

Abstract
Sickle cell disease causes acute and chronic illness, and median life expectancy is reduced by at least 30 years in all countries, with greater reductions in low-income countries. There is a wide spectrum of severity, with some patients having no symptoms and others suffering frequent, life-changing complications. Much of this variability is unexplained, despite increasingly sophisticated genetic studies. Environmental factors, including climate, air quality, socio-economics, exercise and infection, are likely to be important, as demonstrated by the stark differences in outcomes between patients in Africa and USA/Europe. The effects of weather vary with geography, although most studies show that exposure to cold or wind increases hospital attendance with acute pain. Most of the different air pollutants are closely intercorrelated, and increasing overall levels seem to correlate with increased hospital attendance, although higher concentrations of atmospheric carbon monoxide may offer some benefit for patients with sickle cell disease. Exercise causes some adverse physiological changes, although this may be off-set by improvements in cardiovascular health. Most sickle cell disease patients live in low-income countries and socioeconomic factors are undoubtedly important, but little studied beyond documenting that sickle cell disease is associated with decreases in some measures of social status. Infections cause many of the differences in outcomes seen across the world, but again these effects are relatively poorly understood. All the above factors are likely to account for much of the pathology and variability of sickle cell disease, and large prospective studies are needed to understand these effects better.
Introduction
Sickle cell disease (SCD) is a highly variable condition, with some patients being asymptomatic and others admitted frequently to hospital. Despite its genetic simplicity, a single base change T>A at codon 6 of the β gene, the clinical heterogeneity of the disease has long been recognized.1 The polymerization of deoxygenated hemoglobin S (HbS), formation of irreversibly sickled erythrocytes and vaso-occlusion underlie the pathophysiology of SCD2 and lead to the two hallmarks of the disease: recurrent episodes of acute pain and chronic organ damage.3 The severe pain is ischemic in origin, and these painful episodes are the most common reason for patients seeking medical attention. Hemolysis also plays a central role in the pathophysiology, contributing significantly to anemia, vasculopathy, nitric oxide deficiency and inflammation.4
SCD is endemic in many regions in which malaria is or was prevalent, due to the protective nature of the carrier state. It is estimated that around the world more than 312,000 infants are born with homozygous HbS (sickle cell anaemia, SCA) each year with the vast majority of births occurring in the developing world, and an estimated 230,000 such births annually in Sub-Saharan Africa.5 However, migration has led to an increase in the prevalence and genetic heterogeneity of hemoglobinopathies across the world, including many European countries and other non-endemic regions,6 with SCD accounting for many hospital episodes in some cities such as London and Paris.7 A similar pattern is seen in the USA, where many emergency department attendances and inpatient hospitalizations are related to SCD.8 There has been a steady improvement in the early mortality caused by SCD in high-income countries as a result of early diagnosis by newborn screening and improved clinical care. This is in contrast to low-income countries in which early mortality due to SCD is still strikingly high.9,10
The role of genetic factors in SCD has been extensively investigated but only explains a small amount of the observed phenotypic variability to date. The two best-characterized genetic modifiers are determinants of hemoglobin F (HbF) levels,11 and the co-inheritance of alpha-thalassemia.12 Increasing numbers of genetic studies have been published to try and explain the phenotypic variability, including many genome-wide association studies.13 These have identified various loci of interest, but overall have explained very little of the clinical variability, such as differences in the frequency of acute pain, chronic complications and mortality. Environmental factors are likely to explain much of this variability, but have not been studied in depth, at least in part because of the logistical difficulties in conducting these studies. For example, the importance of climatic factors as precipitants of acute pain has been recognized for many years, particularly an increased frequency of pain in cold and rainy seasons,14,15 although this is not a consistent finding.16,17 Other potentially relevant environmental factors include air quality, housing, socio-economic status, physical activities and infections. A better understanding of the relationship between patients and their environment may allow significant improvements in health, by giving simple advice and facilitating the development of appropriate public health policies; genetic factors are generally less amenable to modification.
SCD can be regarded as both a qualitative and quantitative genetic disorder. While the causative genotype is a key determinant of disease severity, the frequency and severity of complications vary considerably within each genotypic group. The rate of HbS polymerization is dependent on hypoxia, pH, temperature and the hydration of red blood cells, all of which are potentially altered by environmental factors. The clinical variability between identical twins demonstrates the important contribution of environmental factors in phenotypic variability.18–20 Similarly, people with SCD have a much more severe course in most of Africa compared to genetically similar patients in the northern hemisphere, and this is likely to be related to environmental factors, including infections, climate and access to healthcare. Equally, there are marked differences in clinical severity between patients with the Arab-Indian βS haplotype in India and the Middle East, with there being both mildly and severely affected patients;21,22 these differences are likely to be at least partly attributable to environmental factors.
This review summarizes published data on the interactions between SCD and environmental factors. Illumination of such modifying factors may guide future areas for research, leading to a better understanding of the natural history of SCD and improved care of patients. Better understanding of environmental factors is also important for studies of pharmaceutical interventions in SCD, because any therapeutic effect may be lost or exaggerated by confounding climatic effects. For example, a particular trial drug used in summer may show far less effect than the same drug used in winter. In this review, we have not considered the effects of infections, including malaria, as this is a very large area and is the subject of several recent reviews.23–25
Climatic variables
Temperature
Extremes of both hot and cold weather have been found to precipitate acute complications.26 The vast majority of patients with SCD live in tropical climates, in which few studies have been conducted. Increasing numbers are managed in temperate areas in western Europe, particularly in larger cities in France and the UK (Figure 1), and North America.27 Anecdotally, many patients with SCD report that exposure to cold results in acute pain, usually starting within a few hours. This association is mentioned in nearly all textbooks and reviews of SCD,28 and patients are routinely advised to avoid getting cold in all parts of the world. This association is very real for patients, although there is surprisingly little evidence supporting such an effect.
Figure 1.
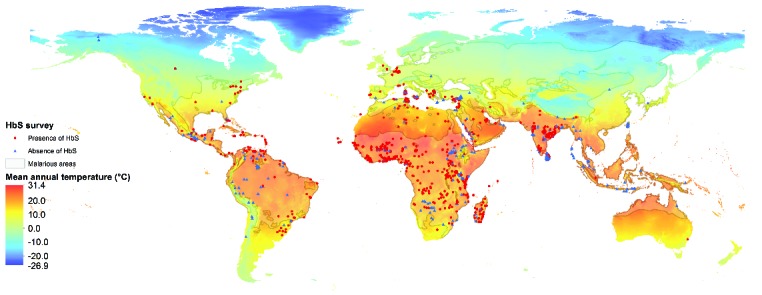
Global map of the presence or absence of the sickle cell mutation in representative population surveys, based on data from Piel et al.20, overlaying the mean annual temperature (in °C) based on data from WorldClim (http://www.worldclim.org/), last accessed on 15 April 2015 and described in detail in Hijmans RJ, et al. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005;25:1965–1978.
The pathophysiology of cold exposure is potentially fairly simple, but not supported by robust experimental data. On cold exposure, peripheral vasoconstriction occurs and the velocity of blood flowing through the vasculature of these areas is reduced. This results in greater deoxygenation of blood in the peripheries, and red cells spending a longer period of time in the deoxygenated part of the circulation. HbS polymerization therefore occurs more readily, and more rigid, sickled red cells are produced which subsequently cause vaso-occlusion, presumably in tissues near to the areas exposed to cold. Countering this adverse effect of cold exposure is the fact that the rate of HbS polymerization decreases at lower temperatures; at 20–30°C, a 1°C increase in temperature halves the delay time before hemoglobin gelation starts to occur.29 Experiments in Jamaica also showed that some patients responded to skin cooling of one arm with marked, reflex vasoconstriction of the other, and that patients showing this response had more episodes of acute pain, suggesting that vascular dysfunction may be part of the explanation for cold exposure causing acute pain.30 Other experimental observations have also suggested that patients with SCD have increased sensitivity to temperature changes.31,32 Patients with SCD exhibit hypersensitivity to thermal stimuli33 and often report cooler weather or exposure to cold as the most important precipitating factor for vaso-occlusion.34,35 Consequent to vaso-constrictive reduction in blood flow, a resulting decline in capillary pressure gradients produces lower shear force across the microvasculature. Studies have suggested that such decreases in pressure gradients and shear flow may play a significant role in the precipitation of acute pain.36–38 Another possibility is that vasoconstriction may be associated with diversion of blood (vascular steal) away from the active bone marrow and cause avascular necrosis and precipitate acute vaso-occlusion.39 The balance between the different effects of cold exposure is complex, and likely to depend on a wide range of factors including the degree of vasoconstriction, hematocrit, and the temperature.
Epidemiological studies have yielded inconsistent results on the effects of temperature on acute pain and other complications in SCD. The link between acute pain and cold weather was first noted in a case report of a patient with SCD by George Graham in the USA in 1924, with the author speculating that cold may ‘light-up’ a focus of infection and thus precipitate pain.40 In 1953, Edington reported that attacks of joint pain were linked to cold weather in ten of 33 patients in the Gold Coast (Ghana), and speculated that cold coincided with the rainy season and increased malarial infections, and that this was the main cause of increased pain.41 Konotey-Ahulu, also drawing on his experience in Ghana, suggested that the peak incidence of pain occurred just before the cold rainy season, when the weather was still hot, and that this was due to the high levels of water vapor in the air causing a small, but critical, reduction in the partial pressure of oxygen.42 A study in New York conducted in the 1970s showed increased admissions to hospital with sickle pain in cold winter months even when episodes with overt infection were excluded, and speculated that this may be due to increased blood viscosity and cold diuresis.14 A 10-year retrospective study in Jamaica, involving 161 hospital admissions, also showed a significant negative correlation between the monthly number of admissions with severe pain in SCD and monthly minimum temperature over a temperature range of 20–25°C.15 Other studies showing a link between acute pain and cold temperatures have been published from Kuwait,43 Virginia, USA44 and Canada.45 A review of data from the Multicenter Study of Hydroxyurea from across the USA suggested that cold seasons were associated with increased pain severity but not frequency, and the same study also found more frequent and severe pain in hot months.44 In France, a recent study showed that both high and low temperatures were associated with higher numbers of emergency attendances, although temperature drop was most significant on multivariate analysis; this study included acute pain and chest disease, in all forms of SCD.46
Other studies have failed to provide evidence that temperature is an important determinant of acute pain or other complications of SCD. Seeler found no effect of season on admission to hospital over a 4-year period in Chicago,16 and a study of 71 patients over 13 months in Georgia, USA also showed no evidence of temperature effects.17 Two separate studies from London, UK showed no link between temperature and hospital admissions with acute pain.47,48
The effects of temperature are complex and seem to vary across the world depending on the range of temperatures, housing, clothing and other environmental factors; for example, the inside environment may negate the effects of temperature changes in countries in which life is largely spent in heated and air-conditioned buildings. Equally, problems could be precipitated by rapid temperature changes, such as leaving a heated house in cold, winter weather. However, in most studies a pattern emerges of increased episodes of acute pain following exposure to cold; this perhaps is more marked as a seasonal effect, which may be related to increased infections, rather than a more acute effect, which may be caused by vasoconstriction leading to increased HbS polymerization.
Wind speed
Wind speed is less seasonal than temperature, with high winds occurring sporadically throughout the year. To detect the effects of wind speed on complications in SCD it is, therefore, necessary to analyze patterns of clinical complications on a daily basis, rather than looking, for example, purely at seasonal effects. A study in London of 1,047 hospital admissions with acute pain of adults and children with SCD over 1,400 days found that high wind speeds (26–57 knots) were associated with significantly more admissions than low wind speed (4–16 knots), both on the day of high wind speed and the following day48 (Figure 2A). This finding has been reproduced in the USA,49 Canada,45 and France.46 There is no established mechanism by which increased wind speed might provoke acute pain or other severe complications requiring hospital admission, although the effects of skin cooling seem the most likely to be relevant.30,31 Wind speed also correlates with temperature, humidity and some air pollutants, which may explain some of the associations.
Figure 2.
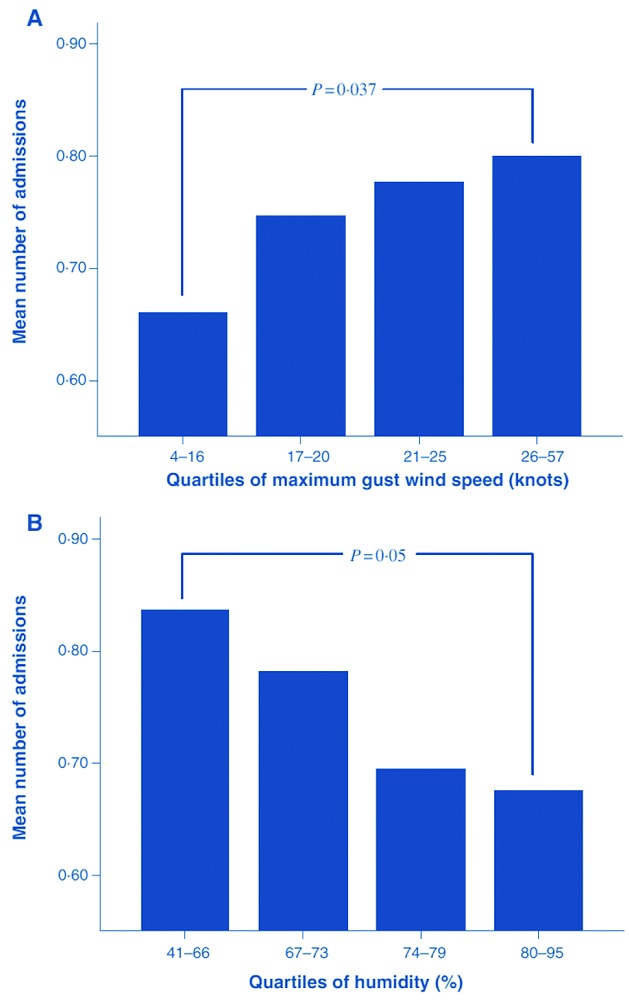
Bar graphs showing the mean number of admissions for the different quartiles of (A) maximum gust wind speed and (B) mean humidity. P values are shown for a comparison of mean values of the first and fourth quartiles (Mann-Whitney U-test) [Reproduced with permission from Jones et al. 200548).
Humidity
Studies examining the effects of humidity have produced mixed results. A study in Canada found higher pain scores in more humid months,45 and acute painful episodes were more common in humid weather in Kuwait.43 The higher incidence of acute pain during the rainy season in Nigeria has also been attributed to the higher humidity causing a lower partial pressure of oxygen.42 Low humidity (41–66%) was associated with increased admissions to hospital in London with acute pain when compared to high humidity (80–95%)48 (Figure 2B), and a French study found that both high and low humidity were associated with increased emergency admissions, with the fewest admissions occurring at around 70% relative humidity.46
Other climatic factors
Rainfall does not seem to influence acute complications in temperate European countries including France46 and the UK,48 although in countries with a tropical rainy season this is reported as a cause of increased pain.15 In London, no association was found between barometric pressure or wind-chill factor, and the strongest predictor of increased hospital attendance with acute pain was a composite factor derived from maximum wind speed divided by humidity.48
There are no studies or case histories reporting the effects of snow on complications in SCD, although anecdotally children are sometimes admitted to hospital after playing in the snow, as might be expected. There are case reports of gangrene and amputation of limbs following the use of ice to treat pain in SCD, including the amputation of two fingers from a 7-month old boy,50 and a below the elbow amputation of a 31-year old man with HbSC disease,51 illustrating the potentially serious effects of near freezing temperatures.
Air quality
Most people with SCD in high-income countries live in capital cities and large urban centers, with variable, high levels of pollutants, as illustrated by the distribution of patients within England and France (Figure 2). The association between daily variations in the level of urban air pollution and adverse health has been well documented in the non-sickle population.52 In 2005 it was estimated that pollution caused 800,000 premature deaths annually,53 and is 13th in the list of causes of mortality in the world.54 Adults inhale about 20 m3 air per day, which in cities will contain significant quantities of pollutant chemicals,55 and may have implications for both acute and chronic complications of SCD. Inhalation of pollutants may have direct toxic effects, and additionally there is some evidence that this may alter gene expression through epigenetic mechanisms such as decreased DNA methylation.56 Analysis of the effects of air quality on health is complicated by the fact that most of the pollutants are closely intercorrelated, and also correlated with climatic factors, making it difficult to establish which are primary effects.
Ozone
Ozone in the troposphere is a secondary pollutant formed by reactions between primary pollutants released from fuel combustion and industrial processes, with peak levels occurring on hot and sunny days. Ozone has been strongly implicated in exacerbation of asthma, most notably by the reduction in acute asthma attacks during the 1996 Atlanta Olympics, when traffic was excluded from the city.57 Asthma and wheezing are both strongly linked to increased acute complications in SCD.58,59 Ozone is, therefore, potentially relevant in SCD, with harmful effects mediated through pulmonary disease. A study in London of more than 1,000 hospital admissions showed that there were more admissions with acute pain in SCD on days with higher ozone levels.55 A study in Brazil found a similar link between high ozone levels and increased emergency hospital attendance in children with SCD.60 However, a large French study of adults and children with all types of SCD demonstrated the opposite effect, of decreased hospital admissions with higher ozone levels.46 There are no studies looking at potential long-term effects of exposure to increased ozone levels. It has even been suggested that ozone might increase the rate and capacity of oxygen absorption in erythrocytes, making it a potential treatment for the prevention or shortening of acute complications in SCD.61
Nitric oxide
Nitric oxide has been strongly implicated in the pathophysiology of SCD. Various studies suggest that increased hemolysis results in functional nitric oxide deficiency, which contributes to vasculopathy62 and might be particularly important in the development of some complications including pulmonary hypertension, priapism, leg ulcers, and possibly cerebrovascular disease.4 Low-dose nitric oxide has been used to treat some complications of SCD including acute chest syndrome, although a placebo-controlled trial showed no evidence of benefit for acute pain.63 Theoretically, exposure to atmospheric nitric oxide might be of therapeutic benefit in SCD, and higher levels of nitric oxide were associated with decreased admissions in the retrospective study in London,55 although this was not assessed in the studies in Brazil and France. Interestingly, a study in south London looking at the effects of air quality on chronic complications suggested that increased nitric oxide exposure was also associated with lower rates of hemolysis, although the same correlation also applied to nitrogen dioxide and dust.64
Carbon monoxide
As for nitric oxide, exposure to low levels of carbon monoxide is potentially therapeutic in SCD, with cytoprotective, vasodilator and anti-inflammatory properties, together with a left shift of the hemoglobin-oxygen dissociation curve.65 Early observations suggested that carbon monoxide prolonged the half-life of red blood cells in SCD.66 In transgenic sickle mice, pegylated carboxyhemoglobin reduced microvascular stasis, mainly by its anti-inflammatory effects mediated by reduced nuclear factor-κB expression;65 based on these studies, clinical trials of pegylayted carboxyhemoglobin clinical trials as a treatment for tissue ischemia, in conditions such as SCD and myocardial ischemia, are currently underway. Higher levels of atmospheric carbon monoxide were also associated with lower numbers of admissions to hospital with acute pain due to SCD in London55 and Paris,46 but increased emergency room attendances in Sao Paulo, Brazil, although this last finding was only for children, and the attendances were for all complications, not just pain.60
Particulate matter
Exposure to particulate matter, both fine (PM2.5) and coarse (PM10), has been strongly linked to a number of health complications in the non-sickle population. It is estimated to be responsible for 3,000,000 deaths per year,67 and is particularly problematic in cities, in which the incidence of SCD is highest in many non-endemic countries (Figure 1). One of the strongest associations is with stroke, with an increased risk following exposure in the short term and possibly longer term.67 The mechanism is thought to be the absorption of fine particles into the blood stream through the lungs, which can then trigger cardiovascular problems. Such a mechanism is particularly relevant in SCD in which vasculopathy plays a central role, and there is increased risk of stroke. Exposure to increased PM10 or PM2.5 was associated with increased daily admissions in the short-term in Paris,46 with PM10 showing an equivalent effect in the Sao Paulo study;60 however, no acute effects of PM10 exposure were seen in London. A second study in London mapping concentrations of PM10 to individuals’ postcodes suggested that long-term exposure to increased particulate material was associated with increased blood velocity in the extracranial carotid artery, hinting that chronic exposure may increase the risk of vasculopathy.64
Other gaseous pollutants
Increasing nitrogen dioxide and sulfur dioxide levels were both associated with increased hospital attendances in the studies in Brazil,60 and France,46 but not England.55 In London, chronic exposure to nitrogen dioxide was nevertheless associated with increased hemolysis.64 However, nitrogen and sulfur dioxide levels correlate closely with each other and most other gaseous pollutants and it is unclear if either has any significant direct effects.
High altitude
There has been concern about the adverse effects of high altitude in SCD for many years, principally because of the potential problems associated with low oxygen partial pressures. Patients with SCD are increasingly exposed to high altitudes on aircraft, which are pressurized to an altitude equivalent of about 2000 m, or when visiting mountainous regions such as the Alps (highest point 4800 m) or the Rocky Mountains (highest point 4400 m). The most frequently reported complication is acute splenic infarction in people with sickle cell trait, occurring both in aircraft and mountainous areas, particularly Denver, Colorado (altitude 1600 m).68 Splenic sequestration has been reported in adults and children with HbSC disease on ascent to high altitude, including a paper describing this complication in four children with HbSC disease who visited mountains at altitudes greater than 2700 m.69 The third complication associated with altitude is the development of acute vaso-occlusive pain in patients with sickle cell anemia. This has been described both in Colorado70 and Saudi Arabia;71 in the latter country the authors found that the rate of acute vaso-occlusive complications in patients with SCD living in the highlands was about double that of lowlanders, associated with a 5% increase in hemoglobin levels. Overall, these data suggest that patients with SCD should be cautious when visiting altitudes higher than 2000 m.
Initial reports suggested that patients with SCD had increased complications associated with air travel. A study from the USA found that patients with sickle cell anemia had a 6.5% risk of developing acute vaso-occlusive complications on commercial airplanes, and recommended that they use oxygen when flying.72 Current guidelines from the International Airtransport Association, Aerospace Medical Association and UK Civil Aviation Authority all recommend that patients with SCD should travel with oxygen on air flights. However, it is now increasingly accepted, without much evidence, that patients SCD rarely suffer complications when flying in commercial airlines, and preflight transfusions or oxygen supplementation are only recommended if a patient has pre-existing complications, such as lung or cerebrovascular disease. Further research is needed in this area.
The home environment
Any effects of climate and air quality are likely to be influenced by, and possibly less significant than, the home environment. In northern countries the effects of cold weather will be modified by the quality of housing, and the presence of central heating and insulation, for example. Conversely, cool accommodation may be important in tropical countries by protecting patients from the effects of extreme heat. Patients and families frequently feel that inadequate housing contributes to acute and chronic complications in SCD. It is plausible that damp, cold houses, with increased exposure to dust and fungal spores would have adverse health effects on SCD, although there is very little evidence in this area.
There is evidence that active and passive smoking are harmful in SCD. The adverse pulmonary effects of environmental tobacco exposure in the general population have been well described.73 Tobacco smoke may have particularly adverse consequences for children with SCD because research in otherwise healthy individuals has demonstrated a relationship between tobacco smoke and inflammation,74 oxidative stress75,76 and endothelial dysfunction.77,78 A study in Jamaica followed 75 adults with SCD for 10 years and found that smoking was significantly associated with increased mortality (hazard ratio 2.7, 95% confidence interval: 1.3 5.5),79 and studies in the USA showed an association between smoking and acute chest syndrome.80,81 Children and adolescents with SCD who were exposed to passive smoking were found to have increased frequencies of acute pain and hospitalization.82
Socio-economic factors
Socio-economic factors are important determinants of health in all people. Numerous studies have shown that increased poverty is associated with worse health.83 This is particularly relevant to SCD which is most prevalent in the poorest countries84 (Figure 3). Additionally, studies suggest that socio-economic status is different in families with SCD compared to matched families in the same country; the Cooperative Study of Sickle Cell Disease in the USA compared 3,538 SCD patients with the US black population and found higher rates of single-parent families and single female heads-of-household, although the percentage of high school graduates and salaries were similar.85
Figure 3.
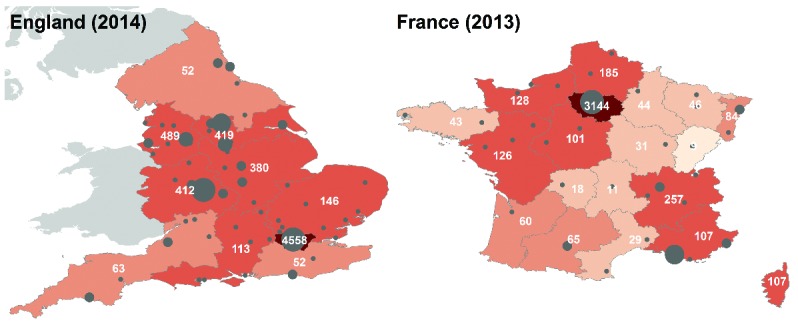
Maps of the distribution of patients with sickle cell disease in England and France. (A). Number of sickle cell patients by commissioning hub, based on data from the 2013/2014 annual report of the National Haemoglobinopathy Registry (NHR), available at http://www.nhr.nhs.uk/docs/annualreport/NHR_AnnualReport_2014.pdf (last accessed on 21 April 2015); (B). Sickle cell births by French region since the implementation of the targeted newborn screening, based on data from the annual report of the Association Française pour le Dépistage et la Prévention des Handicaps de l’Enfant (AFDPHE), available at: http://www.afdphe.org/sites/default/files/bilan_activite_2013.pdf (last accessed on 21 April 2015). Gray circles indicate major cities and are proportional to population size.
Figure 4.
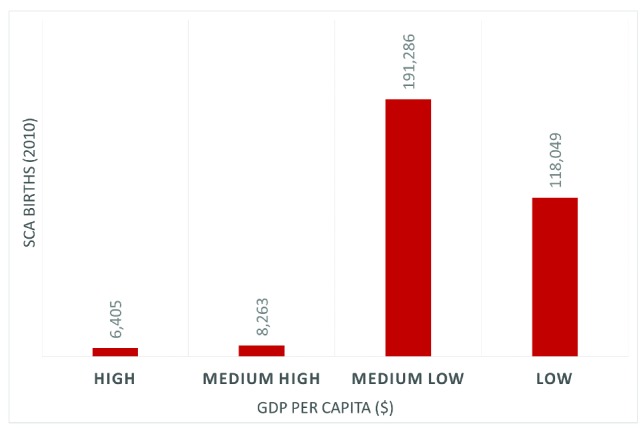
Estimates of annual births with sickle cell anemia (SCA) in 2010 according to the gross national income (GNI) per capita, divided into four classes according to the World Bank income classification (2010): low income: US$1,005 or less; medium low income: US$1,006 to US$3,975; medium-high income: $3,976 to $12,275; and high income: $12,276 or more (derived from Piel et al.84].
Physical activity and sickle cell disease
Patients with SCD often ask whether they can participate in sports and other strenuous physical activities. Anecdotally, many patients report acute pain following certain types of exercise, particularly in cold weather. There is not much evidence on which to base any advice. Exercise and physical activity are known to induce marked metabolic changes, including lactic acidosis, tissue hypoxia and dehydration, all of which predispose towards HbS polymerization and vaso-occlusion. Studies suggest that even moderate exercise can induce significant hypoxia in children with SCD,86 impaired heart rate recovery,87 abnormal increases in pulmonary artery pressure88 and increased oxidative stress.89 The clinical significance of these effects is unclear and studies in sickle mice suggested decreased oxidative stress with exercise.90 Particular issues arise with some sports, such as swimming and skiing. Swimming can be associated with rapid skin cooling on emerging from the pool,91 and patients are typically advised to dry themselves very rapidly, although some choose to avoid the activity, particularly if it has previously caused problems. Skiing involves exposure to cold, lower atmospheric oxygen, and exertion, and in general patients are advised to avoid it.3 The potential adverse effects of exercise have to be balanced against the probable cardiovascular and social benefits, and in general individuals with SCD should be encouraged to participate in exercises and sporting activities.
More studies have been performed to assess the effects of exercise on sickle cell carriers, looking at inflammatory and oxidative stress markers during exercise. One study reported that the risk of death in athletes with sickle cell trait was increased 10 to 30 times.92 Several studies have reported episodes of sudden death after intense exercise in people with sickle cell trait.93–95 Causes of death include rhabdomyolysis, exertional heat stroke and sudden cardiac arrhythmias. Risk factors include dehydration, extreme heat and exercise at high altitude. In 2012, whether athletes in the USA should be systematically screened became the focus of an intense debate following a study analyzing data from the National Collegiate Athletic Association showing a risk ratio of exertional death 37 times higher in football athletes with SCT than in those without SCT.96
Conclusions
Environmental factors have a significant effect on the natural history of SCD, which is demonstrated most graphically by the differences in outcomes between high-and low-income countries. However, it is very difficult to establish a clear link between specific environmental factors and pathophysiological events, and the evidence for these effects is confusing and often contradictory. Although there are biological consequences which follow from environmental changes, such as slower HbS polymerization with lower temperature, these may not be the explanation for any correlation with clinical events. These environmental effects are likely to be far greater than those attributable to genetic factors. Relatively few studies have been published in this area and the results are confusing and sometimes conflicting, partly because of the intricate relationships between climate, pollutants, socioeconomic factors and infections. Overall, cold or windy weather seem to be associated with increased hospital attendance; air quality is more difficult to assess as there is close correlation between many of the pollutants, although most seem to be associated with increased complications, apart from carbon monoxide which may be protective. Global warming, increased urbanization and decreasing air quality make it important to better understand environmental effects on SCD, and further, long-term studies need to be conducted in this area, particularly to assess effects on chronic complications and drive public health policies. Further research could include prospective studies of a cohort of patients measuring a full range of environmental factors, and pathophysiological experiments to characterize the effects of climatic changes and air quality.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010; 376(9757):2018–2031. [DOI] [PubMed] [Google Scholar]
- 2.Inati A, Koussa S, Taher A, Perrine S. Sickle cell disease: new insights into pathophysiology and treatment. Pediatr Ann. 2008;37(5): 311–321. [DOI] [PubMed] [Google Scholar]
- 3.Brousse V, Makani J, Rees DC. Management of sickle cell disease in the community. BMJ. 2014;348:g1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol. 2009;84(9):618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGann PT. Sickle cell anemia: an underappreciated and unaddressed contributor to global childhood mortality. J Pediatr. 2014; 165(1):18–22. [DOI] [PubMed] [Google Scholar]
- 6.Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pizzo E, Laverty AA, Phekoo KJ, et al. A retrospective analysis of the cost of hospitalizations for sickle cell disease with crisis in England, 2010/11. J Public Health (Oxf). 2014. May 5 [E-pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84(6):323–327. [DOI] [PubMed] [Google Scholar]
- 9.Williams TN, Uyoga S, Macharia A, et al. Bacteraemia in Kenyan children with sickle-cell anaemia: a retrospective cohort and case-control study. Lancet. 2009;374(9698): 1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6 Suppl 4):S398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menzel S, Thein SL. Genetic architecture of hemoglobin F control. Curr Opin Hematol. 2009;16(3):179–186. [DOI] [PubMed] [Google Scholar]
- 12.Higgs DR, Aldridge BE, Lamb J, et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N Engl J Med. 1982;306(24):1441–1446. [DOI] [PubMed] [Google Scholar]
- 13.Thein SL. Genetic association studies in beta-hemoglobinopathies. Hematol Am Soc Hematol Educ Program. 2013;2013:354–361. [DOI] [PubMed] [Google Scholar]
- 14.Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. BMJ. 1974;2(5909):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Redwood AM, Williams EM, Desal P, Serjeant GR. Climate and painful crisis of sickle-cell disease in Jamaica. BMJ. 1976;1(6001):66–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seeler RA. Non-seasonality of sickle-cell crisis. Lancet. 1973;2(7831):743. [DOI] [PubMed] [Google Scholar]
- 17.Slovis CM, Talley JD, Pitts RB. Non relationship of climatologic factors and painful sickle cell anemia crisis. J Chronic Dis. 1986;39(2):121–126. [DOI] [PubMed] [Google Scholar]
- 18.Amin BR, Bauersachs RM, Meiselman HJ, et al. Monozygotic twins with sickle cell anemia and discordant clinical courses: clinical and laboratory studies. Hemoglobin. 1991;15(4):247–256. [DOI] [PubMed] [Google Scholar]
- 19.Joishy SK, Griner PF, Rowley PT. Sickle beta-thalassemia: identical twins differing in severity implicate nongenetic factors influencing course. Am J Hematol. 1976;1(1):23–33. [DOI] [PubMed] [Google Scholar]
- 20.Weatherall MW, Higgs DR, Weiss H, Weatherall DJ, Serjeant GR. Phenotype/genotype relationships in sickle cell disease: a pilot twin study. Clin Lab Haematol. 2005;27(6):384–390. [DOI] [PubMed] [Google Scholar]
- 21.Italia K, Kangne H, Shanmukaiah C, Nadkarni AH, Ghosh K, Colah RB. Variable phenotypes of sickle cell disease in India with the Arab-Indian haplotype. Br J Haematol. 2015;168(1):156–159. [DOI] [PubMed] [Google Scholar]
- 22.Alsultan A, Alabdulaali MK, Griffin PJ, et al. Sickle cell disease in Saudi Arabia: the phenotype in adults with the Arab-Indian haplotype is not benign. Br J Haematol. 2014;164(4):597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol. 2014;166(2):165–176. [DOI] [PubMed] [Google Scholar]
- 24.Bunn HF. The triumph of good over evil: protection by the sickle gene against malaria. Blood. 2013;121(1):20–25. [DOI] [PubMed] [Google Scholar]
- 25.Ramakrishnan M, Moisi JC, Klugman KP, et al. Increased risk of invasive bacterial infections in African people with sickle-cell disease: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10(5):329–337. [DOI] [PubMed] [Google Scholar]
- 26.Adekile AD. Sickle cell disease in Kuwait. Hemoglobin. 2001;25(2):219–225. [DOI] [PubMed] [Google Scholar]
- 27.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. Oxford New York: Oxford University Press, 2001. [Google Scholar]
- 29.Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci USA. 1974;71(12):4864–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohan J, Marshall JM, Reid HL, Thomas PW, Hambleton I, Serjeant GR. Peripheral vascular response to mild indirect cooling in patients with homozygous sickle cell (SS) disease and the frequency of painful crisis. Clin Sci. 1998;94(2):111–120. [DOI] [PubMed] [Google Scholar]
- 31.Mohan JS, Marshall JM, Reid HL, Thomas PW, Hambleton I, Serjeant GR. Comparison of responses evoked by mild indirect cooling and by sound in the forearm vasculature in patients with homozygous sickle cell disease and in normal subjects. Clin Auton Res. 1998;8(1):25–30. [DOI] [PubMed] [Google Scholar]
- 32.Rubenstein E. Studies on the relationship of temperature to sickle cell anemia. Am J Med. 1961;30:95–98. [DOI] [PubMed] [Google Scholar]
- 33.Brandow AM, Stucky CL, Hillery CA, Hoffmann RG, Panepinto JA. Patients with sickle cell disease have increased sensitivity to cold and heat. Am J Hematol. 2013;88(1): 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baum KF, Dunn DT, Maude GH, Serjeant GR. The painful crisis of homozygous sickle cell disease. A study of the risk factors. Arch Intern Med. 1987;147(7):1231–1234. [PubMed] [Google Scholar]
- 35.Serjeant GR, Ceulaer CD, Lethbridge R, Morris J, Singhal A, Thomas PW. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol. 1994;87(3):586–591. [DOI] [PubMed] [Google Scholar]
- 36.Berger SA, Carlson BE. Sickle cell blood flow in the microcirculation. Conf Proc IEEE Eng Med Biol Soc. 2004;7:5057–5060. [DOI] [PubMed] [Google Scholar]
- 37.Berger SA, King WS. Diffusion and convection in the capillaries in sickle-cell disease. Blood Cells. 1982;8(1):153–161. [PubMed] [Google Scholar]
- 38.Lipowsky HH, Williams ME. Shear rate dependency of red cell sequestration in skin capillaries in sickle cell disease and its variation with vasoocclusive crisis. Microcirculation. 1997;4(2):289–301. [DOI] [PubMed] [Google Scholar]
- 39.Serjeant GR, Chalmers RM. Current concerns in haematology. 1. Is the painful crisis of sickle cell disease a “steal” syndrome¿ J Clin Pathol. 1990;43(10):789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham GS. A case of sickle cell anemia with necropsy. Arch Intern Med. 1924;34(6): 778–800. [Google Scholar]
- 41.Edington GM. Sickle-cell anaemia in the Accra district of the Gold Coast; a review of twenty cases. BMJ. 1953;2(4843):957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konotey-Ahulu FI. Sicklaemic Human Hygrometers. Lancet. 1965;1(7393):1003–1004. [DOI] [PubMed] [Google Scholar]
- 43.Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg. 1980;74(2):159–161. [DOI] [PubMed] [Google Scholar]
- 44.Smith WR, Coyne P, Smith VS, Mercier B. Temperature changes, temperature extremes, and their relationship to emergency department visits and hospitalizations for sickle cell crisis. Pain Manag Nurs. 2003;4(3):106–111. [DOI] [PubMed] [Google Scholar]
- 45.Rogovik AL, Persaud J, Friedman JN, Kirby MA, Goldman RD. Pediatric vasoocclusive crisis and weather conditions. J Emerg Med. 2011;41(5):559–565. [DOI] [PubMed] [Google Scholar]
- 46.Mekontso Dessap A, Contou D, Dandine-Roulland C, et al. Environmental influences on daily emergency admissions in sickle-cell disease patients. Medicine (Baltimore). 2014;93(29):e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kehinde MO, Marsh JC, Marsh GW. Sickle cell disease in North London. Br J Haematol. 1987. August;66(4):543–7. [DOI] [PubMed] [Google Scholar]
- 48.Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol. 2005;131(4): 530–533. [DOI] [PubMed] [Google Scholar]
- 49.Nolan VG, Zhang Y, Lash T, Sebastiani P, Steinberg MH. Association between wind speed and the occurrence of sickle cell acute painful episodes: results of a case-crossover study. Br J Haematol. 2008;143(3):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Michaels LA, Maraventano MF, Drachtman RA. Thrombosis and gangrene in a patient with sickle cell disease and dactylitis. J Pediatr. 2003;142(4):449. [DOI] [PubMed] [Google Scholar]
- 51.Saleem S, Rice L. Limb amputation in hemoglobin SC disease after application of ice and elevation. Am J Hematol. 2007;82(1):53–54. [DOI] [PubMed] [Google Scholar]
- 52.Spix C, Anderson HR, Schwartz J, et al. Short-term effects of air pollution on hospital admissions of respiratory diseases in Europe: a quantitative summary of APHEA study results. Air Pollution and Health: a European Approach. Arch Environ Health. 1998;53(1):54–64. [DOI] [PubMed] [Google Scholar]
- 53.Cohen AJ, Ross Anderson H, Ostro B, et al. The global burden of disease due to outdoor air pollution. J Toxicol Environ Health A. 2005;68(13–14):1301–1307. [DOI] [PubMed] [Google Scholar]
- 54.World Health Organization. The World Health Report: 2002: Reducing the risks, promoting healthy life. 2002. [DOI] [PubMed] [Google Scholar]
- 55.Yallop D, Duncan ER, Norris E, et al. The associations between air quality and the number of hospital admissions for acute pain and sickle-cell disease in an urban environment. Br J Haematol. 2007; 136(6):844–848. [DOI] [PubMed] [Google Scholar]
- 56.De Prins S, Koppen G, Jacobs G, et al. Influence of ambient air pollution on global DNA methylation in healthy adults: a seasonal follow-up. Environ Int. 2013;59:418–424. [DOI] [PubMed] [Google Scholar]
- 57.Friedman MS, Powell KE, Hutwagner L, Graham LM, Teague WG. Impact of changes in transportation and commuting behaviors during the 1996 Summer Olympic Games in Atlanta on air quality and childhood asthma. JAMA. 2001; 285(7):897–905. [DOI] [PubMed] [Google Scholar]
- 58.Glassberg JA, Chow A, Wisnivesky J, Hoffman R, Debaun MR, Richardson LD. Wheezing and asthma are independent risk factors for increased sickle cell disease morbidity. Br J Haematol. 2012;159(4):472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diep RT, Busani S, Simon J, Punzalan A, Skloot GS, Glassberg JA. Cough and wheeze events are temporally associated with increased pain in individuals with sickle cell disease without asthma. Br J Haematol. March 2015. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barbosa SM, Farhat SC, Martins LC, et al. Air pollution and children’s health: sickle cell disease. Cad Saude Publica. 2015; 31(2):265–275. [DOI] [PubMed] [Google Scholar]
- 61.Bocci V, Aldinucci C. Rational bases for using oxygen-ozonetherapy as a biological response modifier in sickle cell anemia and beta-thalassemia: a therapeutic perspective. J Biol Regul Homeost Agents. 2004; 18(1):38–44. [PubMed] [Google Scholar]
- 62.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–1389. [DOI] [PubMed] [Google Scholar]
- 63.Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011; 305(9):893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mittal H, Roberts L, Fuller GW, et al. The effects of air quality on haematological and clinical parameters in children with sickle cell anaemia. Ann Hematol. 2009; 88(6):529–533. [DOI] [PubMed] [Google Scholar]
- 65.Belcher JD, Young M, Chen C, et al. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood. 2013; 122(15):2757–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beutler E. The effect of carbon monoxide on red cell life span in sickle cell disease. Blood. 1975;46(2):253–259. [PubMed] [Google Scholar]
- 67.Shah AS, Lee KK, McAllister DA, et al. Short term exposure to air pollution and stroke: systematic review and meta-analysis. BMJ. 2015;350:h1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goodman J, Hassell K, Irwin D, Witkowski EH, Nuss R. The splenic syndrome in individuals with sickle cell trait. High Alt Med Biol. 2014;15(4):468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Githens JH, Gross GP, Eife RF, Wallner SF. Splenic sequestration syndrome at mountain altitudes in sickle/hemoglobin C disease. J Pediatr. 1977;90(2):203–206. [DOI] [PubMed] [Google Scholar]
- 70.Mahony BS, Githens JH. Sickling crises and altitude. Occurrence in the Colorado patient population. Clin Pediatr. 1979; 18(7):431–438. [DOI] [PubMed] [Google Scholar]
- 71.Addae S, Adzaku F, Mohammed S, Annobil S. Sickle cell disease in permanent residents of mountain and low altitudes in Saudi Arabia. Trop Geogr Med. 1990;42(4):342–348. [PubMed] [Google Scholar]
- 72.Claster S, Godwin MJ, Embury SH. Risk of altitude exposure in sickle cell disease. West J Med. 1981;135(5):364–367. [PMC free article] [PubMed] [Google Scholar]
- 73.Office on Smoking and Health (US). The health consequences of involuntary exposure to tobacco smoke: a report of the Surgeon General. Atlanta, Georgia: US Department of Health and Human Services, Centers for Disease Control and Prevention. Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2006:1988–2002. [PubMed] [Google Scholar]
- 74.Feleszko W, Zawadzka-Krajewska A, Matysiak K, et al. Parental tobacco smoking is associated with augmented IL-13 secretion in children with allergic asthma. J Allergy Clin Immunol. 2006;117(1):97–102. [DOI] [PubMed] [Google Scholar]
- 75.Noakes PS, Thomas R, Lane C, et al. Association of maternal smoking with increased infant oxidative stress at 3 months of age. Thorax. 2007;62(8):714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ahmadzadehfar H, Oguogho A, Efthimiou Y, Kritz H, Sinzinger H. Passive cigarette smoking increases isoprostane formation. Life Sci. 2006;78(8):894–897. [DOI] [PubMed] [Google Scholar]
- 77.Kallio K, Jokinen E, Raitakari OT, et al. Tobacco smoke exposure is associated with attenuated endothelial function in 11-year-old healthy children. Circulation. 2007;115(25):3205–3212. [DOI] [PubMed] [Google Scholar]
- 78.Kato T, Inoue T, Morooka T, Yoshimoto N, Node K. Short-term passive smoking causes endothelial dysfunction via oxidative stress in nonsmokers. Can J Physiol Pharmacol. 2006;84(5):523–529. [DOI] [PubMed] [Google Scholar]
- 79.Knight-Madden JM, Barton-Gooden A, Weaver SR, Reid M, Greenough A. Mortality, asthma, smoking and acute chest syndrome in young adults with sickle cell disease. Lung. 2013;191(1):95–100. [DOI] [PubMed] [Google Scholar]
- 80.Cohen RT, DeBaun MR, Blinder MA, Strunk RC, Field JJ. Smoking is associated with an increased risk of acute chest syndrome and pain among adults with sickle cell disease. Blood. 2010;115(18):3852–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Young RC, Jr, Rachal RE, Hackney RL, Jr, Uy CG, Scott RB. Smoking is a factor in causing acute chest syndrome in sickle cell anemia. J Natl Med Assoc. 1992;84(3):267–271. [PMC free article] [PubMed] [Google Scholar]
- 82.West DC, Romano PS, Azari R, Rudominer A, Holman M, Sandhu S. Impact of environmental tobacco smoke on children with sickle cell disease. Arch Pediatr Adolesc Med. 2003;157(12):1197–1201. [DOI] [PubMed] [Google Scholar]
- 83.Marmot M. Social determinants of health inequalities. Lancet. 2005;365(9464):1099–1104. [DOI] [PubMed] [Google Scholar]
- 84.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farber MD, Koshy M, Kinney TR. Cooperative Study of Sickle Cell Disease: demographic and socioeconomic characteristics of patients and families with sickle cell disease. J Chronic Dis. 1985;38(6):495–505. [DOI] [PubMed] [Google Scholar]
- 86.Halphen I, Elie C, Brousse V, et al. Severe nocturnal and postexercise hypoxia in children and adolescents with sickle cell disease. PloS One. 2014;9(5):e97462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alvarado AM, Ward KM, Muntz DS, et al. Heart rate recovery is impaired after maximal exercise testing in children with sickle cell anemia. J Pediatr. 2015;166(2):389–393 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Lima-Filho NN, Figueiredo MS, Vicari P, et al. Exercise-induced abnormal increase of systolic pulmonary artery pressure in adult patients with sickle cell anemia: an exercise stress echocardiography study. echocardiography. 2014. December 18 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 89.Faes C, Balayssac-Siransy E, Connes P, et al. Moderate endurance exercise in patients with sickle cell anaemia: effects on oxidative stress and endothelial activation. Br J Haematol. 2014;164(1):124–130. [DOI] [PubMed] [Google Scholar]
- 90.Charrin E, Aufradet E, Douillard A, et al. Oxidative stress is decreased in physically active sickle cell SAD mice. Br J Haematol. 2015;168(5):747–756. [DOI] [PubMed] [Google Scholar]
- 91.Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr. 1991;118(3):407–409. [DOI] [PubMed] [Google Scholar]
- 92.Mitchell BL. Sickle cell trait and sudden death–bringing it home. J Natl Med Assoc. 2007;99(3):300–305. [PMC free article] [PubMed] [Google Scholar]
- 93.Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med. 1987;317(13):781–787. [DOI] [PubMed] [Google Scholar]
- 94.Pretzlaff RK. Death of an adolescent athlete with sickle cell trait caused by exertional heat stroke. Pediatr Crit Care Med. 2002; 3(3):308–310. [DOI] [PubMed] [Google Scholar]
- 95.Rosenthal MA, Parker DJ. Collapse of a young athlete. Ann Emerg Med. 1992; 21(12):1493–1498. [DOI] [PubMed] [Google Scholar]
- 96.Harmon KG, Drezner JA, Klossner D, Asif IM. Sickle cell trait associated with a RR of death of 37 times in National Collegiate Athletic Association football athletes: a database with 2 million athlete-years as the denominator. Br J Sports Med. 2012; 46(5):325–330. [DOI] [PubMed] [Google Scholar]