

Abstract
Quinoxaline 1,4-di-N-oxides (QdNOs) have manifold biological properties, including antimicrobial, antitumoral, antitrypanosomal and antiinflammatory/antioxidant activities. These diverse activities endow them broad applications and prospects in human and veterinary medicines. As QdNOs arouse widespread interest, the evaluation of their medicinal chemistry is still in progress. In the meantime, adverse effects have been reported in some of the QdNO derivatives. For example, genotoxicity and bacterial resistance have been found in QdNO antibacterial growth promoters, conferring urgent need for discovery of new QdNO drugs. However, the modes of actions of QdNOs are not fully understood, hindering the development and innovation of these promising compounds. Here, QdNOs are categorized based on the activities and usages, among which the antimicrobial activities are consist of antibacterial, antimycobacterial and anticandida activities, and the antiprotozoal activities include antitrypanosomal, antimalarial, antitrichomonas, and antiamoebic activities. The structure-activity relationship and the mode of actions of each type of activity of QdNOs are summarized, and the toxicity and the underlying mechanisms are also discussed, providing insight for the future research and development of these fascinating compounds.
Keywords: quinoxaline 1, 4-di-N-oxides, antimicrobial, antitumoral, antiprotozoal, antiinflammatory, antioxidant, structure-activity relationship, mode of action
Introduction
Quinoxaline is a heterocyclic compound containing a benzene ring and a pyrazine ring. Oxidation of both nitrogens of the pyrazine ring to obtain QdNO offers it variety of biological properties, including antitumoral, antibacterial, anticandida, antitrypanosomal, antiinflammatory/antioxidant, and mutagenic properties. The versatile activities of QdNOs have aroused worldwide interests and endowed them potential application in human and veterinary medicines. For example, the QdNO antibacterial growth promoters have been widely used as feed additives to prevent infectious disease and improve animal growth since 1970s (Carta et al., 2005), and the antitumoral drug, TPZ (3-amino-BTO 1,4 dioxide, SR4233), has been subjected to phase II clinical trial (Covens et al., 2006; Maluf et al., 2006; Cohen et al., 2007). Although QdNOs with other activities are still in the research stage, they have exhibited great application prospects.
With the wide use of QdNOs, the toxicity and drug resistance gradually have become the disadvantages for further application of these promising compounds. The molecular targets of quinoxaline derivatives, which is a step beyond simply looking at their activities, should be analyzed. Nevertheless, knowledge about the mode of actions of QdNOs is far from clear, hindering the development of this kind of drugs. Therefore, a detail study on the mode of actions of QdNOs will provide information about the drug targets and the drug action pathway(s) and be helpful to construct models to screen new drugs. Meanwhile, the study of SAR combined with the study of drug action will explain the reason for that different structures of QdNOs exhibit different activities. Furthermore, since most of the drugs have more than one mechanism of action, the deeper study of the drug actions may discover new drug target(s) and provide potential approaches to conquer the problem of drug resistance and to mitigate or avoid toxicity. In this review, the mode of actions of QdNOs and the SAR analysis are elucidated and updated in sort of the different biological properties, giving insight to the future development of these fascinating compounds.
Antimicrobial Activities of QdNOs
Antibacterial Activity and Bacterial Resistance
The antibacterial activity of QdNOs was first reported in McIlwain (1943), and subtherapeutic levels of antibacterial QdNOs have been used for nearly 50 years to promote the growth of animals and improve efficiency of feed conversion in animal husbandry (Carta et al., 2005). As shown in Figure 1, veterinary-used QdNO derivatives include (QdNOs), OLA [2-(N-2′-hydroxyethyl-carbamoyl)-3-methyl quinoxaline 1,4-di-N-oxide firstly synthesized by Bayer in 1967], CBX [hydrazine carboxylic acid (2-quinoxalinyl-methylene) methyl ester 1,4-di-N-oxide firstly synthesized by Pfizer in 1968]. MEQ (3-methyl-2- quinoxalinacetyl-1,4-di-N-oxide) and QCT (3-methyl-2-quinoxalinbenzenevinylketo-1,4-di-N-oxide) are novel synthetic QdNO derivatives developed by Lanzhou Institute of Animal Husbandry and Veterinary Drugs, Chinese Academy of Agricultural Sciences (Lanzhou, China). CYA [cyan acetic acid (1,4-di-N-oxy-quinoxalin-2-ylmethylene] hydrazide firstly designed by Chemapol Benelux) is a new QdNO member which is being evaluated in clinical trials for animals in China.
FIGURE 1.
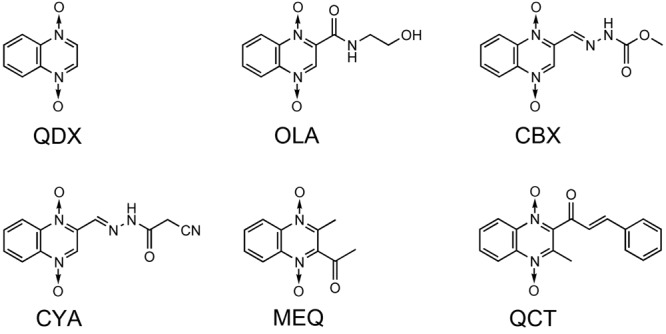
Chemical structures of veterinary-used QdNO antibacterials.
Olaquindox exhibits wide spectrum of antibacterial effect, strongly inhibiting the growth of Escherichia coli, Proteus, Pasteurella, and dysentery spirochete (Drumev, 1981). Because of its genotoxic potential, OLA is not permitted to use in food-producing animals in the European Union and Canada (WHO, 1994; European Union [EU], 1998; Standing Committee for Animal Nutrition [SCAN], 1998). CBX shows good therapeutic effect on swine dysentery caused by Brachyspira hyodysenteriae (Rainier et al., 1973a,b; Downing, 1974) and also has good antibacterial effects against Salmonella, E. coli, and other Gram-negative bacteria (Troutt et al., 1974; Das, 1984). CBX was once used as growth-promoting feed additives for piglets or pigs at growing phase (Yen and Pond, 1993) before it was prohibited by the European Union [EU] (1998) because of its mutagenic effects, developmental and reproductive toxicity and carcinogenicity (WHO, 1991; European Union [EU], 1998). In view of the significant effect of CBX on swine dysentery and bacterial enteritis, the United States, Canada, and other countries still allow it used as therapeutic agents. MEQ, which shows good antibacterial activity against Gram-negative bacteria, especially Salmonella, has been widely used in China as an animal feed additive and veterinary medication for diseases, such as swine dysentery and piglet white diarrhea (Liu et al., 2012). QCT is active against B. hyodysenteriae, and is also effective against Salmonella, E. coli, and other Gram-negative bacilli. In China, QCT is used as a growth promoting agent for pigs, poultry, and aquatic and has been approved as an animal growth promoter in China since 2003 (Ministry of Agriculture of P. R. China, 2003). CYA shows good antibacterial activity against Staphylococcus hyicus, Pasteurella multocida, and E. coli (Ding et al., 2006a,b), and also exhibits good growth-promoting effect on broilers and swine. With good clinical safety, CYA has been regarded as a potential replacement of OLA and CBX (Cihak and Srb, 1983).
Only a few studies have investigated the mechanism of antibacterial action of QdNOs. Suter et al. (1978) first found the synthesis of DNA (but not RNA and protein) was completely inhibited by QDX in the absence of oxygen. QdNOs also induced degradation of DNA in both proliferating and non-proliferating cells, while strains were more resistant in the presence of oxygen. QDX was reduced to quinoxaline-N-oxide by the intact E. coli cells or the cell-free extract. EPR measurements demonstrated the generation of free radicals during the reduction of QDX. Oxygen or deficiency of energy sources impaired the antibacterial activity and the reduction of QDX. In consistence with Suter’s result, our group recently found that CYA also had anaerobe-selective activity, and losing one or two of the oxygen’s of CYA exhibited no antibacterial activity (Cheng et al., 2015).
The ability of QDX to cleave DNA was explicitly characterized using in vitro assays by Ganley et al. (2001). The results evidenced that QDX was a hypoxia-selective, redox-activated DNA-cleaving agent. The action of QDX on DNA yielded direct strand breaks with almost no sequence specificity, consistent with the involvement of radical species. In the absence of oxygen, QDX (a) received an electron and a hydrogen ion to form a neutral radical (b) (Figure 2). They considered two possibilities about the DNA-cleaving radical resulting from redox activation of QDX (a). First, the radical b might directly abstract hydrogen atoms from the DNA backbone, followed by elimination of water to yield the quinoxaline monoxide (c). Alternatively, radical b might fragment to form the known DNA-cleaving agent ∙OH and monoxide c.
FIGURE 2.
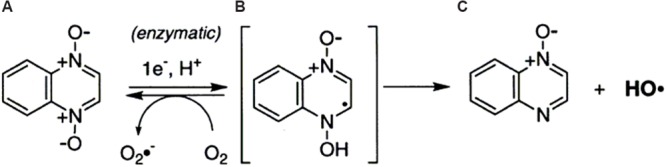
One-electron reduction of quinoxaline 1,4-dioxide by reductase (Ganley et al., 2001).
Early study showed that polA, recA, recB, recC, exrA, and uvrA E. coli mutants were more susceptible to QdNOs than the corresponding repair-proficient strains, while the QdNO reductase activity was demonstrated to be lower in QdNO-resistant mutants than in the susceptible parent strain (Suter et al., 1978), indicating that QdNOs were reduced and caused extensive DNA damage in bacterial. Recently, our group investigated the transcriptomic and proteomic profiles of E. coli exposed to QdNOs and found that QdNOs mainly induced SOS response and oxidative stress (Cheng et al., 2015). We also confirmed that ROS were induced in the QdNO-treated bacteria and the free radical scavengers attenuated the antibacterial action of QdNOs and DNA damage, suggesting an oxidative-DNA-damage action of QdNOs. The QdNO radical intermediates, likely carbon-centered and aryl-type radicals, as identified by EPR, were the major radicals induced by QdNOs, and xanthine oxidase was identified as one of the QdNO-activating enzymes by using specific enzyme inhibitors (Cheng et al., 2015).
Tirapazamine is a prodrug undergoing clinical trials for various types of cancers (discussed in details in section “Antitumor activity of QdNOs”). Shah et al. (2013) showed that TPZ has antibacterial activity against E. coli, S. aureus, and Clostridium difficile, particularly at low oxygen levels. E. coli mutants deficient in HR were hypersusceptible to TPZ, suggesting that drug toxicity may be due to DNA damage. Moreover, E. coli strains deleted for genes encoding putative reductases were resistant to TPZ, implying that these enzymes are responsible for conversion of the prodrug to a toxic compound.
Though the two N-oxide groups are necessary for the antibacterial activity of QdNOs, some reduced form of quinoxaline compounds were still reported with antibacterial activity (Refaat et al., 2004; Singh et al., 2010, 2011). For example, when the C2 chlorine of 2-Chloro-3-methylquinoxaline was replaced with a benzene ring harboring an aldehyde or a free amino group which can be further reacted with aromatic amines and aromatic aldehydes, this compounds also show antimicrobial activity (Singh et al., 2010). Therefore, in addition to the N-oxide group, the side chain of quinoxaline is another determinant of activity.
Hansen et al. (2004) first reported the gene-encoded resistance mechanism to OLA. In their study, two genes of oqxA and oqxB, encoding for proteins homologous to resistance-nodulation-cell-division family efflux systems, were cloned from a conjugative plasmid isolated from E. coli. Plasmids containing the oqxAB genes yielded high resistance to OLA and chloramphenicol in E. coli, indicating oqxAB encodes a multidrug efflux pump. Later, they demonstrated the prevalence of the OqxAB efflux pump by horizontal transfer of OLA resistance from OLA-resistant isolates using an OLA-sensitive E. coli as recipient (Hansen et al., 2005). In addition to OLA and chloramphenicol, the OqxAB pump conferred antimicrobial resistance or reduced susceptibility toward a variety of substrates in E. coli, including animal growth promoters, antimicrobials, disinfectants and detergents (Hansen et al., 2007). Interestingly, oqxA gene was not detected in the CYA/OLA- resistant E. coli induced in vitro (Guo et al., 2012), suggesting there are other mechanisms conferring the bacterial resistance.
Antimycobacterial Activity
Tuberculosis is a common, and in many cases lethal, infectious disease caused by various strains of mycobacteria, usually Mycobacterium tuberculosis. TB has been a companion of mankind since the beginning of human history. In 2010, there where 8.8 million new cases, and 1.5 million deaths, mostly in developing countries (WHO, 2011), where more people contract TB because of compromised immunity due to high rates of AIDS (Lawn and Zumla, 2011). Drug-resistant TB is a public health issue in many developing countries, as treatment of it is longer and requires more expensive drugs. MDR TB is defined as resistance to the two most effective first-line TB drugs, rifampicin and isoniazid. Over the past years, QdNO derivatives as antitubercular drug candidates hold promise in the treatment of TB and its resistant form.
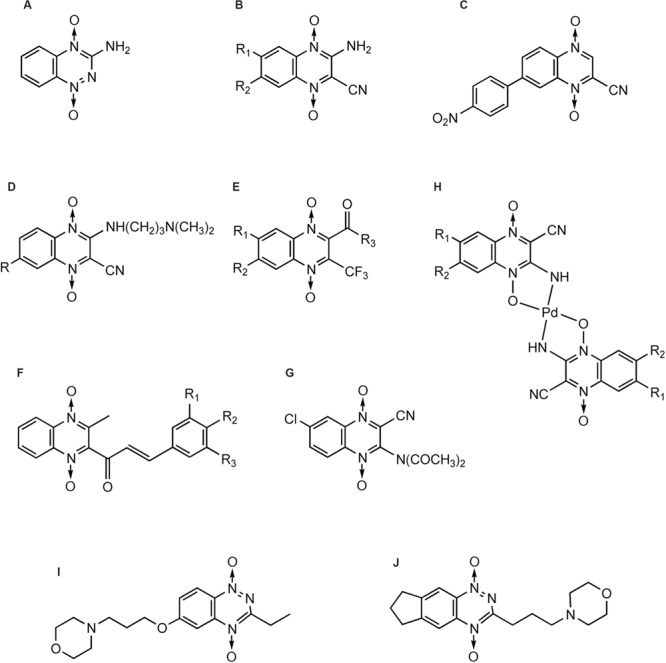
The first publication of QdNO derivatives as antimycobacterium agents dates back to the end of the 1990s by Monge’s team (Montoya et al., 1998). Continued identification of novel antitubercular candidates based on their potency (MIC to H37Rv M. tb), selectivity [SI = IC50(to VERO cell)/MIC(to H37Rv M. tb)] and low cytotoxicity make them valid new leads for synthesizing additional analogs with improved antitubercular activity both in vitro and in vivo. A comprehensive review on the properties of QdNO derivatives developed as potential antitubercular agents by Monge’s team was reported recently (Vicente et al., 2011). As shown in Figure 3, the QdNO antitubercular compounds can be divided into categories based on their structures, including amide derivatives (1–3), ketone derivatives (4–6), ester derivatives (7–10) and other derivatives (11–12). Compounds 8 and 10, proved efficacious in vivo in a murine model of low dose aerosol infection. Moreover, these two compounds also showed activity against non-replicating bacteria, indicating that QdNOs might lead to shortened therapy, because non-replicating bacteria is believed to be a major factor responsible for the prolonged nature of antitubercular therapy. Compound 8 is also active on single-drug resistant and MDR clinical isolates.
FIGURE 3.
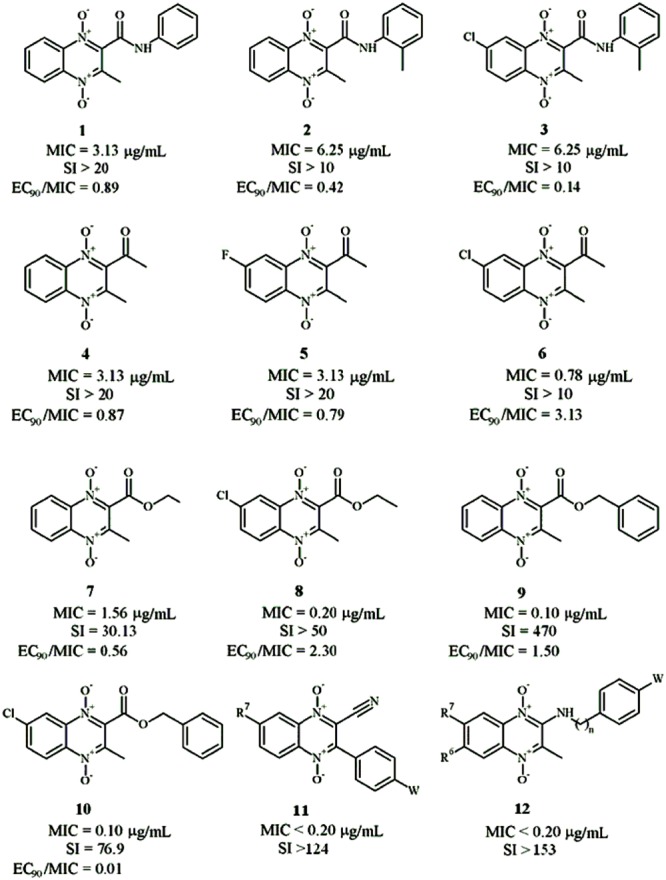
General structures of QdNO derivatives with antimycobacterial activity published by Monge’s team [modified from Vicente et al. (2011)].
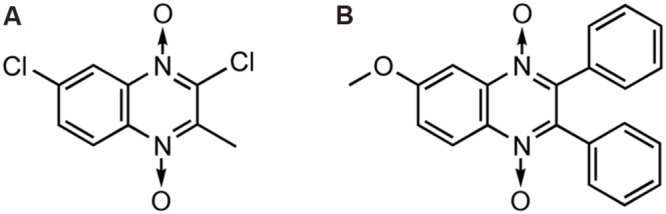
In the meantime, there were other research groups studying the antimycobacterial activity of QdNOs. As shown in Figure 4, Zanetti’s team reported a series of 3-methylquinoxaline 1,4-di-N-oxide derivatives with a phenylthio, phenylsulfonyl, or phenylsulfinyl linked in R2 position of quinoxaline subunit (Figure 4A). The series of 3-methyl-2-phenyl-thioquinoxaline 1,4-di-N-oxide derivatives presented the best MIC data, ranging between 0.39 and 0.78 g/mL, whereas the oxidation of sulfur bridge to yield phenylsulfinyl and phenylsulfonyl derivatives or its replacement with benzylamino (Figure 4B) or phenylamino group (Figure 4C) slightly reduces its activity (Carta et al., 2002, 2004). Furthermore, 1 mg/L of 6,7-difluoro-3-methyl-2-(3,4-dimethoxyphenylthio) quinoxaline 1,4-dioxide (Figure 4D) exhibited good activity in mouse macrophages infected by M. avium paratuberculosis (Zanetti et al., 2005).
FIGURE 4.
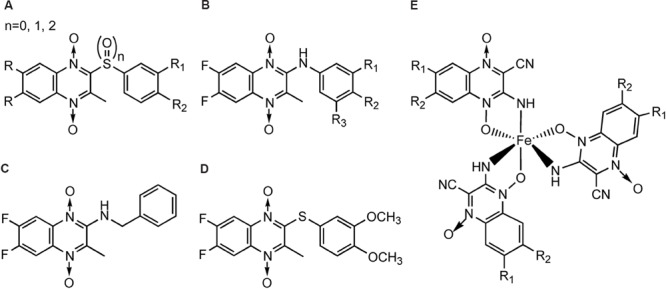
General structures of QdNOs with antimycobacterial activity published by Zanetti’s (A–D) and Gambino’s teams (E).
Gambino’s team has devoted to the synthesis and biological assessment of a large amount of QdNO derivatives complexed with metals, including palladium (Pd; Urquiola et al., 2009) and copper (Cu; Torre et al., 2005; Urquiola et al., 2008) with antitumoral activity, vanadium (Vo) with antitrypanosomal (Urquiola et al., 2006), insulin-mimetic (Noblia et al., 2006), and antitumoral activities (Vieites et al., 2006), and iron (Fe) with antimycobacterial activity (Tarallo et al., 2008, 2010). Two novel iron complexes, [Fe(L3)(3)] (L3: R1 = Cl, R2 = OCH3) and [Fe(L4)(3)] (L4: R1 = H, R2 = CF3; Figure 4E) showed in vitro growth inhibitory activity on M. tuberculosis H(37)Rv (MIC = 0.78 μg/mL), together with very low unspecific cytotoxicity on murine cell line J774. Both complexes showed higher inhibitory effects on M. tuberculosis than the second-line therapeutic drugs (Tarallo et al., 2010).
Structure-activity relationship suggests that the 1,4-di-N-oxide groups in quinoxaline ring are important to increase the antimycobacterial activity (Montoya et al., 1998; Ortega et al., 1999; Sainz et al., 1999). Quinoxaline-2-carbonitrile derivatives has good antimycobacterial activity but appeared to be quite toxic (Ortega et al., 2001, 2002); thus, the replacement of the carbonitrile group with a carboxamide (Zarranz et al., 2003), acetyl, benzoyl (Jaso et al., 2003), or carboxylate groups (Jaso et al., 2005) was proposed. Among the 6(7)-substituted quinoxaline-2-carboxylate 1,4-dioxide derivatives, anti-TB activity principally depends on the substituents in the carboxylate group, improving in the following order: benzyl > ethyl > 2-methoxyethyl > allyl > tert-butyl, and the presence of a chloro, methyl, or methoxy group in position 7 of the benzene moiety reduces the MIC and IC(50) values (Jaso et al., 2005). Recently, a research employed 3D-QSAR and docking analysis to identify molecular structural features required for effective antimycobacterial activity of quinoxaline-2-carboxamide 1,4-di-N-oxide derivatives inside the active site of the Mycobacterium DNA gyrase B subunit, and the obtained binding mode was as same as that of the novobiocin X-ray structure (Radwan and Abdel-Mageed, 2014).
Antifungal Activity
Candida is a genus of yeasts, some of which can cause disease, for example, Candida albicans cause infections (candidiasis or thrush) in humans and other animals. In healthy individuals, these infections can be cured with topical or systemic antifungal medications (commonly over-the-counter treatments like miconazole or clotrimazole), while in debilitated or immunocompromised patients, candidiasis may become a systemic disease.
Carta et al. (2002, 2004) reported the in vitro anticandida activity (against C. albicans, C. glabrata, C. krusei, and C. parapsilosis clinical isolates) of 3-methylquinoxaline 1,4-dioxide derivatives, in particular 2,7-dichloro-3-methyl quinoxaline 1,4-dioxide (Figure 5A) and 2-chloro-7-ethoxy-6-fluoro-3-methylquinoxaline 1,4-dioxide were the most active against C. Krusei, exhibiting MICs of 0.4 and 1.9 g/mL, respectively (miconazole MIC = 0.9 g/mL). Murthy et alreported one of the 2,3-diphenyl quinoxaline 1,4-dioxide derivatives (Figure 5B) as a fruitful matrix for further biological evaluation based on its wide zones of inhibition against C. albicans and S. cerevisiae (Murthy et al., 2011).
FIGURE 5.
General structures of QdNOs with antifungal activity.
Antitumor Activity Of QdNOs
It has been recognized for more than 50 years that very low levels of oxygenation (hypoxia) protect cells from killing by X-irradiation. Hypoxic cells, which are resistant to radiotherapy, are present in solid tumors but not in normal tissues, therefore become a major problem for radiation therapy of tumors. Additionally, hypoxic cells are non-proliferating which do not respond to drugs active against only proliferating cells, and are distant from the blood vessels carrying the drug. Thus the development of drugs with selective toxicity toward hypoxic cells is a key objective in anticancer chemotherapy. The recent development of QdNOs that are non-toxic until they are activated in the hypoxic cell opens a new era.
Tirapazamine (Zeman et al., 1986) (Figure 6A), which actually belongs to BTOs, is the first drug that has been shown to be an efficient and selective cytotoxin for hypoxic cells. TPZ has been subjected to phase II testing in patients with head, neck, and gynecological cancers (Covens et al., 2006; Maluf et al., 2006; Cohen et al., 2007).
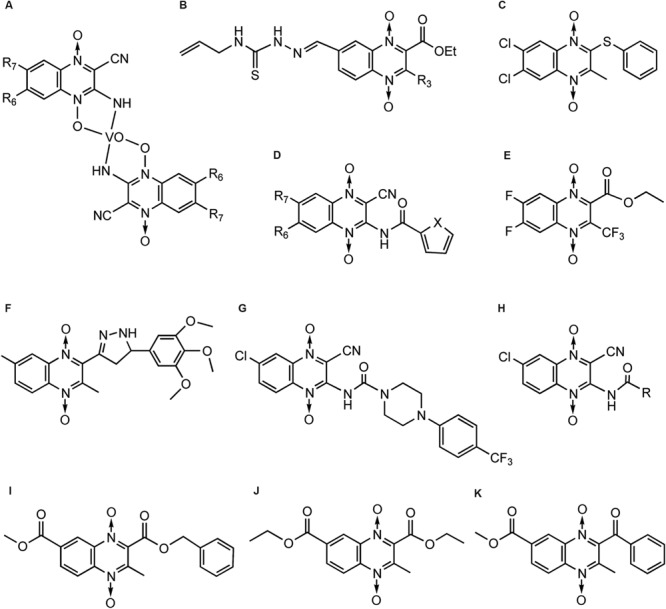
FIGURE 6.
QdNOs with hypoxia-selective and antitumoral activities.
In addition to TPZ, many research teams have been devoted to the synthesis and development of QdNO antitumoral drugs. Monge et al. (1995b) synthesized a series of 3-aminoquinoxaline-2-carbonitrile 1,4-di-N-oxides (Figure 6B) with a range of electron-donating and -withdrawing substituents in the 6- and/or 7- positions and evaluated for toxicity to hypoxic cells. As the electron-withdrawing nature of the 6(7)-substituent increases, the reduction potential becomes more positive and the compound is more readily reduced. The most potent cytotoxins were the 6,7-dichloro and 6,7-difluoro derivatives, which were 30-fold more potent than TPZ. The compound 7-(4-nitrophenyl)-2-quinoxalinecarbonitrile 1,4-di-N-oxide (Figure 6C) is 150-fold more potent than TPZ, demonstrating that the amino group in three position is not necessary for activity (Monge et al., 1995a). Meanwhile, they developed two derivatives bearing a basic side chain. The 7-choloro- and 7-trifluoromethyl 3-(dimethylaminopropyl) amino-2-quinoxalinecarbonitrile 1,4-dioxide (Figure 6D) showed high potency and excellent selectivity (Monge et al., 1995a). They further studied the activity of the basic chain in 3 position of the 2-quinoxalinecarbonitrile 1,4-dioxides and showed that the replacement of (N,N-dialkyl amino) alkyl amino chain with aromatic rigid moieties (anilines and arylpiperazines) in three position held the potency but reduced the hypoxia-selectivity (Ortega et al., 2000). Continuingly, they synthesized a new series of 2-alkylcarbonyl and 2-benzoyl-3-trifluoromethylquinoxaline 1,4-di-N-oxide (Figure 6E) and evaluated for in vitro antitumor activity against MCF7 (breast), NCI-H460 (lung), and SF-268 (central nervous system) cells. In general, anticancer activity depends on the substituents in the carbonyl group, improving in the order: ethyl < isopropyl < tert-butyl < phenyl-ones (Zarranz et al., 2004). By comparison of the 1H NMR spectra, Solano et al. (2007) also showed that the best activity was observed in derivatives with electron-withdrawing groups in position 6 or 7 on the quinoxaline ring whereas loss of one or two oxygens reduced the cytotoxicity.
Besides Monge’s team, Das et al. (2009) reported a series of 2-(3-aryl-2-propenoyl)-3-methylquinoxaline-1,4-dioxides (Figure 6F) could reverse the MDR properties of murine L-5178Y leukemic cells which were transfected with the human MDR1 gene. Ismail et al. (2010) evaluated antitumor activity of a new series of QdNOs against liver carcinoma (Hepg2) and brain tumor (U251) human cell line, and compound 4 (Figure 6G) was the most potent hypoxia selective-cytotoxin on EAC cell line (Ismail et al., 2010). Urquiola et al. (2009) synthesized four new palladium(II) complexes with the formula Pd(L)(2) (Figure 6H), where L were quinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives, and evaluated their cytotoxicity on V79 cells. Pd(L1)(2) and Pd(L2)(2), where L1 was 3-aminoquinoxaline-2-carbonitrile 1,4-di-N-oxide and L2 was 3-amino-6(7)-methylquinoxaline-2-carbonitrile 1,4-di-N-oxide, showed non-selective cytotoxicity. Pd(L3)(2), where L3 was 3-amino-6(7)-chloroquinoxaline-2-carbonitrile 1,4-di-N-oxide, resulted in vitro more potent cytotoxin in hypoxia than the corresponding free ligand and TPZ. Pd(L2)(2) introduced a scission event in supercoiled DNA, yielding the circular relaxed form. Meanwhile, both Pd (L1)(2) and Pd(L3)(2) produced the loss of negative supercoils, rendering a family of topoisomers with reduced electrophoretic mobility. For the highest doses assayed, Pd(L3)(2) was even able to introduce positive supercoils on the plasmid DNA (Urquiola et al., 2009).
Although TPZ has attractive features of targeting hypoxic cells in tumors, it has limited clinical activity, in part because of poor extravascular penetration. Hicks et al. (2010) used a spatially resolved pharmacokinetic/pharmacodynamic model to guide the progression of 281 TPZ analogs through a hierarchical screen. SN29751 (Figure 6I) and SN30000 (Figure 6J) were identified as the most promising hypoxic cytotoxins, and SN30000, in particular, showed higher hypoxic potency and selectivity than TPZ in tumor cell cultures and faster diffusion through HT29 and SiHa multicellular layers.
QdNO-Induced Radicals
The mode of action of QdNO antitumoral drugs was most comprehensively studied in TPZ. As shown in Figure 7, it is known that TPZ (1) is an excellent substrate for a variety of intracellular reductases that add an electron to the drug to form an radical anion (2) (Brown, 1999), which then receives a proton to form a neutral radical (3). This neutral radical itself or produces other active radicals to abstract hydrogen from DNA, producing both SSBs and DSBs, resulting in chromosome breaks (Wang et al., 1992). Under aerobic conditions, oxygen can remove the additional electron from the radical anion (2), thereby back-oxidizing it to the non-toxic parent with a concomitant production of (Lloyd et al., 1991). Thus, the differential hypoxic cytotoxicity results from the fact that the TPZ radical is much more cytotoxic than the .
FIGURE 7.

Bioreduction of TPZ with produced free radicals based on Shinde et al. (2010) and Yin et al. (2012).
There have been a number of suggestions about the identity of active radicals produced by TPZ. An early proposal was that the ∙OH radical was released from the protonated anion (3), and the monoxide of TPZ (4) was produced simultaneously (Laderoute and Rauth, 1986), but this was later rejected in favor of the protonated radical anion itself as being the active species (Laderoute et al., 1988). Release of an ∙OH radical has been favored by other research workers, based on the similarity in the spectrum of products arising from TPZ-mediated damage to both purine and pyrimidine bases and deoxyribose sugars when compared with damage arising from the ∙OH radical, although TPZ-mediated damage exhibited some preference for purine over pyrimidine damage (Kotandeniya et al., 2002; Birincioglu et al., 2003; Chowdhury et al., 2007). Additionally, an EPR study using DMPO as a spin trap reported a composite spectrum of both a C-centered and an OH-adduct, DMPO-OH (Patterson and Taiwo, 2000), supporting evidence for the release of an ∙OH radical.
In contrast, Shinde et al. (2009, 2010) could not find any evidence that ∙OH is eliminated from TPZ following enzymatic reduction. They presented that the protonated radical anion (3) undergoes dehydration to the BTZ (5) (Shinde et al., 2009), causing oxidative damage to DNA and to oxidize TPZ itself (Anderson et al., 2003a,b, 2006). In situ EPR measurement confirmed that a multi N-centered radical, consistent with the BTZ radical (5), was formed (Shinde et al., 2009). Similarly, the one electron reduction potentials [E(1)R] of the BTZ radicals (5) also tracked the hypoxic cytotoxicity to human tumor cells in a series of BTO analogs of TPZ (3-amino BTO; Anderson et al., 2005). However, although the E(1)R value for the BTZ radical (5) of TPZ, 1.31 V (Anderson et al., 2003b), is sufficient to oxidize the purine bases of DNA, but not to directly oxidize the pyrimidine bases of DNA. Later, they trapped TPZ and a series of 3-substituted analogs by N-tert-butyl-R-phenylnitrone and observed by EPR the formation of their aryl (phenyl) radicals (6), which is stronger oxidants than the BTZ radical (5) (Shinde et al., 2010).
There are still some questions whether protonated radical anion (3) will undergo N–OH homolysis to its monoxide (4) and ∙OH, or alternatively, radical anion (2) may react by dehydration and form aminyl radicals (5) and H2O or phenyl radicals (6) and H2O. Recently, Yin et al. (2012) discovered that dehydration might be the result of a two-step sequence that involved N–OH homolysis and formation of ∙OH aggregates of the monoxide (4) followed by H-abstraction within the ∙OH aggregates to form hydrates of aminyl (5) or of phenyl (6) radicals (Figure 7, dashed arrows).
Electron paramagnetic resonance experiments indicate both an aryl-type radical and an oxidizing radical, trapped as a carbon-centered radical, are formed from the protonated radical anion of the bioreductive anticancer prodrug, SN30000 (Anderson et al., 2014). The carbon-centered radical, produced upon the one-electron oxidation of the 2-electron reduced metabolite of SN30000, oxidizes 2-deoxyribose, leads to double strand breaks. Hunter et al. (2014a) found hypoxia-activated prodrugs of DNA-damaging cytotoxins (TPZ, SN30000) might inhibit growth of triple-negative breast cancer (TNBC) by simultaneously addressing the two targets, tumor hypoxia and derangement of HR repair.
Recently, Yadav et al. (2014) found The ∙OH radical was released from 3-trifluoromethyl-quinoxaline 1,4-dioxides upon one-electron reduction by cytochrome P450 oxidoreductase. This process effectively competes with back oxidation of the intermediate radical anion by oxygen and underlies the increased aerobic cytotoxicity of such compounds compared to that seen for, TPZ.
Reductase
Another major unresolved question is the identity of the enzyme(s) that activates a QdNO to cause cell death. Numerous enzymes, including xanthine oxidase (EC 1.2.3.1; Laderoute et al., 1988), cytochrome P450 (Wang et al., 1993), DT-diaphorase (EC 1.6.99.2; Patterson et al., 1994), and NADPH:cytochrome P450 reductase (EC 1.6.2.4; Patterson et al., 1995), are able to metabolize TPZ in vitro under hypoxic conditions. A correlation between NADPH:cytochrome P450 reductase level (Patterson et al., 1997) or NOS (Chinje et al., 2003) level and sensitivity to TPZ has been reported. For a long time, there was no agreement as to which enzyme(s) are involved in the DNA damage until Evans et al. (1998) showed that TPZ was metabolized to DNA-damaging radicals by intra-nuclear enzymes. TPZ radicals formed outside nuclei do not contribute to intranuclear DNA damage, and the 80% of the drug metabolism that occurs in the cytoplasm is probably irrelevant for hypoxic killing effect of this drug. Later, Delahoussaye et al. (2001) demonstrated that multiple reductases in the nuclear matrix metabolized TPZ under hypoxia. DNA SSBs were probably caused by the most abundant source of reductase in the nucleus, while DNA DSBs were formed by an unknown nuclear reductase requiring only NADPH for its activity.
Hunter et al. (2014b) observed that forced expression of FAD-dependent oxidoreductase domain containing 2 (FOXRED2) increased activation of hypoxia-targeted prodrugs TPZ and SN30000. They also identified the flavoprotein P450 (cytochrome) oxidoreductase, which is responsible for prodrug activation during hypoxia, as the predominant determinant of sensitivity to SN30000 (Hunter et al., 2015).
Topoisomerase and Polymerase β
Topoisomerase II is essential in mammalian cells, because it resolves the unfavorable topological structures in DNA during replication and transcription. Drugs that stabilize Topo II with a DNA DSB to form cleavable complex are termed Topo II poisons (Pommier et al., 2010). Since TPZ produces a marked inhibition of DNA replication in the nuclear matrix (Peters et al., 2001), and TPZ-induced DNA DSBs are protein-associated (Olive, 1995; Siim et al., 1996), Peters and Brown (2002) considered TPZ might poison Topo II. They found under hypoxic conditions, the nuclear extracts from LXFL 529 human lung carcinoma cells treated with TPZ reduced the activity of Topo II. Inhibitors of the Topo II catalytic cycle abrogated TPZ-generated DNA DSBs and cytotoxicity, and TPZ stabilized DNA Topo II cleavable complexes (Peters and Brown, 2002). Using Saccharomyces cerevisiae as a model, overexpression of TOP2 (encoding Topo II) leads to hypersensitivity to TPZ, suggesting that Topo II is also a target of TPZ in yeast (Hellauer et al., 2005). XK469 (NSC 697887) and CQS (NSC 339004), two synthetic quinoxaline derivatives without two oxygens on the quinoxaline ring, also show solid tumor selectivity. XK469 and CQS have entered Phase I and Phase II clinical study, respectively (Miller et al., 1997; Bekaii-Saab et al., 2006; Alousi et al., 2007). Gao et al. (1999) reported the primary target of XK469 is Topo IIβ, and CQS was found to be both a Topo-IIα and a Topo-IIβ poison (Gao et al., 2000). The large aromatic side chains of XK469 and CQS can not be ignored to the drug action. Except for TopII, TPZ also induces other DNA-protein cross-links, including DNA-Topo I cleavable complexes (Evans et al., 2008) and DNA-Polβ cross-link with the lesion (Sung and Demple, 2006).
Evans et al. (2008) further showed an overall model of TPZ damage in which DNA SSBs, base damage, and DNA-protein cross-links (including Topo I and II cleavable complexes) produced stalling and collapse of replication forks. The resolution of the complex required HR and XPF/ERCC1 protein for their repair. Cells defective in HR proteins were particularly sensitive to TPZ, and extensive sister chromatid exchanges occurred after treatment with TPZ. In addition, TPZ preferentially kills mutants both with defects in XPF/ERCC1 and base excision repair. H2AX, an indicator of DNA DSBs, is induced preferentially in cells in the S phase of the cell cycle.
In a dose-dependent fashion, polymerase β DNA-protein cross-links (Polβ-DPC) were detected in MDA-MB-231 human cells treated with the antitumor drug TPZ (much more Polβ-DPC under 1% O2 than under 21% O2; Quinones et al., 2015). Mouse embryonic fibroblasts challenged with TPZ also incurred Polβ-DPC.
Hypoxia-Inducible Factor 1
Hypoxia-inducible factor 1, a heterodimeric transcription factor that mediates the adaptation of tumor cells and tissues to the hypoxic environment, has attracted considerable interest as a potential therapeutic target. The subunit 1α of HIF plays an essential role in the transcriptional activation of genes involved in tumor angiogenesis invasiveness and metastasis with its downstream target, VEGF (Carmeliet et al., 1998).
Quinoxaline 1,4-di-N-oxides could reduce the expression of HIF-1α mRNA in T-84 cells, and 2-benzoyl-3-phenyl-6,7-dichloroquinoxaline 1,4-dioxide (DCQ) is shown most effective in decreasing the HIF-1α mRNA and protein levels (Diab-Assef et al., 2002). A hypoxic cytotoxin, 3-amino-2-quinoxalinecarbonitrile 1,4-dioxide (TX-402), which is an improved analog of TPZ, has been shown to inhibit HIF-1α expression (Nagasawa et al., 2003). Recently, it is found that TPZ acts in a novel manner to inhibit HIF-1α accumulation, in which HIF-1α translational regulation is involving, dependent on the phosphorylation of translation initiation factor 2a (eIF2a; Zhang et al., 2010).
Weng et al. (2011) showed that suppression of HIF-1α by 3-(4-Bromophenyl)-2-(ethylsulfonyl)-6-methyl-quinoxaline 1,4-dioxide (Q39) resulted in a drastic decrease in VEGF expression. Unlike TPZ, suppression of HIF-1α accumulation by Q39 correlated with prominent dephosphorylation of mTOR (mammalian target of rapamycin) and initiation factor 4E-binding protein 1 at the translational level. 3-[2-hydroxyethyl(methyl)amino]-2-quinoxalinecarbonitrile 1,4-dioxide (TX-2098) was also shown to have antitumor effect in pancreatic cancer, through inhibiting VEGF and HIF-1α targeted gene expression (Miyake et al., 2012). Recently, it is demonstrated that DCQ blocks breast cancer metastasis by targeting the HIF-1 pathway (Ghattass et al., 2014). Cancer cell death was associated with an increase in ROS independently of p53 and was inhibited by antioxidants. DCQ-induced ROS was associated with DNA damage, the downregulation of HIF-1α, and inhibition of VEGF secretion. In MCF-7 (p53 wildtype), HIF-1α inhibition was partially via p53-activation and was accompanied by a decrease in p-mTOR protein, suggesting interference with HIF-1α translation. In MDA-MB-231 (p53 mutant), DCQ reduced HIF-1α through proteasomal-dependent degradation mechanisms.
Combinational use of TPZ with other inhibitors represents a novel mechanism for targeting tumor. In combination with TPZ, topoisomerase I inhibitors exhibited synergistic cytotoxicity and induced significant apoptosis in several hepatocellular carcinoma cell lines (Cai et al., 2014). The enhanced apoptosis induced by TPZ plus SN-38 (the active metabolite of irinotecan) was accompanied by increased mitochondrial depolarization and caspase pathway activation. The combination treatment dramatically inhibited the accumulation of HIF-1α protein, decreased the HIF-1α transcriptional activation, and impaired the phosphorylation of proteins involved in the HR repair pathway, ultimately resulting in the synergism of these two drugs. TPZ mediates central vascular dysfunction in tumors and is also a competitive inhibitor of NOS. Baker et al. (2013) further investigated the vascular-targeting activity of TPZ by combining it with NOS inhibitor L-NNA, or with low oxygen content gas breathing. Irreversible loss of perfusion and enhanced tumor cell death was observed when TPZ was combined with either low oxygen or a NOS inhibitor, illustrating a novel use of hypoxia-activated cytotoxic prodrugs as vascular targeting agents.
Antiprotozoal Activities Of QdNOs
Antitrypanosomal Activity
Trypanosoma cruzi is the haemoflagellate protozoan that causes the Human American trypanosomiasis, or Chagas disease, representing a relevant health problem in Central and South America. The acute form of Chagas disease usually goes unnoticed and may present as a localized swelling at the site of entry. The chronic form may develop 10 to 20 years after infection, which affects internal organs (e.g., the heart, the esophagus, the colon, and the peripheral nervous system), sometimes causing death to affected people from heart failure. The first line of treatment is nifurtimox and benznidazole.
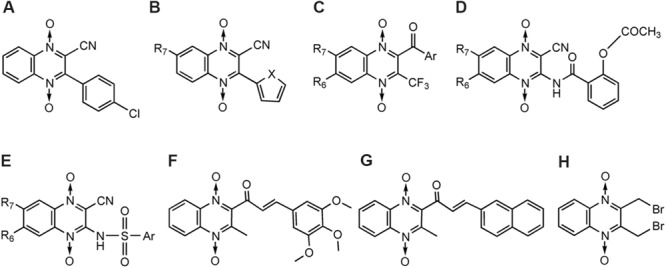
Cerecetto’s team (Cerecetto et al., 1999) firstly noticed QdNO derivatives had antitrypanosomal activity against epimastigote forms of T. cruzi. They reported a vanadium complexe of QdNO, [V(IV)O(L)(2)], where L were 3-aminoquinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives (Figure 8A), was provide with excellent antitrypanosomal activity, similar to that of the reference drugs nifurtimox and benznidazole and higher than that of the corresponding free ligands. The antitrypanosomal activity of these vanadium complexes could be explained on the basis of their lipophilicity and the electronic characteristics of the quinoxaline substituents (Urquiola et al., 2006). They also reported two QdNO compound, R3 = methyl (SI > 53.3, selectivity index = ID50(macrophage)/ID50(T. cruzi)) and R3 = phenyl (SI > 33.3; Figure 8B), displayed excellent parasite/mammal selectivity indexes. Those compounds are able to accumulate squalene suggesting that anti-T. cruzi mechanism of action is not involved the inhibition of sterol biosynthesis (Gerpe et al., 2010).
FIGURE 8.
QdNOs with antitrypanosomal activity.
Monge’s team also found that QdNO derivatives presented good inhibitor activity to the growth of T. cruzi in in vitro assays, among which a QdNO derivative (Figure 8C) were the most cytotoxic compounds against the protozoan, and the IC50 were of the same order of that of nifurtimox (Aguirre et al., 2004). The authors reported some SAR deductions: the presence of a halo-substituent (electron-withdrawing) at benzene moiety of quinoxaline produces more active compounds; more hydrophilic compounds decrease the antitrypanosomal activity; a reductive metabolism could be implicated in the mechanism of action. Later, they prepared a series of heterocyclic-2-carboxylic acid (3-cyano-1,4-di-N-oxidequinoxalin-2-yl)amide derivatives (Figure 8D), among which compounds (R6 = F, R7 = F, X = O) and (R6 = F, R7 = F, X = S; Figure 8D) presented IC50 values in the same order as nifurtimox (Ancizu et al., 2009). Recently, they synthesized a series of 3-trifluoromethyl QdNOs, among which derivatives possessing electron-withdrawing substituents in the 2-, 3-, 6-, and 7-positions were the most active compounds (Benitez et al., 2011). One 3-trifluoromethyl QdNO compound (Figure 8E), which was substituted with fluoro groups at the 6- and 7-positions of the quinoxaline ring, was the most active (IC50= 0.4 μM) and selective (SI = 10) in the cytotoxicity assay (Torres et al., 2013). It is demonstrated that inhibition of mitochondrial dehydrogenases are involved in the anti-T. cruzi activity of the most active derivatives (Benitez et al., 2011; Torres et al., 2013).
Leishmaniasis is a parasitic disease which appears in visceral, cutaneous and mucocutaneous forms affecting millions of people throughout the world. Monge’s team tested pyrazole quinoxaline derivatives against Leishmania peruviana. 2,6-dimethyl-3-f-quinoxaline 1,4-dioxide (Figure 8F) was the most active compound of this series against L. peruviana (IC50= 8.9 μM; Estevez et al., 2011). This compound resulted non-toxic for Vero and LLc-Mk2 cells, and was almost 6 to 13 times more active on Leishmania than on THP-1 or MPM. According to the study of Barea et al. (2012), piperazine linked QdNO (Figure 8G) emerged as the best leishmanicidal agent against L. infantum (IC50= 5.7 μM). Their later study indicated that the R = cyclohexyl derivative (Figure 8H) had the best antileishmanial activity against L. infantum (IC50= 2.5 μM) while the R = 3-chloropropyl derivative (Figure 8H) was the best against L. amazonensis (IC50= 0.7 μM; Barea et al., 2013). Villalobos-Rocha et al. (2014) evaluated the in vitro biological activity of 33 ethyl and methyl quinoxaline-7-carboxylate 1,4-di-N-oxide derivatives on T. cruzi and L. mexicana, of which M7 (Figure 8I) and E4 (Figure 8J) displayed activity against both parasites. Compound M2 (Figure 8K) was predicted in the docking procedure as a potential T. cruzi trypanothione reductase inhibitor by its interaction with five residues close to the active site of the enzyme.
Antimalarial Activity
Plasmodium falciparum is one of the species of Plasmodium that cause malaria in humans. Malaria caused by this species is the most dangerous form of malaria, with the highest rates of complications and mortality. For nearly half a century, chloroquine has been the primary therapy of choice. However, chloroquine-resistant P. falciparum is now observed in nearly all of the malaria-endemic regions and causes the most deadly form of malaria. Therefore, it is necessary to develop cheaper and more effective drugs against the parasite.
Zarranz et al. (2005) synthesized new series of 3-arylquinoxaline- carbonitrile derivatives (Figure 9A) and tested for their in vitro and in vivo activity against the erythrocytic development of P. falciparum with different chloroquine-resistance status. These series showed superior antimalarial activity in respect to reduced quinoxaline analogs. The best activity was observed with non-substituted QdNOs in positions 6 and 7 of the aromatic ring and with a hydrogen or chloro substituent in para position of the phenyl group (Figure 9A). Vicente et al. (2008) identified new compounds structurally based on 3-phenyl-quinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives (Figure 9B) active against P. falciparum. Derivative 1 (Figure 9B, X = O, R7 = H) demonstrated high potency [IC(50) = 0.63 mM] and good selectivity (SI = 10.35), thereby becoming a new lead-compound. Marin et al. (2008) synthesized derivatives of 3-trifluoromethyl-2-arylcarbonylquinoxaline 1,4-di-N-oxide (Figure 9C) and evaluated for their capacity to inhibit the growth of chloroquine-resistant P. falciparum FCB1 strain in culture. Compound 7-chloro-2-(2-furylcarbonyl)-3-trifluoromethyl-1,4-quinoxaline di-N-oxide (Figure 8C, Ar = 2-Furyl, R1/R2 = Cl/H) was the most active, being almost five times more active than chloroquine. It was also 50 times more active against P. falciparum than toxic toward MCF7 cells. SAR showed that bioisosteric modification of phenyl group by 2-thienyl or 2-furyl subunits, R2 position must be free or occupied by a methyl group and R1 position must be free or occupied by Cl, CH3, OCH3 or CF3. Barea et al. (2011) synthesized new 3-amino-1,4-di-N-oxide quinoxaline-2-carbonitrile derivatives as acetoxybenzamides (Figure 9D) and sulfonamides (Figure 9E), and evaluated for their in vitro antimalarial and antileishmanial activity. Compounds with one halogenous group substituted in position 6 and 7 provide an efficient approach for further development of antimalarial and antileishmanial agents. From a series of quinoxaline analogs of chalcones in another study, compounds 1a (IC50= 6.2 μM; Figure 9F) and 2a (IC50= 5.8 μM; Figure 9G) were the most active against FCR-3 P. falciparum (Gil et al., 2014). SAR demonstrated the importance of an enone moiety linked to the quinoxaline ring in the search for antimalarial ligands.
FIGURE 9.
QdNOs with antimalarial activity.
The intracellular mechanism of the antiparasitic action of QdNOs is barely elucidated. Recently, Brizuela et al. (2014) shows that one QdNO-derived compound, Conoidin A [2,3-bis(bromomethyl)-1,4-dioxide-quinoxaline] (Figure 9H), exhibites potent lytic activity against P. falciparum and constitutes an irreversible Peroxiredoxin-2 (Prx2) inhibitor. Conoidin A is first shown as an inhibitor of host cell invasion by the human pathogen T. gondii (Carey et al., 2004), and this compound is a covalent inhibitor of T. gondii peroxiredoxin II (Haraldsen et al., 2009). Conoidin A can also inactivate the peroxiredoxin-1 from the human hookworm Ancylostoma ceylanicum by alkylating or crosslinking the catalytic cysteines, while maintaining the enzyme in the “locally unfolded” conformation (Nguyen et al., 2013). Peroxiredoxin, a thiol-dependent peroxidase, serves a critical role in converting ROS signals into a cellular response. When Plasmodium sp. invades red cells, it imports Prx2 from the host cell to the parasite cytosol during intraerythrocytic development in an attempt to make up for degradation of peroxides generated during cell metabolism. Therefore, treatment of erythrocytes with Conoidin A produces an unviable growth of the parasite inside, and enhances parasite sensitivity to chloroquine (Brizuela et al., 2014).
Antitrichomonas Activity
Trichomoniasis is a protozoan infection of the human and bovine urogenital tracts. Metronidazole, Tinidazole and other nitroimidazoles are the most effective drugs, currently available for treatment. In the early 1980s, Glazer and Chappel (1982) reported the synthesis and the activity of a novel series of pyrido[2,3-b]quinoxaline 5-oxides against Trichomonas foetus. Carta et al. (2004) reported the synthesis and antitrichomonas activity of a series of 6,7-difluoro-3-methylquinoxaline 1,4-dioxides (Figure 10A). In particular, several 2-phenylthio derivatives resulted 20- to 30-fold more potent than the reference drug Metronidazole activity against T. vaginalis (SS22) in vitro, isolated in Italy from a case of acute vaginal trichomoniasis. For example, sulfoxide derivative (Figure 10B) was reported to be more effective than the reference drug metronidazole against T. vaginalis while one compound (Figure 10C) inhibited the growth of T. vaginalis at MIC value of 6.25 mg/mL, after 24 h of incubation.
FIGURE 10.
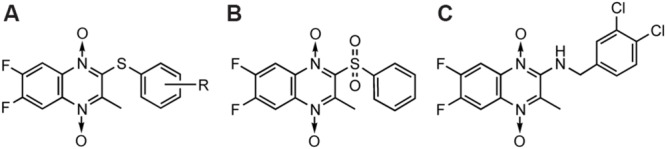
QdNOs with antitrichomonas activity.
Antiamoebic Activity
Amebiasis is a protozoan infection of human gastrointestinal tract caused by Entamoeba histolytica which results in 500 million cases and approximately 110,000 deaths annually. Out of a new series of ethyl and methyl quinoxaline-7-carboxylate 1,4-di-N-oxide derivatives synthesized by Duque-Montano et al. (2013), thiophene bearing motif (Figure 11A; IC50= 0.35 μM) stood out, having enhanced biological activity which was 11-fold higher than others but having selectivity index [IC50 (VERO cells)/IC50 (E. histolytica)] of 16.74. Compounds T-001 (Figure 11B) and T-016 (Figure 11C) showed IC50 values of 1.41 and 1.47 μM, respectively, with a value of selectivity index > 60.
FIGURE 11.
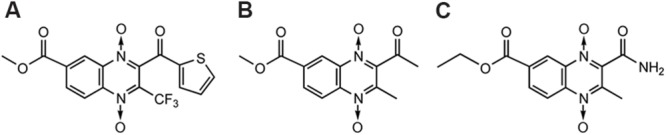
QdNOs with antiamoebic activity.
Antiinflammatory And Antioxidant Activities Of QdNOs
Arachidonic acid metabolism, mediated by the LOX enzyme family, leads to the generation of leukotrienes, a type of pro-inflammatory mediator involved in processes such as fever, asthma, or cardiovascular disease. Additionally, aberrant arachidonic acid metabolism is related to carcinogenesis. For example, increased LOX expression levels have been found in a wide range of cancers, including pancreatic, bladder, and breast cancer (Hofmann and Steinhilber, 2013). During the inflammation process, phagocytic leukocytes produce ROS. A number of commercially available non-steroidal antiinflammatory drugs, such as acetaminophen, salicylates, indomethacin, and nimesulide, have been shown to possess radical scavenging properties. Therefore, the development of new compounds having both antiinflammatory and antioxidant activities and being LOX inhibitors is an interesting approach for cancer prevention, treatment of chronic inflammation and other related pathological conditions.
3-phenyl-1-(1,4-di-N-oxide quinoxalin-2-yl)-2-propen-1-one derivatives and their 4,5-dihydro-(1H)-pyrazole analogs were discovered to show very interesting antioxidant and antiinflammatory properties. Compound 2a (Figure 12, series 2) displayed an in vivo antiinflammatory effect (56.1%) higher than the reference drug, indomethacin, and promising in vitro inhibition values of LOX (IC50 < 1 μM; Burguete et al., 2007). Compound 7b (Figure 12, series 7) showed significant protection against carrageenan-induced paw edema, in which the in vivo antiinflammatory effect (41%) was similar to that of indomethacin (47%; Burguete et al., 2011).
FIGURE 12.
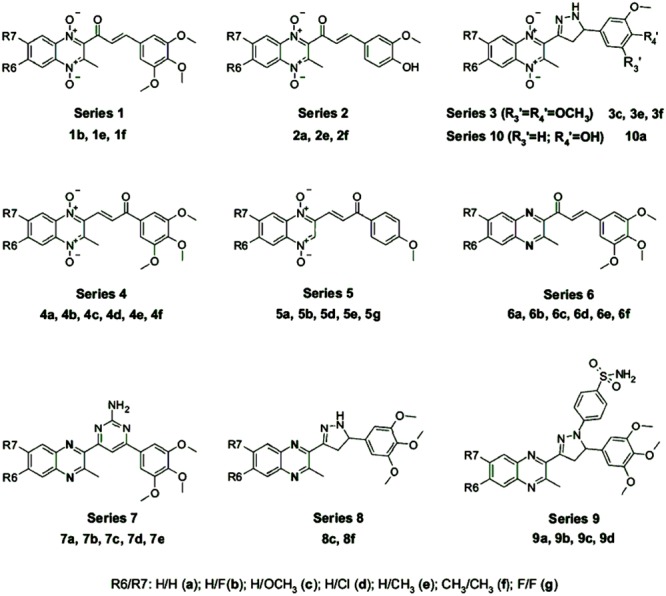
QdNOs with antiinflammatory and antioxidant activities [adapted from Burguete et al. (2011)].
Burguete et al. (2011) has studied the SAR which demonstrates that the radical scavenging ability needs a phenolic group (series 2) and/or a free amino pyrazoline ring (series 3, 8, and 10) in the structure (Figure 12). In the presence of the pyrazoline moiety, those with N-oxide groups in the quinoxaline ring (series 3 and 10) exhibited significantly increased reducing activity compared to their reduced analogs (series 8), resulting in increased scavenging activity. Compounds with an α, β-unsaturated ketone system (1b, 2e, 4f, 5a, 5b, 5g, and 6a–f) presented the best scavenging activities, indicating that the olefinic moiety might play an important role in the activity by trapping the . The derivatives are also good ∙OH scavengers. In the inhibition of LOX, the best IC50 values were shown by compounds of series 7, 9, and compound 8f. Although compounds that displayed good activities of inhibition of lipid peroxidation also presented good values of inhibition of LOX, the best inhibitors of lipid peroxidation were those compounds without any substitution in the quinoxaline ring while the best inhibitors of LOX were obtained by fluoro- and methyl-substituted derivatives.
Toxicological Effects Of QdNOs
The toxicology effects, including short- and long-term toxicities (Fang et al., 2006; Ihsan et al., 2010, 2011; Wang et al., 2010, 2011a,b, 2012), genotoxicity (Ihsan et al., 2013a,b), and photoallergic toxicity (He et al., 2006), have been extensively studied in the veterinary-used QdNOs. The liver is the main target organ of QdNOs, and OLA and MEQ show obvious toxic effect on the kidney and the adrenal gland. High dose of OLA, MEQ, CBX, and QCT can significantly inhibit the growth and development, reproductive function and embryonic development of rats, whereas CYA has minor effect. OLA, CBX, and MEQ have genotoxicity, and CBX shows obvious carcinogenic effects. QCT has certain genetic toxicity, and no genotoxicity has been detected in CYA.
Quinoxaline 1, 4-di-N-oxides show mutagenic and DNA-damaging effects on various organisms. Voogd et al. (1980) reported that QDX, CAX, and OLA were mutagenic on Saccharomyces cerevisiae, Klebsiella pneumoniae, S. typhimurium and E. coli, causing base-pair substitutions and frame-shift mutations. Beutin et al. (1981) found that mutagenicity of QdNOs was dependent on the presence of N-oxide groups, since quinoxaline, a completely reduced derivative of QDX, was not mutagenic, whereas the partially reduced quinoxaline-N-oxide exhibited a lower mutagenic activity than QDX. It was suggested that the mutagenicity of QdNOs resulted from the error-prone repair involved in SOS responses (Nunoshiba and Nishioka, 1989). Ihsan et al. (2013a,b) compared the genotoxic potential of QdNOs in Ames test, HGPRT gene mutation test in V79 cells, unscheduled DNA synthesis assay in human peripheral lymphocytes, chromosome aberration test, and micronucleus test in mice bone, and found that OLA, CBX, and MEQ were more genotoxic than QCT and CYA. Using alkalic comet assay, pronounced increase of DNA fragmentation were observed in Vero cells treated with CBX, OLA, and QCT (Chen et al., 2009). In contrast, DNA damage was significantly decreased after incubation with S9 mix, suggesting that the intermediate metabolites of these compounds exerted lower genotoxicity than their parent drugs. Jin et al. (2009) also found that QCT caused significant DNA fragment migration in a dose-dependent manner in human hepatoma (HepG2).
The mutagenicity of QdNOs with antiparasitic activity has also been studied. Introduction of electron-withdrawing substituents at C-6 and/or C-7 of the quinoxaline ring enhanced in vitro biological activity against T. cruzi. In addition, this led to the obtainment of non-mutagenic derivatives in both the Ames assays that were performed or their mutagenicity disappeared when performing the assay using metabolic activation (Torres et al., 2013). Using Ames test, Gabay et al. (2014) investigated the mutagenicity of an in-house chemical library of eighty five N-oxide containing heterocycles, and they found that in some cases, a relationship was found between the presence of N-oxide and mutagenicity, and in other cases, such as quinoxaline dioxides with antiparasitic activity, mutagenicity was substituent dependent.
Quinoxaline 1, 4-di-N-oxides can produce ROS and cause oxidative cell damage. It was found that OLA induced the increased levels of ROS and 8-OHdG in HepG2 cells, inferring that OLA exerts genotoxic effects in HepG2 cells probably through the ROS-induced oxidative DNA damage (Zou et al., 2009). Using γ-H2AX as a surrogate marker for DNA damage, Liu et al. (2012) found that MEQ treatment induced cellular DNA damage, which paralleled the chemical-induced elevation of ROS levels, and expression of the antioxidant enzyme catalase partially alleviated these MEQ-associated effects. Huang et al. (2010b) found that OLA irritated a persistent and utmost release of ROS while MEQ made a similar but weaker reaction. CYA, however, had a short and unstable release of intracellular ROS. On the other hand, quinoxalinine-2-carboxylie acid, one of the metabolites of OLA and MEQ, did not cause any significant production of ROS and showed relatively lower toxicity than its parents. Zhang et al. (2014) reported that QCT damaged the antioxidant defense abilities of HepG2 cells by reducing the activities of endogenous antioxidant enzymes, lowering glutathione concentration, and elevating malondialdehyde level. One hundred and sixty QCT-responsive genes were found to be associated with cell proliferation, glucose metabolism, oxidative stress, and apoptosis, such as NAD(P)H dehydrogenase. However, QCT metabolites (1,4-bisdesoxyquinocetone and 3-methylquinoxaline-2-carboxylic acid) showed little effects on HepG2 cells. Wang et al. (2015b) demonstrated that the rank orders of the desoxy and bidesoxy rates in rat and pig liver microsomes were QCT < CBX < MEQ < OLA < CYA and QCT < MEQ < CBX < OLA < CYA, respectively. In rats, porcine primary hepatocytes, and HepG2 cells, oxidative stress indices and DNA damage showed inverse relationships with the deoxidation rate, indicating that faster deoxidation of QdNOs results in lower DNA-damage-induced toxicity.
The molecular mechanism of cell cycle arrest and apoptosis induced by QdNOs were explored. Liu et al. (2012) report that MEQ inhibited cell proliferation by arresting cells at the G2/M phase of the cell cycle. Zou et al. (2011) demonstrated that OLA induced cell cycle arrest to the S phase and dose-dependent apoptotic cell death in HepG2 cells through a caspase-9 and -3 dependent mitochondrial pathway. Zhang et al. (2013) found that QCT induced apoptosis in HepG2 cells via activation of caspase, interaction of TNF-α and TNFR1 and modulation of the protein levels of Bid, Bax, and Bcl-2, involving the participation of p53, p38, and JNK. c-MYC-dependent activation of the mitochondrial apoptotic pathway may be associated with QCT-induced toxicity (Zhang et al., 2014). Dysregulated or excessive autophagy may lead to ‘type II programmed cell death,’ which is closely associated with apoptosis. Zhao et al. (2015) showed that OLA-induced autophagy in HepG2 cells is upregulated by Beclin 1 but downregulated by ROS-dependent c-Jun N-terminal protein kinase (JNK).
The organ toxicities of QdNOs have also been studied. Huang et al. (2009) reported the dose-dependent long-term toxicity of MEQ on adrenal gland in male rats, and its mechanism may involve in oxidative stress and steroid hormone biosynthesis pathway. Then, they found the complex interactions of MEQ metabolism, renin–angiotensin–aldosterone system, NADPH oxidase and oxidative stress in response to MEQ-induced tissue toxicity and aldosterone secretion (Huang et al., 2010a). Wang et al. (2015a) estimated the adrenal cell damage induced by QCT and its bidesoxy-QCT (B-QCT) metabolite, and suggested that its toxic effects resulted from N-oxide groups, and its toxic mechanism might involve the interference of the steroid hormone biosynthesis pathway. They also found ROS, the Janus kinase-signal transducer and activator of transcription (JAK/STAT) pathway, suppressors of cytokine signaling and inflammatory cytokines (TNF-α and IL-6) were involved in the liver and spleen toxicities of MEQ (Wang et al., 2011c). Ihsan et al. (2011) verified that MEQ exerted testicular toxicity by causing oxidative stress and steroidal gene expression profiles.
Perspectives
The versatile activities of QdNOs are close related to their chemical structures. The two N-oxide groups in the quinoxaline ring are necessary for the antibacterial (Montoya et al., 1998; Ortega et al., 1999; Sainz et al., 1999; Zhang, 2012), antitumor (Solano et al., 2007), anti-Trypanosoma (Aguirre et al., 2004), and antimalaria (Zarranz et al., 2005) properties of QdNOs. Some reduced forms of QdNOs, in which the two N-oxide groups are absent, still possess biological activities. We assume the large side chains of these reduced quinoxaline compounds contribute more to the activities, based on the fact that in the 2-quinoxalinecarbonitrile 1,4-dioxides, the replacement of (N,N-dialkyl amino) alkyl amino chain with aromatic rigid moieties (anilines and arylpiperazines) in three position held the potency but reduced the hypoxia-selectivity (Ortega et al., 2000). The different side chains probably affect the desoxy and bidesoxy rates of QdNOs (Wang et al., 2015b), which may result in different levels of activities and toxicities.
For QdNO antitumoral compounds, when the electron-withdrawing nature of the 6(7)-substituent increases, the compound is more readily reduced and show the best activity (Monge et al., 1995b; Solano et al., 2007). This also is the case for antimycobacterial (Jaso et al., 2005), antitrypanosomal (Aguirre et al., 2004; Benitez et al., 2011), and antimalarial (Zarranz et al., 2005) activities. Interestingly, among 2-alkylcarbonyl-3- trifluoromethylquinoxaline 1,4-di-N-oxide derivatives, anticancer activity depends on the substituents in the carbonyl group, improving in the order: ethyl < isopropyl < tert-butyl < phenyl-ones (Zarranz et al., 2004), while among 6(7)- substituted quinoxaline-2-carboxylate 1,4-dioxide derivatives, anti-TB activity depends on the substituents in the carboxylate group, improving in the following order: tert-butyl < allyl < 2-methoxyethyl < ethyl < benzyl (Jaso et al., 2005). We speculate that the activity of alkyl group substitution must be mainly affected by the substituted carbonyl or carboxylate group, but the large benzene ring itself may contribute more to the anticancer or anti-TB activities.
In summary, SAR studies for QdNOs suggest that the position C(2) of quinoxaline should be substituted by electron-withdrawing moieties, preferably by a nitrile or an aroyl, an ester, an N-substituted amide group or a short brominated alkyl chain (Jampilek, 2014). The position C(3) is most often substituted by short alkyl chain (e.g., CH3, CF3) or phenyl. Substitution of C(6) or C(7) of QdNOs by Cl, F, CF3, or OCH3 is also advantageous. When the nitrile moiety is C(2) substituent, substituents in other positions should not be so much electron-withdrawing. It can be concluded that generally lipophilic and mostly electron-withdrawing substituents are preferred.
Although it has been generally accepted that QdNOs are prodrugs, transport forms of agents that are reduced to active metabolites in the body, the modes of actions of QdNOs are still indistinct. The research of the mechanism of actions of QdNOs is in progress, pushing the development of new compounds with more efficient potentials and less harmful effects. Though modes of antitumoral and antibacterial actions of QdNOs are comparatively clear, there are still some puzzles need to be explained. First, the enzyme(s), reducing and activating QdNOs to produce the free radicals, need to be identified. Since free radicals are short-lived, the location of enzyme(s) might indicate the subcellular or molecular target(s) of the drugs. Second, the type of free radical(s) and how they attack DNA or other target(s) should be identified and elucidated. Third, the network of the drug action and the interactions between drugs and organisms should be fully studied. Fortunately, the genomic and proteomic techniques have become effective ways to investigate drug action pathway and discover new drug targets. Furthermore, the research of the mode of action of QdNOs will provide clues to the study of drug resistant and toxicological mechanism, and guide more effective medication of QdNO drugs, e.g., drug combination therapy. For those modes of actions of QdNOs which are still vague, such as antiprotozoal and anticandida activity, researchers are suggested to investigate them according to the already known mechanisms and the SAR of QdNOs combined with new research tools, such as omics technology and 3D-QSAR. A deeper knowledge of the molecular targets of QdNOs is required for the development of new and more specific drugs through a rational design strategy to avoid undesirable side effects.
Author Contributions
GC contributed to the design of the review and wrote the review. WS, CC, LG, HH, and ZL revised the review. XW and ZY contributed to the conception of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31502115 and 31272614), and the National Program for Risk Assessment of Quality and Safety of Livestock and Poultry Products (GJFP2016008).
Abbreviations
- ADG
average daily gain
- BTO
1,2,4-benzotriazine 1,4-dioxide
- BTZ
benzotriazinyl radical
- CBX
carbadox
- CYA
cyadox
- DMPO
5,5′-dimethylpyrroline 1-N-oxide
- DSBs
double-strand breaks
- E(1)R
one electron reduction potential
- EPR
electron paramagnetic resonance
- HIF
hypoxia-inducible factor
- HR
homologous recombination
- LOX
lipoxygenase
- MDR
multidrug resistant
- MEQ
mequindox
- MIC
minimum inhibitory concentration
- NOAEL
no observed adverse effect level
- NOS
nitric oxide synthase
- ∙OH
hydroxyl radical
- OLA
olaquindox
superoxide radical
- QCT
quinocetone
- QdNOs
quinoxaline 1,4-di-N-oxides
- QDX
quindoxin
- ROS
reactive oxygen species
- SAR
structure-activity relationship
- SSBs
single-strand breaks
- TB
Tuberculosis
- Topo II
topoisomerase II
- TPZ
tirapazamine
- VEGF
vascular endothelial growth factor
- 2DE-MS
two-dimensional polyacrylamide gel electrophoresis combined with mass spectrometry
References
- Aguirre G., Cerecetto H., Di Maio R., Gonzalez M., Alfaro M. E., Jaso A., et al. (2004). Quinoxaline N,N′-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi. Structure-activity relationships. Bioorg. Med. Chem. Lett. 14 3835–3839. 10.1016/j.bmcl.2004.04.088 [DOI] [PubMed] [Google Scholar]
- Alousi A. M., Boinpally R., Wiegand R., Parchment R., Gadgeel S., Heilbrun L. K., et al. (2007). A phase 1 trial of XK469: toxicity profile of a selective topoisomerase IIbeta inhibitor. Invest. New Drugs 25 147–154. 10.1007/s10637-006-9024-5 [DOI] [PubMed] [Google Scholar]
- Ancizu S., Moreno E., Torres E., Burguete A., Perez-Silanes S., Benitez D., et al. (2009). Heterocyclic-2-carboxylic acid (3-cyano-1,4-di-N-oxidequinoxalin-2-yl)amide derivatives as hits for the development of neglected disease drugs. Molecules 14 2256–2272. 10.3390/molecules14062256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. F., Harris T. A., Hay M. P., Denny W. A. (2003a). Enhanced conversion of DNA radical damage to double strand breaks by 1,2,4-benzotriazine 1,4-dioxides linked to a DNA binder compared to tirapazamine. Chem. Res. Toxicol. 16 1477–1483. 10.1021/tx034116v [DOI] [PubMed] [Google Scholar]
- Anderson R. F., Shinde S. S., Hay M. P., Gamage S. A., Denny W. A. (2003b). Activation of 3-amino-1,2,4-benzotriazine 1,4-dioxide antitumor agents to oxidizing species following their one-electron reduction. J. Am. Chem. Soc. 125 748–756. 10.1021/ja0209363 [DOI] [PubMed] [Google Scholar]
- Anderson R. F., Shinde S. S., Hay M. P., Denny W. A. (2006). Potentiation of the cytotoxicity of the anticancer agent tirapazamine by benzotriazine N-oxides: the role of redox equilibria. J. Am. Chem. Soc. 128 245–249. 10.1021/ja0559101 [DOI] [PubMed] [Google Scholar]
- Anderson R. F., Shinde S. S., Hay M. P., Gamage S. A., Denny W. A. (2005). Radical properties governing the hypoxia-selective cytotoxicity of antitumor 3-amino-1,2,4-benzotriazine 1,4-dioxides. Org. Biomol. Chem. 3 2167–2174. 10.1039/b502586a [DOI] [PubMed] [Google Scholar]
- Anderson R. F., Yadav P., Patel D., Reynisson J., Tipparaju S. R., Guise C. P., et al. (2014). Characterisation of radicals formed by the triazine 1,4-dioxide hypoxia-activated prodrug, SN30000. Org. Biomol. Chem. 12 3386–3392. 10.1039/c4ob00236a [DOI] [PubMed] [Google Scholar]
- Baker J. H., Kyle A. H., Bartels K. L., Methot S. P., Flanagan E. J., Balbirnie A., et al. (2013). Targeting the tumour vasculature: exploitation of low oxygenation and sensitivity to NOS inhibition by treatment with a hypoxic cytotoxin. PLoS ONE 8:e76832 10.1371/journal.pone.0076832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barea C., Pabon A., Castillo D., Zimic M., Quiliano M., Galiano S., et al. (2011). New salicylamide and sulfonamide derivatives of quinoxaline 1,4-di-N-oxide with antileishmanial and antimalarial activities. Bioorg. Med. Chem. Lett. 21 4498–4502. 10.1016/j.bmcl.2011.05.125 [DOI] [PubMed] [Google Scholar]
- Barea C., Pabon A., Galiano S., Perez-Silanes S., Gonzalez G., Deyssard C., et al. (2012). Antiplasmodial and leishmanicidal activities of 2-cyano-3-(4-phenylpiperazine-1-carboxamido) quinoxaline 1,4-dioxide derivatives. Molecules 17 9451–9461. 10.3390/molecules17089451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barea C., Pabon A., Perez-Silanes S., Galiano S., Gonzalez G., Monge A., et al. (2013). New amide derivatives of quinoxaline 1,4-di-N-oxide with leishmanicidal and antiplasmodial activities. Molecules 18 4718–4727. 10.3390/molecules18044718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekaii-Saab T. S., Mortazavi A., Hicks L. G., Zalupski M., Pelley R. J., Chan K. K., et al. (2006). A phase II study of chloroquinoxaline sulfonamide (CQS) in patients with metastatic colorectal carcinoma (MCRC). Invest. New Drugs 24 343–346. 10.1007/s10637-005-4827-3 [DOI] [PubMed] [Google Scholar]
- Benitez D., Cabrera M., Hernandez P., Boiani L., Lavaggi M. L., Di Maio R., et al. (2011). 3-Trifluoromethylquinoxaline N,N’-dioxides as anti-trypanosomatid agents. Identification of optimal anti-T. cruzi agents and mechanism of action studies. J. Med. Chem. 54 3624–3636. 10.1021/jm2002469 [DOI] [PubMed] [Google Scholar]
- Beutin L., Preller E., Kowalski B. (1981). Mutagenicity of quindoxin, its metabolites, and two substituted quinoxaline-di-N-oxides. Antimicrob. Agents Chemother. 20 336–343. 10.1128/AAC.20.3.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birincioglu M., Jaruga P., Chowdhury G., Rodriguez H., Dizdaroglu M., Gates K. S. (2003). DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J. Am. Chem. Soc. 125 11607–11615. 10.1021/ja0352146 [DOI] [PubMed] [Google Scholar]
- Brizuela M., Huang H. M., Smith C., Burgio G., Foote S. J., McMorran B. J. (2014). Treatment of erythrocytes with the 2-cys peroxiredoxin inhibitor, Conoidin A, prevents the growth of Plasmodium falciparum and enhances parasite sensitivity to chloroquine. PLoS ONE 9:e92411 10.1371/journal.pone.0092411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. M. (1999). The hypoxic cell: a target for selective cancer therapy–eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 59 5863–5870. [PubMed] [Google Scholar]
- Burguete A., Pontiki E., Hadjipavlou-Litina D., Ancizu S., Villar R., Solano B., et al. (2011). Synthesis and biological evaluation of new quinoxaline derivatives as antioxidant and anti-inflammatory agents. Chem. Biol. Drug Des. 77 255–267. 10.1111/j.1747-0285.2011.01076.x [DOI] [PubMed] [Google Scholar]
- Burguete A., Pontiki E., Hadjipavlou-Litina D., Villar R., Vicente E., Solano B., et al. (2007). Synthesis and anti-inflammatory/antioxidant activities of some new ring substituted 3-phenyl-1-(1,4-di-N-oxide quinoxalin-2-yl)-2-propen-1-one derivatives and of their 4,5-dihydro-(1H)-pyrazole analogues. Bioorg. Med. Chem. Lett. 17 6439–6443. 10.1016/j.bmcl.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Cai T. Y., Liu X. W., Zhu H., Cao J., Zhang J., Ding L., et al. (2014). Tirapazamine sensitizes hepatocellular carcinoma cells to topoisomerase I inhibitors via cooperative modulation of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 13 630–642. 10.1158/1535-7163.MCT-13-0490 [DOI] [PubMed] [Google Scholar]
- Carey K. L., Westwood N. J., Mitchison T. J., Ward G. E. (2004). A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 101 7433–7438. 10.1073/pnas.0307769101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Dor Y., Herbert J. M., Fukumura D., Brusselmans K., Dewerchin M., et al. (1998). Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394 485–490. 10.1038/28867 [DOI] [PubMed] [Google Scholar]
- Carta A., Corona P., Loriga M. (2005). Quinoxaline 1,4-dioxide: a versatile scaffold endowed with manifold activities. Curr. Med. Chem. 12 2259–2272. 10.2174/0929867054864831 [DOI] [PubMed] [Google Scholar]
- Carta A., Loriga M., Paglietti G., Mattana A., Fiori P. L., Mollicotti P., et al. (2004). Synthesis, anti-mycobacterial, anti-trichomonas and anti-candida in vitro activities of 2-substituted-6,7-difluoro-3-methylquinoxaline 1,4-dioxides. Eur. J. Med. Chem. 39 195–203. 10.1016/j.ejmech.2003.11.008 [DOI] [PubMed] [Google Scholar]
- Carta A., Paglietti G., Rahbar Nikookar M. E., Sanna P., Sechi L., Zanetti S. (2002). Novel substituted quinoxaline 1,4-dioxides with in vitro antimycobacterial and anticandida activity. Eur. J. Med. Chem. 37 355–366. 10.1016/S0223-5234(02)01346-6 [DOI] [PubMed] [Google Scholar]
- Cerecetto H., Di Maio R., Gonzalez M., Risso M., Saenz P., Seoane G., et al. (1999). 1,2,5-Oxadiazole N-oxide derivatives and related compounds as potential antitrypanosomal drugs: structure-activity relationships. J. Med. Chem. 42 1941–1950. 10.1021/jm9805790 [DOI] [PubMed] [Google Scholar]
- Chen Q., Tang S., Jin X., Zou J., Chen K., Zhang T., et al. (2009). Investigation of the genotoxicity of quinocetone, carbadox and olaquindox in vitro using Vero cells. Food Chem. Toxicol. 47 328–334. 10.1016/j.fct.2008.11.020 [DOI] [PubMed] [Google Scholar]
- Cheng G., Li B., Wang C., Zhang H., Liang G., Weng Z., et al. (2015). Systematic and molecular basis of the antibacterial action of quinoxaline 1,4-Di-N-oxides against Escherichia coli. PLoS ONE 10:e0136450 10.1371/journal.pone.0136450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinje E. C., Cowen R. L., Feng J., Sharma S. P., Wind N. S., Harris A. L., et al. (2003). Non-nuclear localized human NOSII enhances the bioactivation and toxicity of tirapazamine (SR4233) in vitro. Mol. Pharmacol. 63 1248–1255. 10.1124/mol.63.6.1248 [DOI] [PubMed] [Google Scholar]
- Chowdhury G., Junnotula V., Daniels J. S., Greenberg M. M., Gates K. S. (2007). DNA strand damage product analysis provides evidence that the tumor cell-specific cytotoxin tirapazamine produces hydroxyl radical and acts as a surrogate for O(2). J. Am. Chem. Soc. 129 12870–12877. 10.1021/ja074432m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihak R., Srb V. (1983). Cytogenetic effects of quinoxaline-1,4-dioxide-type growth-promoting agents. I. Micronucleus test in rats. Mutat. Res. 116 129–135. [DOI] [PubMed] [Google Scholar]
- Cohen E. E., Rosine D., Haraf D. J., Loh E., Shen L., Lusinchi A., et al. (2007). Phase I trial of tirapazamine, cisplatin, and concurrent accelerated boost reirradiation in patients with recurrent head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 67 678–684. 10.1016/j.ijrobp.2006.09.056 [DOI] [PubMed] [Google Scholar]
- Covens A., Blessing J., Bender D., Mannel R., Morgan M. (2006). A phase II evaluation of tirapazamine plus cisplatin in the treatment of recurrent platinum-sensitive ovarian or primary peritoneal cancer: a Gynecologic Oncology Group study. Gynecol. Oncol. 100 586–590. 10.1016/j.ygyno.2005.09.032 [DOI] [PubMed] [Google Scholar]
- Das N. K. (1984). In vitro susceptibility of Escherichia coli of swine origin to carbadox and other antimicrobials. Am. J. Vet. Res. 45 252–254. [PubMed] [Google Scholar]
- Das U., Pati H. N., Panda A. K., De Clercq E., Balzarini J., Molnar J., et al. (2009). 2-(3-Aryl-2-propenoyl)-3-methylquinoxaline-1,4-dioxides: a novel cluster of tumor-specific cytotoxins which reverse multidrug resistance. Bioorg. Med. Chem. 17 3909–3915. 10.1016/j.bmc.2009.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahoussaye Y. M., Evans J. W., Brown J. M. (2001). Metabolism of tirapazamine by multiple reductases in the nucleus. Biochem. Pharmacol. 62 1201–1209. 10.1016/S0006-2952(01)00784-5 [DOI] [PubMed] [Google Scholar]
- Diab-Assef M., Haddadin M. J., Yared P., Assaad C., Gali-Muhtasib H. U. (2002). Quinoxaline 1,4-dioxides: hypoxia-selective therapeutic agents. Mol. Carcinog. 33 198–205. 10.1002/mc.10036 [DOI] [PubMed] [Google Scholar]
- Ding M. X., Wang Y. L., Zhu H. L., Yuan Z. H. (2006a). Effects of cyadox and olaquindox on intestinal mucosal immunity and on fecal shedding of Escherichia coli in piglets. J. Anim. Sci. 84 2367–2373. 10.2527/jas.2005-564 [DOI] [PubMed] [Google Scholar]
- Ding M. X., Yuan Z. H., Wang Y. L., Zhu H. L., Fan S. X. (2006b). Olaquindox and cyadox stimulate growth and decrease intestinal mucosal immunity of piglets orally inoculated with Escherichia coli. J. Anim. Physiol. Anim. Nutr. 90 238–243. 10.1111/j.1439-0396.2005.00593.x [DOI] [PubMed] [Google Scholar]
- Downing J. C. (1974). Carbadox for control of swine dysentery. Mod. Vet. Pract. 55 167–168 passim. [PubMed] [Google Scholar]
- Drumev D. (1981). [Current animal feeds with antimicrobial activity]. Vet. Med. Nauki 18 10–25. [PubMed] [Google Scholar]
- Duque-Montano B. E., Gomez-Caro L. C., Sanchez-Sanchez M., Monge A., Hernandez-Baltazar E., Rivera G., et al. (2013). Synthesis and in vitro evaluation of new ethyl and methyl quinoxaline-7-carboxylate 1,4-di-N-oxide against Entamoeba histolytica. Bioorg. Med. Chem. 21 4550–4558. 10.1016/j.bmc.2013.05.036 [DOI] [PubMed] [Google Scholar]
- Estevez Y., Quiliano M., Burguete A., Cabanillas B., Zimic M., Malaga E., et al. (2011). Trypanocidal properties, structure-activity relationship and computational studies of quinoxaline 1,4-di-N-oxide derivatives. Exp. Parasitol. 127 745–751. 10.1016/j.exppara.2011.01.009 [DOI] [PubMed] [Google Scholar]
- European Union [EU] (1998). Commission Regulation (EC) No. 2788/98. Off. J. Eur. Commun. L347:3. [Google Scholar]
- Evans J. W., Chernikova S. B., Kachnic L. A., Banath J. P., Sordet O., Delahoussaye Y. M., et al. (2008). Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res. 68 257–265. 10.1158/0008-5472.CAN-06-4497 [DOI] [PubMed] [Google Scholar]
- Evans J. W., Yudoh K., Delahoussaye Y. M., Brown J. M. (1998). Tirapazamine is metabolized to its DNA-damaging radical by intranuclear enzymes. Cancer Res. 58 2098–2101. [PubMed] [Google Scholar]
- Fang G., He Q., Zhou S., Wang D., Zhang Y., Yuan Z. (2006). Subchronic oral toxicity study with cyadox in Wistar rats. Food Chem. Toxicol. 44 36–41. 10.1016/j.fct.2005.06.003 [DOI] [PubMed] [Google Scholar]
- Gabay M., Cabrera M., Maio R. D., Paez J. A., Campillo N., Lavaggi M. L., et al. (2014). Mutagenicity of N-oxide containing heterocycles and related compounds: experimental and theoretical studies. Curr. Top. Med. Chem. 14 1374–1387. 10.2174/1568026614666140506123235 [DOI] [PubMed] [Google Scholar]
- Ganley B., Chowdhury G., Bhansali J., Daniels J. S., Gates K. S. (2001). Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide. Bioorg. Med. Chem. 9 2395–2401. 10.1016/S0968-0896(01)00163-8 [DOI] [PubMed] [Google Scholar]
- Gao H., Huang K. C., Yamasaki E. F., Chan K. K., Chohan L., Snapka R. M. (1999). XK469 a selective topoisomerase IIbeta poison. Proc. Natl. Acad. Sci. U.S.A. 96 12168–12173. 10.1073/pnas.96.21.12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Yamasaki E. F., Chan K. K., Shen L. L., Snapka R. M. (2000). Chloroquinoxaline sulfonamide (NSC 339004) is a topoisomerase IIalpha/beta poison. Cancer Res. 60 5937–5940. [PubMed] [Google Scholar]
- Gerpe A., Boiani L., Hernandez P., Sortino M., Zacchino S., Gonzalez M., et al. (2010). Naftifine-analogues as anti-Trypanosoma cruzi agents. Eur. J. Med. Chem. 45 2154–2164. 10.1016/j.ejmech.2010.01.052 [DOI] [PubMed] [Google Scholar]
- Ghattass K., El-Sitt S., Zibara K., Rayes S., Haddadin M. J., El-Sabban M., et al. (2014). The quinoxaline di-N-oxide DCQ blocks breast cancer metastasis in vitro and in vivo by targeting the hypoxia inducible factor-1 pathway. Mol. Cancer 13 12 10.1186/1476-4598-13-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A., Pabon A., Galiano S., Burguete A., Perez-Silanes S., Deharo E., et al. (2014). Synthesis, biological evaluation and structure-activity relationships of new quinoxaline derivatives as anti-Plasmodium falciparum agents. Molecules 19 2166–2180. 10.3390/molecules19022166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazer E. A., Chappel L. R. (1982). Pyridoquinoxaline N-oxides. 1. A new class of antitrichomonal agents. J. Med. Chem. 25 766–769. 10.1021/jm00349a021 [DOI] [PubMed] [Google Scholar]
- Guo W., Hao H., Dai M., Wang Y., Huang L., Peng D., et al. (2012). Development of quinoxaline 1, 4-dioxides resistance in Escherichia coli and molecular change under resistance selection. PLoS ONE 7:e43322 10.1371/journal.pone.0043322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen L. H., Jensen L. B., Sorensen H. I., Sorensen S. J. (2007). Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria. J. Antimicrob. Chemother. 60 145–147. 10.1093/jac/dkm167 [DOI] [PubMed] [Google Scholar]
- Hansen L. H., Johannesen E., Burmolle M., Sorensen A. H., Sorensen S. J. (2004). Plasmid-encoded multidrug efflux pump conferring resistance to olaquindox in Escherichia coli. Antimicrob. Agents Chemother. 48 3332–3337. 10.1128/AAC.48.9.3332-3337.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen L. H., Sorensen S. J., Jorgensen H. S., Jensen L. B. (2005). The prevalence of the OqxAB multidrug efflux pump amongst olaquindox-resistant Escherichia coli in pigs. Microbiol. Drug Resist. 11 378–382. 10.1089/mdr.2005.11.378 [DOI] [PubMed] [Google Scholar]
- Haraldsen J. D., Liu G., Botting C. H., Walton J. G., Storm J., Phalen T. J., et al. (2009). Identification of conoidin a as a covalent inhibitor of peroxiredoxin II. Org. Biomol. Chem. 7 3040–3048. 10.1039/b901735f [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q., Fang G., Wang Y., Wei Z., Wang D., Zhou S., et al. (2006). Experimental evaluation of cyadox phototoxicity to Balb/c mouse skin. Photodermatol. Photoimmunol. Photomed. 22 100–104. 10.1111/j.1600-0781.2006.00199.x [DOI] [PubMed] [Google Scholar]
- Hellauer K., Lesage G., Sdicu A. M., Turcotte B. (2005). Large-scale analysis of genes that alter sensitivity to the anticancer drug tirapazamine in Saccharomyces cerevisiae. Mol. Pharmacol. 68 1365–1375. 10.1124/mol.105.012963 [DOI] [PubMed] [Google Scholar]
- Hicks K. O., Siim B. G., Jaiswal J. K., Pruijn F. B., Fraser A. M., Patel R., et al. (2010). Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin. Cancer Res. 16 4946–4957. 10.1158/1078-0432.CCR-10-1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann B., Steinhilber D. (2013). 5-Lipoxygenase inhibitors: a review of recent patents (2010-2012). Expert Opin. Ther. Pat. 23 895–909. 10.1517/13543776.2013.791678 [DOI] [PubMed] [Google Scholar]
- Huang X. J., Ihsan A., Wang X., Dai M. H., Wang Y. L., Su S. J., et al. (2009). Long-term dose-dependent response of Mequindox on aldosterone, corticosterone and five steroidogenic enzyme mRNAs in the adrenal of male rats. Toxicol. Lett. 191 167–173. 10.1016/j.toxlet.2009.08.021 [DOI] [PubMed] [Google Scholar]
- Huang X. J., Wang X., Ihsan A., Liu Q., Xue X. J., Su S. J., et al. (2010a). Interactions of NADPH oxidase, renin-angiotensin-aldosterone system and reactive oxygen species in mequindox-mediated aldosterone secretion in Wistar rats. Toxicol. Lett. 198 112–118. 10.1016/j.toxlet.2010.05.013 [DOI] [PubMed] [Google Scholar]
- Huang X. J., Zhang H. H., Wang X., Huang L. L., Zhang L. Y., Yan C. X., et al. (2010b). ROS mediated cytotoxicity of porcine adrenocortical cells induced by QdNOs derivatives in vitro. Chem. Biol. Interact. 185 227–234. 10.1016/j.cbi.2010.02.030 [DOI] [PubMed] [Google Scholar]
- Hunter F. W., Hsu H. L., Su J., Pullen S. M., Wilson W. R., Wang J. (2014a). Dual targeting of hypoxia and homologous recombination repair dysfunction in triple-negative breast cancer. Mol. Cancer Ther. 13 2501–2514. 10.1158/1535-7163.MCT-14-0476 [DOI] [PubMed] [Google Scholar]
- Hunter F. W., Jaiswal J. K., Hurley D. G., Liyanage H. D., McManaway S. P., Gu Y., et al. (2014b). The flavoprotein FOXRED2 reductively activates nitro-chloromethylbenzindolines and other hypoxia-targeting prodrugs. Biochem. Pharmacol. 89 224–235. 10.1016/j.bcp.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Hunter F. W., Young R. J., Shalev Z., Vellanki R. N., Wang J., Gu Y., et al. (2015). Identification of P450 oxidoreductase as a major determinant of sensitivity to hypoxia-activated prodrugs. Cancer Res. 75 4211–4223. 10.1158/0008-5472.CAN-15-1107 [DOI] [PubMed] [Google Scholar]
- Ihsan A., Wang X., Huang X. J., Liu Y., Liu Q., Zhou W., et al. (2010). Acute and subchronic toxicological evaluation of Mequindox in Wistar rats. Regul. Toxicol. Pharmacol. 57 307–314. 10.1016/j.yrtph.2010.03.011 [DOI] [PubMed] [Google Scholar]
- Ihsan A., Wang X., Liu Z., Wang Y., Huang X., Liu Y., et al. (2011). Long-term mequindox treatment induced endocrine and reproductive toxicity via oxidative stress in male Wistar rats. Toxicol. Appl. Pharmacol. 252 281–288. 10.1016/j.taap.2011.02.020 [DOI] [PubMed] [Google Scholar]
- Ihsan A., Wang X., Tu H. G., Zhang W., Dai M. H., Peng D. P., et al. (2013a). Genotoxicity evaluation of Mequindox in different short-term tests. Food Chem. Toxicol. 51 330–336. 10.1016/j.fct.2012.10.003 [DOI] [PubMed] [Google Scholar]
- Ihsan A., Wang X., Zhang W., Tu H., Wang Y., Huang L., et al. (2013b). Genotoxicity of quinocetone, cyadox and olaquindox in vitro and in vivo. Food Chem. Toxicol. 59 207–214. 10.1016/j.fct.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Ismail M. M., Amin K. M., Noaman E., Soliman D. H., Ammar Y. A. (2010). New quinoxaline 1, 4-di-N-oxides: anticancer and hypoxia-selective therapeutic agents. Eur. J. Med. Chem. 45 2733–2738. 10.1016/j.ejmech.2010.02.052 [DOI] [PubMed] [Google Scholar]
- Jampilek J. (2014). Recent advances in design of potential quinoxaline anti-infectives. Curr. Med. Chem. 21 4347–4373. 10.2174/0929867321666141011194825 [DOI] [PubMed] [Google Scholar]
- Jaso A., Zarranz B., Aldana I., Monge A. (2003). Synthesis of new 2-acetyl and 2-benzoyl quinoxaline 1,4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents. Eur. J. Med. Chem. 38 791–800. 10.1016/S0223-5234(03)00137-5 [DOI] [PubMed] [Google Scholar]
- Jaso A., Zarranz B., Aldana I., Monge A. (2005). Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 48 2019–2025. 10.1021/jm049952w [DOI] [PubMed] [Google Scholar]
- Jin X., Chen Q., Tang S. S., Zou J. J., Chen K. P., Zhang T., et al. (2009). Investigation of quinocetone-induced genotoxicity in HepG2 cells using the comet assay, cytokinesis-block micronucleus test and RAPD analysis. Toxicol. In Vitro 23 1209–1214. 10.1016/j.tiv.2009.07.038 [DOI] [PubMed] [Google Scholar]
- Kotandeniya D., Ganley B., Gates K. S. (2002). Oxidative DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). Bioorg. Med. Chem. Lett. 12 2325–2329. 10.1016/S0960-894X(02)00468-7 [DOI] [PubMed] [Google Scholar]
- Laderoute K. R., Rauth A. M. (1986). Identification of two major reduction products of the hypoxic cell toxin 3-amino-1,2,4-benzotriazine-1,4-dioxide. Biochem. Pharmacol. 35 3417–3420. 10.1016/0006-2952(86)90448-X [DOI] [PubMed] [Google Scholar]
- Laderoute K., Wardman P., Rauth A. M. (1988). Molecular mechanisms for the hypoxia-dependent activation of 3-amino-1,2,4-benzotriazine-1,4-dioxide (SR 4233). Biochem. Pharmacol. 37 1487–1495. 10.1016/0006-2952(88)90010-X [DOI] [PubMed] [Google Scholar]
- Lawn S. D., Zumla A. I. (2011). Tuberculosis. Lancet 378 57–72. 10.1016/S0140-6736(10)62173-3 [DOI] [PubMed] [Google Scholar]
- Liu J., Ouyang M., Jiang J., Mu P., Wu J., Yang Q., et al. (2012). Mequindox induced cellular DNA damage via generation of reactive oxygen species. Mutat. Res. 741 70–75. 10.1016/j.mrgentox.2011.10.012 [DOI] [PubMed] [Google Scholar]
- Lloyd R. V., Duling D. R., Rumyantseva G. V., Mason R. P., Bridson P. K. (1991). Microsomal reduction of 3-amino-1,2,4-benzotriazine 1,4-dioxide to a free radical. Mol. Pharmacol. 40 440–445. [PubMed] [Google Scholar]
- Maluf F. C., Leiser A. L., Aghajanian C., Sabbatini P., Pezzulli S., Chi D. S., et al. (2006). Phase II study of tirapazamine plus cisplatin in patients with advanced or recurrent cervical cancer. Int. J. Gynecol. Cancer 16 1165–1171. 10.1111/j.1525-1438.2006.00454.x [DOI] [PubMed] [Google Scholar]
- Marin A., Moreira Lima L., Solano B., Vicente E., Perez Silanes S., Maurel S., et al. (2008). Antiplasmodial structure-activity relationship of 3-trifluoromethyl-2-arylcarbonylquinoxaline 1,4-di-N-oxide derivatives. Exp. Parasitol. 118 25–31. 10.1016/j.exppara.2007.05.009 [DOI] [PubMed] [Google Scholar]
- McIlwain H. J. (1943). Bacterial inhibition by metabolite analogues. Part V. Reactions and antibacterial properties of p-diazine di-n-oxides. J. Chem. Soc. 1943 322–325. 10.1039/jr9430000322 [DOI] [Google Scholar]
- Miller V. A., Rigas J. R., Tong W. P., Reid J. R., Pisters K. M., Grant S. C., et al. (1997). Phase II trial of chloroquinoxaline sulfonamide (CQS) in patients with stage III and IV non-small-cell lung cancer. Cancer Chemother. Pharmacol. 40 415–418. 10.1007/s002800050679 [DOI] [PubMed] [Google Scholar]
- Ministry of Agriculture of P. R. China (2003). Notice No. 295. Beijing: Ministry of Agriculture. [Google Scholar]
- Miyake K., Nishioka M., Imura S., Batmunkh E., Uto Y., Nagasawa H., et al. (2012). The novel hypoxic cytotoxin, TX-2098 has antitumor effect in pancreatic cancer; possible mechanism through inhibiting VEGF and hypoxia inducible factor-1alpha targeted gene expression. Exp. Cell Res. 318 1554–1563. 10.1016/j.yexcr.2012.03.013 [DOI] [PubMed] [Google Scholar]
- Monge A., Martinez-Crespo F. J., Lopez de Cerain A., Palop J. A., Narro S., Senador V., et al. (1995a). Hypoxia-selective agents derived from 2-quinoxalinecarbonitrile 1,4-di-N-oxides. 2. J. Med. Chem. 38 4488–4494. 10.1021/jm00010a023 [DOI] [PubMed] [Google Scholar]
- Monge A., Palop J. A., Lopez de Cerain A., Senador V., Martinez-Crespo F. J., Sainz Y., et al. (1995b). Hypoxia-selective agents derived from quinoxaline 1,4-di-N-oxides. J. Med. Chem. 38 1786–1792. 10.1021/jm00010a023 [DOI] [PubMed] [Google Scholar]
- Montoya M. E., Sainz Y., Ortega M. A., Lopez De Cerain A., Monge A. (1998). Synthesis and antituberculosis activity of some new 2-quinoxalinecarbonitriles. Farmaco 53 570–573. 10.1016/S0014-827X(98)00067-6 [DOI] [PubMed] [Google Scholar]
- Murthy Y. L. N., Mani P., Govindh B., Diwakar B. S., Karthikeyan N., Rao T. R., et al. (2011). Synthesis and characterization of 2,3-diphenyl quinoxaline 1,4-di-N-oxide derivatives and study of their antimicrobial activities. Res. J. Pharm. Biol. Chem. Sci. 2 553–560. [Google Scholar]
- Nagasawa H., Mikamo N., Nakajima Y., Matsumoto H., Uto Y., Hori H. (2003). Antiangiogenic hypoxic cytotoxin TX-402 inhibits hypoxia-inducible factor 1 signaling pathway. Anticancer Res. 23 4427–4434. [PubMed] [Google Scholar]
- Nguyen J. B., Pool C. D., Wong C. Y., Treger R. S., Williams D. L., Cappello M., et al. (2013). Peroxiredoxin-1 from the human hookworm Ancylostoma ceylanicum forms a stable oxidized decamer and is covalently inhibited by conoidin A. Chem. Biol. 20 991–1001. 10.1016/j.chembiol.2013.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noblia P., Vieites M., Torre M. H., Costa-Filho A. J., Cerecetto H., Gonzalez M., et al. (2006). Novel vanadyl complexes with quinoxaline N(1),N(4)-dioxide derivatives as potent in vitro insulin-mimetic compounds. J. Inorg. Biochem. 100 281–287. 10.1016/j.jinorgbio.2005.11.012 [DOI] [PubMed] [Google Scholar]
- Nunoshiba T., Nishioka H. (1989). Genotoxicity of quinoxaline 1,4-dioxide derivatives in Escherichia coli and Salmonella typhimurium. Mutat. Res. 217 203–209. 10.1016/0921-8777(89)90072-4 [DOI] [PubMed] [Google Scholar]
- Olive P. L. (1995). Detection of hypoxia by measurement of DNA damage in individual cells from spheroids and murine tumours exposed to bioreductive drugs. II. RSU 1069. Br. J. Cancer 71 537–542. 10.1038/bjc.1995.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M. A., Montoya M. E., Jaso A., Zarranz B., Tirapu I., Aldana I., et al. (2001). Antimycobacterial activity of new quinoxaline-2-carbonitrile and quinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives. Pharmazie 56 205–207. [PubMed] [Google Scholar]
- Ortega M. A., Morancho M. J., Martinez-Crespo F. J., Sainz Y., Montoya M. E., Lopez de Cerain A., et al. (2000). New quinoxalinecarbonitrile 1,4-di-N-oxide derivatives as hypoxic-cytotoxic agents. Eur. J. Med. Chem. 35 21–30. 10.1016/S0223-5234(00)00112-4 [DOI] [PubMed] [Google Scholar]
- Ortega M. A., Sainz Y., Montoya M. E., Jaso A., Zarranz B., Aldana I., et al. (2002). Anti-Mycobacterium tuberculosis agents derived from quinoxaline-2-carbonitrile and quinoxaline-2-carbonitrile 1,4-di-N-oxide. Arzneimittelforschung 52 113–119. [DOI] [PubMed] [Google Scholar]
- Ortega M. A., Sainz Y., Montoya M. E., Lopez De Cerain A., Monge A. (1999). Synthesis and antituberculosis activity of new 2-quinoxalinecarbonitrile 1,4-di-N-oxides. Pharmazie 54 24–25. [PubMed] [Google Scholar]
- Patterson A. V., Barham H. M., Chinje E. C., Adams G. E., Harris A. L., Stratford I. J. (1995). Importance of P450 reductase activity in determining sensitivity of breast tumour cells to the bioreductive drug, tirapazamine (SR 4233). Br. J. Cancer 72 1144–1150. 10.1038/bjc.1995.478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson A. V., Robertson N., Houlbrook S., Stephens M. A., Adams G. E., Harris A. L., et al. (1994). The role of DT-diaphorase in determining the sensitivity of human tumor cells to tirapazamine (SR 4233). Int. J. Radiat. Oncol. Biol. Phys. 29 369–372. 10.1016/0360-3016(94)90291-7 [DOI] [PubMed] [Google Scholar]
- Patterson A. V., Saunders M. P., Chinje E. C., Talbot D. C., Harris A. L., Strafford I. J. (1997). Overexpression of human NADPH:cytochrome c (P450) reductase confers enhanced sensitivity to both tirapazamine (SR 4233) and RSU 1069. Br. J. Cancer 76 1338–1347. 10.1038/bjc.1997.558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson L. H., Taiwo F. A. (2000). Electron paramagnetic resonance spectrometry evidence for bioreduction of tirapazamine to oxidising free radicals under anaerobic conditions. Biochem. Pharmacol. 60 1933–1935. 10.1016/S0006-2952(00)00487-1 [DOI] [PubMed] [Google Scholar]
- Peters K. B., Brown J. M. (2002). Tirapazamine: a hypoxia-activated topoisomerase II poison. Cancer Res. 62 5248–5253. [PubMed] [Google Scholar]
- Peters K. B., Wang H., Brown J. M., Iliakis G. (2001). Inhibition of DNA replication by tirapazamine. Cancer Res. 61 5425–5431. [PubMed] [Google Scholar]
- Pommier Y., Leo E., Zhang H. L., Marchand C. (2010). DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17 421–433. 10.1016/j.chembiol.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones J. L., Thapar U., Yu K., Fang Q., Sobol R. W., Demple B. (2015). Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase beta in vivo. Proc. Natl. Acad. Sci. U.S.A. 112 8602–8607. 10.1073/pnas.1501101112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwan A. A., Abdel-Mageed W. M. (2014). In silico studies of quinoxaline-2-carboxamide 1,4-di-n-oxide derivatives as antimycobacterial agents. Molecules 19 2247–2260. 10.3390/molecules19022247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainier R. H., Chalquest R. R., Babcock W. E., Thrasher G. W. (1973a). Efficacy of carbadox in prevention of field outbreaks of swine dysentery. Vet. Med. Small Anim. Clin. 68 171–175. [PubMed] [Google Scholar]
- Rainier R. H., Chalquest R. R., Babcock W. E., Thrasher G. W. (1973b). Evaluation of carbadox for prophylaxis and treatment of induced swine dysentery. J. Am. Vet. Med. Assoc. 163 457–461. [PubMed] [Google Scholar]
- Refaat H. M., Moneer A. A., Khalil O. M. (2004). Synthesis and antimicrobial activity of certain novel quinoxalines. Arch. Pharm. Res. 27 1093–1098. 10.1007/BF02975110 [DOI] [PubMed] [Google Scholar]
- Sainz Y., Montoya M. E., Martinez-Crespo F. J., Ortega M. A., Lopez de Cerain A., Monge A. (1999). New quinoxaline 1,4-di-N-oxides for treatment of tuberculosis. Arzneimittelforschung 49 55–59. [DOI] [PubMed] [Google Scholar]
- Shah Z., Mahbuba R., Turcotte B. (2013). The anticancer drug tirapazamine has antimicrobial activity against Escherichia coli, Staphylococcus aureus and Clostridium difficile. FEMS Microbiol. Lett. 347 61–69. 10.1111/1574-6968.12223 [DOI] [PubMed] [Google Scholar]
- Shinde S. S., Hay M. P., Patterson A. V., Denny W. A., Anderson R. F. (2009). Spin trapping of radicals other than the *OH radical upon reduction of the anticancer agent tirapazamine by cytochrome P450 reductase. J. Am. Chem. Soc. 131 14220–14221. 10.1021/ja906860a [DOI] [PubMed] [Google Scholar]
- Shinde S. S., Maroz A., Hay M. P., Patterson A. V., Denny W. A., Anderson R. F. (2010). Characterization of radicals formed following enzymatic reduction of 3-substituted analogues of the hypoxia-selective cytotoxin 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J. Am. Chem. Soc. 132 2591–2599. 10.1021/ja908689f [DOI] [PubMed] [Google Scholar]
- Siim B. G., van Zijl P. L., Brown J. M. (1996). Tirapazamine-induced DNA damage measured using the comet assay correlates with cytotoxicity towards hypoxic tumour cells in vitro. Br. J. Cancer 73 952–960. 10.1038/bjc.1996.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D. C. P., Hashim S. R., Singhal R. G. (2011). Synthesis and antimicrobial activity of some new thioether derivatives of quinoxaline. J. Chem. 8 635–642. 10.1155/2011/482831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D. P., Deivedi S. K., Hashim S. R., Singhal R. G. (2010). Synthesis and antimicrobial activity of some new quinoxaline derivatives. Pharmaceuticals 3 2416–2425. 10.3390/ph3082416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solano B., Junnotula V., Marin A., Villar R., Burguete A., Vicente E., et al. (2007). Synthesis and biological evaluation of new 2-arylcarbonyl-3-trifluoromethylquinoxaline 1,4-di-N-oxide derivatives and their reduced analogues. J. Med. Chem. 50 5485–5492. 10.1021/jm0703993 [DOI] [PubMed] [Google Scholar]
- Standing Committee for Animal Nutrition [SCAN] (ed.) (1998). Short Report of the Meeting of the Standing Committee for Animal Nutrition, SCAN. Brussels: European Commission, Health and Food Safety. [Google Scholar]
- Sung J. S., Demple B. (2006). Analysis of base excision DNA repair of the oxidative lesion 2-deoxyribonolactone and the formation of DNA-protein cross-links. Methods Enzymol. 408 48–64. 10.1016/S0076-6879(06)08004-9 [DOI] [PubMed] [Google Scholar]
- Suter W., Rosselet A., Knusel F. (1978). Mode of action of quindoxin and substituted quinoxaline-di-N-oxides on Escherichia coli. Antimicrob. Agents Chemother. 13 770–783. 10.1128/AAC.13.5.770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarallo M. B., Urquiola C., Monge A., Costa B. P., Ribeiro R. R., Costa-Filho A. J., et al. (2010). Design of novel iron compounds as potential therapeutic agents against tuberculosis. J. Inorg. Biochem. 104 1164–1170. 10.1016/j.jinorgbio.2010.07.005 [DOI] [PubMed] [Google Scholar]
- Tarallo M. B., Urquiola C., Monge A., Pavan F. R., Leite C. Q., Torre M. H., et al. (2008). Novel iron complexes with quinoxaline N-1, N-4-dioxide derivatives: synthesis, characterization and antimycobacterial activity. Met. Ions Biol. Med. 10 867–872. [Google Scholar]
- Torre M. H., Gambino D., Araujo J., Cerecetto H., Gonzalez M., Lavaggi M. L., et al. (2005). Novel Cu(II) quinoxaline N1,N,4-dioxide complexes as selective hypoxic cytotoxins. Eur. J. Med. Chem. 40 473–480. 10.1016/j.ejmech.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Torres E., Moreno-Viguri E., Galiano S., Devarapally G., Crawford P. W., Azqueta A., et al. (2013). Novel quinoxaline 1,4-di-N-oxide derivatives as new potential antichagasic agents. Eur. J. Med. Chem. 66 324–334. 10.1016/j.ejmech.2013.04.065 [DOI] [PubMed] [Google Scholar]
- Troutt H. F., Hooper B. E., Harrington R. (1974). Effect of carbadox in experimentally induced salmonellosis of swine. J. Am. Vet. Med. Assoc. 164 402–404. [PubMed] [Google Scholar]
- Urquiola C., Gambino D., Cabrera M., Lavaggi M. L., Cerecetto H., Gonzalez M., et al. (2008). New copper-based complexes with quinoxaline N,1,N,4-dioxide derivatives, potential antitumoral agents. J. Inorg. Biochem. 102 119–126. 10.1016/j.jinorgbio.2007.07.028 [DOI] [PubMed] [Google Scholar]
- Urquiola C., Vieites M., Aguirre G., Marin A., Solano B., Arrambide G., et al. (2006). Improving anti-trypanosomal activity of 3-aminoquinoxaline-2-carbonitrile N1,N,4-dioxide derivatives by complexation with vanadium. Bioorg. Med. Chem. 14 5503–5509. 10.1016/j.bmc.2006.04.041 [DOI] [PubMed] [Google Scholar]
- Urquiola C., Vieites M., Torre M. H., Cabrera M., Lavaggi M. L., Cerecetto H., et al. (2009). Cytotoxic palladium complexes of bioreductive quinoxaline N,1,N,4-dioxide prodrugs. Bioorg. Med. Chem. 17 1623–1629. 10.1016/j.bmc.2008.12.064 [DOI] [PubMed] [Google Scholar]
- Vicente E., Charnaud S., Bongard E., Villar R., Burguete A., Solano B., et al. (2008). Synthesis and antiplasmodial activity of 3-furyl and 3-thienylquinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives. Molecules 13 69–77. 10.3390/molecules13010069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente E., Villar R., Perez-Silanes S., Aldana I., Goldman R. C., Mong A. (2011). Quinoxaline 1,4-di-N-oxide and the potential for treating tuberculosis. Infect. Disord. Drug Targets 11 196–204. 10.2174/187152611795589735 [DOI] [PubMed] [Google Scholar]
- Vieites M., Noblia P., Torre M. H., Cerecetto H., Laura Lavaggi M., Costa-Filho A. J., et al. (2006). Selective hypoxia-cytotoxins based on vanadyl complexes with 3-aminoquinoxaline-2-carbonitrile-N,1,N,4-dioxide derivatives. J. Inorg. Biochem. 100 1358–1367. 10.1016/j.jinorgbio.2006.03.012 [DOI] [PubMed] [Google Scholar]
- Villalobos-Rocha J. C., Sanchez-Torres L., Nogueda-Torres B., Segura-Cabrera A., Garcia-Perez C. A., Bocanegra-Garcia V., et al. (2014). Anti-Trypanosoma cruzi and anti-leishmanial activity by quinoxaline-7-carboxylate 1,4-di-N-oxide derivatives. Parasitol. Res. 113 2027–2035. 10.1007/s00436-014-3850-8 [DOI] [PubMed] [Google Scholar]
- Voogd C. E., van der Stel J. J., Jacobs J. J. (1980). The mutagenic action of quindoxin, carbadox, olaquindox and some other N-oxides on bacteria and yeast. Mutat. Res. 78 233–242. 10.1016/0165-1218(80)90104-4 [DOI] [PubMed] [Google Scholar]
- Wang J., Biedermann K. A., Brown J. M. (1992). Repair of DNA and chromosome breaks in cells exposed to SR 4233 under hypoxia or to ionizing radiation. Cancer Res. 52 4473–4477. [PubMed] [Google Scholar]
- Wang J., Biedermann K. A., Wolf C. R., Brown J. M. (1993). Metabolism of the bioreductive cytotoxin SR 4233 by tumour cells: enzymatic studies. Br. J. Cancer 67 321–325. 10.1038/bjc.1993.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Fang G. J., Wang Y. L., Ihsan A., Huang L. L., Zhou W., et al. (2011a). Two generation reproduction and teratogenicity studies of feeding cyadox in Wistar rats. Food Chem. Toxicol. 49 1068–1079. 10.1016/j.fct.2011.01.014 [DOI] [PubMed] [Google Scholar]
- Wang X., He Q. H., Wang Y. L., Ihsan A., Huang L. L., Zhou W., et al. (2011b). A chronic toxicity study of cyadox in Wistar rats. Regul. Toxicol. Pharmacol. 59 324–333. 10.1016/j.yrtph.2010.11.004 [DOI] [PubMed] [Google Scholar]
- Wang X., Huang X. J., Ihsan A., Liu Z. Y., Huang L. L., Zhang H. H., et al. (2011c). Metabolites and JAK/STAT pathway were involved in the liver and spleen damage in male Wistar rats fed with mequindox. Toxicology 280 126–134. 10.1016/j.tox.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Wang X., Wan D., Ihsan A., Liu Q., Cheng G., Li J., et al. (2015a). Mechanism of adrenocortical toxicity induced by quinocetone and its bidesoxy-quinocetone metabolite in porcine adrenocortical cells in vitro. Food Chem. Toxicol. 84 115–124. 10.1016/j.fct.2015.08.016 [DOI] [PubMed] [Google Scholar]
- Wang X., Zhang H., Huang L., Pan Y., Li J., Chen D., et al. (2015b). Deoxidation rates play a critical role in DNA damage mediated by important synthetic drugs, quinoxaline 1,4-dioxides. Chem. Res. Toxicol. 28 470–481. 10.1021/tx5004326 [DOI] [PubMed] [Google Scholar]
- Wang X., Zhang W., Wang Y. L., Ihsan A., Dai M. H., Huang L. L., et al. (2012). Two generation reproduction and teratogenicity studies of feeding quinocetone fed to Wistar rats. Food Chem. Toxicol. 50 1600–1609. 10.1016/j.fct.2011.12.039 [DOI] [PubMed] [Google Scholar]
- Wang X., Zhang W., Wang Y., Peng D., Ihsan A., Huang X., et al. (2010). Acute and sub-chronic oral toxicological evaluations of quinocetone in Wistar rats. Regul. Toxicol. Pharmacol. 58 421–427. 10.1016/j.yrtph.2010.08.008 [DOI] [PubMed] [Google Scholar]
- Weng Q., Zhang J., Cao J., Xia Q., Wang D., Hu Y., et al. (2011). Q39 a quinoxaline 1,4-Di-N-oxide derivative, inhibits hypoxia-inducible factor-1alpha expression and the Akt/mTOR/4E-BP1 signaling pathway in human hepatoma cells. Invest. New Drugs 29 1177–1187. 10.1007/s10637-010-9462-y [DOI] [PubMed] [Google Scholar]
- WHO (1991). Toxicological evaluation of certain veterinary drug residues in food Carbadox. WHO Food Addit. Ser. 27 694. [Google Scholar]
- WHO (1994). Toxicological evaluation of certain veterinary drug residues in food. WHO Food Addit. Ser. 33 804–817. [Google Scholar]
- WHO (2011). “Global tuberculosis control 2011” in The Sixteenth Global Report on Tuberculosis (Geneva: World Health Organization; ). [Google Scholar]
- Yadav P., Marshall A. J., Reynisson J., Denny W. A., Hay M. P., Anderson R. F. (2014). Fragmentation of the quinoxaline N-oxide bond to the OH radical upon one-electron bioreduction. Chem. Commun. (Cambridge, Engl.) 50 13729–13731. 10.1039/c4cc05657d [DOI] [PubMed] [Google Scholar]
- Yen J. T., Pond W. G. (1993). Effects of carbadox, copper, or Yucca schidigera extract on growth performance and visceral weight of young pigs. J. Anim. Sci. 71 2140–2146. [DOI] [PubMed] [Google Scholar]
- Yin J., Glaser R., Gates K. S. (2012). On the reaction mechanism of tirapazamine reduction chemistry: unimolecular N-OH homolysis, stepwise dehydration, or triazene ring-opening. Chem. Res. Toxicol. 25 634–645. 10.1021/tx200546u [DOI] [PubMed] [Google Scholar]
- Zanetti S., Sechi L. A., Molicotti P., Cannas S., Bua A., Deriu A., et al. (2005). In vitro activity of new quinoxalin 1,4-dioxide derivatives against strains of Mycobacterium tuberculosis and other mycobacteria. Int. J. Antimicrob. Agents 25 179–181. 10.1016/j.ijantimicag.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Zarranz B., Jaso A., Aldana I., Monge A. (2003). Synthesis and antimycobacterial activity of new quinoxaline-2-carboxamide 1,4-di-N-oxide derivatives. Bioorg. Med. Chem. 11 2149–2156. 10.1016/S0968-0896(03)00119-6 [DOI] [PubMed] [Google Scholar]
- Zarranz B., Jaso A., Aldana I., Monge A. (2004). Synthesis and anticancer activity evaluation of new 2-alkylcarbonyl and 2-benzoyl-3-trifluoromethyl-quinoxaline 1,4-di-N-oxide derivatives. Bioorg. Med. Chem. 12 3711–3721. 10.1016/j.bmc.2004.04.013 [DOI] [PubMed] [Google Scholar]
- Zarranz B., Jaso A., Aldana I., Monge A., Maurel S., Deharo E., et al. (2005). Synthesis and antimalarial activity of new 3-arylquinoxaline-2-carbonitrile derivatives. Arzneimittelforschung 55 754–761. [DOI] [PubMed] [Google Scholar]
- Zeman E. M., Brown J. M., Lemmon M. J., Hirst V. K., Lee W. W. (1986). SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int. J. Radiat. Oncol. Biol. Phys. 12 1239–1242. 10.1016/0360-3016(86)90267-1 [DOI] [PubMed] [Google Scholar]
- Zhang C., Wang C., Tang S., Sun Y., Zhao D., Zhang S., et al. (2013). TNFR1/TNF-alpha and mitochondria interrelated signaling pathway mediates quinocetone-induced apoptosis in HepG2 cells. Food Chem. Toxicol. 62 825–838. 10.1016/j.fct.2013.10.022 [DOI] [PubMed] [Google Scholar]
- Zhang H. (2012). The Study of Antibacterial Mechanism of Cyadox on Escherichia coli. Wuhan: Huazhong Agricultural University. [Google Scholar]
- Zhang J., Cao J., Weng Q., Wu R., Yan Y., Jing H., et al. (2010). Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) by tirapazamine is dependent on eIF2alpha phosphorylation rather than the mTORC1/4E-BP1 pathway. PLoS ONE 5:e13910 10.1371/journal.pone.0013910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Zheng W., Zheng H., Wang C., Wang M., Li T., et al. (2014). Identification of oxidative stress and responsive genes of HepG2 cells exposed to quinocetone, and compared with its metabolites. Cell Biol. Toxicol. 30 313–329. 10.1007/s10565-014-9287-0 [DOI] [PubMed] [Google Scholar]
- Zhao D., Wang C., Tang S., Zhang C., Zhang S., Zhou Y., et al. (2015). Reactive oxygen species-dependent JNK downregulated olaquindox-induced autophagy in HepG2 cells. J. Appl. Toxicol. 35 709–716. 10.1002/jat.3022 [DOI] [PubMed] [Google Scholar]
- Zou J., Chen Q., Jin X., Tang S., Chen K., Zhang T., et al. (2011). Olaquindox induces apoptosis through the mitochondrial pathway in HepG2 cells. Toxicology 285 104–113. 10.1016/j.tox.2011.04.010 [DOI] [PubMed] [Google Scholar]
- Zou J., Chen Q., Tang S., Jin X., Chen K., Zhang T., et al. (2009). Olaquindox-induced genotoxicity and oxidative DNA damage in human hepatoma G2 (HepG2) cells. Mutat. Res. 676 27–33. 10.1016/j.mrgentox.2009.03.001 [DOI] [PubMed] [Google Scholar]