

Clinical outcome in anaplastic large cell lymphoma (ALCL), especially in systemic anaplastic lymphoma kinase (ALK)-negative ALCL is frequently poor, despite intensive therapy regimens including CHOP (vincristine, doxorubicin, cyclophosphamide and prednisone), with or without etoposide. Although promising results have been obtained with targeted therapies such as brentuximab vedotin, alemtuzumab and/or stem cell transplantation, alternative therapies are needed which can improve patient outcome.1,2
Recent studies have demonstrated that expression levels of many NF-κB regulated anti-apoptotic proteins are elevated in ALCL.3–5 NF-κB activity can be inhibited by the proteasome inhibitor bortezomib, resulting in induction of apoptosis.6 Bortezomib has shown remarkable preclinical anti-tumor activity in various types of cancer and was approved by the FDA for the treatment of multiple myeloma and mantle cell lymphoma. In the present study, we therefore investigated if bortezomib can induce apoptosis of primary lymphoma cells from ALK-negative and ALK-positive ALCL patients and in ALCL cell lines.
Cultured lymphoma cells7 of five patients with systemic ALCL were exposed to increasing concentrations of bortezomib for 24 hours. Treatment with bortezomib resulted in cell death in all ALCL patient samples tested (Figure 1A). Bortezomib induced a significant concentration-dependent induction of cell death in most cases. ALK-negative ALCL were relatively more sensitive to bortezomib than ALK-positive ALCL cases. In ALK-negative ALCL patient samples the lethal dose (LD50) values ranged from 56.3 to 76.5nM, whereas both ALK-positive cases demonstrated a LD50 of >100nM.
Figure 1.
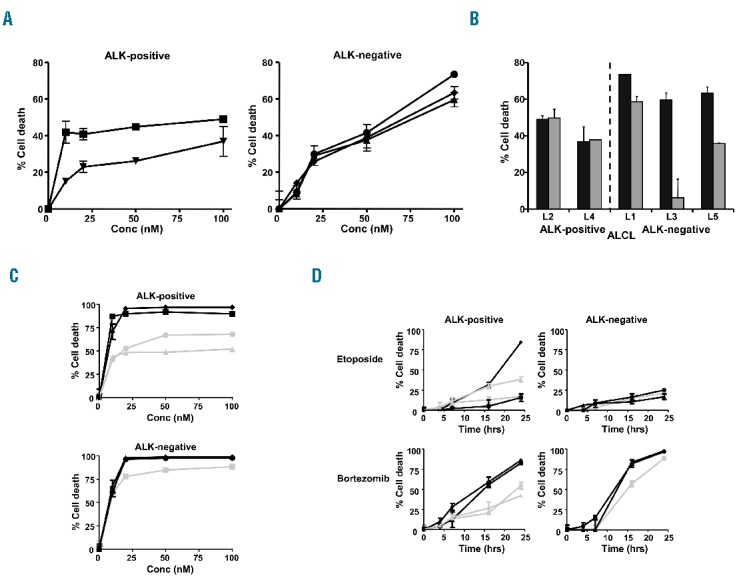
Bortezomib induces cell death of ALCL cells, including etoposide-resistant tumor cells. (A) Dose-response curves of ALK-positive L2 (■) and L4 (▼) and ALK-negative L1(•), L3 (▲) and L5 (♦) ALCL patient samples after 24 hours of treatment with bortezomib. (B) Detection of cell death in ALCL patient cells treated with 100nM bortezomib (■) or 500nM etoposide (■) for 24 hours. (C) Dose-response curves of ALK-positive ALCL cell lines: SUDHL1 (•), SUPM2 (■), L82 (♦) and Karpas 299 (▲) and ALK-negative ALCL cell lines: JK (■), Mac1 (▲) and Mac2A (•) with bortezomib. (D) Detection of cell death in ALCL cell lines: SUDHL1 (•), SUPM2 (■), L82 (♦), Karpas 299 (▲), JK (■), Mac1 (▲) and Mac2A (•) treated with 20nM bortezomib or 500nM etoposide for increasing periods of time.
Most chemotherapeutic drugs (CHOP and etoposide) used in the treatment of ALCL induce apoptosis via the intrinsic apoptosis pathway. Sensitivity to bortezomib was compared with sensitivity to the conventional chemotherapeutic agent etoposide. All ALK-negative ALCL patient samples were highly sensitive to bortezomib and showed relatively lower levels of etoposide-induced cell death. In the ALK-positive samples, no clear difference in sensitivity to bortezomib or etoposide was observed (Figure 1B).
The cell death inducing effect of bortezomib was further investigated in four ALK-positive ALCL cell lines: SUDHL1, SUPM2, L82, Karpas 299 and three ALK-negative ALCL cell lines: JK, Mac1 and Mac2A. All ALCL cell lines tested were sensitive to bortezomib (Figure 1C). Mean LD50 values for ALK-positive and ALK-negative ALCL cell lines were 19nM (range, < 0.01–55.8nM) and 1nM (range, 0.6–2.0nM), respectively, which suggests that also the ALK-negative ALCL cell lines were relatively more sensitive to bortezomib than the ALK-positive ALCL cell lines (Figure 1C). Compared with etoposide, bortezomib induced cell death in four out of five etoposide-resistant cell lines, including one ALK-positive and all ALK-negative ALCL cell lines (Figure 1D).
Upon entering the cell, bortezomib reversibly binds to the threonine hydroxyl group in the chymotrypsin-like active site of the proteasome, thereby blocking its activity. Treatment with bortezomib resulted in proteasome inhibition of more than 80% in all primary ALCL cells and ALCL cell lines tested. The reduction in proteasome activity was similar in ALK-positive and -negative ALCL cells (Online Supplementary Figure S1).
The addition of the pan caspase inhibitor z-VAD-fmk largely blocked the killing of cells induced by bortezomib, indicating that the induced cell death is caspase-dependent (Figure 2A). Moreover, treatment with bortezomib resulted in clearly detectable caspase-3/7 activity and loss of mitochondrial membrane potential (ψm), (Online Supplementary Figure S2, Figure 2B). Experiments with specific caspase-9 inhibitor, LEHD-FMK demonstrated that the observed cell death was caspase-9 dependent in most ALCL cell lines, except for Karpas 299 (Figure 2A). Sensitivity to bortezomib was not, or was less, dependent on caspase-8 activation (data not shown). These findings suggest that bortezomib-induced apoptosis in ALCL mainly involves activation of the intrinsic apoptosis pathway which is consistent with a report in four ALCL cell lines.8
Figure 2.
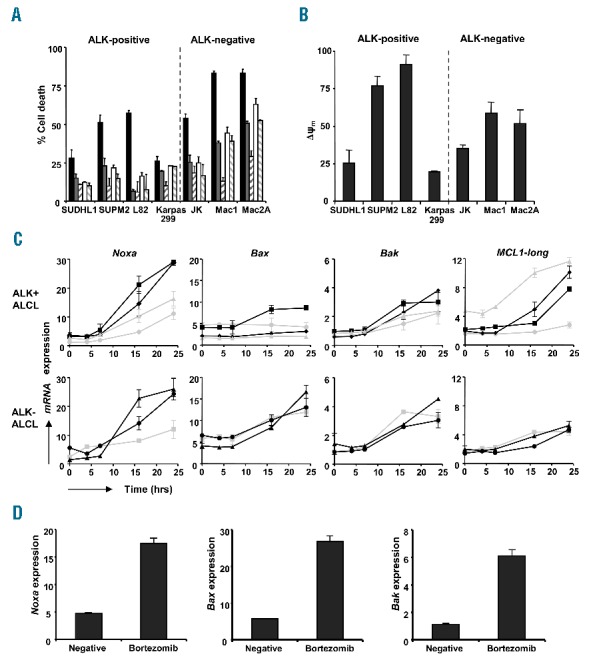
Bortezomib induces caspase-9-mediated apoptosis through modulation of Bcl-2 family members in ALCL patient cells and cell lines. (A) Cell death detected in ALK-positive and ALK-negative ALCL cell lines after treatment with 20nM bortezomib in combination with increasing concentrations of the pancaspase inhibitor z-VAD-fmk or the caspase-9 inhibitor LEHD-fmk for 16 hours. (■) 20nM bortezomib, (■) bortezomib + 20μM zVAD-FMK, (/) bortezomib + 40μM zVAD-FMK, () bortezomib + 20μM LEHD-FMK, (\) bortezomib + 40μM LEHD-FMK. (B) Cell lines were treated with or without 20nM bortezomib for 24 hours and stained with TMRE. ΔΨ was determined as the % unstained cells in bortezomib treated samples – the % unstained cells in untreated samples. (C) Increased mRNA expression of the pro-apoptotic Bcl-2 family members Noxa, Bax and Bak and the anti-apoptotic gene MCL1-long in ALCL cell lines SUDHL1 (•), SUPM2 (■), L82 (♦), Karpas 299 (▲), JK (■), Mac1 (▲) and Mac2A (•) by RT-MLPA analysis after incubation with 20nM bortezomib for increasing periods of time. (D) Increased Noxa, Bax and Bak expression in ALCL patient L3 after treatment with 20nM bortezomib for 24 hours, using RT-MLPA analysis.
Subsequently, we analyzed the effect of bortezomib on the levels of 34 apoptosis-inhibiting and apoptosis-inducing genes implicated in the intrinsic apoptosis pathway using RT-MLPA analysis, (see Online Supplementary Table SI). The most striking accumulation observed was of the BH3-only gene Noxa, which was already detectable after 7 hours of incubation (Figure 2C). Bortezomib-induced upregulation of Noxa seemed to be greater in highly bortezomib-sensitive ALCL cells compared to less sensitive ALCL cells (P=0.007, at 24 hours). Furthermore, considerable rises in gene expression levels of Bax and Bak were detected after incubation (Figure 2C). Evident upregulation of the gene expression of Noxa and Bax, and to a lesser extent of Bak, was also detected in ALCL patient cells (Figure 2D). ALK-negative ALCL cells demonstrated strongly upregulated levels of bortezomib-induced Bax activation compared to ALK-positive ALCL cells (P=0.007, at 24 hours). This difference is probably caused by expression in ALK-positive ALCL of heat shock protein 72 (HSP72) that prevents Bax activation, inhibits the release of cytochrome c from the mitochondria and limits downstream caspase-3 activity.9 HSP72 is involved in the folding of the NPM-ALK kinase and is overexpressed in ALK-positive ALCL, whereas it is underexpressed in ALK-negative ALCL, which might explain the difference in bortezomib-sensitivity between ALK-positive and ALK-negative ALCL. After bortezomib incubation, expression levels of Bcl-2, Bcl-W, Bcl-2A1 and IAP family members were not affected in either ALCL patient cells or cell lines. Bcl-XL expression levels decreased slightly in the highly bortezomib-sensitive ALCL cells. Moreover, bortezomib induced upregulation of the anti-apoptotic gene MCL1-long, and an increase of the pro-apoptotic gene MCL1-short was also found.
Using western blot analysis we could confirm the mRNA expression levels with actual protein expression levels (Figure 3A). Bortezomib increased the expression of MCL1-long, however the expression of the different pro-apoptotic proteins is apparently sufficient for the triggering of apoptosis. This is in agreement with previous studies in mantle cell lymphoma and cutaneous T-cell lymphoma.10,11 MCL1 has a short half-life and is rapidly degraded by the proteasome. Accumulation of MCL1 in ALCL cells might be due to the decrease of proteasomal degradation and/or caspase-3-dependent MCL1 protein cleavage.12 However in our study, a clear increase in MCL1 mRNA expression was also observed, primarily in ALK-positive ALCL cells, indicating that the induction of MCL1 transcription plays an important role in upregulation of MCL1 in these cells. The activation of the transcription of MCL1 might be stimulated by ALK regulated Janus kinase (Jak)/signal transducer and activator of transcription (STAT) dependent pathways.5
Figure 3.
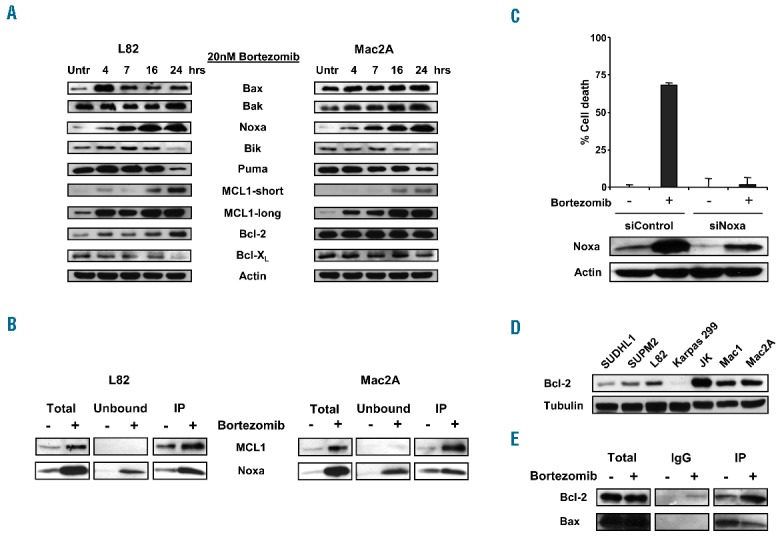
ALK-negative ALCL is sensitive to bortezomib through Noxa upregulation and release of Bax from Bcl-2. (A) Protein expression of pro- and anti-apoptotic Bcl-2 family members from a time-course treatment of ALK-positive L82 ALCL cells and ALK-negative Mac2A ALCL cells with 20nM bortezomib. (B) ALK-positive L82 ALCL cells and ALK-negative Mac2A ALCL cells were treated with 20nM bortezomib for 16 hours. Whole-cell lysates were immunoprecipitated with anti-MCL1 antibody. Total extracts, IP (bound) and unbound fractions were analyzed by western blot analysis for the presence of MCL1 and Noxa expression. (C) Percentage cell death after 16 hours of incubation with 20nM bortezomib in Mac2A-transfected cells with siControl or siNoxa RNA. Expression of Noxa in Mac2A cells after transfection with siControl or siNoxa RNA was analyzed using western blot analysis. (D) Expression of Bcl-2 in ALK-positive and ALK-negative ALCL cell lines using western blot analysis. (E) ALK-negative Mac2A ALCL cells were incubated with 20nM bortezomib for 16 hours. Whole-cell lysates were immunoprecipitated with anti-Bcl-2 antibody. Expression levels of co-precipitating Bax were analyzed using western blot analysis.
Pro- and anti-apoptotic Bcl-2 family members interact with each other and the balance between these proteins can determine cell fate. Immunoprecipitation with MCL1 in bortezomib-treated ALK-positive L82 and ALK-negative Mac2A ALCL cells revealed that Noxa partly co-immunoprecipitated with MCL1 (Figure 3B). These data demonstrate that upregulated Noxa is able to bind to MCL1 and antagonizes the anti-apoptotic effect of MCL1 in both ALK-positive and ALK-negative ALCL cells after bortezomib treatment. The formation of this Noxa-MCL1 complex probably allows the release of Bak from MCL1, resulting in the triggering of apoptosis.
To further substantiate if induction of Noxa expression is involved in sensitivity to bortezomib in ALCL cells, siRNA analysis was used to knockdown Noxa expression. A reduction of almost 60% in Noxa protein levels was observed after transfection with siNoxa (Figure 3C). Mac2A cells transfected with siNoxa showed a decrease of 98% in bortezomib-induced cell death compared with control-transfected cells. Thus, inhibition of Noxa almost completely blocks bortezomib cytotoxicity, suggesting that Noxa is a key protein in bortezomib-induced apoptosis of ALCL cells. Upregulation of Noxa by bortezomib is not caused by proteasome inhibition, but due to activation of transcription. Initially, it was reported that Noxa was activated directly by p53, however several investigators have shown p53-independent induction of Noxa following exposure to bortezomib.13,14 The present study demonstrates that bortezomib is able to trigger Noxa induction in ALCL cells independent of p53 status, although lower levels of upregulated Noxa were observed in ALK-positive SUDHL1 and Karpas 299 ALCL cells carrying mutated p53. In addition, wild-type p53 ALCL cells showed an increase in p53 protein expression after treatment with bortezomib (data not shown), suggesting that p53 is partly involved in upregulation of Noxa.
Similar to previous reports, relatively high expression levels of the anti-apoptotic regulator Bcl-2 were observed in ALK-negative ALCL cells, in contrast with ALK-positive ALCL cells.3 (Figure 3D). Despite this Bcl-2 expression, ALK-negative ALCL cells were sensitive to bortezomib. Since Bax was strongly upregulated after treatment with bortezomib in ALK-negative ALCL cells, we explored if bortezomib would affect the binding of Bcl-2 to Bax. Bcl-2-Bax complexes were found in abundance in untreated Mac2A ALCL cells. However, after treatment with bortezomib, Bax was partly released from Bcl-2, probably due to binding of BH3-only proteins to Bcl-2 (Figure 3E). A recent study in lymphoid cells has shown that Noxa might be an eligible candidate for this interaction, since it is also able to bind to Bcl-2.15
In conclusion, our preclinical data support the potential use of bortezomib as a therapeutic agent in ALK-negative ALCL and might also provide a rationale for the design of new bortezomib combination therapies for the treatment of ALCL patients, in order to improve their prognosis. In vivo studies in human ALCL xenografts and clinical trials in patients with ALCL need to confirm that bortezomib is effective in ALCL, as the microenvironment of ALCL tumor cells might contribute to sensitivity to bortezomib. Moreover, mechanisms by which bortezomib upregulates Noxa expression must be elucidated in further studies, and might provide clinically available biomarkers which can predict sensitivity to bortezomib in each individual patient.
Acknowledgments
the authors would like to thank prof.dr. M. Kadin at the Roger Williams Medical Center, Providence, USA and dr. J. Eberle at the Charité-University Medical Center, Berlin, Germany for providing us the ALK-negative ALCL cell lines used in this study.
Footnotes
Funding: this work was supported by grant VU 2010-4704 from the Dutch Cancer Society. SAGM Cillessen and LM Moesbergen were funded by grant VU 2010-4704 from the Dutch Cancer Society.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Ferreri AJ, Govi S, Pileri SA, Savage KJ. Anaplastic large cell lymphoma, ALK-negative. Crit Rev Oncol Hematol. 2013;85(2):206–215. [DOI] [PubMed] [Google Scholar]
- 2.Ferreri AJ, Govi S, Pileri SA, Savage KJ. Anaplastic large cell lymphoma, ALK-positive. Crit Rev Oncol Hematol. 2012;83(2):293–302. [DOI] [PubMed] [Google Scholar]
- 3.Rassidakis GZ, Sarris AH, Herling M, et al. Differential expression of bcl-2 family proteins in ALK-positive and ALK-negative anaplastic large cell lymphoma of T/null-cell lineage. Am J Pathol. 2001; 159(2):527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlette EJ, Medeiros LJ, Goy A, Lai R, Rassidakis GZ. Survivin expression predicts poorer prognosis in anaplastic large-cell lymphoma. J Clin Oncol. 2004;22(9):1682–1688. [DOI] [PubMed] [Google Scholar]
- 5.Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protect hematopoietic cells from cell death. Oncogene. 2002;21(7):1038–1047. [DOI] [PubMed] [Google Scholar]
- 6.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001; 61(7):3071–3076. [PubMed] [Google Scholar]
- 7.Cillessen SA, Meijer CJ, Ossenkoppele GJ, et al. Human soluble TRAIL/Apo2L induces apoptosis in a subpopulation of chemotherapy refractory nodal diffuse large B-cell lymphomas, determined by a highly sensitive in vitro apoptosis assay. Br J Haematol. 2006; 134(3):283–293. [DOI] [PubMed] [Google Scholar]
- 8.Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007; 21(4):838–842. [DOI] [PubMed] [Google Scholar]
- 9.Bonvini P, Zorzi E, Mussolin L, et al. Consequences of heat shock protein 72 (hsp72) expression and activity on stress-induced apoptosis in CD30+ NPM-ALK+ anaplastic large-cell lymphomas. Leukemia. 2012;26(6):1375–1382. [DOI] [PubMed] [Google Scholar]
- 10.Ri M, Iida S, Ishida T, et al. Bortezomib-induced apoptosis in mature T-cell lymphoma cells partially depends on upregulation of Noxa and functional repression of Mcl-1. Cancer Sci. 2009;100(2):341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pérez-Galán P, Roué G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107(1):257–264. [DOI] [PubMed] [Google Scholar]
- 12.Nencioni A, Hua F, Dillon CP, et al. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood. 2005; 105(8):3255–3262. [DOI] [PubMed] [Google Scholar]
- 13.Qin JZ, Stennett L, Bacon P, et al. P53-independent NOXA induction overcomes apoptotic resistance of malignant melanomas. Mol Cancer Ther. 2004;3(8):895–902. [PubMed] [Google Scholar]
- 14.Nikiforov MA, Riblett M, Tang WH, et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc Natl Acad Sci USA. 2007;104(49):19488–19493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith AJ, Dai H, Correia C, et al. Noxa/Bcl-2 interactions contribute to bortezomib resistance in human lymphoid cells. J Biol Chem. 2011;286(20):17682–17692. [DOI] [PMC free article] [PubMed] [Google Scholar]