

Abstract
Angiotensin II (AngII) is a pivotal peptide implicated in the regulation of blood pressure. In addition to its systemic vascular and renal effects, AngII acts centrally to modulate the activities of neuroendocrine and sympathetic neuronal networks, influencing in turn sympatho‐humoral outflows to the circulation. Moreover, a large body of evidence supports AngII signalling dysregulation as a key mechanism contributing to exacerbated sympathoexcitation during hypertension. Due to its hydrophilic actions, circulating AngII does not cross the blood–brain barrier (BBB), signalling to the brain via the circumventricular organs which lack a tight BBB. In this review, we present and discuss recent studies from our laboratory showing that elevated circulating levels of AngII during hypertension result in disruption of the BBB integrity, allowing access of circulating AngII to critical sympathoexcitatory brain centres such as the paraventricular nucleus of the hypothalamus and the rostral ventrolateral medulla. We propose the novel hypothesis that AngII‐driven BBB breakdown constitutes a complementary mechanism by which circulating AngII, working in tandem with the central renin–angiotensin system, further exacerbates sympatho‐humoral activation during hypertension. These results are discussed within the context of a growing body of evidence in the literature supporting AngII as a pro‐inflammatory signal, and brain microglia as key cell targets mediating central AngII actions during hypertension.
Abbreviations
- 2K1C
Goldblatt hypertensive model; two‐kidneys, one‐clip
- AngII
angiotensin II
- AT1R
angiotensin type 1 receptor
- BBB
blood–brain barrier
- BP
blood pressure
- CVOs
circumventricular organs
- NTS
nucleus of the tractus solitarius
- PVN
paraventricular nucleus of the hypothalamus
- ROS
reactive oxygen species
- RVLM
rostral ventrolateral medulla
- SFO
subfornical organ
- SHR
spontaneous hypertensive rats
- WKY
Wistar Kyoto rats
Introduction
Hypertension remains a global public health challenge. It affects one‐third of US adults, being a key risk factor for stroke, myocardial infarction, vascular disease and chronic kidney disease. A large proportion of the hypertensive population is categorized as ‘neurogenic’, displaying an increased activation of the sympathetic nervous system during the initiation and maintenance phases of the disease (Parati & Esler, 2012). Importantly, sympathoexcitation is a major determinant of morbidity and mortality in hypertensive patients (Mancia et al. 1999; Parati & Esler, 2012). Thus, understanding the precise neuroanatomical pathways and neurobiological mechanisms underlying increased sympathetic outflow in hypertension is of critical physiological and clinical importance.
Regulation of blood pressure (BP) by the central nervous system involves coordinated activities among highly interconnected neuronal networks distributed throughout the spinal cord, brainstem and forebrain, including, among others, the nucleus of the tractus solitarius (NTS), the rostral ventrolateral medulla (RVLM) and the paraventricular nucleus of the hypothalamus (PVN) (Swanson & Sawchenko, 1983; Guyenet, 2006). Within these centres, numerous chemical signals have been identified to play critical roles in regulating sympathoexcitatory outflow to the cardiovascular system (Gabor & Leenen, 2012). Among them, the neuropeptide angiotensin II (AngII), which also acts as a circulating hormone, constitutes a pivotal signal modulating the activities of both central neuroendocrine and sympathetic neuronal networks. Moreover, a large body of evidence supports AngII signalling dysregulation as one of the key mechanisms involved in stimulating the sympathetic nervous system within the brain (Fink, 1997; Leenen, 2014). There are excellent reviews in the literature covering the contribution of central and systemic AngII to hypertension (Ferrario, 1983; Osborn et al. 2007; Paton et al. 2008; Coble et al. 2015).
Recent findings from our and other laboratories, however, provide support for a novel mechanism by which the peripheral and central renin–angiotensin systems, via disruption of the blood–brain barrier (BBB), may act in tandem to promote exacerbated sympathoexcitatory activity, contributing in turn to neurogenic hypertension (Biancardi et al. 2014). We summarize and review here these novel findings, discussing them in the context of previous literature in the field.
AngII brain signalling and the regulation of blood pressure
AngII is the major effector peptide of the renin–angiotensin systems. It is produced by cleavage of angiotensinogen by the proteolytic enzyme renin into angiotensin I, which is in turn converted to AngII by the angiotensin‐converting enzyme. Besides its classical peripheral actions, including vasoconstriction and kidney sodium reabsorption (Cogan, 1990), AngII has been implicated in the regulation of the cardiovascular system via actions within the brain. Central AngII actions include stimulation of fluid intake, sodium appetite, secretion of hormones (adrenocorticotropic hormone, oxytocin and vasopressin), modulation of baroreflex and activation of the sympathetic nervous system (Lang et al. 1981; Iovino & Steardo, 1984; Ganong & Murakami, 1987; McKinley et al. 1996, 2001, 2003; Paton et al. 2001; Grippo et al. 2002; Ito et al. 2002; Sanderford & Bishop, 2002; Guyenet, 2006; Tan et al. 2007; Coble et al. 2015). Because AngII is highly hydrophilic, the general consensus is that circulating AngII triggers changes in the brain through actions on the circumventricular organs (CVOs), which have an incomplete BBB, consisting of fenestrated capillaries and an incomplete basal membrane and astrocyte ensheathment (Broadwell & Brightman, 1976; Shaver et al. 1992; Daneman, 2012). This particular topographical arrangement creates a large perivascular space that allows passage of large molecules from the circulation into the brain parenchyma. The CVOs are divided into secretory and sensory groups. The secretory group includes the median eminence, the neurohypophysis, the intermediate lobe of the pituitary gland and the pineal gland, while the sensory group includes the subfornical organ (SFO), the organum vasculosum lamina terminalis and the area postrema (Fry & Ferguson, 2007). Circulating AngII signalling through the sensory CVOs is then integrated and conveyed to major brain autonomic and neurosecretory centres, including the PVN, which through descending projections to the RVLM and the NTS mediate the sympathoexcitatory, neurosecretory and modulation‐of‐baroreflex effects of the circulating peptide (Bains et al. 1992; Ferguson & Bains, 1997; Anderson et al. 2001; Dampney et al. 2007; Tan et al. 2007; Ferguson, 2009). Indeed, elevated circulating levels of AngII, resulting in over‐activation of the PVN and RVLM, is recognized as a critical factor contributing to excessive sympathetic activation and vasopressin outflow in neurogenic hypertension in different experimental animal models (Ferguson & Bains, 1997; Bergamaschi et al. 2002; de Oliveira‐Sales et al. 2010; Chen et al. 2011; Huber & Schreihofer, 2011; Qi et al. 2013).
Most of the central AngII actions are mediated by activation of the AngII type 1 receptor (AT1R), a G protein‐coupled receptor (McKinley et al. 1996). AT1Rs have been shown to be present within the sensory CVOs, the hypothalamus and the brainstem in several mammals, including the human (Mendelsohn et al. 1984; Song et al. 1992; MacGregor et al. 1995; McKinley et al. 1996; Lenkei et al. 1997). In fact, AngII microinjected into those nuclei causes dose‐dependent pressor responses (Casto & Phillips, 1984; Muratani et al. 1991; Jensen et al. 1992; Toney & Porter, 1993 b), while AT1R blockade with losartan prevents the increase in BP and vasopressin secretion following intracerebroventricular injections of AngII (Toney & Porter, 1993 a).
In addition to circulating AngII, all the components of the renin–angiotensin system are also present within the brain, including the newly discovered (pro)renin receptor, allowing local formation of AngII and its use as a central neurotransmitter (Ganten & Speck, 1978; McKinley et al. 2003; Li et al. 2012). For example, AngII has been identified as a key neurotransmitter in the SFO–PVN pathway (Osborn et al. 2007; Ferguson, 2009; Coble et al. 2015). Ferguson and collaborators have shown that either electrical stimulation of the SFO, or AngII directly applied within the PVN, increased the firing activity of PVN neurons, responses that were attenuated by the AT1R blocker losartan (Li & Ferguson, 1993; Bains & Ferguson, 1995). Brain AngII has also been shown to be functionally relevant within the RVLM, where an injection of AT1R antagonist decreased mean arterial pressure (∼14 mmHg) in animal models with either high or low plasma renin activity, such as transgenic rats with over‐expression of a mouse renin gene (Fontes et al. 2000), the Dahl salt‐sensitive hypertensive rat (∼35 mmHg) (Ito et al. 2003) and the spontaneous hypertensive rat (SHR) (∼35 mmHg) (Ito et al. 2002), with no effect in normotensive animals.
Although local formation has been implicated as the main source of central AngII contributing to sympathoexcitation and hypertension (Fink, 1997), we have obtained recent evidence, as summarized and discussed further below, that circulating AngII, under certain conditions, may gain access to cardiovascular centres within the central nervous system, exerting its own direct actions on sympathoexcitatory‐related nuclei.
AngII and brain inflammation during hypertension
Vascular brain inflammation has emerged as a novel pathophysiological mechanism contributing to neurogenic hypertension (Paton & Waki, 2009; Lazartigues, 2010; Winklewski et al. 2015). A growing body of evidence supports AngII as a pro‐inflammatory molecule, particularly in hypertension. For example, AngII has been shown to be critical for T‐cell activation and development of vascular inflammation during hypertension, via both central and peripheral mechanisms (Marvar et al. 2010, 2011). Moreover, Francis and collaborators have shown that in an AngII‐induced hypertensive model, AngII stimulated production and release of the transcription factor NFκB along with various pro‐inflammatory cytokines, including TNFα, IL‐1β and IL‐6 within the PVN, all factors that contributed to neurohumoral excitation via oxidative stress (Kang et al. 2009; Cardinale et al. 2012; Sriramula et al. 2013). Similar results have been recently reported in renovascular hypertensive rats (Goldblatt model, two‐kidneys, one‐clip (2K1C)), in which mRNA expression of AT1 receptors and NAD(P)H oxidase subunits were shown to be increased within the RVLM and PVN of hypertensive animals, contributing to elevated BP and sympathetic activity (Oliveira‐Sales et al. 2009), effects that were attenuated by AT1R blockade within the RVLM (Nishi et al. 2013). In addition, higher levels of cytokines (TNFα, IL‐1β and IL‐6, and chemokine MCP‐1) have also been reported within the RVLM in this hypertensive model (Li et al. 2014). Likewise, pro‐inflammatory factors such as the junctional adhesion molecule‐1 were found to be upregulated within the NTS of SHR (Waki et al. 2007, 2011).
Microglia cells are the primary resident immune cells of the brain (Saijo & Glass, 2011), and thus likely candidates mediating central AngII pro‐inflammatory actions. In fact, several recent papers reported that AngII can turn microglia from a resting state into an activated, inflammatory state (Miyoshi et al. 2008; Rodriguez‐Pallares et al. 2008; Benicky et al. 2009). Moreover, AngII‐mediated microglia cell activation within the PVN has been shown to contribute to high BP in an angiotensin II‐induced hypertensive rat model, via generation of pro‐inflammatory cytokines (IL‐6, IL1β and TNFα) (Shi et al. 2010).
Disruption of the blood–brain barrier (BBB) integrity in neuro‐inflammatory disorders and hypertension
The BBB acts as a dynamic physical barrier at the brain–blood interface, which effectively excludes substances that are lipid insoluble, as well as those of high molecular weight (Abbott et al. 2006, 2010; Obermeier et al. 2013; Wong et al. 2013). The BBB is mainly composed of tight junctions formed between endothelial cells and the enwrapping astrocyte endfeet, which limit the paracellular diffusion of hydrophilic molecules (Stamatovic et al. 2008; Abbott et al. 2010). In addition, BBB endothelial cells, under normal conditions, have minimal vesicle transport activity, which limits transcellular transport (Stamatovic et al. 2008). This process is essential to maintain homeostasis of the brain environment. In general, astrocytes, pericytes and neurons that are in direct physical contact with the capillary endothelium play an important role in the regulation of BBB integrity (Abbott et al. 2010).
Breakdown of the BBB, resulting in increased permeability and access to the brain of circulating substances normally excluded, is a common feature in neuro‐inflammatory disorders, including ischaemia and multiple sclerosis (Waubant, 2006; Yang & Rosenberg, 2011). An altered BBB state has also been shown in hypertension (Mayhan et al. 1989; Ueno et al. 2004; Vital et al. 2010; Pelisch et al. 2011, 2013). Still, the precise underlying mechanisms contributing to BBB disruption in hypertension, and the potential consequences in terms of brain access and actions of circulating AngII within central nervous system pathways involved in BP regulation have not yet been thoroughly assessed.
To further study changes in BBB integrity and function, and to determine whether circulating AngII gains access to the brain during hypertension, we performed a study in which we used a combination of fluorescent dyes of different sizes that were injected into the systemic circulation of Wistar Kyoto (WKY) and SHR rats. This approach enabled us to perform a quantitative assessment of the degree of intravascular and extravascular dyes within the parenchyma of specific brain nuclei, specifically within hypothalamus and brainstem regions, crucial to BP regulation and baroreceptor function (Biancardi et al. 2014). As previously reported (Mayhan et al. 1989), the presence of extravasated dye within the brain parenchyma was considered indicative of BBB increased permeability. To validate this approach, we compared first in control rats the presence or absence of extravasated dyes in brain areas known to reside within and outside the BBB. As shown in Fig. 1 A–D, we found large amounts of the intravascularly injected dye dextran‐fluorescein isothiocyanate (FITC) 10 kDa (FITC10) extravasated in the parenchyma of the SFO (Fig. 1 A) and the area postrema (Fig. 1 B), both brain regions known to lack a tight BBB. Conversely, minimal extravasation was observed in areas known to possess a tight BBB, such as the PVN (Fig. 1 C) and the supraoptic nucleus of the hypothalamus (Fig. 1 D). When we applied this approach to SHR, we observed a large degree of FITC10 extravasation within the PVN (∼85% increase), which was significantly more abundant compared to aged‐matched WKYs (Fig. 1 E–G). Importantly, similar results were observed in a renovascular hypertensive model (the 2K1C model of hypertension) (Fig. 1 H–J), indicating that BBB disruption and increased permeability is not a unique feature of the SHR model. Similar to the PVN, the brainstem (RVLM and the NTS) also displayed disrupted BBB in both hypertensive models (SHR and 2K1C) (Biancardi et al. 2014).
Figure 1. Extravasation of FITC10 within the brain in different conditions .
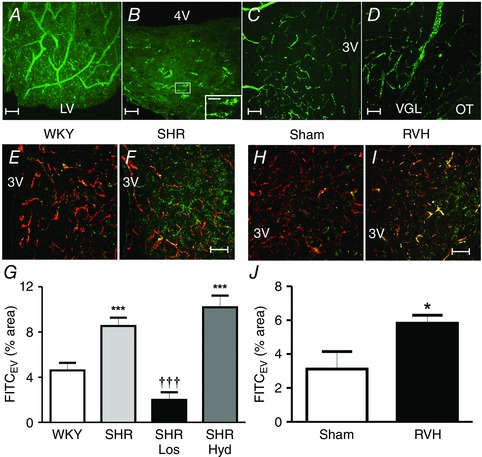
A–D, representative images showing a comparison of extravasated (EV) FITC10 in control rats in brain areas lacking (A, subfornical organ; B, area postrema) or possessing (C, PVN; D, supraoptic hypothalamic nuclei) an intact BBB. The inset in B shows a higher magnification of the squared area. Note the presence of extravascular FITC10 in A and B, but not C or D. E–J shows increased extravasated small size FITC10 (green dye) but not large size dextran‐rhodamine 70 kDA (RHO70) (red dye), indicative of increased BBB permeability, within the PVN of spontaneous (SHR, E–G) and renovascular hypertensive rats (RVH, H–J) as indicated in the respective summary. G, summary data showing that leakage of FITC10 is blunted in SHR treated with losartan (Los) but not hydralazine (Hyd). Scale bars: 25 μm for A, C and D and inset in B; 50 μm for B, E, F, H and I. ***P < 0.001 and *P < 0.05 vs. WKY or Sham; ††† P < 0.001 vs. SHR. n = 8 SHR/WKY in G; n = 4 SHR‐Los/SHR‐Hyd in G; n = 3 Sham/RVH in J. 3V and 4V, third and fourth ventricle; LV, lateral ventricle; VGL, ventral glial lamina; OT, optic tract (modified from Biancardi et al. 2014).
To determine whether in addition to the inert dextran dyes, physiologically relevant molecules could also leak through the disrupted BBB during hypertension, we repeated the experiments using systemic infusions of a fluorescently labelled form of AngII (AngII‐FITC). Similar to FITC10, we found large levels of extravasated AngII‐FITC within the PVN of hypertensive animals (∼44%) (Fig. 2 A–C). These results indicate that the increased BBB permeability observed during hypertension allows the leakage and access of circulating factors, namely AngII, directly into key sympathoexcitatory brain areas, such as the PVN and the RVLM. These results are in line with a recent study by Yao & May, who showed that when the BBB was disrupted with hypertonic mannitol, systemically applied AngII activated tyrosine hydroxylase‐expressing RVLM neurons, indicative of leakage of AngII into the RVLM (Yao & May, 2013).
Figure 2. Circulating angiotensin II leaks into the PVN parenchyma in SHR, targeting neurons and microglia .
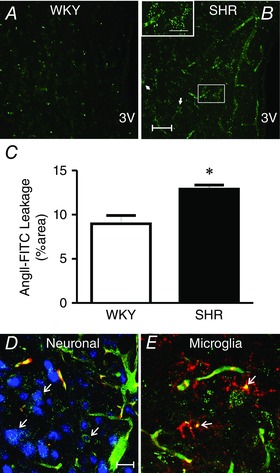
A–C, images showing increased extravasated levels of AngII‐FITC (intravascularly delivered) within the PVN of hypertensive (SHR, B) compared with normotensive (WKY, A) rats, as indicated in the summary data (C). D and E, extravasated AngII‐FITC (green) is co‐localized with neurons (D, neuronal marker NeuN (blue) and the RHO70 dye contained in the vasculature, red) and with microglia (E, microglia marker CD11b, red). Scale bars: 50 μm. *P < 0.05 vs. WKY. n = 4 WKY/SHR. 3V: third ventricle (modified from Biancardi et al. 2014).
We found the extravasated AngII‐FITC in the PVN of hypertensive animals to be associated both with neurons and microglia cells (Fig. 2 D and E), supporting these two cell types as likely targets for circulating AngII actions within the brain during hypertension. These results are thus in agreement with several other reports, as discussed above, supporting microglia as likely mediators of pro‐inflammatory actions of AngII within the CNS.
Mechanisms contributing to BBB disruption during hypertension
Endothelial dysfunction associated with high BP conditions is a likely underlying factor contributing to vascular inflammation and downstream BBB disruption during hypertension (Ueno et al. 2004; Pires et al. 2013). Alternatively, direct AT1R‐mediated signalling could also contribute to this phenomenon. For example, AT1R activation has been shown to affect BBB permeability in cultured microvessels (Fleegal‐DeMotta et al. 2009). Likewise, chronic infusion of AngII, in an AT1R‐dependent manner, has been implicated in increased BBB permeability, when measured in whole mouse brain homogenates (Vital et al. 2010). Moreover, the AT1R blocker olmesartan was shown to prevent altered BBB permeability within the hippocampus of AngII‐induced and Dahl salt‐sensitive hypertensive rats (Pelisch et al. 2011, 2013).
Thus, to gain more insights into the specific mechanisms contributing to altered BBB permeability during hypertension, we experimentally assessed the relative contribution of high blood pressure itself vs. AngII‐AT1R signalling. To this end, we evaluated the effect of treating SHR with either the vasodilator hydralazine (which lowered BP independently of AngII signalling) or the AT1R blocker losartan, which also lowered BP by blocking AngII actions. We found that the disrupted BBB in SHR was prevented by the treatment with losartan (decrease of ∼76% in dye extravasation), but not with hydralazine (Fig. 1 G), despite similar decreases in BP obtained with the two treatments (Biancardi et al. 2014). These results support a major contribution of AT1R‐mediated signalling to altered BBB permeability during hypertension, rather than high BP itself.
One technical aspect of these studies that needs to be taken into consideration is that any residual free fluorescein‐isothiocyanate present in our FITC‐dextran preparations could potentially be transported across the BBB via organic anion transporters (Sun et al. 2001), resulting in false positive signals. However, the facts that (a) we observed a significantly larger amount of leaked FITC10 in hypertensive rats, (b) that only the FITC‐dextran of smaller molecular size leaked, and (c) that these differences were largely prevented in rats treated with AT1 receptor blockers, would argue against a positive signal due to transport of residual free FITC10. Still, the precise route by which the FITC‐dextran influx accessed these areas of the brain, and particularly whether it occurs via the paracellular space, remains unknown.
Do active microglia contribute to AngII‐driven reactive oxygen species production?
As summarized above, it is now generally recognized that AngII‐mediated pro‐inflammatory actions constitute a critical mechanism contributing to sympathoexcitation in hypertension, and a growing body of evidence supports microglia as key cellular targets mediating central AngII pro‐inflammatory effects. Moreover, our recent studies also support this mechanism to contribute to BBB disruption during hypertension (Biancardi et al. 2014).
AngII‐mediated generation of reactive oxygen species (ROS) within the SFO–PVN–RVLM pathway has also been implicated as an important contributor to sympathoexcitation in hypertension (Zimmerman et al. 2002,2004; Braga et al. 2011; Capone et al. 2012). Despite the robust evidence supporting this mechanism, the precise cellular targets and sources of ROS remain largely unknown. In this sense, activated microglia, besides releasing a variety of pro‐inflammatory cytokines, also generate and release ROS (Saijo & Glass, 2011). However, whether AngII‐dependent, microglia‐derived ROS contribute to altered BBB permeability during hypertension remains to be determined.
Final remarks
AngII and its AT1Rs is one of the most important and most widely studied signalling pathways contributing to the central regulation of blood pressure, both in health and disease conditions. We believe that our recent studies summarized above further our current understanding of the mechanisms by which circulating AngII exerts its central effects. While the general consensus in the field is that circulating AngII accesses the central nervous system through the CVOs that reside outside of the BBB, our recent data suggest, that under pathological conditions such as hypertension, an additional route for AngII signalling in the brain is gated. Thus, we propose that a compromised BBB facilitates the direct access of circulating AngII to critical sympathoexcitatory brain centres that are normally protected, constituting a complementary mechanism that, working in tandem with the local central renin–angiotensin system, further exacerbates AngII‐driven neurohumoral activation during hypertension. The facts that elevated circulating levels of AngII at the onset of hypertension (a) contribute to BBB integrity, (b) facilitate their own access to brain sympathoexcitatory centres, and (c) contribute to further increasing BP thus support a highly deleterious AngII‐mediated feedforward mechanism during hypertension.
AT1R blockade, as well as angiotensin‐converting enzyme inhibitors, are widely used therapeutic agents for the treatment of cardiovascular diseases (Perret‐Guillaume et al. 2009). Moreover, AT1R blockade has been shown to have neuroprotective effects when used for inflammatory brain disorders that accompany BBB disruption such as traumatic brain injury, stroke, dementia, Alzheimer's and Parkinson's diseases (Villapol & Saavedra, 2015). Thus, AngII and its central AT1 receptors stand as novel therapeutic targets that may help prevent and/or rescue an altered BBB status in numerous inflammatory diseases, including hypertension.
Additional information
Competing interests
None declared.
Funding
This work was supported by a National Heart, Lung, and Blood Institute Grant (NIH HL112225; J.E.S.) and a Scientific Development Grant (AHA 14SDG20400015; V.C.B.).
Acknowledgements
We would like to acknowledge the contribution of Dr J. A. Filosa (Georgia Regents University), Ms S. J. Son and Ms S. Ahmadi for their valuable contribution to different aspects of this work.
Biographies
Vinicia Biancardi did her PhD with Ruy R. Campos at Federal University of Sao Paulo, Brazil and a postdoctoral fellowship with Javier Stern at the University of Cincinnati and Georgia Regents University before becoming an Assistant Research Scientist at Georgia Regents University.
Javier Stern started at the University of Tennessee, Memphis, as a postdoc in William E. Armstrong's lab in 1994. He became Assistant Professor at Wright State University in 1999, and was then recruited as an Associate Professor at the University of Cincinnati in 2004. He is currently a Professor in the Department of Physiology at Georgia Regents University. We share a common interest in understanding hypothalamic neurobiological mechanisms underlying the central control of cardiovascular and neuroendocrine function both in health and disease states, including hypertension, heart failure and the metabolic syndrome.

This review was presented at the symposium “Bristol‐São Paulo Research Summit” ‐ Autonomic and neuroendocrine dysfunction in chronic diseases, which took place at the University of São Paulo, Brasil, between 7–10 August 2014.
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR & Begley DJ (2010). Structure and function of the blood‐brain barrier. Neurobiol Dis 37, 13–25. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L & Hansson E (2006). Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci 7, 41–53. [DOI] [PubMed] [Google Scholar]
- Anderson JW, Smith PM & Ferguson AV (2001). Subfornical organ neurons projecting to paraventricular nucleus: whole‐cell properties. Brain Res 921, 78–85. [DOI] [PubMed] [Google Scholar]
- Bains JS & Ferguson AV (1995). Paraventricular nucleus neurons projecting to the spinal cord receive excitatory input from the subfornical organ. Am J Physiol 268, R625–R633. [DOI] [PubMed] [Google Scholar]
- Bains JS, Potyok A & Ferguson AV (1992). Angiotensin II actions in paraventricular nucleus: functional evidence for neurotransmitter role in efferents originating in subfornical organ. Brain Res 599, 223–229. [DOI] [PubMed] [Google Scholar]
- Benicky J, Sanchez‐Lemus E, Pavel J & Saavedra JM (2009). Anti‐inflammatory effects of angiotensin receptor blockers in the brain and the periphery. Cell Mol Neurobiol 29, 781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi CT, Biancardi VC, Lopes OU & Campos RR (2002). Effects of angiotensin blockade in the rostral ventrolateral medulla on maintenance of hypertension induced by chronic l‐NAME treatment. Brain Res 927, 195–199. [DOI] [PubMed] [Google Scholar]
- Biancardi VC, Son SJ, Ahmadi S, Filosa JA & Stern JE (2014). Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood‐brain barrier. Hypertension 63, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VA, Medeiros IA, Ribeiro TP, França‐Silva MS, Botelho‐Ono MS & Guimarães DD (2011). Angiotensin‐II‐induced reactive oxygen species along the SFO‐PVN‐RVLM pathway: implications in neurogenic hypertension. Braz J Med Biol Res 44, 871–876. [DOI] [PubMed] [Google Scholar]
- Broadwell RD & Brightman MW (1976). Entry of peroxidase into neurons of the central and peripheral nervous systems from extracerebral and cerebral blood. J Comp Neurol 166, 257–283. [DOI] [PubMed] [Google Scholar]
- Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, Pickel VM, Davisson RL & Iadecola C (2012). Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci 32, 4878–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale JP, Sriramula S, Mariappan N, Agarwal D & Francis J (2012). Angiotensin II‐induced hypertension is modulated by nuclear factor‐κB in the paraventricular nucleus. Hypertension 59, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casto R & Phillips MI (1984). Mechanism of pressor effects by angiotensin in the nucleus tractus solitarius of rats. Am J Physiol 247, R575–R581. [DOI] [PubMed] [Google Scholar]
- Chen AD, Zhang SJ, Yuan N, Xu Y, De W, Gao XY & Zhu GQ (2011). Angiotensin AT1 receptors in paraventricular nucleus contribute to sympathetic activation and enhanced cardiac sympathetic afferent reflex in renovascular hypertensive rats. Exp Physiol 96, 94–103. [DOI] [PubMed] [Google Scholar]
- Coble JP, Grobe JL, Johnson AK & Sigmund CD (2015). Mechanisms of brain renin angiotensin system‐induced drinking and blood pressure: importance of the subfornical organ. Am J Physiol Regul Integr Comp Physiol 308, R238–R249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan MG (1990). Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension 15, 451–458. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Tan PS, Sheriff MJ, Fontes MA & Horiuchi J (2007). Cardiovascular effects of angiotensin II in the rostral ventrolateral medulla: the push‐pull hypothesis. Curr Hypertens Rep 9, 222–227. [DOI] [PubMed] [Google Scholar]
- Daneman R (2012). The blood‐brain barrier in health and disease. Ann Neurol 72, 648–672. [DOI] [PubMed] [Google Scholar]
- de Oliveira‐Sales EB, Nishi EE, Boim MA, Dolnikoff MS, Bergamaschi CT & Campos RR (2010). Upregulation of AT1R and iNOS in the rostral ventrolateral medulla (RVLM) is essential for the sympathetic hyperactivity and hypertension in the 2K‐1C Wistar rat model. Am J Hypertens 23, 708–715. [DOI] [PubMed] [Google Scholar]
- Ferguson AV (2009). Angiotensinergic regulation of autonomic and neuroendocrine outputs: critical roles for the subfornical organ and paraventricular nucleus. Neuroendocrinology 89, 370–376. [DOI] [PubMed] [Google Scholar]
- Ferguson AV & Bains JS (1997). Actions of angiotensin in the subfornical organ and area postrema: implications for long term control of autonomic output. Clin Exp Pharmacol Physiol 24, 96–101. [DOI] [PubMed] [Google Scholar]
- Ferrario CM (1983). Neurogenic actions of angiotensin II. Hypertension 5, V73–79. [DOI] [PubMed] [Google Scholar]
- Fink GD (1997). Long‐term sympatho‐excitatory effect of angiotensin II: a mechanism of spontaneous and renovascular hypertension. Clin Exp Pharmacol Physiol 24, 91–95. [DOI] [PubMed] [Google Scholar]
- Fleegal‐DeMotta MA, Doghu S & Banks WA (2009). Angiotensin II modulates BBB permeability via activation of the AT1 receptor in brain endothelial cells. J Cereb Blood Flow Metab 29, 640–647. [DOI] [PubMed] [Google Scholar]
- Fontes MA, Baltatu O, Caligiorne SM, Campagnole‐Santos MJ, Ganten D, Bader M & Santos RA (2000). Angiotensin peptides acting at rostral ventrolateral medulla contribute to hypertension of TGR(mREN2)27 rats. Physiol Genomics 2, 137–142. [DOI] [PubMed] [Google Scholar]
- Fry M & Ferguson AV (2007). The sensory circumventricular organs: brain targets for circulating signals controlling ingestive behavior. Physiol Behav 91, 413–423. [DOI] [PubMed] [Google Scholar]
- Gabor A & Leenen FH (2012). Central neuromodulatory pathways regulating sympathetic activity in hypertension. J Appl Physiol (1985) 113, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganong WF & Murakami K (1987). The role of angiotensin II in the regulation of ACTH secretion. Ann NY Acad Sci 512, 176–186. [DOI] [PubMed] [Google Scholar]
- Ganten D & Speck G (1978). The brain renin‐angiotensin system: a model for the synthesis of peptides in the brain. Biochem Pharmacol 27, 2379–2389. [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Kirby RF, Beltz TG & Johnson AK (2002). Angiotensin II‐induced drinking and pressor responses to central or systemic irbesartan and losartan. Pharmacol Biochem Behav 71, 139–146. [DOI] [PubMed] [Google Scholar]
- Guyenet PG (2006). The sympathetic control of blood pressure. Nat Rev Neurosci 7, 335–346. [DOI] [PubMed] [Google Scholar]
- Huber DA & Schreihofer AM (2011). Altered regulation of the rostral ventrolateral medulla in hypertensive obese Zucker rats. Am J Physiol Heart Circ Physiol 301, H230–H240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino M & Steardo L (1984). Vasopressin release to central and peripheral angiotensin II in rats with lesions of the subfornical organ. Brain Res 322, 365–368. [DOI] [PubMed] [Google Scholar]
- Ito S, Hiratsuka M, Komatsu K, Tsukamoto K, Kanmatsuse K & Sved AF (2003). Ventrolateral medulla AT1 receptors support arterial pressure in Dahl salt‐sensitive rats. Hypertension 41, 744–750. [DOI] [PubMed] [Google Scholar]
- Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K & Sved AF (2002). Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension 40, 552–559. [DOI] [PubMed] [Google Scholar]
- Jensen LL, Harding JW & Wright JW (1992). Role of paraventricular nucleus in control of blood pressure and drinking in rats. Am J Physiol 262, F1068–F1075. [DOI] [PubMed] [Google Scholar]
- Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM & Francis J (2009). Brain nuclear factor‐kappa B activation contributes to neurohumoral excitation in angiotensin II‐induced hypertension. Cardiovasc Res 82, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RE, Rascher W, Heil J, Unger T, Wiedemann G & Ganten D (1981). Angiotensin stimulates oxytocin release. Life Sci 29, 1425–1428. [DOI] [PubMed] [Google Scholar]
- Lazartigues E (2010). Inflammation and neurogenic hypertension: a new role for the circumventricular organs? Circ Res 107, 166–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenen FH (2014). Actions of circulating angiotensin II and aldosterone in the brain contributing to hypertension. Am J Hypertens 27, 1024–1032. [DOI] [PubMed] [Google Scholar]
- Lenkei Z, Palkovits M, Corvol P & Llorens‐Cortes C (1997). Expression of angiotensin type‐1 (AT1) and type‐2 (AT2) receptor mRNAs in the adult rat brain: a functional neuroanatomical review. Front Neuroendocrinol 18, 383–439. [DOI] [PubMed] [Google Scholar]
- Li HB, Qin DN, Ma L, Miao YW, Zhang DM, Lu Y, Song XA, Zhu GQ & Kang YM (2014). Chronic infusion of lisinopril into hypothalamic paraventricular nucleus modulates cytokines and attenuates oxidative stress in rostral ventrolateral medulla in hypertension. Toxicol Appl Pharmacol 279, 141–149. [DOI] [PubMed] [Google Scholar]
- Li W, Peng H, Seth DM & Feng Y (2012). The prorenin and (pro)renin receptor: new players in the brain renin‐angiotensin system? Int J Hypertens 2012, 290635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z & Ferguson AV (1993). Subfornical organ efferents to paraventricular nucleus utilize angiotensin as a neurotransmitter. Am J Physiol 265, R302–R309. [DOI] [PubMed] [Google Scholar]
- MacGregor DP, Murone C, Song K, Allen AM, Paxinos G & Mendelsohn FA (1995). Angiotensin II receptor subtypes in the human central nervous system. Brain Res 675, 231–240. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, Oldfield BJ, Mendelsohn FA & Chai SY (2003). The brain renin‐angiotensin system: location and physiological roles. Int J Biochem Cell Biol 35, 901–918. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Allen AM, Mathai ML, May C, McAllen RM, Oldfield BJ & Weisinger RS (2001). Brain angiotensin and body fluid homeostasis. Jpn J Physiol 51, 281–289. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, McAllen RM, Pennington GL, Smardencas A, Weisinger RS & Oldfield BJ (1996). Physiological actions of angiotensin II mediated by AT1 and AT2 receptors in the brain. Clin Exp Pharmacol Physiol 23, Suppl. 3, S99–104. [PubMed] [Google Scholar]
- Mancia G, Grassi G, Giannattasio C & Seravalle G (1999). Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 34, 724–728. [DOI] [PubMed] [Google Scholar]
- Marvar PJ, Lob H, Vinh A, Zarreen F & Harrison DG (2011). The central nervous system and inflammation in hypertension. Curr Opin Pharmacol 11, 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ & Harrison DG (2010). Central and peripheral mechanisms of T‐lymphocyte activation and vascular inflammation produced by angiotensin II‐induced hypertension. Circ Res 107, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhan WG, Faraci FM, Siems JL & Heistad DD (1989). Role of molecular charge in disruption of the blood‐brain barrier during acute hypertension. Circ Res 64, 658–664. [DOI] [PubMed] [Google Scholar]
- Mendelsohn FA, Quirion R, Saavedra JM, Aguilera G & Catt KJ (1984). Autoradiographic localization of angiotensin II receptors in rat brain. Proc Natl Acad Sci USA 81, 1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi M, Miyano K, Moriyama N, Taniguchi M & Watanabe T (2008). Angiotensin type 1 receptor antagonist inhibits lipopolysaccharide‐induced stimulation of rat microglial cells by suppressing nuclear factor κ B and activator protein‐1 activation. Eur J Neurosci 27, 343–351. [DOI] [PubMed] [Google Scholar]
- Muratani H, Averill DB & Ferrario CM (1991). Effect of angiotensin II in ventrolateral medulla of spontaneously hypertensive rats. Am J Physiol 260, R977–R984. [DOI] [PubMed] [Google Scholar]
- Nishi EE, Bergamaschi CT, Oliveira‐Sales EB, Simon KA & Campos RR (2013). Losartan reduces oxidative stress within the rostral ventrolateral medulla of rats with renovascular hypertension. Am J Hypertens 26, 858–865. [DOI] [PubMed] [Google Scholar]
- Obermeier B, Daneman R & Ransohoff RM (2013). Development, maintenance and disruption of the blood‐brain barrier. Nat Med 19, 1584–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Nishi EE, Carillo BA, Boim MA, Dolnikoff MS, Bergamaschi CT & Campos RR (2009). Oxidative stress in the sympathetic premotor neurons contributes to sympathetic activation in renovascular hypertension. Am J Hypertens 22, 484–492. [DOI] [PubMed] [Google Scholar]
- Osborn JW, Fink GD, Sved AF, Toney GM & Raizada MK (2007). Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9, 228–235. [DOI] [PubMed] [Google Scholar]
- Parati G & Esler M (2012). The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J 33, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Paton JF, Deuchars J, Ahmad Z, Wong LF, Murphy D & Kasparov S (2001). Adenoviral vector demonstrates that angiotensin II‐induced depression of the cardiac baroreflex is mediated by endothelial nitric oxide synthase in the nucleus tractus solitarii of the rat. J Physiol 531, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF & Waki H (2009). Is neurogenic hypertension related to vascular inflammation of the brainstem? Neurosci Biobehav Rev 33, 89–94. [DOI] [PubMed] [Google Scholar]
- Paton JF, Wang S, Polson JW & Kasparov S (2008). Signalling across the blood brain barrier by angiotensin II: novel implications for neurogenic hypertension. J Mol Med (Berl) 86, 705–710. [DOI] [PubMed] [Google Scholar]
- Pelisch N, Hosomi N, Mori H, Masaki T & Nishiyama A (2013). RAS inhibition attenuates cognitive impairment by reducing blood‐ brain barrier permeability in hypertensive subjects. Curr Hypertens Rev 9, 93–98. [DOI] [PubMed] [Google Scholar]
- Pelisch N, Hosomi N, Ueno M, Nakano D, Hitomi H, Mogi M, Shimada K, Kobori H, Horiuchi M, Sakamoto H, Matsumoto M, Kohno M & Nishiyama A (2011). Blockade of AT1 receptors protects the blood‐brain barrier and improves cognition in Dahl salt‐sensitive hypertensive rats. Am J Hypertens 24, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perret‐Guillaume C, Joly L, Jankowski P & Benetos A (2009). Benefits of the RAS blockade: clinical evidence before the ONTARGET study. J Hypertens Suppl 27, S3–7. [DOI] [PubMed] [Google Scholar]
- Pires PW, Dams Ramos CM, Matin N & Dorrance AM (2013). The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol 304, H1598–H1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Zhang DM, Suo YP, Song XA, Yu XJ, Elks C, Lin YX, Xu YY, Zang WJ, Zhu Z & Kang YM (2013). Renin‐angiotensin system modulates neurotransmitters in the paraventricular nucleus and contributes to angiotensin II‐induced hypertensive response. Cardiovasc Toxicol 13, 48–54. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Pallares J, Rey P, Parga JA, Munoz A, Guerra MJ & Labandeira‐Garcia JL (2008). Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH‐derived ROS. Neurobiol Dis 31, 58–73. [DOI] [PubMed] [Google Scholar]
- Saijo K & Glass CK (2011). Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 11, 775–787. [DOI] [PubMed] [Google Scholar]
- Sanderford MG & Bishop VS (2002). Central mechanisms of acute ANG II modulation of arterial baroreflex control of renal sympathetic nerve activity. Am J Physiol Heart Circ Physiol 282, H1592–H1602. [DOI] [PubMed] [Google Scholar]
- Shaver SW, Wall KM, Wainman DS & Gross PM (1992). Regional quantitative permeability of blood‐brain barrier lesions in rats with chronic renal hypertension. Brain Res 579, 99–106. [DOI] [PubMed] [Google Scholar]
- Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C & Raizada MK (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Allen AM, Paxinos G & Mendelsohn FA (1992). Mapping of angiotensin II receptor subtype heterogeneity in rat brain. J Comp Neurol 316, 467–484. [DOI] [PubMed] [Google Scholar]
- Sriramula S, Cardinale JP & Francis J (2013). Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II‐induced hypertension. PLoS One 8, e63847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF & Andjelkovic AV (2008). Brain endothelial cell‐cell junctions: how to ‘open’ the blood brain barrier. Curr Neuropharmacol 6, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Miller DW & Elmquist WF (2001). Effect of probenecid on fluorescein transport in the central nervous system using in vitro and in vivo models. Pharm Res 18, 1542–1549. [DOI] [PubMed] [Google Scholar]
- Swanson LW & Sawchenko PE (1983). Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Ann Rev Neurosci 6, 269–324. [DOI] [PubMed] [Google Scholar]
- Tan PS, Killinger S, Horiuchi J & Dampney RA (2007). Baroreceptor reflex modulation by circulating angiotensin II is mediated by AT1 receptors in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 293, R2267–R2278. [DOI] [PubMed] [Google Scholar]
- Toney GM & Porter JP (1993. a). Effects of blockade of AT1 and AT2 receptors in brain on the central angiotensin II pressor response in conscious spontaneously hypertensive rats. Neuropharmacology 32, 581–589. [DOI] [PubMed] [Google Scholar]
- Toney GM & Porter JP (1993. b). Functional role of brain AT1 and AT2 receptors in the central angiotensin II pressor response. Brain Res 603, 57–63. [DOI] [PubMed] [Google Scholar]
- Ueno M, Sakamoto H, Liao YJ, Onodera M, Huang CL, Miyanaka H & Nakagawa T (2004). Blood‐brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem Cell Biol 122, 131–137. [DOI] [PubMed] [Google Scholar]
- Villapol S & Saavedra JM (2015). Neuroprotective effects of angiotensin receptor blockers. Am J Hypertens 28, 289–299. [DOI] [PubMed] [Google Scholar]
- Vital SA, Terao S, Nagai M & Granger DN (2010). Mechanisms underlying the cerebral microvascular responses to angiotensin II‐induced hypertension. Microcirculation 17, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki H, Gouraud SS, Maeda M, Raizada MK & Paton JF (2011). Contributions of vascular inflammation in the brainstem for neurogenic hypertension. Respir Physiol Neurobiol 178, 422–428. [DOI] [PubMed] [Google Scholar]
- Waki H, Liu B, Miyake M, Katahira K, Murphy D, Kasparov S & Paton JF (2007). Junctional adhesion molecule‐1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension 49, 1321–1327. [DOI] [PubMed] [Google Scholar]
- Waubant E (2006). Biomarkers indicative of blood‐brain barrier disruption in multiple sclerosis. Dis Markers 22, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklewski PJ, Radkowski M, Wszedybyl‐Winklewska M & Demkow U (2015). Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation 12, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE & Searson PC (2013). The blood‐brain barrier: an engineering perspective. Front Neuroeng 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y & Rosenberg GA (2011). Blood‐brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42, 3323–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao ST & May CN (2013). Intra‐carotid angiotensin II activates tyrosine hydroxylase‐expressing rostral ventrolateral medulla neurons following blood‐brain barrier disruption in rats. Neuroscience 245, 148–156. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR & Davisson RL (2002). Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91, 1038–1045. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV & Davisson RL (2004). Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95, 210–216. [DOI] [PubMed] [Google Scholar]