

Abstract
Multiple myeloma is preceded by an asymptomatic phase, comprising monoclonal gammopathy of uncertain significance and smoldering myeloma. Compared to the former, smoldering myeloma has a higher and non-uniform rate of progression to clinical myeloma, reflecting a subset of patients with higher risk. We evaluated the gene expression profile of smoldering myeloma plasma cells among 105 patients enrolled in a prospective observational trial at our institution, with a view to identifying a high-risk signature. Baseline clinical, bone marrow, cytogenetic and radiologic data were evaluated for their potential to predict time to therapy for symptomatic myeloma. A gene signature derived from four genes, at an optimal binary cut-point of 9.28, identified 14 patients (13%) with a 2-year therapy risk of 85.7%. Conversely, a low four-gene score (<9.28) combined with baseline monoclonal protein <3 g/dL and albumin ≥3.5 g/dL identified 61 patients with low-risk smoldering myeloma with a 5.0% chance of progression at 2 years. The top 40 probe sets showed concordance with indices of chromosome instability. These data demonstrate high discriminatory power of a gene-based assay and suggest a role for dysregulation of mitotic checkpoints in the context of genomic instability as a hallmark of high-risk smoldering myeloma.
Introduction
Models of development of multiple myeloma (MM) involve the transformation of a normal plasma cell through a series of related precursor stages, including monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM).1,2 Using current definitions, proposed by the International Myeloma Working Group, SMM is distinguished from MGUS by the presence of >10% marrow plasmacytosis and/or ≥3 g/dL serum M-protein, together with the absence of the so-called CRAB criteria (hypercalcemia, renal failure, anemia and bony lesions).3 MGUS is essentially a benign clinical condition with an annual risk of approximately 1% of transformation into MM.4 In comparison to MGUS, SMM has a greater risk of progression to MM with approximately 10% of patients progressing to MM each year for the first 5 years, after which the progression rate is more similar to that of MGUS.5 It would appear clinically important to distinguish a high-risk subset of patients with SMM with a greater potential for progression from those patients with a more MGUS-like pattern of progression. Early intervention in the high-risk SMM group would avoid development of CRAB criteria and thus improve patients’ survival. The current practice guidelines of withholding treatment until end-organ damage develops put patients at risk of renal failure6 and adverse skeletal events.7 This strategy of delaying treatment does not only influence patients’ morbidity and overall costs, but also mortality, since renal insufficiency at diagnosis of MM has been shown to predict for inferior survival8 except when bortezomib was included in the upfront management.9
Several groups have attempted to risk-stratify precursor states of MM by using clinical laboratory data such as presence of serum free light chains (FLC),10–12 >95% aberrant/bone marrow plasma cells, DNA aneuploidy and immunoparesis,13 plasma cell proliferation,14 circulating plasma cells15 and imaging.16 Some of these findings led to a recently adopted International Myeloma Working Group proposal that patients with bone marrow plasmacytosis >60%, involved/uninvolved light chain ratio >100 and more than one focal lesion on magnetic resonance imaging (MRI) should be considered for anti-MM therapy.17 While both Mayo and Spanish prognostic models identified patients with high-risk MM, the risk factors emerged in multivariate analysis as independent prognostic factors, even though they did not overlap in the two models.18 The development of an objective and reproducible tool that can consistently identify high-risk SMM is, therefore, urgently needed. This issue is particularly relevant given the evidence from a Spanish trial showing improved survival with early initiation of therapy in high-risk SMM.19
We explored the use of gene expression profiling (GEP) to risk-stratify SMM. In an analysis of South West Oncology Group (SWOG) trial S0120, we evaluated GEP70, developed for defining high-risk behavior in symptomatic MM,20,21 for its predictive value for high-risk MM.22 A GEP70 score ≤−0.26), together with a serum M-protein >3g/dL and serum FLC >25 mg/dL, identified SMM patients with a 70% 2-year risk of progression to MM requiring therapy.22 In order to improve on the GEP70 approach, we developed a new, optimized signature based on a novel analysis using all the differentially expressed genes present on the Affymetrix platform.
Methods
Eligibility criteria and study design
Patients with SMM seen at the Myeloma Institute for Research and Therapy were included in a prospective, observational clinical trial (SWOG S0120) as part of a national cooperative group trial to identify biological correlates that may relate to progression to symptomatic disease. Other eligibility criteria included no prior therapy for the plasma cell disorder and willingness to submit samples for research. Diagnostic criteria for SMM were based on the International Myeloma Working Group convention.3 All patients signed informed consent, in keeping with the Declaration of Helsinki and federal and institutional guidelines. The protocol was approved by the National Cancer Institute and all participating centers’ internal review boards.
All patients underwent detailed clinical staging at initial registration, including full blood count, analysis of blood chemistry, and standard MM-related serological and urinary measurements. Nephelometric analysis was performed to determine serum immunoglobulin levels. Immunofixation analyses of serum and urine were performed to define the nature of the monoclonal protein present in serum and/or urine. Bone marrow aspirates and biopsies were obtained for cytological and histopathological evaluation of the degree of plasma cell infiltration, including immunohistochemical clonality assessment. Metaphase karyotyping was performed on at least 20 Giemsa-stained metaphases.23 In most patients, serum FLC assays were used to quantify κ and λ light chains. Imaging studies involved standard metastatic bone surveys by X-ray examination, and, when possible, MRI of the entire spine was used to identify focal lesions. The minimum follow-up of patients involved MM-related laboratory studies at 3, 6, and 12 months in the first year, and then every 6 to 12 months.
Gene expression profiling and statistical methods
A sample of bone marrow aspirate was collected to isolate CD138+ plasma cells with immunomagnetic bead selection (autoMACS; Miltenyi Biotec) as described elsewhere.24 The purity of the plasma cells was monitored by flow cytometry and >85% purity was used as a criterion for inclusion of GEP data. Total RNA from these plasma cells was used for the GEP with Affymetrix U133 Plus 2.0 microarrays. We identified patients with SMM and baseline GEP data who were cared for at this institution. We evaluated 54,675 Affymetrix gene probes for their potential to predict time to myeloma therapy (TTT). Probes were ranked by their q-values;25 we used 10-fold cross validation to identify the number of genes at the top of this list, which collectively maximized the concordance between risk score and survival. Gene scores were computed by subtracting the sum of the expressions of the up-regulated probes from the sum of the expressions of the down-regulated probes, then dividing by the total number of probes. A running log-rank test was used to identify statistically optimal binary cut-points for the gene scores within the S0210 population for TTT analysis.
To determine how many probe sets from the ranked probe set list were needed to create a score, we performed 10-fold cross-validation. We divided our data into ten folds of approximately equal size, using nine folds at a time to create a score with the top n probe sets, where n takes integer values from 1 to 100. We used the remaining fold for validation and to calculate the c-index, a measure of the proportion of agreement between observed and predicted responses.26 We repeated this for ten iterations per value of n; the c-index for each 10-fold cross-validation was the average value of those obtained using each fold as the validation fold, and our final c-index was the mean c-index from the ten iterations.
Cumulative incidence analysis was used to model TTT with death as a competing risk. Univariate and multivariate Cox proportional hazards regression analyses were used to identify factors significantly associated with TTT.27 Recursive partitioning was used to build regression trees based on statistically optimal binary cut-points for continuous baseline covariates.28 This method was used in order to observe which of the many covariates considered would best define the classification of patients for being at high, intermediate, or low risk of progression requiring therapy. The R2 statistic was used to quantify the percentage of total variation explained by the models considered.29 The c-index was used to measure the performance of various models with TTT as response.
Results
Identifying gene probes linked to time to treatment for myeloma
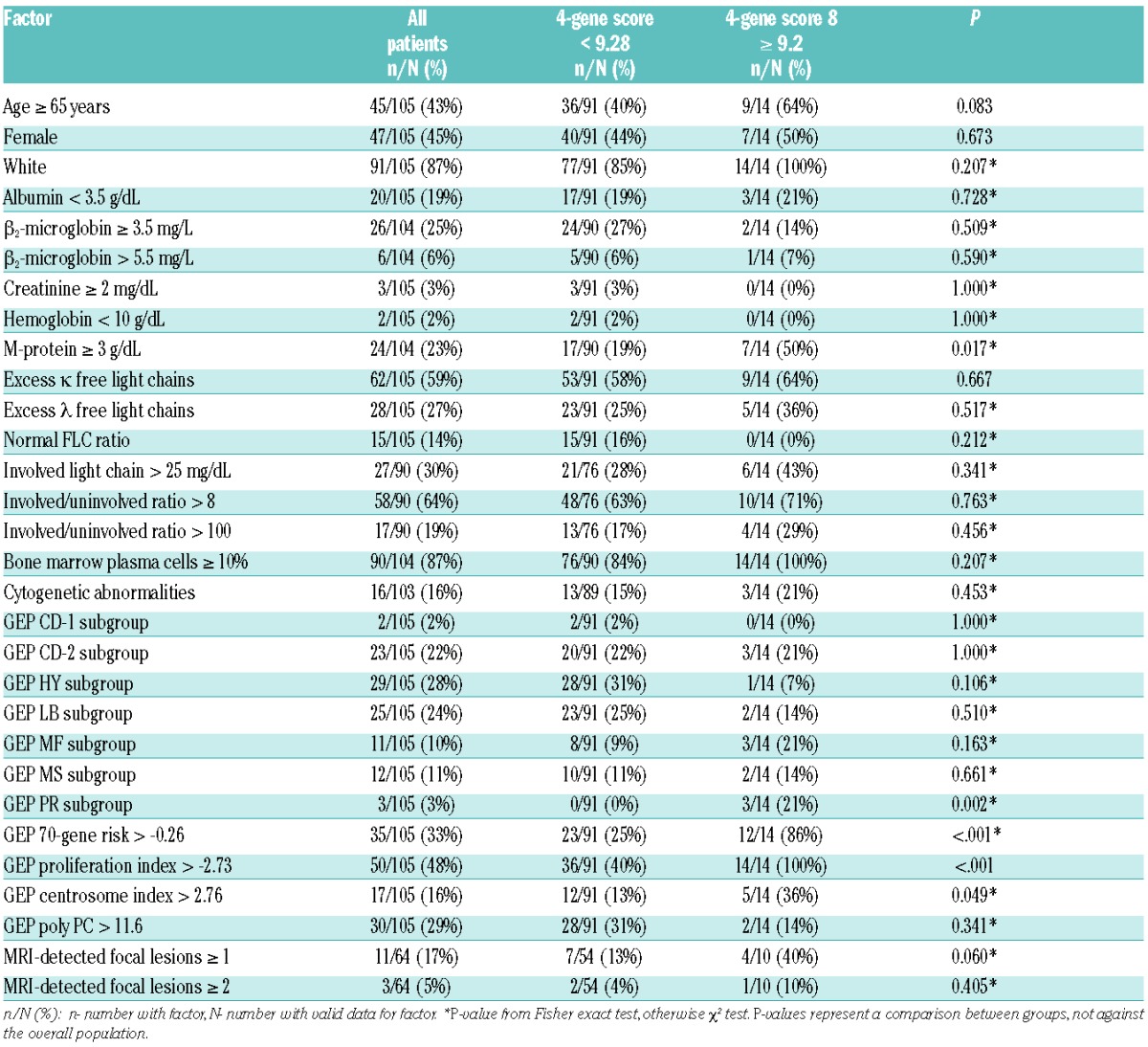
There were 105 SMM patients with baseline GEP data available for analyses: the baseline characteristics of these patients are presented in Table 1. Bone marrow plasma cell data were available for 104 (99.0%) and staging MRI was performed in 64 (60.9%) patients. Of the 54,675 Affymetrix probes, using q statistics at <0.05 and <0.10, 40 and 81 probes, respectively, were significantly associated with TTT. Of all gene list sizes considered by 10-fold cross-validation, the list using the top four probe sets (GEP4) maximized the concordance between risk score and TTT. The top four genes in descending order of predictive power were RRM2 (2p25-p24), DTL (1q32), TMEM48 (1p32.3) and ASPM (1q31). An optimal cut-point for risk of progression by the four-gene score was found to be 9.28, which identified a subset of 14 (13.3%) patients with a 2-year progression probability of 85.7%, with the remainder of the 91 patients (87%) have a probability of requiring therapy of only 17.8% (Figure 1A). The criteria for clinical MM were recently revised to include a FLC ratio >100 (with involved FLC >100 mg/L) and more than one focal lesion on MRI.17 We, therefore, reanalyzed the impact of GEP4 after excluding the nine patients who were re-categorized as having MM based on the new criteria. Exclusion of these patients did not alter the prognostic impact of GEP4, with a 2-year estimate of 81.8% for high-risk patients and 15.5% for low-risk ones (Figure 1B). By comparison, patients classified as high-risk by the GEP70 cut-point had a 2-year progression probability of 49.7% (Figure 1C).24 The patients’ characteristics that varied between GEP4-defined high-risk and low-risk SMM included M-protein ≥3 g/dL, GEP-PR molecular sub-group, GEP70-gene high-risk, GEP-based high proliferation index, and GEP-based high centrosome index (Table 2).
Table 1.
Patients’ characteristics.
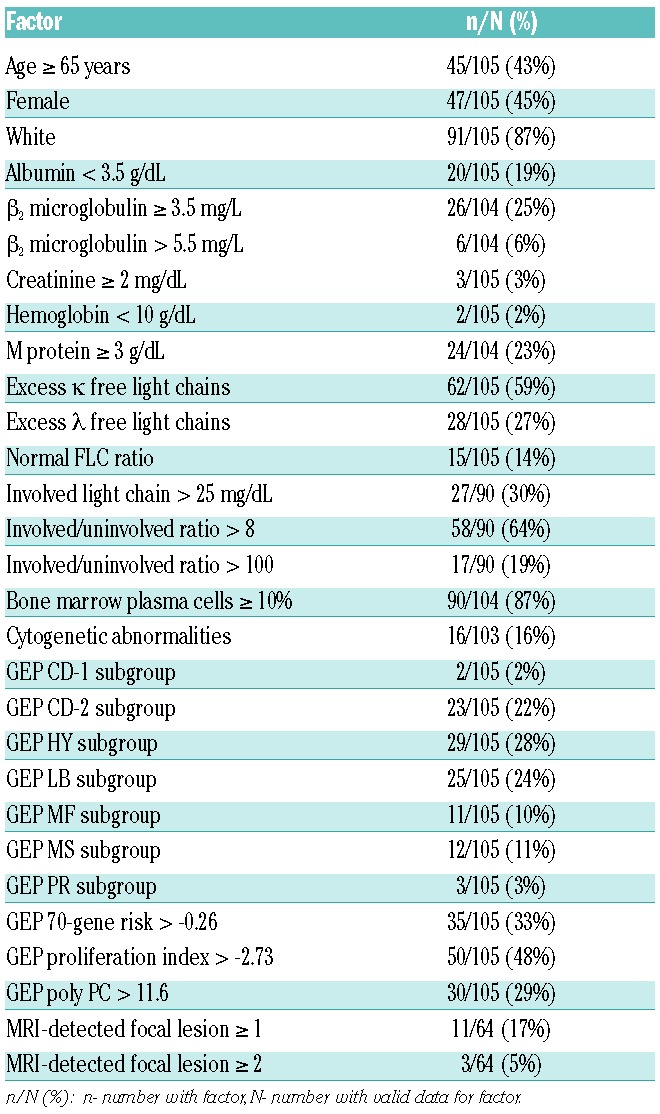
Figure 1.

(A). Time to progression requiring myeloma therapy (TTT). The four-gene score identified a high-risk group with an estimated probability of 2-year progression to myeloma of 85.7%. (B) Limiting the four-gene score to 96 patients with SMM, as defined by the recent International Myeloma Working Group criteria yielded a 2-year risk of treatment of 81.8%. (C). By comparison, the GEP70 identified a high-risk group with an estimated 2-year progression probability of 49.7%
Table 2.
Patients’ characteristics by GEP4 classification.
Online Supplementary Table S1 lists the top 40 most significant (q<0.05) probe sets relating to 34 unique genes, 11 of which were located on chromosome 1q, six on chromosome 15q and four on chromosome 1p. Cellular processes affected included cell cycle checkpoint control and apoptosis, DNA repair, biosynthesis and metabolism, cellular assembly and organization, RNA transport and processing, and interleukin-6 signaling (Online Supplementary Table S2).
Identifying predictors of time to myeloma therapy
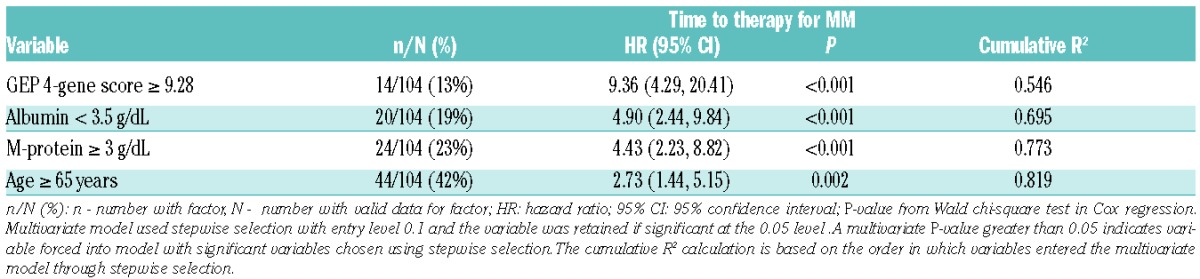
We next evaluated other baseline factors for their univariate potential to predict TTT (Online Supplementary Table S3). TTT was linked to age (>65 years), albumin (<3.5 g/dL), serum M-protein (≥3 g/dL), bone marrow plasma cells (>10%), excess λ FLC, involved light chains >25 mg/dL and involved/uninvolved FLC ratio >8. The presence of at least one focal lesion on MRI, available in 64 patients, was also significant (hazard ratio, HR=2.59). In contrast to the Mayo dataset on which the International Myeloma Working Group guidelines were constructed, in our cohort TTT was not affected by FLC ratio >100,11 pertaining to 17 of 90 evaluable patients. It is notable that while FLC ratio is a risk factor for disease progression in the SWOG dataset,22 the hazard ratio for FLC ratio >100 (HR 1.53, P=0.243) is lower than in the Mayo dataset. None of our patients had >60% bone marrow plasma cells. Time-dependent analyses of changes in serum M-protein were found to be predictive for progression with a serum M-protein increase from baseline of 1 g/dL carrying a 21-fold higher risk of earlier TTT. Among GEP parameters associated with shorter TTT, we identified GEP4 score >9.28, GEP70 score > −0.26, GEP PR subgroup, GEP proliferation index > −2.73, and GEP centrosome index >2.76. In contrast, GEP poly PC >11.6, indicating a higher proportion of normal plasma cells, was TTT-protective. On multivariate analysis of clinical variables, age >65 years, albumin <3.5 g/dL and M-protein ≥3 g/dL were independently correlated with TTT, with a cumulative R2 value of 66.0%. Consideration of GEP parameters revealed that GEP4 ≥9.28 provided the highest hazard ratio value of 9.36, although not displacing the three aforementioned standard variables (Table 3). As reflected by cumulative R2 statistics, GEP4 high risk alone accounted for 54.6% of all variability encountered, to which albumin, serum M-protein and age added a cumulative 27.3% (cumulative total R2=81.9%).
Table 3.
Multivariate Cox proportional hazards analysis of risk factors of progression to clinical MM requiring therapy with cumulative R2 for each variable.
Recursive partitioning model of time to myeloma therapy
In order to refine the identification of patients with SMM at highest risk of progression requiring therapy, we combined standard clinical and GEP-derived variables to model for high-risk SMM using recursive partitioning (Figure 2). GEP4 ≥9.28, pertaining to 13% of patients, was linked to a 2-year estimate of therapy requirement of 85.7%. GEP4 <9.28 with either serum M-protein ≥3 g/dL or albumin <3.5g/dL (28% of patients) was associated with an intermediate probability of progression (45% at 2 years). The lowest probability of requiring myeloma therapy was seen among the 59% of patients with GEP4 <9.28, serum M-protein <3 g/dL and albumin ≥3.5 g/dL (5% at 2 years).
Figure 2.
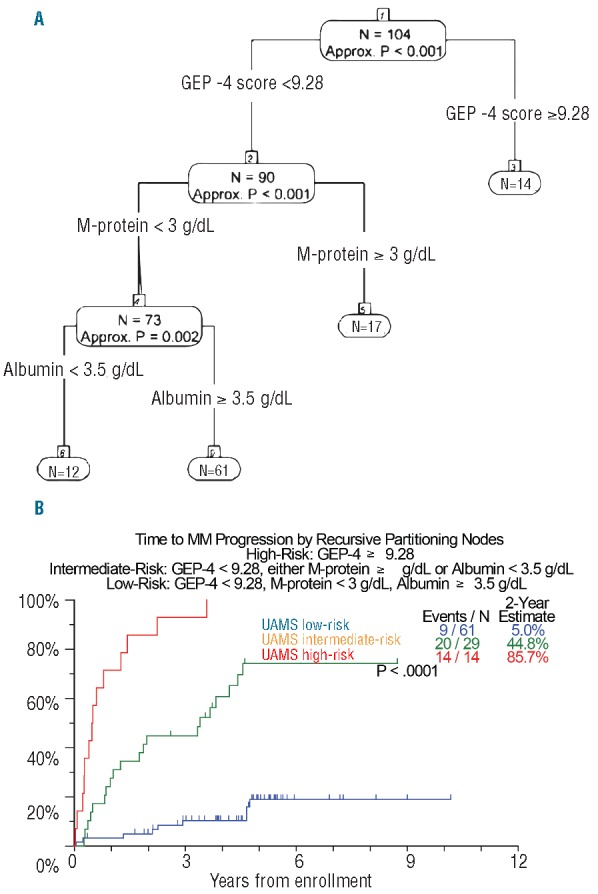
Recursive partitioning for TTT. (A). A recursive partitioning algorithm used GEP and clinical variables to define distinct clinically relevant risk groups. (B). A high-risk group is defined by GEP4 ≥9.28 and has a 2-year progression probability of 85.7%; a low-risk group comprises patients with GEP4 <9.28, M-protein <3 g/dL and albumin ≥3.5 g/dL and has a 5% probability of progression at 2 years; and an intermediate-risk group featured patients not in the high-risk or low-risk group and has a 2-year progression probability of 44.8%, UAMS: University of Arkansas for Medical Sciences.
Comparison with the Mayo model
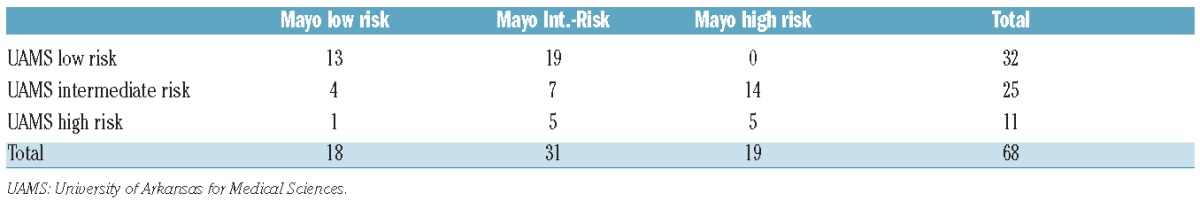
There were 68 patients at the University of Arkansas for Medical Sciences with complete data sets for GEP, M-protein, age, albumin, bone marrow plasma cells and involved/uninvolved FLC allowing for comparison with a published risk stratification model utilizing FLC ratio >8 (Figure 3). The c-index, a measure of correlation between the test and survival, was marginally higher with the GEP-based model (0.753 versus 0.658) (Table 4A). Overall, the GEP4 model identified 47% of patients as having low-risk SMM compared to 26% using the FLC model, resulting in 2-year therapy requirements in 9.4% and 16.7%, respectively. With regards to high-risk SMM, GEP identified 11% and FLC 19% of such patients, with 2-year therapy requirement estimates of 81.8% and 63.2%, respectively. There was minimal concordance between the two models (Table 4B), as 54.5% of GEP4-defined high-risk SMM patients were classified as either low or intermediate risk by the FLC ratio model; furthermore, the majority of patients (56%) in the GEP4-defined intermediate-risk group were at high risk according to their FLC ratio, and those with GEP4 low risk were split between intermediate- and low-risk subsets according to the model based on FLC ratio.
Figure 3.
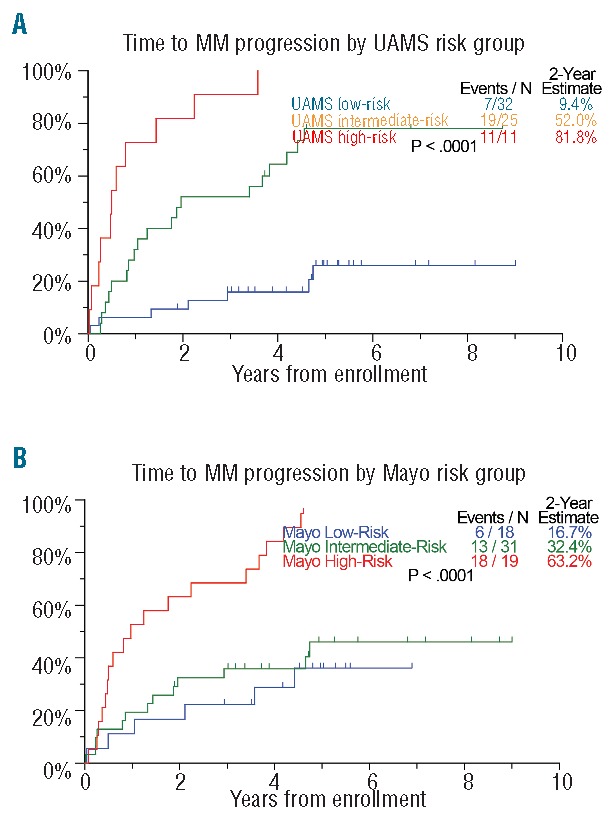
TTT comparison on University of Arkansas for Medical Sciences (UAMS) and Mayo risk models. Patients with complete data for model comparison were classified and compared for TTT using (A) the classification scheme driven by the four-gene score and (B) the Mayo FLC-ratio >8 risk stratification model.
Table 4A.
C-index comparison of the GEP4 and the FLC-R >8 model.

Table 4B.
A 3 × 3 table to evaluate overlap between the two models showing minimal concordance.
Discussion
GEP was developed and has been validated as a potent prognostic tool in clinically symptomatic MM such that 15% of patients with high-risk MM defined by GEP continue with a median progression-free survival of only 2 years or less.20 In the current study, we generated a high-risk SMM signature based on only four genes that, alone, predicted a 2-year probability of therapy requirement of 85.7%. When combined with standard clinical tests (serum M-protein <3 g/dL, albumin ≥3.5 g/dL), a powerful high-fidelity risk stratification model in SMM was generated, in which absence of risk factors successfully selected for an indolent MGUS-like population of 59% of patients with a low 2-year estimated rate of therapy requirement of 5%.
Previously we have reported on the utility of GEP70 in identifying patients with high-risk SMM.22 Here we show that GEP4 outperforms GEP70. Thus, GEP70-defined high-risk SMM predicted a 2-year risk of progression to requiring therapy of 49.7% while GEP4-defined high-risk SMM raised that estimate to 85.7%. Significantly, and shown previously, serum M-protein kinetics was the most predictive marker of progression,30 superseding GEP variables, hence justifying close monitoring of MM parameters in the first 3 months in all patients.
Previous high-risk SMM models have identified FLC ratio,10–12 bone marrow plasma cells >60%,12 percent of bone marrow plasma cell aberrancy on multiparametric flow cytometry,13 presence of more than one focal lesion on whole-body MRI,16 and presence of circulating plasma cells by slide-based immunofluorescence (Online Supplementary Table S4).15 Cross-comparison of our GEP-based strategy with these models was limited by sample size. We did not perform peripheral blood plasma cell immunofluorescence, and bone marrow plasma cell counts >60% were not observed in our cohort. The combination of FLC ratio, bone marrow plasma cells and serum M-protein has been shown to segregate risk of progression to MM, so that patients exhibiting all three risk features had a probability of progression of 76% at 5 years.10 Although the Mayo model performed well in predicting progression in our dataset, there was significant discordance between risk classification subsets, as seen previously in comparing the Mayo Clinic and Spanish PETHEMA models.18 GEP-defined molecular sub-groups, associated with superior progression-free and overall survival (HY, CD-2, LB) in MM, were more common in SMM than were the MF, MS and PR subtypes.24 While the t(4:14) translocation, identified by fluorescence in situ hybridization, predicted earlier progression in SMM,31 the GEP-based MS molecular subtype was not linked to pro gression. Although only three patients were assigned to the PR molecular subtype, the hazard for TTT was 9-fold the average, identifying a subset of patients with imminent progression, as also reported by Hose et al.32
The top gene in the GEP4 model, RRM2, is the beta subunit of ribonucleotide reductase (RNR). RRM2 overexpression is associated with cellular invasiveness, metastasis and tumor angiogenesis33 via activation of the ERK1/2 signaling pathway34 in cancer. RRM2 expression independently identified 15.2% patients with a 2-year TTT estimate of 75% (Online Supplementary Figure S1). Baseline RRM2 was found to be differentially overexpressed in the IFM GEP15 highest risk (quartile 4) group of MM patients35 and is prognostic in patients treated on our TT2 and TT3 protocols (unpublished data). Paired analyses of RRM2 expression in patients with clinical MM at baseline and in the relapsed setting showed overexpression with disease progression.36 Other constituents of GEP4 included DTL (also called retinoic acid-regulated nuclear matrix-associated protein – RAMP), which has been implicated in oncogenesis of solid tumors via its role in apoptosis and cell cycle control,37 and ASPM, shown previously to be a marker of poor prognosis in MM.38
A significant number of genes (15 genes) within the top 40 probe sets were located on chromosome 1, corroborating its previously reported importance in MM disease progression.39 Since there are no comparable GEP-based datasets with mature follow up in SMM, we compared the top 40 top probe sets with pre-existing GEP-based MM prognostic models, which revealed minimal overlap (Online Supplementary Table S5). Interestingly, there was significant overlap with several published GEP-based indices of chromosomal instability.40–42 This concordance suggests dysregulation of mitotic checkpoints in the setting of genomic instability as an important hallmark of high-risk SMM.
As the current study is the first to prospectively analyze baseline GEP in the setting of SMM, it will be critical to evaluate this signature in other datasets. In an institutional, observational, follow-up study of 76 cases, the 2-year progression estimate was 45% for patients with GEP4-defined high-risk SMM as opposed to 5% in the remaining patients (P<0.05), but longer follow-up is needed. Our data corroborate the recent finding that baseline genomic variations, such as the pattern of mutations,43 may differ between patients with MGUS/SMM who progress and those who do not. In order to generate a cogent hypothesis, it would, in addition, be important to perform serial clinical and GEP analyses on patients with SMM as they progress to clinical disease, as well as incorporate additional aspects such as epigenetic and bone marrow microenvironmental factors in future studies. While our GEP-based models are highly quantitative and reproducible, GEP analysis is limited to a few specialized centers. Once the GEP4 model has been validated in independent datasets, we envision centralized laboratories could offer such a service.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749–757. [PubMed] [Google Scholar]
- 4.Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–569. [DOI] [PubMed] [Google Scholar]
- 5.Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–2590. [DOI] [PubMed] [Google Scholar]
- 6.D’Arena G, Gobbi PG, Broglia C, et al. Pamidronate versus observation in asymptomatic myeloma: final results with long-term follow-up of a randomized study. Leuk Lymphoma. 2011;52(5):771–775. [DOI] [PubMed] [Google Scholar]
- 7.Wisloff F, Andersen P, Andersson TR, et al. Incidence and follow-up of asymptomatic multiple myeloma. The myeloma project of health region I in Norway. II. Eur J Haematol. 1991;47(5):338–341. [DOI] [PubMed] [Google Scholar]
- 8.Winearls CG. Acute myeloma kidney. Kidney Int. 1995;48(4):1347–1361. [DOI] [PubMed] [Google Scholar]
- 9.Scheid C, Sonneveld P, Schmidt-Wolf IG, et al. Bortezomib before and after autologous stem cell transplantation overcomes the negative prognostic impact of renal impairment in newly diagnosed multiple myeloma: a subgroup analysis from the HOVON-65/GMMG-HD4 trial. Haematologica. 2014;99(1):148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111(2):785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen JT, Kumar SK, Dispenzieri A, Kyle RA, Katzmann JA, Rajkumar SV. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma. Leukemia. 2013;27(4):941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kastritis E, Terpos E, Moulopoulos L, et al. Extensive bone marrow infiltration and abnormal free light chain ratio identifies patients with asymptomatic myeloma at high risk for progression to symptomatic disease. Leukemia. 2013;27(4):947–953. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Persona E, Vidriales MB, Mateo G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–2592. [DOI] [PubMed] [Google Scholar]
- 14.Madan S, Kyle RA, Greipp PR. Plasma cell labeling index in the evaluation of smoldering (asymptomatic) multiple myeloma. Mayo Clin Proc. 2010;85(3):300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bianchi G, Kyle RA, Larson DR, et al. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia. 2013;27(3):680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hillengass J, Weber MA, Kilk K, et al. Prognostic significance of whole-body MRI in patients with monoclonal gammopathy of undetermined significance. Leukemia. 2014;28(1):174–178. [DOI] [PubMed] [Google Scholar]
- 17.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–548. [DOI] [PubMed] [Google Scholar]
- 18.Cherry BM, Korde N, Kwok M, et al. Modeling progression risk for smoldering multiple myeloma: results from a prospective clinical study. Leuk Lymphoma. 2013;54(10):2215–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mateos MV, Hernandez MT, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438–447. [DOI] [PubMed] [Google Scholar]
- 20.Shaughnessy JD, Jr, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Barlogie B, Shaughnessy JD., Jr The molecular characterization and clinical management of multiple myeloma in the post-genome era. Leukemia. 2009;23(11):1941–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhodapkar MV, Sexton R, Waheed S, et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood. 2014;123(1):78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawyer JR. Metaphase cytogenetic techniques in multiple myeloma. Methods Mol Biol. 2011;730:149–158. [DOI] [PubMed] [Google Scholar]
- 24.Zhan F, Huang Y, Colla S, Stewart JP, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrell FE, Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15(4):361–387. [DOI] [PubMed] [Google Scholar]
- 27.Cox DR. Regression models and life-tables. J R Stat Soc. 1972;34(2):187–220. [Google Scholar]
- 28.LeBlanc M, Crowley J. Survival trees by goodness of split. Am Stat Ass. 1993;38:457–467. [Google Scholar]
- 29.O’Quigley J, Xu R, Stare J. Explained randomness in proportional hazards models. Stat Med. 2005;24(3):479–489. [DOI] [PubMed] [Google Scholar]
- 30.Rosinol L, Blade J, Esteve J, et al. Smoldering multiple myeloma: natural history and recognition of an evolving type. Br J Haematol. 2003;123(4):631–636. [DOI] [PubMed] [Google Scholar]
- 31.Rajkumar SV, Gupta V, Fonseca R, et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27(8):1738–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hose D, Reme T, Hielscher T, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011;96(1):87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K, Hu S, Wu J, et al. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: implication of RRM2 in angiogenesis. Mol Cancer. 2009;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang N, Zhan T, Ke T, et al. Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br J Cancer. 2014; 110(4):1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Decaux O, Lode L, Magrangeas F, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. J Clin Oncol. 2008;26(29):4798–4805. [DOI] [PubMed] [Google Scholar]
- 36.Zhou W, Yang Y, Xia J, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23(1):48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Ng EK, Ng YP, et al. Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br J Cancer. 2009;101(4):691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan F, Hardin J, Kordsmeier B, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99(5):1745–1757. [DOI] [PubMed] [Google Scholar]
- 39.Hanamura I, Huang Y, Zhan F, Barlogie B, Shaughnessy J. Prognostic value of cyclin D2 mRNA expression in newly diagnosed multiple myeloma treated with high-dose chemotherapy and tandem autologous stem cell transplantations. Leukemia. 2006;20(7): 1288–1290. [DOI] [PubMed] [Google Scholar]
- 40.Chung TH, Mulligan G, Fonseca R, Chng WJ. A novel measure of chromosome instability can account for prognostic difference in multiple myeloma. PLoS ONE. 2013; 8(6):e66361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38(9):1043–1048. [DOI] [PubMed] [Google Scholar]
- 42.Chibon F, Lagarde P, Salas S, et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med. 2010;16(7): 781–787. [DOI] [PubMed] [Google Scholar]
- 43.Zhao S, Choi M, Heuck C, et al. Serial exome analysis of disease progression in premalignant gammopathies. Leukemia. 2014;28(7):1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]