forskolin‐stimulated fluid in hypercapnia (P > 0.05; n = 3). Together, these findings suggest that CFTR‐dependent electrogenic Cl− secretion is CO2‐sensitive, whilst pendrin‐dependent HCO3 − secretion is CO2‐insenstive, and supports the findings from I sc and pHi measurements (Figs 2, 6 and 7). In addition as mucin secretion has been shown to be dependent on [HCO3 −] (Garcia et al. 2009; Chen et al. 2010; Gustafsson et al. 2012; Ridley et al. 2014), we also analysed the glycoprotein content of the secreted fluid by the PAS assay. In normocapnia, forskolin did not alter the amount of glycoproteins detected relative to unstimulated cells (18.5 ± 0.5 vs. 18.2 ± 1.0 μg ml−1, respectively; P > 0.05; n = 3; Fig. 8 C). Furthermore, hypercapnia had no effect on glycoprotein secretion from Calu‐3 cells relative to normocapnia in either basal or forskolin‐stimulated cells. Unstimulated cells secreted 19.2 ± 0.1 μg ml−1 glycoprotein (P > 0.05 vs. unstimulated cells in normocapnia; n = 3), which was unchanged in response to forskolin stimulation (24.0 ± 4.0 μg ml−1; P > 0.05 vs. unstimulated cells in hypercapnia; P > 0.05 vs. stimulated cells in normocapnia; n = 3; Fig. 8 C). Therefore, hypercapnia modulated transporters involved in regulating the volume of secreted fluid but not those involved in mediating its composition.
Figure 8. Hypercapnia reduces the volume of forskolin‐stimulated fluid secretion in Calu‐3 cells .

Cells were stimulated with forskolin (Fsk; 5 μm) and incubated for 24 h in either 5% CO2 (v/v) in air or 10% CO2 (v/v) in air in high Cl− Krebs solution at 37°C. A, the effect of chronic hypercapnia on the volume of fluid secreted over 24 h. **Significant effect of forskolin stimulation compared to unstimulated control cells (P < 0.01; ***P < 0.001); †significant effect of 10% CO2 (P < 0.05). Data represent mean ± SEM; n = 3 for each. B, the increase in pH of forskolin‐stimulated secreted fluid relative to unstimulated control cells. Data represent mean ± SEM; n = 3 for each. C, the effects of forskolin and hypercapnia on the amount of glycoprotein present in the secreted fluid, quantified by the PAS assay. Data represent mean ± SEM; n = 3 for each.
Hypercapnia reduces forskolin‐stimulated increases in I sc across primary human bronchial epithelial cells
To assess whether hypercapnia elicited similar effects in primary airway epithelia as it did in an airway epithelial cell line, I sc measurements were made on fully differentiated primary human bronchial epithelial cells (HBECs) grown under ALI. Figure 9 A and B shows representative experiments performed in conditions of normocapnia and hypercapnia, respectively. Hypercapnia had no effect on basal I sc (4.3 ± 1.1 μA cm−2 in normocapnia and 3.8 ± 0.5 μA cm−2 in acute hypercapnia; P > 0.05 vs. normocapnia; n = 6; Fig. 9 C). However, it was found that the basal I sc was sensitive to apical amiloride (10 μm), which reduced basal I sc by 5.0 ± 0.9 μA cm−2 in normocapnia (n = 6) and 4.4 ± 0.6 μA cm−2 in hypercapnia (P > 0.05 vs. normocapnia; n = 6), indicating that these cells expressed functional epithelial Na+ channels (ENaC). Stimulation of cells with forskolin in normocapnia induced a maximal increase in I sc of 13.9 ± 1.8 μA cm−2 (n = 6) which was significantly reduced to 8.8 ± 1.3 μA cm−2 in cells that had been exposed to acute hypercapnia (P < 0.05 vs. normocapnia; n = 6; Fig. 9 D). Furthermore, the rate of forskolin‐stimulated I sc increase was also significantly reduced from 31.3 ± 4.4 μA cm−2 min−1 (n = 6) in normocapnia to 18.1 ± 2.6 μA cm−2 min−1 in hypercapnia (P < 0.05 vs. normocapnia; n = 6; Fig. 9 E). These data are consistent with the findings from Calu‐3 cells and suggest that hypercapnia reduces cAMP‐stimulated CFTR‐dependent anion transport in primary human airway epithelial cells as well as in an airway epithelia cell line. When measuring the amount of CFTRinh‐172‐sensitive current, it was again found that there was a clear trend for this to be lower in acute hypercapnia, supporting the findings that CFTR activity was reduced by 10% CO2. As shown in Fig. 9 F, in normocapnia, forskolin‐stimulated CFTRinh‐172‐sensitive current was 8.3 ± 1.6 μA cm−2 and was reduced in hypercapnia to 4.4 ± 0.9 μA cm−2 (n = 6; P > 0.05 vs. normocapnia; Fig. 9 F).
Figure 9. Forskolin‐stimulated transepithelial anion secretion is reduced in conditions of acute hypercapnia in primary human bronchial epithelial cells .
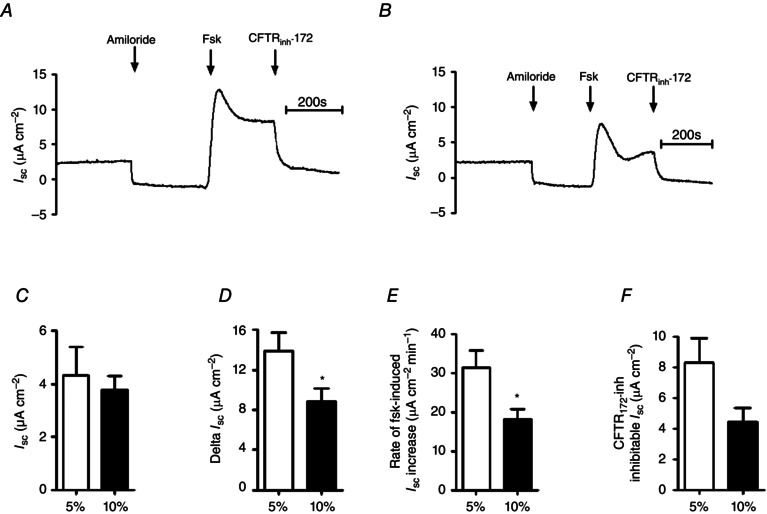
Primary human bronchial epithelial cells were grown on collagen‐coated permeable Snapwell supports and allowed to differentiate at an ALI for 30–35 days before I sc was measured using an Ussing chamber. A, a representative I sc recording of a control experiment in which cells were exposed to 5% (v/v) CO2/95% (v/v) O2; B, a representative recording in which cells were pre‐exposed to 10% (v/v) CO2/90% (v/v) O2 for 20 min prior to being studied. Apical [Cl−] and basolateral [Cl−] were both 124 mm for these experiments. Cells were treated with apical amiloride (Amil; 10 μm) and stimulated with forskolin (Fsk; 10 μm) before addition of apical CFTRinh‐172 (20 μm) as indicated. C–F, basal I sc (C), maximal forskolin‐stimulated increase in I sc (D), rate of increase in forskolin‐stimulated I sc (E) and amount of forskolin‐stimulated current that was inhibitied by CFTRinh‐172 (F). *Significant effect of hypercapnia (P < 0.05). Data represent mean ± SEM; n = 6 for each.
Discussion
The ability of CO2 to act as a cell signalling molecule is currently gaining substantial support within human physiology. Here we show, for the first time, that hypercapnia modulates cAMP‐dependent signalling, as well as cAMP‐dependent ion and fluid transport, in both a human airway epithelial cell line and also in primary HBECs. We found that acute hypercapnia caused a significant reduction in forskolin‐stimulated [cAMP]i levels in Calu‐3 cells – even in the presence of a PDE inhibitor – which was independent of CO2‐induced intracellular or extracellular acidosis (Fig. 1 B). Interestingly, hypercapnia did not affect cAMP levels in cells stimulated with IBMX only (Fig. 1 B), implying that the CO2‐induced attenuation of [cAMP]i was not due to modulation of PDE activity, consistent with our previous results (Townsend et al. 2009; Cook et al. 2012). The apparent lack of effect of hypercapnia in the absence of forskolin suggests that for hypercapnia to alter tmAC activity, the cyclase needs to be in an active state. Zhang et al. (1997) have described the presence of hydrophobic forskolin binding pockets on tmAC, and forskolin binding at these sites induces a conformational change leading to dimerisation of the two catalytic subunits of tmAC. Thus, it seems likely that CO2 can only modulate tmAC activity when it is held within this ‘forskolin‐bound’ state. Similar conformational changes in tmAC are induced when free Gαs bind to the enzyme, implying that CO2 modulates tmAC activity via the same mechanism when cells are stimulated with G‐protein coupled receptor agonists such as adenosine (Tesmer et al. 1997).
The hypercapnic‐induced reduction in forskolin‐stimulated cAMP levels also had significant effects on forskolin‐stimulated transepithelial ion transport in Calu‐3 cells. In the presence of a basolateral to apical Cl− gradient, 10% CO2 caused an ∼45% reduction in the rate of forskolin‐stimulated increase in CFTRinh‐172 and bumetanide‐sensitive I sc (Fig. 2 E). These findings imply that CO2‐induced changes in [cAMP]i were sufficient to reduce CFTR‐dependent electrogenic anion secretion in Calu‐3 cells. Hypercapnia also produced the same effect when cells were stimulated with the physiological cAMP agonist adenosine but did not alter IBMX‐stimulated changes in I sc (Fig. 3). These findings indicated that CO2‐dependent reductions in [cAMP]i were a result of modulations to tmAC‐dependent cAMP production as opposed to PDE‐dependent cAMP breakdown, which supports previous findings from our laboratory (Townsend et al. 2009; Cook et al. 2012). We were also able to conclude that the modulations to cAMP‐regulated anion transport in hypercapnia were not a result of the CO2‐induced intracellular acidosis as mimicking this acid load using sodium acetate did not alter forskolin‐stimulated increases in I sc (Fig. 4).
Biotinylation experiments further showed that the effect of hypercapnia on I sc could not be explained by a reduction in surface levels of CFTR (Fig. 5). These findings support our hypothesis that in cAMP‐stimulated conditions, the effects of CO2 were due to modulation of [cAMP]i as opposed to CO2‐dependent effects on pathways involved in regulating CFTR surface expression, for instance endocytosis. Furthermore, these findings are of particular relevance given that hypercapnia has been shown to modulate the surface expression of the Na+/K+‐ATPase in mammalian alveolar epithelia (Briva et al. 2007), which therefore suggests that CO2 only induces endocytosis of specific ion transporters. Acute hypercapnia also significantly lowered basal I sc in Calu‐3 cells. That a large component of this basal I sc was sensitive to CFTRinh‐172 suggests that hypercapnia also reduced the activity of CFTR under these conditions. However, hypercapnia did not alter levels of [cAMP]i under resting conditions (Fig. 1 B), and hypercapnia did not alter surface CFTR expression (Fig. 5), indicating that the effect of high CO2 on resting CFTR activity was independent of its effects on cAMP and not due to loss of CFTR at the plasma membrane. Therefore, why we observed a decrease in basal I sc in Calu‐3 cells exposed to acute hypercapnia remains unclear, but we cannot exclude the possibility that hypercapnia may have effects on basal [cAMP]i that cannot be detected using our current method of quantification. Note that whilst hypercapnia induces a reversible intracellular acidosis (Fig. 1 A) and that CFTR has been shown to be pH‐sensitive (Reddy et al. 1998; Chen et al. 2009; Melani et al. 2010), the 10% CO2‐induced acidosis of ∼0.2 units is unlikely to significantly alter CFTR activity based on single channel recordings of CFTR expressed in mammalian cells (Chen et al. 2009) and measurements of CFTR‐dependent Cl− conductance made in human sweat ducts (Reddy et al. 1998). Furthermore, the fact that all measurements of cAMP‐stimulated CFTR activity were made after cells had recovered pHi in response to CO2‐induced acidosis also strongly argues against any pHi‐dependent effects on CFTR activity in hypercapnia.
To identify the transport of which anion (Cl− or HCO3 −) hypercapnia was modulating, pHi measurements were performed to indirectly measure HCO3 − transport in real time in polarised cultures of Calu‐3 cells. Importantly, we showed that cAMP‐stimulated, pendrin‐dependent apical HCO3 − secretion and cAMP‐stimulated, NBC‐dependent basolateral HCO3 − influx were both insensitive to hypercapnia (Figs 6 and 7), suggesting that hypercapnia did not alter HCO3 − transport directly in Calu‐3 cells. Thus, the results from the I sc measurements suggested that the CO2‐induced reduction in electrogenic anion secretion was specifically due to a reduction in transepithelial Cl− secretion. Thus, it appears that cAMP‐regulated transporters have different sensitivities to CO2‐induced decreases in [cAMP]i in Calu‐3 cells. Although the reasons for this are unclear at the present, it is known that CFTR exists in a microdomain at the apical membrane of airway epithelial cells, in which cAMP signalling is highly compartmentalised (Barnes et al. 2005; Penmatsa et al. 2010). A decrease in cAMP levels in such a compartmentalised microdomain would have more pronounced effects than in areas of the cell where cAMP signalling is less compartmentalised, for instance at the basolateral subcellular location. Similarly, apical and basolateral microdomains may possess distinct tmAC isoforms that could display differential sensitivities to raised CO2.
We also observed similar results when investigating the effects of hypercapnia on cAMP‐stimulated anion and fluid transport using a different approach. Incubating cells for 24 h in hypercapnia enabled us to assess the effect of hypercapnia on the volume, as well as the composition, of the secreted fluid (Fig. 8). We found that hypercapnia did not affect the amount of fluid secreted under basal conditions. This is consistent with results from Fig. 1 B that demonstrated cAMP levels in non‐stimulated Calu‐3 cells were insensitive to hypercapnia. However, the fluid secretion data do contradict our I sc measurements in which CFTRinh‐172‐sensitive basal I sc was reduced in hypercapnia, suggesting that CFTR may be altered by hypercapnia through a cAMP‐independent mechanism. Nonetheless, hypercapnia caused a significant reduction in the amount of secreted fluid under forskolin‐stimulated conditions (Fig. 8 A). We have previously shown that the volume of forskolin‐stimulated fluid secretion is predominantly mediated by electrogenic CFTR‐dependent Cl− secretion (Garnett et al. 2011), strongly suggesting that hypercapnia reduced fluid secretion via an effect on CFTR‐dependent Cl− transport. This was probably due to the CO2‐induced reduction in forskolin‐stimulated cAMP levels (Fig. 1 B). Although we demonstrated chronic hypercapnia did not affect the transepithelial resistance of Calu‐3 monolayers, indicating paracellular ion and fluid transport was not altered by 10% CO2, one cannot rule out the possibility that hypercapnia may alter the water permeability of the epithelial monolayer, which would be another interesting effect of elevated CO2. However, unpublished findings from our laboratory have found that the osmolarity of secreted fluid in Calu‐3 cells is unchanged in forskolin‐stimulated cells compared to control cells. Thus, as we know forskolin to increase ion and fluid secretion in Calu‐3 cells, these findings demonstrate that changes in transepithelial ion secretion do not alter water permeability and thus are unlikely to contribute to the changes in fluid secretion observed in hypercapnia. Kim et al. (2014) also suggest water permeability is unchanged in Calu‐3 cells even in conditions where ion secretion is stimulated. Interestingly, the [HCO3 −] of forskolin‐stimulated fluid secretion was unaffected by chronic hypercapnia (Fig. 8 B). Garnett et al. (2011) demonstrated that the pH of forskolin‐secreted fluid was predominately regulated by the Cl−/HCO3 − exchanger pendrin, and not directly by CFTR, as fluid pH was insensitive to GlyH‐101 or genetic knockdown of CFTR, but was reduced by pendrin K D. Thus, our results demonstrate that CFTR and pendrin exhibit differential sensitivities to CO2. In addition, neither forskolin nor hypercapnia had any effect on the amount of glycoprotein detected in apical secretions from Calu‐3 cells, suggesting that neither treatment modified mucus secretion. Kreda et al. (2007) demonstrated that secretion of mucins by Calu‐3 cells, including MUC5AC, was a result of Ca2+‐dependent exocytosis of mucin granules, probably explaining why forskolin did not alter mucus secretion. Furthermore, these findings also imply that hypercapnia does not alter Ca2+‐dependent mucin secretion and therefore only modulates cAMP‐regulated responses.
Finally, the findings of acute hypercapnia on CFTR‐dependent I sc in Calu‐3 cells were also replicated in fully differentiated HBECs. In these cells, 10% CO2 also significantly reduced cAMP‐stimulated CFTR‐dependent anion transport (Fig. 9). Although we did not measure [cAMP]i in response to hypercapnia in HBECs, the ∼42% decrease in the rate of forskolin‐stimulated I sc increase in HBECs was comparable to the ∼45% decrease observed in Calu‐3 cells, and thus suggests CO2 elicited its effects via similar mechanisms in both cell types. However, one interesting difference was the fact that hypercapnia had no effect on basal I sc in HBECs whereas it did in Calu‐3 monolayers (see Figs 2 C and 9 C), suggesting that basal CFTR activity is less sensitive to CO2 in primary airway epithelia. However, basal I sc in Calu‐3 cells was amiloride‐insensitive (our unpublished observations), as opposed to the large component of basal I sc in HBECs that was inhibited by amiloride, suggesting different transporters regulate basal I sc in the two cell types and probably explaining the differences in response to hypercapnia. Furthermore, given there was no effect of CO2 on amiloride‐sensitive I sc in HBECs, ENaC activity was probably insensitive to acute hypercapnia. This reinforces the findings that acute hypercapnia mediates specific effects on CFTR as opposed to other membrane ion transporters.
In summary, we have shown for the first time that acute hypercapnia reduced cAMP production as well as cAMP‐stimulated, CFTR‐dependent Cl−, but not HCO3 −, secretion in human airway epithelial cells. We propose that CO2‐induced reductions in cytosolic cAMP inhibit CFTR activity and thus CFTR‐dependent Cl− secretion. However, the lack of an effect on pendrin‐dependent HCO3 − secretion implies that there was sufficient residual CFTR activity to maintain Cl−/HCO3 − exchange by pendrin, and thus efficient HCO3 − secretion persisted. This is consistent with our previous results in which we showed significant pendrin‐mediated anion exchange activity was still present in Calu‐3 cells where CFTR levels were knocked down by ∼ 75% (Garnett et al. 2011). However, dysregulation of CFTR‐dependent Cl− and fluid secretion would be predicted to reduce airways hydration and compromise the innate defence mechanisms of the lungs (Pezzulo et al. 2012) predisposing the airways to bacterial colonisation. These findings are of particular relevance to patients suffering from chronic lung diseases, such as chronic obstructive pulmonary disease (COPD) or severe CF, in which bacterial infection is a major problem and hypercapnia is a complication. Thus, based on our findings, hypercapnia may be an additional contributing factor to airways pathophysiology in these situations (Lourenco & Miranda, 1968; Holland et al. 2003; Sheikh et al. 2011). However, the effects of hypercapnia that we have reported should also be considered for those patients receiving treatment for acute respiratory distress syndrome (ARDS) who suffer from pulmonary oedema due to increased permeability of the alveolar epithelium (Grommes & Soehnlein, 2011). These patients become hypercapnic as a consequence of their clinical treatment (Prin et al. 2002) and it has been postulated that it is the elevated CO2 that provides the beneficial effects of the treatment. We suggest that a potential protective role of hypercapnia for ARDS patients could be in the reduction in the amount of cAMP‐stimulated fluid secretion in the airways, which would help to minimise the extent of the oedema without compromising the pH‐dependent components of the airway innate defence mechanisms. Interestingly, our findings somewhat contradict those published by the Snzajder group who demonstrated that (i) hypercapnia reduced alveolar fluid reabsorption and thus increased pulmonary oedema in rat alveolar cells (Briva et al. 2007; Vadasz et al. 2008) and (ii) high CO2 increased apical [cAMP]i in both A549 cells and rat alveolar type II cells (Lecuona et al. 2013). The findings reported here highlight potential differences in CO2 signalling between rat and humans as well as suggest that secretory cells of the conducting airways respond differently to hypercapnia compared to absorptive cells of the respiratory airways. Several studies have also implicated CO2 as an anti‐inflammatory agent (Laffey et al. 2000; Sinclair et al. 2002; De Smet et al. 2007; Contreras et al. 2012; Oliver et al. 2012) whilst hypercapnia has also been shown to attenuate ventilator‐induced lung injury in mice (Otulakowski et al. 2014). Our findings may suggest another possible protective role of hypercapnia in ARDS patients which would complement the other reported benefits of hypercapnia.
Additional information
Competing interests
None declared.
Author Contributions
M.J.T., M.J.C. and M.A.G. conceived and designed the experiments. M.J.T., V.S., W.P., S.I. and B.V. conducted experiments and collected data. M.J.T., V.S. and W.P. performed data analysis. J.P.G. and C.W. provided resources. M.J.T., C.W., R.T., M.J.C. and M.A.G. drafted the article or revised it critically for important intellectual content.
Funding
This work was supported by an MRC Studentship awarded to M.J.T., a BBSRC studentship to W.P. and an overseas studentship to S.H.I. funded by the Higher Committee for Education Development (HCED), Iraq. Additional funding was also provided by the Cystic Fibrosis Trust (Grant SRC003).
Acknowledgements
The authors acknowledge the technical expertise of Yishan Luo in assisting with cell surface biotinylation experiments.
References
- Adijanto J, Banzon T, Jalickee S, Wang NS & Miller SS (2009). CO2‐induced ion and fluid transport in human retinal pigment epithelium. J Gen Physiol 133, 603–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard ST, Trout L, Garrison J & Inglis SK (2006). Ionic mechanism of forskolin‐induced liquid secretion by porcine bronchi. Am J Physiol Lung Cell Mol Physiol 290, L97–104. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Livera G, Huang P, Sun C, O'Neal WK, Conti M, Stutts MJ & Milgram SL (2005). Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem 280, 7997–8003. [DOI] [PubMed] [Google Scholar]
- Boucher RC (2007). Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med 261, 5–16. [DOI] [PubMed] [Google Scholar]
- Bouyer P, Zhou Y & Boron WF (2003). An increase in intracellular calcium concentration that is induced by basolateral CO2 in rabbit renal proximal tubule. Am J Physiol Renal Physiol 285, F674–687. [DOI] [PubMed] [Google Scholar]
- Briva A, Santos C, Malacrida L, Rocchiccioli F, Soto J, Angulo M, Batthyany C, Cairoli E & Piriz H (2011). Adenosine triphosphate‐dependent calcium signaling during ventilator‐induced lung injury is amplified by hypercapnia. Exp Lung Res 37, 471–481. [DOI] [PubMed] [Google Scholar]
- Briva A, Vadasz I, Lecuona E, Welch LC, Chen J, Dada LA, Trejo HE, Dumasius V, Azzam ZS, Myrianthefs PM, Batlle D, Gruenbaum Y & Sznajder JI (2007). High CO2 levels impair alveolar epithelial function independently of pH. PLoS One 2, e1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck J, Sinclair ML, Schapal L, Cann MJ & Levin LR (1999). Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci USA 96, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann M (2004). Bicarbonate stimulated adenylyl cyclases. IUBMB Life 56, 529–534. [DOI] [PubMed] [Google Scholar]
- Cann MJ, Hammer A, Zhou J & Kanacher T (2003). A defined subset of adenylyl cyclases is regulated by bicarbonate ion. J Biol Chem 278, 35033–35038. [DOI] [PubMed] [Google Scholar]
- Chen EY, Yang N, Quinton PM & Chin WC (2010). A new role for bicarbonate in mucus formation. Am J Physiol Lung Cell Mol Physiol 299, L542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Cai Z & Sheppard DN (2009). Direct sensing of intracellular pH by the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem 284, 35495–35506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR & Buck J (2000). Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 289, 625–628. [DOI] [PubMed] [Google Scholar]
- Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O'Neal WK & Boucher RC (2003). Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA 100, 16083–16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BR, Fan L, Kovacs TE, Sorscher EJ & Clancy JP (2003). Adenosine receptors and phosphodiesterase inhibitors stimulate Cl− secretion in Calu‐3 cells. Am J Respir Cell Mol Biol 29, 410–418. [DOI] [PubMed] [Google Scholar]
- Contreras M, Ansari B, Curley G, Higgins BD, Hassett P, O'Toole D & Laffey JG (2012). Hypercapnic acidosis attenuates ventilation‐induced lung injury by a nuclear factor‐kappaB‐dependent mechanism. Crit Care Med 40, 2622–2630. [DOI] [PubMed] [Google Scholar]
- Cook ZC, Gray MA & Cann MJ (2012). Elevated carbon dioxide blunts mammalian cAMP signaling dependent on inositol 1,4,5‐triphosphate receptor‐mediated Ca2+ release. J Biol Chem 287, 26291–26301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins EP, Selfridge AC, Sporn PH, Sznajder JI & Taylor CT (2014). Carbon dioxide‐sensing in organisms and its implications for human disease. Cell Mol Life Sci 71, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert AW, Supuran CT & MacVinish LJ (2003). Bicarbonate‐dependent chloride secretion in Calu‐3 epithelia in response to 7,8‐benzoquinoline. J Physiol 551, 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet HR, Bersten AD, Barr HA & Doyle IR (2007). Hypercapnic acidosis modulates inflammation, lung mechanics, and edema in the isolated perfused lung. J Crit Care 22, 305–313. [DOI] [PubMed] [Google Scholar]
- Devor DC, Singh AK, Lambert LC, DeLuca A, Frizzell RA & Bridges RJ (1999). Bicarbonate and chloride secretion in Calu‐3 human airway epithelial cells. J Gen Physiol 113, 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest IA, Murphy DM, Ward C, Jones D, Johnson GE, Archer L, Gould FK, Cawston TE, Lordan JL & Corris PA (2005). Primary airway epithelial cell culture from lung transplant recipients. Eur Respir J 26, 1080–1085. [DOI] [PubMed] [Google Scholar]
- Fulcher ML, Gabriel S, Burns KA, Yankaskas JR & Randell SH (2005). Well‐differentiated human airway epithelial cell cultures. Methods Mol Med 107, 183–206. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Yang N & Quinton PM (2009). Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator‐dependent bicarbonate secretion. J Clin Invest 119, 2613–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett JP, Hickman E, Burrows R, Hegyi P, Tiszlavicz L, Cuthbert AW, Fong P & Gray MA (2011). Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)‐expressing airway serous cells. J Biol Chem 286, 41069–41082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett JP, Hickman E, Tunkamnerdthai O, Cuthbert AW & Gray MA (2013). Protein phosphatase 1 coordinates CFTR‐dependent airway epithelial HCO3 − secretion by reciprocal regulation of apical and basolateral membrane Cl−–HCO3 − exchangers. Br J Pharmacol 168, 1946–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommes J & Soehnlein O (2011). Contribution of neutrophils to acute lung injury. Mol Med 17, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson JK, Ermund A, Ambort D, Johansson ME, Nilsson HE, Thorell K, Hebert H, Sjovall H & Hansson GC (2012). Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J Exp Med 209, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Abbott SB, Depuy SD, Fortuna MG & Kanbar R (2010). Central CO2 chemoreception and integrated neural mechanisms of cardiovascular and respiratory control. J Appl Physiol (1985) 108, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer A, Hodgson DR & Cann MJ (2006). Regulation of prokaryotic adenylyl cyclases by CO2 . Biochem J 396, 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi P, Gray MA & Argent BE (2003). Substance P inhibits bicarbonate secretion from guinea pig pancreatic ducts by modulating an anion exchanger. Am J Physiol Cell Physiol 285, C268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AE, Wilson JW, Kotsimbos TC & Naughton MT (2003). Metabolic alkalosis contributes to acute hypercapnic respiratory failure in adult cystic fibrosis. Chest 124, 490–493. [DOI] [PubMed] [Google Scholar]
- Huang J, Shan J, Kim D, Liao J, Evagelidis A, Alper SL & Hanrahan JW (2012). Basolateral chloride loading by the anion exchanger type 2: role in fluid secretion by the human airway epithelial cell line Calu‐3. J Physiol 590, 5299–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT & Dale N (2011). CO2‐dependent opening of an inwardly rectifying K+ channel. Pflugers Arch 461, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Eason R, Sachdev A & Dale N (2010. a). CO2‐dependent opening of connexin 26 and related beta connexins. J Physiol 588, 3921–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, id Bihi R, Eason R, Spyer KM, Dicke N, Willecke K, Marina N, Gourine AV & Dale N (2010. b). Connexin hemichannel‐mediated CO2‐dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J Physiol 588, 3901–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianowski JP, Choi JY, Wine JJ & Hanrahan JW (2007). Mucus secretion by single tracheal submucosal glands from normal and cystic fibrosis transmembrane conductance regulator knockout mice. J Physiol 580, 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Alvarez R & Salomon Y (1994). Determination of adenylyl cyclase catalytic activity using single and double column procedures. Methods Enzymol 238, 31–56. [DOI] [PubMed] [Google Scholar]
- Joo NS, Irokawa T, Wu JV, Robbins RC, Whyte RI & Wine JJ (2002). Absent secretion to vasoactive intestinal peptide in cystic fibrosis airway glands. J Biol Chem 277, 50710–50715. [DOI] [PubMed] [Google Scholar]
- Kim D, Liao J & Hanrahan JW (2014). The buffer capacity of airway epithelial secretions. Front Physiol 5, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreda SM, Okada SF, van Heusden CA, O'Neal W, Gabriel S, Abdullah L, Davis CW, Boucher RC & Lazarowski ER (2007). Coordinated release of nucleotides and mucin from human airway epithelial Calu‐3 cells. J Physiol 584, 245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreda SM, Seminario‐Vidal L, van Heusden CA, O'Neal W, Jones L, Boucher RC & Lazarowski ER (2010). Receptor‐promoted exocytosis of airway epithelial mucin granules containing a spectrum of adenine nucleotides. J Physiol 588, 2255–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krouse ME, Talbott JF, Lee MM, Joo NS & Wine JJ (2004). Acid and base secretion in the Calu‐3 model of human serous cells. Am J Physiol Lung Cell Mol Physiol 287, L1274–1283. [DOI] [PubMed] [Google Scholar]
- Laffey JG, Tanaka M, Engelberts D, Luo X, Yuan S, Tanswell AK, Post M, Lindsay T & Kavanagh BP (2000). Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am J Respir Crit Care Med 162, 2287–2294. [DOI] [PubMed] [Google Scholar]
- Lecuona E, Sun H, Chen J, Trejo HE, Baker MA & Sznajder JI (2013). Protein kinase A‐Iα regulates Na,K‐ATPase endocytosis in alveolar epithelial cells exposed to high CO2 concentrations. Am J Respir Cell Mol Biol 48, 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Penland CM, Widdicombe JH & Wine JJ (1998). Evidence that Calu‐3 human airway cells secrete bicarbonate. Am J Physiol 274, L450‐453. [DOI] [PubMed] [Google Scholar]
- Lee RJ & Foskett JK (2010). cAMP‐activated Ca2+ signaling is required for CFTR‐mediated serous cell fluid secretion in porcine and human airways. J Clin Invest 120, 3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco RV & Miranda JM (1968). Drive and performance of the ventilatory apparatus in chronic obstructive lung disease. N Engl J Med 279, 53–59. [DOI] [PubMed] [Google Scholar]
- Marques PA, Magalhaes MC & Correia RN (2003). Inorganic plasma with physiological CO2/HCO3 − buffer. Biomaterials 24, 1541–1548. [DOI] [PubMed] [Google Scholar]
- Masereel B, Pochet L & Laeckmann D (2003). An overview of inhibitors of Na+/H+ exchanger. Eur J Med Chem 38, 547–554. [DOI] [PubMed] [Google Scholar]
- Meigh L, Greenhalgh SA, Rodgers TL, Cann MJ, Roper DI & Dale N (2013). CO2 directly modulates connexin 26 by formation of carbamate bridges between subunits. eLife 2, e01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani R, Tomati V, Galietta LJ & Zegarra‐Moran O (2010). Modulation of cystic fibrosis transmembrane conductance regulator (CFTR) activity and genistein binding by cytosolic pH. J Biol Chem 285, 41591–41596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio K, Suzuki Y, Takeshita K, Aoki T, Kudo H, Sato N, Naoki K, Miyao N, Ishii M & Yamaguchi K (2001). Effects of hypercapnia and hypocapnia on [Ca2+]i mobilization in human pulmonary artery endothelial cells. J Appl Physiol (1985) 90, 2094–2100. [DOI] [PubMed] [Google Scholar]
- Oliver KM, Lenihan CR, Bruning U, Cheong A, Laffey JG, McLoughlin P, Taylor CT & Cummins EP (2012). Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J Biol Chem 287, 14004–14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otulakowski G, Engelberts D, Gusarova GA, Bhattacharya J, Post M & Kavanagh BP (2014). Hypercapnia attenuates ventilator‐induced lung injury via a disintegrin and metalloprotease‐17. J Physiol 592, 4507–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa H, Zhang W, Yarlagadda S, Li C, Conoley VG, Yue J, Bahouth SW, Buddington RK, Zhang G, Nelson DJ, Sonecha MD, Manganiello V, Wine JJ & Naren AP (2010). Compartmentalized cyclic adenosine 3′,5′‐monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol Biol Cell 21, 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford‐Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB, Jr , Welsh MJ & Zabner J (2012). Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prin S, Chergui K, Augarde R, Page B, Jardin F & Vieillard‐Baron A (2002). Ability and safety of a heated humidifier to control hypercapnic acidosis in severe ARDS. Intensive Care Med 28, 1756–1760. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Kopito RR & Quinton PM (1998). Cytosolic pH regulates GCl through control of phosphorylation states of CFTR. Am J Physiol 275, C1040–1047. [DOI] [PubMed] [Google Scholar]
- Ridley C, Kouvatsos N, Raynal BD, Howard M, Collins RF, Desseyn JL, Jowitt TA, Baldock C, Davis CW, Hardingham TE & Thornton DJ (2014). Assembly of the respiratory mucin MUC5B: a new model for a gel‐forming mucin. J Biol Chem 289, 16409–16420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos A & Boron WF (1981). Intracellular pH. Physiol Rev 61, 296–434. [DOI] [PubMed] [Google Scholar]
- Shan J, Liao J, Huang J, Robert R, Palmer ML, Fahrenkrug SC, O'Grady SM & Hanrahan JW (2012). Bicarbonate‐dependent chloride transport drives fluid secretion by the human airway epithelial cell line Calu‐3. J Physiol 590, 5273–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh HS, Tiangco ND, Harrell C & Vender RL (2011). Severe hypercapnia in critically ill adult cystic fibrosis patients. J Clin Med Res 3, 209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen BQ, Finkbeiner WE, Wine JJ, Mrsny RJ & Widdicombe JH (1994). Calu‐3: a human airway epithelial cell line that shows cAMP‐dependent Cl− secretion. Am J Physiol 266, L493–501. [DOI] [PubMed] [Google Scholar]
- Sinclair SE, Kregenow DA, Lamm WJ, Starr IR, Chi EY & Hlastala MP (2002). Hypercapnic acidosis is protective in an in vivo model of ventilator‐induced lung injury. Am J Respir Crit Care Med 166, 403–408. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC & Abboud FM (1989). Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J Appl Physiol (1985) 67, 2101–2106. [DOI] [PubMed] [Google Scholar]
- Song Y, Salinas D, Nielson DW & Verkman AS (2006). Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am J Physiol Cell Physiol 290, C741–749. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Gilman AG & Sprang SR (1997). Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα.GTPγS. Science 278, 1907–1916. [DOI] [PubMed] [Google Scholar]
- Townsend PD, Holliday PM, Fenyk S, Hess KC, Gray MA, Hodgson DR & Cann MJ (2009). Stimulation of mammalian G‐protein‐responsive adenylyl cyclases by carbon dioxide. J Biol Chem 284, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadasz I, Dada LA, Briva A, Trejo HE, Welch LC, Chen J, Toth PT, Lecuona E, Witters LA, Schumacker PT, Chandel NS, Seeger W & Sznajder JI (2008). AMP‐activated protein kinase regulates CO2‐induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K‐ATPase endocytosis. J Clin Invest 118, 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan‐Luijkx IT, van der Laan S, Uglietti C, Schibig MF, Neubert REM, Meijer HAJ, Brand WA, Jordan A, Richter JM, Rothe M & Leuenberger MC (2013). Atmospheric CO2, δ(O2/N2) and δ13CO2 measurements at Jungfraujoch, Switzerland: results from a flask sampling intercomparison program. Atmospheric Measurement Techniques 6, 1805–1815. [Google Scholar]
- Welsh MJ & Smith JJ (2001). cAMP stimulation of HCO3 – secretion across airway epithelia. JOP 2, 291–293. [PubMed] [Google Scholar]
- Yang D, Shcheynikov N, Zeng W, Ohana E, So I, Ando H, Mizutani A, Mikoshiba K & Muallem S (2009). IRBIT coordinates epithelial fluid and HCO3 − secretion by stimulating the transporters pNBC1 and CFTR in the murine pancreatic duct. J Clin Invest 119, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Liu Y, Ruoho AE & Hurley JH (1997). Structure of the adenylyl cyclase catalytic core. Nature 386, 247–253. [DOI] [PubMed] [Google Scholar]