

Mutation in ten-eleven-translocation oncogene family member 2 (TET2) has been extensively investigated in the context of several hematologic malignancies and occurs in 7%–23% of patients with acute myeloid leukemia (AML).1–4 TET2 acts to demethylate DNA, and TET2 mutations are leukemogenic in animal models.5–8 Any prognostic role played by TET2 mutation in a patient with normal karyotype (NK)-AML remains a subject of debate.1,2,9–11 Although the prevalence of homozygous TET2 mutation is 14.8% in patients with TET2-mutated myelodysplastic syndrome, and 9.3% in patients with TET2-mutated AML, the prognostic role played by this genotype in the context of treatment outcomes has not been investigated in depth.9,12
In this study, we evaluated the prevalence of TET2 mutations in patients with NK-AML and tried to clarify the prognostic role played by TET2 mutation in patients with NK-AML, especially in those patients with homozygous mutation.
In total, 407 patients were included in the present study, and all met the following eligibility criteria: i) age ≥15 years; ii) a diagnosis of NK-AML confirmed by conventional cytogenetic analysis; and iii) treatment with induction chemotherapy using a standard protocol (a 3-day course of anthracycline with a 7-day course of cytosine arabinoside). Patients with NK-AML were diagnosed between October 1998 and September 2012 in seven participating institutes. Patients who achieved complete remission (CR) received consolidation chemotherapy with or without allogeneic stem cell transplantation (HCT), depending on the availability of a matched related or unrelated donor. Cryopreserved bone marrow (BM) or peripheral blood samples taken at diagnosis were archived before genomic DNA extraction using QIAamp DNA blood mini-kits (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. Mutation analysis was performed using Sanger sequencing according to PCR methodology. The sequencing was performed using an ABI 3130xl genetic analyzer (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s protocol. TET2, fms-related tyrosine kinase 3-internal tandem duplication (FLT3-ITD), and nucleophosmin1 (NPM1) mutation testing was performed as previously described.9,12,13 Homozygosity of TET2 mutations was considered to be present when the mutant nucleotide signal peak was of a single color and equal in height to adjacent nucleotides. Analysis of the CCAAT/enhancer binding protein α (CEBPA) gene is described in Online Supplementary Table S1. More details of the CR criteria, survival end point, and statistical analysis are provided in the Online Supplementary Appendix.
In total, 407 samples were evaluated in terms of TET2 mutation. Sixty-five different TET2 mutations were detected in 54 patients (13.2%) (Online Supplementary Figure S1). Single and double TET2 mutations were detected in 27 patients, and 14 homozygous mutations were observed in patients with double mutations. Details are shown in Online Supplementary Table S2.
TET2 mutation was associated with poor prognostic features, such as older age (P<0.001) or a high white blood cell (WBC) count (P=0.013) (Online Supplementary Table S3). NPM1 mutation was observed more frequently in patients with TET2 mutations (P=0.017). Of 407 patients receiving induction chemotherapy, CR was achieved by 332 (81.6%); no significant difference in CR rates was noted between groups with and without TET2 mutations (75.9% vs. 82.4%, respectively; P=0.250). Allogeneic HCT was performed in 32.1% of the whole patient study cohort, but there was no difference in TET2 mutation frequency between those who did and those who did not receive allogeneic HCT (P=0.154). At the median follow-up time of 59.4 months (range: 0.9–179.8 months) among survivors, long-term outcomes were analyzed by TET2 mutational status. In all patients (n=407), no difference in relapse incidence (RI; 47.6% vs. 43.3% at 5 years; P=0.717), event-free survival (EFS; 28.0% vs. 34.5% at 5 years; P=0.391), or overall survival (OS; 35.8% vs. 37.4% at 5 years; P=0.581) was noted between patients with or without TET2 mutations (Online Supplementary Table S4 and Online Supplementary Figure S2 and). Upon multivariate analysis of factors affecting RI, EFS, and OS, independent risk factors for RI, EFS, and OS were performance of allogeneic HCT and mutations in NPM1, CEBPA, or FLT3-ITD, but neither a TET2 mutation alone nor older age had any prognostic impact on RI, EFS, or OS (Online Supplementary Table S5).
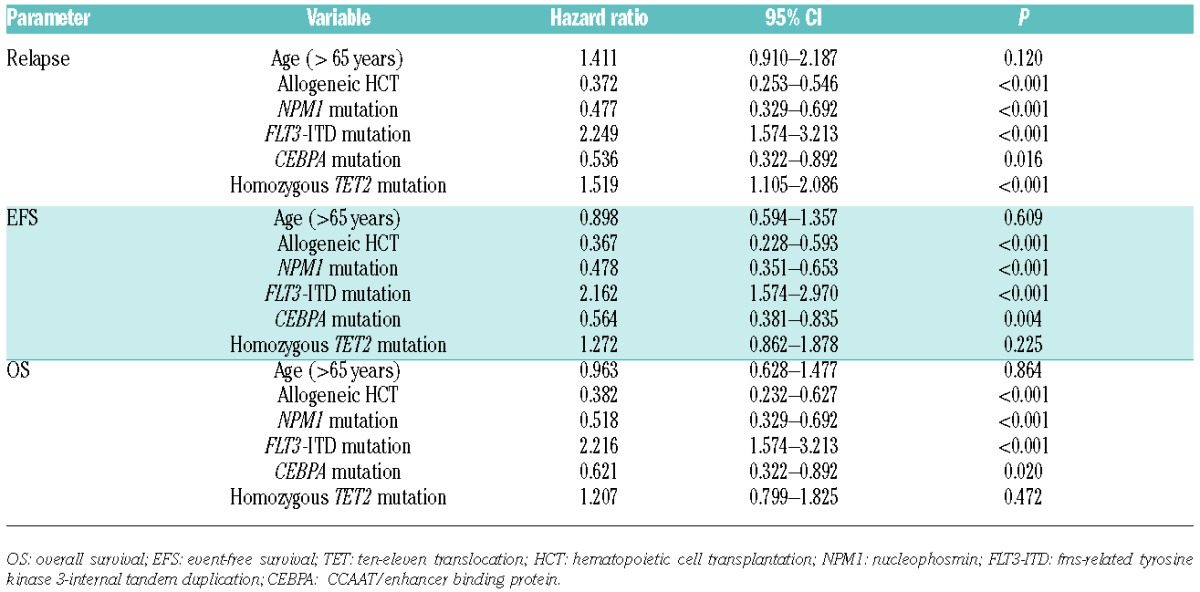
Homozygous TET2 mutations were detected in 14 (25.9%) patients with TET2 mutations. The NPM1 mutation was observed more frequently in patients with non-homozygous TET2 mutations than in patients with homozygous TET2 mutations (P=0.017). However, there was no difference in the clinical features and other molecular mutational status between patients with homozygous and non-homozygous TET2 mutations (Online Supplementary Table S6). We categorized the TET2 mutational status as wild-type, single mutation, and heterozygous double or homozygous mutation; allogeneic HCT was performed in 33.7%, 25.9%, 23.1% and 14.2% of each group without any statistical difference being found (P=0.378). In OS and EFS, there was no statistical difference among TET2 wild-type, TET2 single mutation, and heterozygous double TET2 mutation. However, patients with homozygous TET2 mutation showed significantly inferior EFS compared with those with wild-type TET2 (P=0.048). Importantly, patients with homozygous TET2 mutation showed a higher relapse rate compared with those with wild-type TET2 (RI at 5 years: 100.0% vs. 43.1%; P=0.002) or single TET2 mutations (RI at 5 years: 100.0% vs. 41.1%; P=0.012) or TET2 heterozygous double mutation (RI at 5 years: 100.0% vs. 27.3%; P=0.023). However, the patients with single or heterozygous double TET2 mutations showed similar relapse rates at five years compared with those with wild-type TET2 (36.4% vs. 42.4%; P=0.673) (Online Supplementary Table S7). Homozygous TET2 mutation was an independent adverse prognostic factor for RI in multivariate analysis (HR: 1.519: 95%CI: 1.105–2.089; P<0.001) (Table 1); however, such status appeared not to affect EFS and OS.
Table 1.
Multivariate analysis of overall survival, event-free survival, and relapse incidence in patients with acute myeloid leukemia with risk factors including homozygous TET2 mutations.
Notably, we found that the RI of patients with NK-AML with homozygous TET2 mutations was significantly higher than that of patients with non-homozygous TET2 mutation or wild-type TET2. In animal models, TET2-haploinsufficient mice were similar to mice with homozygous TET2 null mutations, in that both types of animal developed various hematopoietic malignancies.5,6,8 However, also in mouse models, BM cells with heterozygous TET2 null mutations exhibited TET2-encoding mRNA expression levels of 40%–50% those of wild-type animals, whereas no such mRNA was synthesized in homozygous TET2-null mice.8 The 5-hydroxymethylcytosine level reflects dynamic epigenetic changes in DNA, and was reduced dramatically in homozygous TET2-null mice compared to heterozygous TET2-null and wild-type animals. Approximately 33% of homozygous TET2-null and 8% of heterozygous TET2-null mice developed lethal myeloid malignancies in the first year of life, suggesting that disease latency was much longer in heterozygous TET2-null mice than in homozygous TET2-null animals.8 Such results imply that TET2 loss triggers dose-dependent effects on hematopoiesis and myeloid transformation. In other studies, at least one TET2 mutant allele was present in most cells (>70%), and biallelic mutation was also observed frequently in 26%–42%.12,14,15 However, the mutant allele burden or monoallelic versus biallelic TET2 mutation did not significantly correlate with poorer clinical prognosis or OS.9,12,15 We recorded homozygous TET2 mutations in 25.9% of NK-AML patients with TET2 mutations, and this was strongly correlated with higher RI even in comparison with the non-homozygous TET2 mutation group, suggesting that the homozygous TET2 mutation may be associated with a short interval prior to leukemic relapse, thus exhibiting a “threshold effect.”
Our present study had several limitations as regards the interpretation of the clinical significance of homozygous TET2 mutation. First, our study had a methodological limitation in that the detection sensitivity of Sanger fluorescent dideoxynucleotide chain termination sequencing analysis is approximately 10% of mutant alleles. Using sequencing analysis, it is difficult to determine and distinguish biallelic mutations from double mutations because of this relatively low sensitivity, depending on the sequencing trace source. Second, the work was retrospective in nature, included patients treated in several centers, and the consolidative therapies received by patients were not homogeneous. Third, the sample size of the group with homozygous TET2 mutations was small, limiting the power of direct comparisons with patients bearing non-homozygous TET2 mutations or TET2 wild-type. However, our work has significant clinical relevance in that we included a large number of patients with NK-AML only. Our work clarifies the prognostic significance of the TET2 mutation in treated populations, and helps clarify the prognostic significance of such mutation in patients with NK-AML. In addition, the present study is the first to investigate the significance of homozygous TET2 mutational status in patients with NK-AML.
Figure 1.

The prognostic significance according to TET2 mutational status. Overall survival (A), event-free survival (B), and estimated relapse incidence (C).
In conclusion, such non-homozygous TET2 mutations did not influence treatment outcomes. However, homozygous TET2 mutational status was prognostic in terms of a higher RI, suggesting that the TET2 mutation exerts a “threshold effect” in the context of relapse. Further study of larger numbers of patients will yield valuable data on the prognostic role played by the TET2 mutation in terms of treatment outcomes in NK-AML. Additional biological validation work is also required to explore the prognostic role played by homozygous TET2 mutational status in terms of risk of leukemia relapse.
Footnotes
Funding: this study was supported by a grant from the National Project for Personalized Genomic Medicine, Ministry for Health & Welfare, Republic of Korea (A111218-11-GM06). This research was also supported by the Leading Foreign Research Institute Recruitment Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (MEST) (NRF-2011-0030034).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Metzeler KH, Maharry K, Radmacher MD, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29(10):1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaidzik VI, Paschka P, Spath D, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30(12):1350–1357. [DOI] [PubMed] [Google Scholar]
- 3.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28(3):485–496. [DOI] [PubMed] [Google Scholar]
- 6.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko M, Bandukwala HS, An J, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA. 2011; 108(35):14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Cai X, Cai CL, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou WC, Chou SC, Liu CY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–3810. [DOI] [PubMed] [Google Scholar]
- 10.Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. [DOI] [PubMed] [Google Scholar]
- 11.Nibourel O, Kosmider O, Cheok M, et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood. 2010;116(7):1132–1135. [DOI] [PubMed] [Google Scholar]
- 12.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–842. [DOI] [PubMed] [Google Scholar]
- 13.Kim YK, Kim HN, Lee SR, et al. Prognostic significance of nucleophosmin mutations and FLT3 internal tandem duplication in adult patients with cytogenetically normal acute myeloid leukemia. Korean J Hematol. 2010;45(1):36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011; 364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]