

Abstract
MiR-200a has been reported to be able to suppress the epithelial-mesenchymal transition process in pancreatic cancer stem cells, suggesting that miR-200a could suppress the metastasis of pancreatic ductal adenocarcinoma (PDAC). However, its role in proliferation and metastasis of PDAC and the underlying mechanism by which miR-200a works in PDAC have not been elucidated. In our study, we for the first time identified that DEK gene is a direct downstream target of miR-200a. It was found that overexpression of miR-200a decreased DEK expression, suppressing the proliferation, migration, and invasion of PDAC cells. Meanwhile, knockdown of miR-200a can increase DEK level, promoting the proliferation, migration, and invasion of PDAC cells. Our study demonstrated that miR-200a suppresses the metastasis in pancreatic PDAC through downregulation of DEK, suggesting that miR-200a may be used as a novel potential marker in prediction of metastasis of PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a malignancy with the lowest prognosis, with the majority of patients diagnosed with advanced disease that often happened to metastasize from lymph nodes to distant organs [1]. Therefore, there is a great need to understand the biological mechanisms that contribute to pancreatic cancer development and progression so as to develop effective therapies.
MiR-200a has been reported to be able to suppress the epithelial-mesenchymal transition process, which is a critical step for the initiation of cancer metastasis, of pancreatic cancer stem cell [2] and colorectal carcinoma cell [3]. In addition, miR-200a has also been found to suppress cell proliferation and migration in pancreatic cancer [4] and hepatocellular carcinoma [5]. Based on the four relevant literatures, it can be hypothesized that miR-200a could suppress the proliferation and metastasis of PDAC. However, the role of miR-200a in PDAC and the underlying mechanism have not been elucidated.
DEK protein was originally related to chromatin reconstruction [6] and transcription factor involved in stabilization of heterochromatin and cruciform structures [7]. Subsequently, it has been increasingly found to be generally overexpressed in various cancers, being shown to play an important role in the development and progression of different types of cancers [7], [8]. In pancreatic cancer, DEK gene was first mentioned and found to be upregulated among the most significantly differential genes that related to liver metastasis [9], indicating its role as a metastasis associated gene in PDAC. Besides, there has been, however, no more subsequent study to follow the role of DEK in PDAC.
In the present study, we for the first time identified and found that DEK gene is a direct downstream target of miR-200a. Our study demonstrated that miR-200a suppresses both the proliferation and metastasis in PDAC through downregulation of DEK, suggesting that miR-200a may be used as a novel potential marker in prediction of metastasis of PDAC.
Materials and Methods
Clinical Tissues
The present study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University. Tissue microarray used for immunostaining analysis of DEK was purchased from Shanghai Outdo Biotech. Co. Ltd. (Shanghai, China). The array consisted of 81 cases of pancreatic cancer and 79 cases of paired adjacent normal control. Staging and grading were assessed in accordance with the World Health Organization classification and grading system. None of the patients received chemoradiotherapy before operation. Informed consents were obtained for all the subjects involved, as proclaimed by the company. Eighty-one cases of fresh pancreatic cancer tissues and paired normal control were collected in the Department of Pathology, which was reserved in liquid nitrogen until use.
Cell Lines
Pancreatic cancer cell lines PK-1, KLM-1, PK-8, and AsPC-1 were purchased from ATCC and maintained following ATCC guidelines. All the HCC cells were cultured in 5% CO2 at 37°C in RPMI1640 (life Technologies, Inc.) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies, Inc.), 2 mM L-glutamine, 100 U/ml of penicillin G, and 100 mg/ml of streptomycin (Life Technologies, Inc.).
siRNA and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
SiRNA of miR-200a mimics, sense: UAAUACUGCCGGGUAAUGAUGGA; antisense: CAUCAUUACCCGGCAGUAUUAUU; miR-200a inhibitor UCCAUCAUUACCCGGCAGUAUUA; miR-200a scramble sense: GUGGCGAUAGACAAUCGAUGUAU; antisense: ACAUCGAUUGUCUAUCGCCACUU, which was designed and synthesized by GenePharm Company (GenePharm, Shanghai, China). Total RNA was extracted using the TRIzol reagent (Invitrogen). The primers for miR-200a and U6 detection assays were purchased from Shangong (Shangong, Shanghai, China). Total RNAs were reverse-transcribed using a specific stem-loop RT primer (50 nmol/l) and the Kit (Thermo, USA). The RT conditions consisted of 15 minutes at 42°C followed by 5 minutes at 98°C. Levels of mature miRNAs were quantified by qRT-PCR using the SYBR Green Real-time PCR Master Mix (Thermo, USA). The normalized expression of each sample was designated as CT and obtained by dividing the CT value of miR-200a by the U6 CT of the same sample. The relative amount of miRNA in each sample was calculated using the comparative CT method. The results are presented as fold change of expression.
Luciferase Reporter Assay
The psiCHECK-2 dual luciferase vector (Promega Corporation, Madison, WI) was used to construct the plasmid containing the 3-untranslated region (3-UTR) of DEK. The fragments containing the predicted wild and mutant sites were directly synthesized (Genewiz, Inc., Suzhou, China) and then subcloned into the psiCHECK-2 vector following digestion with XhoI and NotI restriction enzymes (Thermo Fisher Scientific, Inc., Pittsburgh, PA) to generate the DEK-3′-UTR-wild and DEK-3-UTR-mutant vectors. Subsequently, PK-1 cells (1 × 105/well) were seeded into a 24-well plate and co-transfected with 50 ng/well DEK-3-UTR-wild or DEK-3-3′-UTR-mutant vector and 50 nM/well miR-200a mimics or scrambled microRNA negative control. Following culture for 48 hours, the PK-1 cells were collected, and the luciferase activities were measured by the Dual-Luciferase Reporter Assay kit (Promega Corporation) on a TD20/20 Luminometer (Turner Designs, Westport, MA). Each experiment was performed in triplicate. The results were expressed as relative Renilla luciferase activities, which were obtained following normalization to firefly luciferase activities. All the transient transfections were performed using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA).
Western Blot Analysis
The cells were lysed after various treatments through RIPA lysis buffer (Bioteke, Beijing) containing 10 mM PMSF for 30 minutes on ice, followed by centrifuge at 12,000 × g for 10 minutes. The protein concentration was determined using the Bradford method. Samples of 40 μg of total protein were subjected to 10% sodium dodecy sulfate polyacrylamide gel electrophoresis and transferred onto PVDF membrane (Millipore). The membranes were incubated with primary antibody against human DEK (dilution at 1:2000, ab166624, Abcam) for overnight at 4°C followed by horseradish peroxidase–conjugated secondary antibodies, the immunoblots were visualized with chemiluminescence with SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific, USA), and images were captured with a Bio-Rad camera system (Bio-Rad, USA). GAPDH and β-actin were used as the internal loading control. Each immunoblot was repeated at least three times, and representative figures were presented.
Cell Proliferation Assay
Cell viability was examined by the methylthiazolyl blue tetrazolium (MTT) assay (Shangon, Shanghai, China) according to the standard protocol after transfection with miR-200a mimics or miR-200a inhibitor for 0, 24, 48, 72, and 96 hours. AsPC-1 and PK-1 cells were plated in 96-well plates at a density of 5 × 103 cells per well. After transfection, cell proliferation was assessed. Cells were incubated for 4 hours in 20 μl of MTT at 37°C. The color was developed by incubating the cells in 150 μl of dimethyl sulfoxide; the absorbance was detected at 490-nm wave length. The data were obtained from three independent experiments.
Migration and Invasion Assays In Vitro
Cell migration ability was calculated by the wound healing assay. AsPC-1 and PK-1 cells were plated in 6-well plate at a concentration of 4 × 105 cells per well and allowed to form a confluent monolayer for 24 hours. After the transfection, the monolayer was scratched with a sterile 10-μl pipette tip, washed with serum free medium to remove floated and detached cells, and photographed (time 0 and 48 hours) by inversion fluorescence microscope (Olympus, Japan). Cell culture inserts (24-well, pore size 8 μm; BD Biosciences) were seeded with 5 × 103 cells in 100 μl of medium with 0.1% FBS. Inserts precoated with Matrigel (40 μl, 1 mg/ml; BD Biosciences) were used for invasion assays. Medium with 10% FBS (400 μl) was added to the lower chamber and served as a chemotactic agent. Noninvasive cells were wiped from the upper side of the membrane, and cells on the lower side were fixed in cold methanol (− 20°C) and air dried. PBS and 10% FBS were served as negative and positive control, respectively. Cell were stained with 0.1% crystal violet (dissolved in methanol) and counted using the inverted microscope. Each individual experiment had triplicate inserts, and four microscopic fields were counted per insert.
Immunohistochemistry
Paraffin-embedded liver were sectioned to 4-μm thickness. Peroxidase blockage and visualization were achieved with Dako Envision + system (Dako, Denmark). Following heat-induced epitope retrieval according to manufacturer’s protocol, incubation with monoclonal antibodies against human DEK (dilution at 1:200, ab166624, Abcam) was performed. The sections were evaluated by light microscopic examination, and cellular localization of the protein and immunostaining level in each section were assessed by two pathologists. The staining patterns were scored as follows: negative, weak (less than 30% of cells with positive staining), moderate (less than 60% but more than 30% of cells with positive staining), and strong positive (more than 60% of cells with positive staining) according to the immunostaining intensity. Both negative immunostaining and weak immunostaining were categorized into the low-expression group; moderate immunostaining and strong immunostaining were categorized into high expression.
Statistical Analysis
Statistical analysis was carried out using SPSS 17.0 version (SPSS, Chicago, IL) and Graphpad Prism 5.0. Data were expressed as mean ± SD and were analyzed by Student's t test and χ2 test as appropriate. Kaplan-Meier survival curves were plotted, and log rank test was done. P value < .05 was defined as statistically significant. *P < .05, **P < .01, and ***P < .001 in comparison with the control group.
Results
Expression of DEK
To detect the DEK expression status in pancreatic cancer tissues as well as paired normal control tissues, immunohistochemistry was carried out with pancreatic cancer tissue microarray that consisted of 81 cases of pancreatic cancer tissues and 79 cases of paired normal control tissues. It can be seen that DEK was mainly immunohistochemically sublocated in nucleus in cells and that DEK was significantly upregulated in pancreatic cancer tissues as compared with paired normal control tissues (P = .000) (Figure 1). Actually, DEK was heterogeneously expressed in pancreatic cancer tissues, with its expression being weakly, moderately, and strongly positive. However, DEK expression was comparatively homogeneously in paired normal control tissues, with its expression being negative or weak. Clinicopathologically, with the exception of pancreatic cancer tissues and paired normal control tissues, there was also statistically significant difference of DEK expression between T classification (P = .033), clinical stage (P = .023), lymph node metastases (P = .001), differentiation degree (P = .009), and M classification (P = .012) (Table 1). However, there was no significant association between DEK expression and demographic parameters, such as gender and age.
Figure 1.

DEK was significantly upregulated in pancreatic cancer in comparison with paired normal control tissue. Immunostaining of DEK in pancreatic cancer tissues and normal pancreatic tissue with tissue microarray by DEK antibody (1:200). Three representative photographs with different expression status of DEK, ranging from low expression and high expression in pancreatic cancer tissues as well as in normal pancreatic tissue, were taken at different magnifications in pancreatic cancer tissues and normal pancreatic tissue, respectively.
Table 1.
The Clinicopathological Significance of DEK Expression in Pancreatic Cancer Tissues
| Clinicopathological Parameters | Total | DEK Expression |
χ2 | P Value | |
|---|---|---|---|---|---|
| High (++, +++) | Low (−, +) | ||||
| Pancreatic cancer | 81 | 46 | 35 | 32.044 | .000 |
| Normal control | 79 | 11 | 68 | ||
| Gender | |||||
| Male | 41 | 26 | 15 | 1.485 | .266 |
| Female | 40 | 20 | 20 | ||
| Age | |||||
| ≤ 60 | 36 | 19 | 17 | 0.425 | .652 |
| > 60 | 45 | 27 | 18 | ||
| T classification | |||||
| T1-2 | 28 | 11 | 17 | 5.343 | .033 |
| T3-4 | 53 | 35 | 18 | ||
| Clinical stage | |||||
| Stage I-II | 32 | 13 | 19 | 5.633 | .023 |
| Stage III-IV | 49 | 33 | 16 | ||
| Lymph node metastases | |||||
| No | 38 | 14 | 24 | 12.662 | .001 |
| N1-2 | 43 | 32 | 11 | ||
| Differentiation degree | |||||
| Well-moderate | 31 | 10 | 21 | 7.491 | .009 |
| Low | 50 | 36 | 14 | ||
| M classification | |||||
| M0 | 33 | 13 | 20 | 6.868 | .012 |
| M1-2 | 48 | 33 | 15 | ||
Clinicopathological significance analysis of DEK
To observe whether or not there was correlation between DEK expression and overall prognosis, Kaplan-Meier survival analysis was carried out. Of 81 cases of pancreatic cancer tissues, 46 cases was classified as having high DEK expression, and the rest (35 cases) had low expression of DEK according to our histopathological evaluation criteria. It was found that there was extremely significant difference of overall prognosis between patients with high DEK expression and with low DEK expression (Figure 2), suggesting that DEK expression could be used as a prognostic marker for patients diagnosed with pancreatic cancer.
Figure 2.
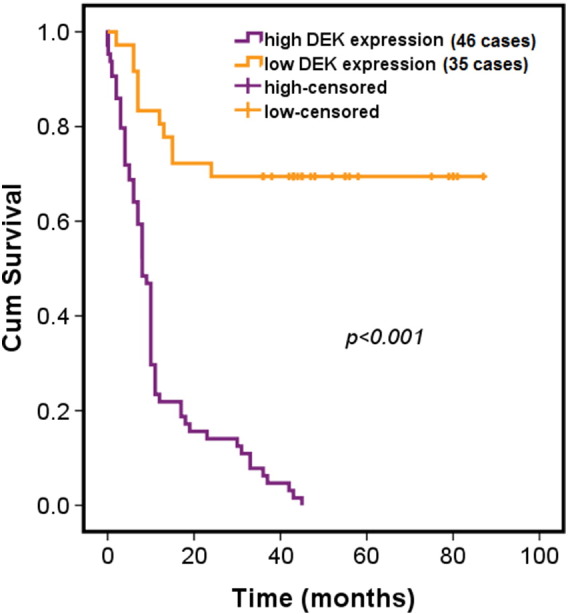
High expression of DEK was significantly associated with poor prognosis. Kaplan-Meier survival analysis of overall prognosis was performed between patients with high DEK expression (35 cases) and patients with low DEK (46 cases). In terms of immunostaining intensity, both negative (−) immunostaining and weak (+) immunostaining were categorized into the low-expression group, whereas medium (++) immunostaining and strong (+++) immunostaining were categorized into the high-expression group. Log-rank test was used to statistically analyze the difference.
Identification of miR-200a downstream target
Having found that DEK was significantly overexpressed in pancreatic cancer tissues, we sought to find out its own upstream regulator from the angle of micoRNA. Using bioinformatics, we predicted that miR-200a was an upstream regulatory miRNA of DEK (Figure 3D). To confirm the prediction we made, luciferase reporter assay was performed to analyze the actual regulation and control between miR-200a and DEK gene. It was found that miR-200a can directly regulate and control the transcriptional activity of DEK genes (Figure 3E). Based on this, we’ve simultaneously detected the basal expression status of miR-200a in clinical pancreatic cancer samples (Figure 3A), and both miR-200a (Figure 3B) and DEK (Figure 3C) gene were also assayed in vitro in pancreatic cancer cell lines. It can be seen that there was a tendency that miR-200a negatively correlated with DEK. To further confirm, we’ve chosen two different kinds of pancreatic cancer cell lines with contrast expression status of miR-200a or DEK. Using transfection with miR-200a mimics or miR-200a inhibitor, it was shown that there was negative regulation between miR-200a and DEK expression (Figure 3F).
Figure 3.

MiR-200a directly and negatively regulated DEK gene. (A) miR-200a was significantly upregulated in normal pancreatic tissues compared with pancreatic tissues. miR-200a was detected using qRT-PCR in 81 cases of fresh pancreatic cancer tissues and its paired normal control tissues. (B) Basal level of miR-200a was detected using qRT-PCR technique in four different pancreatic cancer cell lines (PK-1, KLM-1, PK-8, and AsPC-1). (C) Endogenous expression level of DEK gene was detected using Western blot in four different pancreatic cancer cell lines (PK-1, KLM-1, PK-8, and AsPC-1). (D) DEK gene was bioinformatically predicted as target of miR-200a using online software (http://www.microrna.org/). (E) Luciferase reporter assay was performed to confirm that DEK gene was a direct and downstream target of miR-200a. (F) Western blot was carried out finding that miR-200a negatively regulated DEK gene. Independent-sample t test was carried out to analyze the statistical difference. *P < .05, **P < .01, and ***P < .001 compared with control group.
Functional analysis of miR-200a
To investigate the biological role of miR-200a in pancreatic cancer cells, we’ve firstly chosen two different kinds of pancreatic cancer cell lines with contrast expression status of miR-200a or DEK. Then, we’ve used both knockdown and reexpression strategy on cell lines in vitro to explore the possible role of miR-200a in terms of proliferation, migration, and invasion. MTT assay showed that reexpression of miR-200a could markedly suppress the proliferation of pancreatic cancer cell lines (Figure 4A). In addition, wound-healing assay showed that reexpression of miR-200a could significantly suppress the migration (Figure 4B); Transwell assay showed that reexpression of miR-200a could also remarkably inhibit the invasive ability (Figure 4C), suggesting that miR-200a plays an antitumor role in pancreatic cancer cells.
Figure 4.

MiR-200a can significantly suppress the proliferation, migration, and invasion of pancreatic cancer cell lines. (A) MTT assay was performed to analyze the proliferative variation after transfection with miR-200a mimics into AsPC-1 cells where basal level of miR-200a was lowest among the four different kinds of pancreatic cancer cell lines and transfection with miR-200a-inhibitor into PK-1 cells where basal level of miR-200a was highest among the four different kinds of pancreatic cancer cell lines. The absorbance value was detected at 24, 48, 72, and 96 hours posttransfection. (B) Wound-healing assay was qualitatively and quantitatively carried out to detect the migratory variation in PK-1 and AsPC-1 cells after transfection with miR-200a inhibitor and miR-200a mimics, respectively. (C) Transwell assay was conducted to analyze the invasive variation in PK-1 and AsPC-1 cells after transfection with miR-200a inhibitor and miR-200a mimics, respectively. Independent-sample t test was employed. *P < .05 and **P < .01 in comparison with control group.
Discussion
In our study, we have for the first time identified that DEK gene was a direct downstream target gene of miR-200a in PDAC, finding that miR-200a was significantly overexpressed in PDAC tissues compared with paired normal control. Overexpression of miR-200a can decrease the level of DEK, suppressing the proliferation, migration, and invasion of PDAC cells. Our study suggests that miR-200a may be used as a novel potential marker in prediction of metastasis of PDAC.
PDAC is the fourth most common cause of death from cancer. Its 5-year survival rate is less than 5%. This poor prognosis is mostly due to the cancer's early invasion and metastasis formation, leading to an initial diagnosis at an advanced incurable stage in the majority of patients [10]. The molecular mechanisms underlying the invasion and metastasis of PDAC remain to be clarified. Epithelial to mesenchymal transition (EMT) is a developmental process that leads the phenotype shift from an epithelial morphology to a motile, fibroblast-like morphology [11]. Recent studies showed that EMT is involved in the invasion and metastasis of many types of carcinomas including PDAC [2], [12], [13]. In the regulation of EMT in PDAC, miR-200a has been found to be able to suppress the process by which PDAC cells metastasized further [2]. Based on the discovery, we’ve therefore hypothesized that miR-200a could play an important role in suppressing the metastasis of PDAC cells. To test the hypothesis, we’ve analyzed the role of miR-200a on PDAC cell lines ex vivo. It was found that reexpression of miR-200a using transfection with miR-200a mimics was able to suppress the proliferation, migration, and invasion of PDAC cell lines. Meanwhile, the phenotype was rescued by knocking down of miR-200a with transfection of miR-200a inhibitor, suggesting that miR-200a can suppress the proliferation, migration, and invasion of PDAC cells.
It has been well known that miRNAs regulated gene expression at the posttranscriptional level through both translational inhibition and mRNA destabilization [14]. Using bioinformation prediction and luciferase reporter assay, we’ve identified that DEK gene was a direct, downstream target of miR-200a, which has never been reported ever before. DEK gene has been found to be significantly overexpressed in various cancers [7], [15], [16], [17], but it has been never reported regarding its possible role in PDAC with the exception of one previous study that found and mentioned a potential liver metastasis gene [9], but there has been no follow-up study of DEK’s biological function in PDAC. In our study, we firstly identified the role of DEK gene in PDAC cells as oncoprotein that promotes the proliferation, migration, and invasion of PDAC cells, which is totally in line with and in agreement with the previous definition of DEK in various cancers [16], [18], [19], [20].
In our study, we’ve just only employed the PDAC cell lines to identify that the DEK gene was a direct and down-stream target of miR-200a; it makes sense that we should have confirmed the association between miR-200a expression status and DEK expression level on clinical tissues with PDAC, but given that the PDAC clinical tissues are unavailable at hand, we’ve failed to confirm the relationship between miR-200a and DEK on the tissue level, which is one limitation of our study. Another limitation is that, in consideration that miR-200 family, consisted of miR-200a, miR-200b, and miR-200c, have been reported to play a key role in the regulation (to be exact, suppression) of EMT process, indicating that miR-200b and miR-200c might have the same role as miR-200a did in the suppression of migration and invasion of PDAC cells. We should have meanwhile analyzed the possible role of miR-200b and miR-200c, respectively, in PDAC tissues. Thus, in the following, we are going to overcome the attendant limitations of the present study.
Together, our study demonstrated that miR-200a suppresses the metastasis in pancreatic PDAC through downregulation of DEK, suggesting that miR-200a may be used as a novel potential marker in prediction of metastasis of PDAC.
Conflict of Interests
The authors have declared that they have no conflict of interests in any form with anyone, organization, or company pertaining to the article.
Acknowledgements
The work was technically supported by the staff from the Department of General Surgery, Jiangsu Cancer Hospital, Affiliated Cancer Hospital of Nanjing Medical University.
Footnotes
The work was supported by the National Science Foundation of China (No. 81402523, 81373990 and 81201797), Jiangsu Province "Six Adjults Just" high peak (2009-D-63), Youth Projects of Jiangsu Provincial Health Department (H201065), and the National Science Foundation of Jiangsu Province (BK2009445).
References
- 1.Sclafani F, Iyer R, Cunningham D, Starling N. Management of metastatic pancreatic cancer: current treatment options and potential new therapeutic targets. Crit Rev Oncol Hematol. 2015;95(3):318–336. doi: 10.1016/j.critrevonc.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Lu J, Li X, Zhu H, Fan X, Zhu S, Wang Y, Guo Q, Wang L, Huang Y. MiR-200a inhibits epithelial-mesenchymal transition of pancreatic cancer stem cell. BMC Cancer. 2014;14:85. doi: 10.1186/1471-2407-14-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen A, Lin W, Chen Y, Liu L, Chen H, Zhuang Q, Lin J, Sferra TJ, Peng J. Pien Tze Huang inhibits metastasis of human colorectal carcinoma cells via modulation of TGF-beta1/ZEB/miR-200 signaling network. Int J Oncol. 2015;46(2):685–690. doi: 10.3892/ijo.2014.2772. [DOI] [PubMed] [Google Scholar]
- 4.Soubani O, Ali AS, Logna F, Ali S, Philip PA, Sarkar FH. Re-expression of miR-200 by novel approaches regulates the expression of PTEN and MT1-MMP in pancreatic cancer. Carcinogenesis. 2012;33(8):1563–1571. doi: 10.1093/carcin/bgs189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng J, Wang J, Chen M, Chen G, Wu Z, Ying L, Zhuo Q, Zhang J, Wang W. miR-200a suppresses cell growth and migration by targeting MACC1 and predicts prognosis in hepatocellular carcinoma. Oncol Rep. 2015;33(2):713–720. doi: 10.3892/or.2014.3642. [DOI] [PubMed] [Google Scholar]
- 6.Lin L, Piao J, Gao W, Piao Y, Jin G, Ma Y, Li J, Lin Z. DEK over expression as an independent biomarker for poor prognosis in colorectal cancer. BMC Cancer. 2013;13:366. doi: 10.1186/1471-2407-13-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Useros J, Rodriguez-Remirez M, Borrero-Palacios A, Moreno I, Cebrian A, Gomez del Pulgar T, del Puerto-Nevado L, Vega-Bravo R, Puime-Otin A, Perez N. DEK is a potential marker for aggressive phenotype and irinotecan-based therapy response in metastatic colorectal cancer. BMC Cancer. 2014;14:965. doi: 10.1186/1471-2407-14-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin L, Piao J, Ma Y, Jin T, Quan C, Kong J, Li Y, Lin Z. Mechanisms underlying cancer growth and apoptosis by DEK overexpression in colorectal cancer. PLoS One. 2014;9(10):e111260. doi: 10.1371/journal.pone.0111260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura T, Furukawa Y, Nakagawa H, Tsunoda T, Ohigashi H, Murata K, Ishikawa O, Ohgaki K, Kashimura N, Miyamoto M. Genome-wide cDNA microarray analysis of gene expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene. 2004;23(13):2385–2400. doi: 10.1038/sj.onc.1207392. [DOI] [PubMed] [Google Scholar]
- 10.Honselmann KC, Pross M, Jung CM, Wellner UF, Deichmann S, Keck T, Bausch D. Regulation mechanisms of the hedgehog pathway in pancreatic cancer: a review. JOP. 2015;16(1):25–32. doi: 10.6092/1590-8577/2894. [DOI] [PubMed] [Google Scholar]
- 11.Cano CE, Motoo Y, Iovanna JL. Epithelial-to-mesenchymal transition in pancreatic adenocarcinoma. TheScientificWorldJOURNAL. 2010;10:1947–1957. doi: 10.1100/tsw.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satoh K, Hamada S, Shimosegawa T. Involvement of epithelial to mesenchymal transition in the development of pancreatic ductal adenocarcinoma. J Gastroenterol. 2015;50(2):140–146. doi: 10.1007/s00535-014-0997-0. [DOI] [PubMed] [Google Scholar]
- 13.Wu Q, Miele L, Sarkar FH, Wang Z. The role of EMT in pancreatic cancer progression. Pancreat Disord Ther. 2012;2(3) doi: 10.4172/2165-7092.1000e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabian MR, Sundermeier TR, Sonenberg N. Understanding how miRNAs post-transcriptionally regulate gene expression. Prog Mol Subcell Biol. 2010;50:1–20. doi: 10.1007/978-3-642-03103-8_1. [DOI] [PubMed] [Google Scholar]
- 15.Lin D, Dong X, Wang K, Wyatt AW, Crea F, Xue H, Wang Y, Wu R, Bell RH, Haegert A. Identification of DEK as a potential therapeutic target for neuroendocrine prostate cancer. Oncotarget. 2015;6(3):1806–1820. doi: 10.18632/oncotarget.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Lin L, Ren X, Lin Z, Li Z, Li C, Jin T. High expression of oncoprotein DEK predicts poor prognosis of small cell lung cancer. Int J Clin Exp Pathol. 2014;7(8):5016–5023. [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Wang X, Sun F, Kong J, Li Z, Lin Z. DEK overexpression is correlated with the clinical features of breast cancer. Pathol Int. 2012;62(3):176–181. doi: 10.1111/j.1440-1827.2011.02775.x. [DOI] [PubMed] [Google Scholar]
- 18.Sanden C, Jarvstrat L, Lennartsson A, Brattas PL, Nilsson B, Gullberg U. The DEK oncoprotein binds to highly and ubiquitously expressed genes with a dual role in their transcriptional regulation. Mol Cancer. 2014;13:215. doi: 10.1186/1476-4598-13-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Datta A, Adelson ME, Mogilevkin Y, Mordechai E, Sidi AA, Trama JP. Oncoprotein DEK as a tissue and urinary biomarker for bladder cancer. BMC Cancer. 2011;11:234. doi: 10.1186/1471-2407-11-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanden C, Nilsson HJ, Gullberg U. The DEK oncoprotein is upregulated by multiple leukemia-associated fusion genes. Blood Cells Mol Dis. 2015;54(3):284–285. doi: 10.1016/j.bcmd.2014.11.014. [DOI] [PubMed] [Google Scholar]