

Abstract
In patients with acute coronary syndrome, dual antiplatelet therapy with aspirin and a P2Y12 inhibitor like prasugrel is prescribed for one year. Here, we investigated how the hemostatic function of platelets recovers after discontinuation of prasugrel treatment. Therefore, 16 patients who suffered from ST-elevation myocardial infarction were investigated. Patients were treated with aspirin (100 mg/day, long-term) and stopped taking prasugrel (10 mg/day) after one year. Blood was collected at the last day of prasugrel intake and at 1, 2, 5, 12 and 30 days later. Platelet function in response to ADP was normalized between five and 30 days after treatment cessation and in vitro addition of the reversible P2Y12 receptor antagonist ticagrelor fully suppressed the regained activation response. Discontinuation of prasugrel resulted in the formation of an emerging subpopulation of ADP-responsive platelets, exhibiting high expression of active integrin αIIbβ3. Two different mRNA probes, thiazole orange and the novel 5′Cy5-oligo-dT probe revealed that this subpopulation consisted of juvenile platelets, which progressively contributed to platelet aggregation and thrombus formation under flow. During offset, juvenile platelets were overall more reactive than older platelets. Interestingly, the responsiveness of both juvenile and older platelets increased in time, pointing towards a residual inhibitory effect of prasugrel on the megakaryocyte level. In conclusion, the gradual increase in thrombogenicity after cessation of prasugrel treatment is due to the increased activity of juvenile platelets.
Introduction
The autocrine mediator ADP is a moderately strong platelet agonist, stimulating platelet aggregation and thrombus formation and contributing to thrombus stabilization.1 At present, the ADP receptor P2Y12 is one of the most effective targets for antiplatelet therapy, i.e. by clopidogrel, prasugrel, and ticagrelor.2,3 Clopidogrel in combination with aspirin has been the standard therapy over the last decade to prevent recurrent atherothrombotic complications in patients who had a myocardial infarction.4 More recently, prescription of prasugrel or ticagrelor instead of clopidogrel is increasingly recommended following publication of the TRITON and PLATO studies, which demonstrated a net clinical benefit over clopidogrel on top of aspirin, due to a higher degree of platelet inhibition.4–9 The thienopyridines clopidogrel and prasugrel are both prodrugs, which require metabolic conversion to form an active metabolite that irreversibly interacts with the P2Y12 receptors of circulating platelets.10 In contrast, ticagrelor is a reversible, non-competitive P2Y12 antagonist (belonging to the cyto-pentyl-triazolo class of pyrimidines) interacting with the platelet receptors without metabolic conversion.10–12
Due to the irreversible inhibition of the platelet P2Y12 receptors by the active metabolites of clopidogrel and prasugrel, the formation of new platelets is required to recover platelet function.7,9 After ticagrelor treatment, recovery of platelet function is only determined by the elimination time of the drug (half-life of 8 h).10 There is limited evidence that during the offset period of irreversible P2Y12 inhibitors (usually prescribed for one year) patients may have an increased risk of a recurrent myocardial event.13–17 This may point to hyperactivity of the newly formed platelets in the offset period, although the mechanism is unclear.
Previous experiments with rats have indicated that stopping clopidogrel treatment resulted in the sudden appearance of a population of fully responsive platelets, whereas recovery from ticagrelor treatment led to a more gradual regain in function of all platelets.18 The newly formed, juvenile platelets with active P2Y12 receptors seemed to preferentially incorporate into thrombi generated under flow conditions.18
Juvenile platelets, also described as reticulated platelets because of the presence of reticular-bound mRNA, are those platelets that are shed most recently from the megakaryocytes in the bone marrow.19–21 Due to the gradual degradation of mRNA, they form only a small part of the entire platelet population. Little is known about the properties of reticulated platelets, although incidental studies report on a larger size with more granules,22 and a high reactivity towards platelet agonists.23,24 With a Sysmex analyzer, using cell-permeant fluorescent mRNA dyes containing polymethine and oxazine, or using the mRNA probe thiazole orange, evidence has been obtained that patients with more reticulated platelets respond less effective to clopidogrel or prasugrel medication.25–27
In the present study, we investigated how the hemostatic function of platelets recovers after discontinuation of prasugrel treatment. We hypothesized an immediate recovery of newly formed, juvenile platelets to the level of untreated platelets. Our data provide evidence for a critical role of the juvenile platelets in the regained aggregation of platelets and thrombus formation, and also show that these platelets gradually increase in responsiveness.
Methods
Patients and control subjects
This study was approved by the local medical ethics committee (MEC 12-3-075). All patients and healthy volunteers gave written informed consent to participate in the study according to the Declaration of Helsinki. Sixteen patients were studied who were treated with prasugrel (10 mg/day) for one year and long-term aspirin (80–100 mg/day) due to a myocardial infarction with ST elevation. After one year of prasugrel treatment, blood was collected on the last treatment day, and at 1, 2, 5 and 30 days later. From 2 patients, blood samples were also collected after 12 days to better understand the delayed regain of platelet function. Patients with a malignancy, active infection or a known platelet disorder were not included. Blood was obtained by venipuncture into Vacuette tubes, containing K2-EDTA, for measurement of hemostatic variables and immature platelet fraction (IPF) using a Sysmex XN-9000 analyzer (Sysmex, Chuo-ku Kobe, Japan); 3.2% (w/v) trisodium citrate for platelet function measurements, or hirudin for whole blood platelet aggregation. Control experiments were performed with blood drawn from healthy volunteers collected in trisodium citrate or acidic citrate dextrose.28
Preparation of platelet-rich plasma, platelets and red cells
Platelet-rich plasma (PRP), platelet-free plasma and washed platelets were prepared as described.29 Platelet counts were determined with a thrombocounter XP300 Sysmex analyzer (Sysmex, Chuo-ku Kobe, Japan). Washed red blood cells were prepared as previously shown.30
Irreversible P2Y12 inhibition in vitro
PRP from healthy donors was treated with lysine aspirin.28 The platelets were incubated with the active metabolite of clopidogrel (CAM) or vehicle medium. Mixtures of washed CAM-treated and vehicle-treated platelets were used for measurement of platelet aggregation, integrin αIIbβ3 activation by flow cytometry and perfusion experiments with reconstituted whole blood.
Platelet aggregation
Aggregation of platelets in PRP was measured using a Chronolog aggregometer (Stago, Asnières sur Seine Cedex, France).31 Aggregation of platelets in whole blood was measured by Multiplate impedance aggregometry (Roche Diagnostics, Basel, Switzerland) as described.32 Ticagrelor (1 μM), being more potent than prasugrel,33 was added in vitro to block residual P2Y12 activity, where indicated.
Flow cytometric analysis of platelet subpopulations
Flow cytometry was performed on an Accuri C6 flow cytometer with CFlow Plus software (Becton-Dickinson Bioscience). To check for integrin αIIbβ3 activation, platelets were activated with 2MeS-ADP in the presence of FITC-conjugated PAC-1 antibody against the activated αIIbβ3 integrin. Activated platelets were identified as before.31 Ticagrelor (1 μM) was added, where indicated.
Juvenile platelets were identified using two different methods of mRNA staining, i.e. with thiazole orange34 or by a novel method using Cy5-labeled oligo-dT, which binds to the poly-A tail of mRNA species. Thiazole orange (15% in filtered PBS) was added to PRP, according to established procedures.34 Samples were activated with 2MeS-ADP in the presence of AF647-fibrinogen. For staining with 5′Cy5-oligo-dT, washed platelets were activated with 2MeS-ADP in the presence of OG488-fibrinogen. Samples were fixed with 0.2% formaldehyde, permeabilized with 0.1% saponin and subsequently incubated with 5′-Cy5-oligo-dT at 37°C. For all samples, 5′Cy5-oligo-dA was used as a negative control probe to check for specificity of the staining. Color compensation was not required as fluorescent spectra did not overlap.
The average percentage of juvenile platelets as analyzed by the thiazole orange staining and the oligo-dT staining was 6.7% (±1.9%) and 21.5% (±5.8%), respectively. The discrepancy in the percentage of detected juvenile platelets can be explained by the higher sensitivity of the oligo-dT staining to detect mRNA in comparison to thiazole orange. In order to use a uniform definition of juvenile platelets, the threshold for juvenile platelets was based on the IPF as determined by the Sysmex XN9000 analyzer, which is an internationally validated method in the clinic. An alternative analysis of juvenile platelets, based on the negative controls of both stainings, is presented in Online Supplementary Figure S3.
Thrombus formation in whole blood
Whole blood thrombus formation on microspots in a parallel-plate flow chamber was measured, basically as described before.35 Patient blood samples were perfused through the chamber for 4 min at a wall-shear rate of 1600 s−1, while 2MeS-ADP (0.1 μM, f.c.) was co-perfused with a second pump. Ticagrelor was added where indicated. Thrombi were stained with AF647-labeled fibrinogen and, when indicated, with DiOC6.35 Brightfield and fluorescence microscopic images were captured with an EVOS fluorescence microscope, equipped with a 60x oil objective. Images were analyzed using Metamorph (Molecular Devices, Sunnyvale CA, USA) and ImageJ (open access) software.35
For measurement of thrombus formation of reconstituted blood samples from healthy controls, mixtures of CAM- and vehicle-treated platelets were added to washed red cells and plasma. In these experiments, the CAM-treated and vehicle-treated platelets were pre-labeled with the membrane probes CellVue Maroon and PKH26, respectively. Microscopic DIC and confocal fluorescent images were taken using a Zeiss LSM7 microscope (Oberkochen, Germany).35
Statistical analysis
Statistical analysis was performed using the SPSS Statistics 22 package (Armonk, NY, USA). Statistical analysis was performed using a one-way-repeated-measures-ANOVA or with a Friedman test with a post hoc Wilcoxon signed rank test. Bonferroni correction was applied when comparing multiple groups.
For a detailed description of the methods, please see the Online Supplementary Appendix.
Results
P2Y12-inhibited platelets participate less in thrombus formation
In order to determine how platelets with non-responsive P2Y12 receptors interact with responsive platelets in aggregation and thrombus formation, platelets from control subjects were treated with the clopidogrel active metabolite (CAM) and mixed in various proportions with untreated platelets. All platelets were also treated with aspirin, in order to mimic conditions as in patients. Flow cytometric analysis indicated that, upon stimulation with ADP, these platelet mixtures formed two distinct populations in terms of activation of integrin αIIbβ3 and binding of OG488-fibrinogen. The population of fibrinogen-binding platelets decreased with increasing fractions of CAM-treated platelets (Figure 1A and Online Supplementary Figure S1A). Addition of the P2Y12 receptor antagonist ticagrelor decreased the population of fibrinogen-binding platelets to the level of 100% CAM-treated platelets (Figure 1A), indicating that the CAM treatment had fully blocked the P2Y12 receptors. Light transmission measurements indicated a gradual decrease in ADP-induced platelet aggregation, when the fraction of CAM-treated platelets increased (Figure 1B). Addition of ticagrelor again antagonized the remaining aggregation response. Together, these data suggest that the reduced integrin activation of the CAM-treated platelets prevented their incorporation into aggregates.
Figure 1.
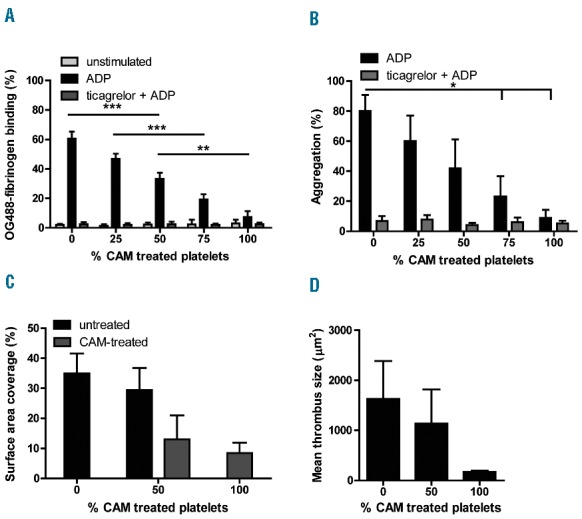
Impaired aggregation and contribution to thrombus formation of P2Y12-inhibited platelets. (A and B) Different mixtures of uninhibited platelets and CAM-treated platelets were pre-incubated with vehicle or 1 μM ticagrelor, and stimulated with 20 μM 2MeS-ADP. (A) Fractions of OG488-fibrinogen binding platelets, representative for uninhibited platelets with activated integrin αIIbβ3 (flow cytometry). (B) Maximal aggregation (% change in light transmission) within 5 minutes. (C and D) Reconstituted blood with different fractions of CAM-treated platelets was perfused 4 minutes over collagen at 1600 s−1 in the presence of 2MeS-ADP. Uninhibited platelets were pre-labeled with PKH26 and CAM-treated platelets with CellVue Maroon; for images see Online Supplementary Figure S2. (C) Quantification of the surface area covered by the populations of labeled platelets. (D) Mean thrombus size. Means ± SD (n=3), *P<0.05, **P<0.01, ***P<0.001 (ANOVA with Bonferroni correction).
To investigate this further, we assessed how the CAM-treated platelets participated in thrombus formation on immobilized collagen under high-shear flow conditions. Therefore, the P2Y12-inhibited platelets were labeled with the red-exciting membrane label CellVue Maroon, whereas the untreated platelets were labeled with the green-exciting membrane label PKH26. This labeling did not affect platelet activation responses (data not shown). Mixtures with 0%, 50% or 100% of CAM-treated platelets were added to red blood cells and plasma from the same donor to obtain reconstituted blood with different proportions of P2Y12 -inhibited platelets. In comparison to reconstituted blood with solely uninhibited platelets, increasing proportions of CAM-treated platelets had limited impact on platelet adhesion to the collagen surface, but markedly suppressed the formation of large platelet aggregates (Online Supplementary Figure S2). As a result, with CAM-treated platelets, surface area coverage (Figure 1C) and mean thrombus size (Figure 1D) on collagen progressively decreased. Strikingly, with 50% of CAM-treated platelets, approximately 30% of the surface area coverage was occupied by the P2Y12 inhibited platelets, while the remaining 70% was occupied by the P2Y12-responsive platelets. Together, these results indicate that P2Y12 -inhibited platelets participate less in platelet aggregation and thrombus formation.
Gradual restoration of platelet aggregation in patients upon prasugrel offset
The offset phase of prasugrel medication was studied in 16 patients. The patients had a mean age of 59±9 years (mean±SD); 3 patients were diagnosed with type II diabetes mellitus (Online Supplementary Table S1). Blood samples taken at day 0 (i.e. last day of prasugrel intake) showed a normal hematocrit of 0.435±0.035 L/L and platelet count of 239±81×109/L. Subsequent blood samples were taken at days 1, 2, 5, and 30, and no noticeable changes in hematocrit or platelet count were observed.
Measurements of whole blood aggregation (Multiplate assay) showed a gradual increase in ADP-induced aggregation upon offset from days 2 to 30 (Figure 2A). Interestingly, the aggregation response further increased at day 30 in comparison to day 5. Whole blood aggregation in response to arachidonic acid remained below the normal range (Figure 2B), thus confirming that all patients still used aspirin during the offset period. The aggregation responses to collagen and thrombin receptor-activating peptide (TRAP) were within the normal ranges, but slightly increased at later sampling points (Figure 2C and D). Similarly, light transmission aggregometry in PRP indicated a restoration in ADP-induced platelet aggregation from day 2 onward (Figure 3A). In this test, the aggregation response with two ADP concentrations was near maximal already at day 5. Collagen-induced platelet aggregation also significantly improved, but only at later time points (Figure 3B). Control experiments in the presence of ticagrelor showed that the increase in aggregation during prasugrel offset was fully antagonized, confirming that the regained platelet reactivity was fully due to increased P2Y12 receptor function (Figure 3A and B).
Figure 2.
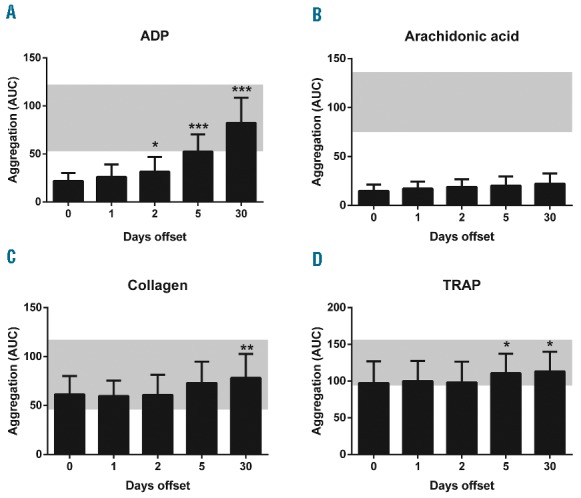
Partial restoration of platelet aggregation in whole blood upon prasugrel offset. Whole blood samples from patients (at indicated days after stopping prasugrel intake) were stimulated with 6.4 μM ADP (A), 0.5 mM arachidonic acid (B), 3.2 μg/mL collagen (C), or 32 μM TRAP (D). Measurements by Multiplate impedance aggregometry; outcome expressed as the area under the impedance curve (AUC). Gray blocks indicate normal ranges, established for healthy subjects. Means ± SD (n = 15–16), *P<0.05, **P<0.01, ***P<0.001 versusday 0 (ANOVA with Bonferroni correction).
Figure 3.
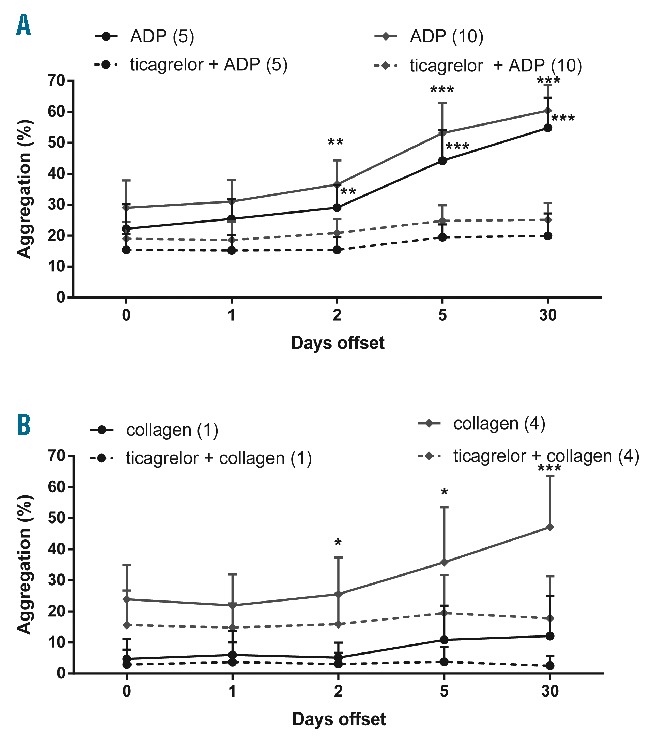
Restored ADP- and collagen-induced aggregation of platelets upon prasugrel offset. Patient PRP (at indicated days after stopping prasugrel intake) activated with 5–10 μM ADP (A) or 1–4 μg/mL collagen (B), in the presence or absence of 1 μM ticagrelor (blocking P2Y12 receptors). Platelet aggregation was assessed by light transmission aggregometry (% change in maximal light transmission). Means ± SD (n = 16), *P<0.05, **P<0.01 and ***P<0.001 versus day 0 (ANOVA with Bonferroni correction).
Formation of a highly reactive population of juvenile platelets upon prasugrel offset
Based on earlier experiments with rats,18 we expected during the offset phase of prasugrel medication the rapid formation of a population of newly formed, fully P2Y12-responsive platelets. A pertinent question was how the regained response in P2Y12 receptor activity was linked to the appearance of this new platelet population. To investigate this, flow cytometry was used to analyze platelets stimulated with the stable (nucleotidase-resistant) ADP analog, 2MeS-ADP, for binding of FITC-labeled PAC-1 antibody, indicative of integrin αIIbβ3 activation. At day 0, a limited fraction of 26±9% of the platelets showed activated αIIbβ3, and this fraction (recognized as a separate peak in the histograms) gradually increased to 56±5% at day 5 and 72±7% at day 30 (Figure 4A and B). In comparison, activation of aspirin-treated platelets from healthy control subjects with 2MeS-ADP resulted in a similar percentage of platelets with activated αIIbβ3 as platelets collected at 30 days after prasugrel cessation (Figure 4C). In platelets from patients (Figure 4B) and healthy controls (Figure 4C), the fraction of platelets binding FITC-PAC1 antibody was greatly, but incompletely reduced by the addition of ticagrelor. Co-incubations with the P2Y1 receptor antagonist MRS-2179 indicated that the residual αIIbβ3 activation was most likely due to activation via P2Y1 receptors.
Figure 4.
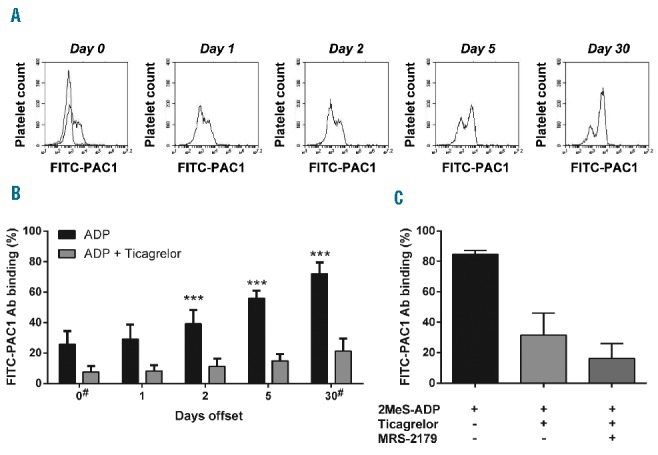
Reappearance of ADP-responsive platelets upon prasugrel offset. Whole blood from patients or healthy controls was preincubated with vehicle, 1 μM ticagrelor or 50 μM MRS-2179 (as indicated), and then stimulated with 1 μM 2MeS-ADP. Activation of αIIbβ3 was assessed by flow cytometric analysis using FITC-labeled PAC-1 mAb. (A) Representative histograms of PAC-1 mAb binding to patient platelets stimulated with ADP (black) or unstimulated (gray) during offset. (B) Quantification of positive platelets. (C) Flow cytometric analysis of healthy control platelets. Means ± SD (n=16), ***P<0.001 versus day 0 ADP (ANOVA with Bonferroni correction); #n=15.
Several assays were performed to determine whether the accumulating platelets with activated αIIbβ3 indeed consisted of newly formed, juvenile platelets. Therefore, platelet mRNA was quantified using two different mRNA probes: thiazole orange as an established, but weak fluorescent mRNA dye;22 and 5′Cy5-labeled oligo-dT, binding to the platelet mRNA poly-A tails,21 which was added to pre-activated and permeabilized platelets. Online Supplementary Figure S3 shows typical dot plots of unstimulated and ADP-activated platelets stained with the 5′Cy5-oligo-dT (Online Supplementary Figure S3A and B) or thiazole orange (Online Supplementary Figure S3C and D). Juvenile and old platelets were discriminated per blood sample, based on the negative control and the immature platelet fraction determined by Sysmex XN-9000 (Figure 5A). For the majority of the patients, with one noticeable exception, the immature platelet fraction was in the normal range, with mean values of 3.6±1.9%, and remaining constant per patient during the study (Figure 5A). Markedly, with either mRNA probe (thiazole orange or 5′Cy5-oligo-dT), the fraction of platelets with positive staining showed increased fibrinogen binding following ADP stimulation from day 2 on (Figure 5B and C). Fibrinogen binding (αIIbβ3 activation) of this juvenile platelet population continued to increase from day 5 to day 30. The difference between 5 and 30 days was significant for thiazole orange (P=0.003), and was borderline significant for 5′Cy5-oligo-dT (P=0.063), thus suggesting partial inhibition of these platelets even after 5 days offset. With either mRNA probe, the fraction of (older) platelets with negative staining was substantially lower in fibrinogen binding, with noticeable increase of activated integrins only from day 5 onwards (Figure 5B and C). The difference in fibrinogen binding between juvenile and older platelets was significant at all days (P<0.001). These results were confirmed using an alternative analysis based on the appropriate negative controls (Online Supplementary Figure S3) Here again, the reactivity of juvenile platelets, expressed as activation ratio, was significantly higher in comparison to mature platelets at all days. Moreover, this analysis also shows the increasing reactivity of juvenile platelets over time for both stainings (Online Supplementary Figure S4A). Additional platelet measurements with a limited number of patients showed that, after 12 days of prasugrel discontinuation, the reactivity of juvenile platelets was in between the day 5 and 30 values (Online Supplementary Figure S4B). Interestingly, in patients with a high IPF (≥7.0%), the population of juvenile platelets showed a faster increase in fibrinogen binding, being near maximal already at day 5 of prasugrel offset (Online Supplementary Figure S4A and B).
Figure 5.
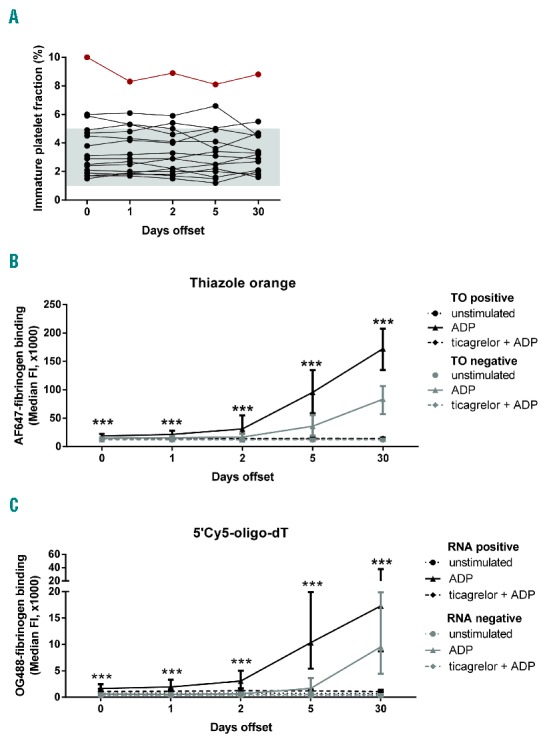
Increased ADP-induced αIIbβ3 activation of juvenile platelets formed upon prasugrel offset. Platelets from 16 patients during offset from prasugrel were activated and analyzed by flow cytometry, with per sample a gating for juvenile platelets based on the immature platelet fraction (Sysmex XN-9000 analyzer). (A) Immature platelet fraction for each of the patients during offset, as determined with a Sysmex XN-9000 analyzer. Red dots are from a patient with a high immature platelet fraction (IPF = 8.8%). Juvenile platelets were identified by staining with thiazole orange or 5′Cy5-oligo-dT. (B) PRP was stained with thiazole orange and activated with 1 μM 2MeS-ADP in the presence of AF647-fibrinogen. Shown is extent of activated αIIbβ3 of thiazole-positive and -negative platelets, as assessed from fibrinogen binding. (C) Washed platelets were activated with 1 μM 2MeS-ADP in the presence of OG488-fibrinogen. The cells were subsequently fixed and permeabilized with saponin to allow staining of mRNA by incubation with 5′Cy5-oligo-dT. Negative control stains were performed with 5′Cy5-oligo-dA. Shown is extent of αIIbβ3 activation of mRNA-positive and -negative platelets, as determined from fibrinogen binding. Medians ± IQR (n = 16), ***P<0.001 versus corresponding mature platelet fraction (Friedman test).
Gradual increase in thrombus size upon prasugrel offset
To investigate whether the increased reactivity of juvenile platelets after prasugrel cessation translates into enhanced thrombus formation, whole blood was perfused over microspots containing vWF/fibrinogen or type I collagen.35 Given the major role of P2Y12 signaling in thrombus buildup,36 we determined thrombus size at the different time points. Regardless of the surface, at day 0 many single platelets and small aggregates were detected, whilst at later offset days larger aggregates were formed. From the recorded brightfield images it was apparent that the thrombi at days 5 and 30 displayed a more contracted morphology (Figure 6A). Quantification of the feature size showed on both microspots a progressive increase in mean thrombus size during offset (Figure 6B and C). On the other hand, platelet adhesion to vWF/fibrinogen or collagen was not changed between day 0 and 30, as surface area coverage (by single platelets and aggregates) remained similar (Online Supplementary Figure S5A and B).
Figure 6.
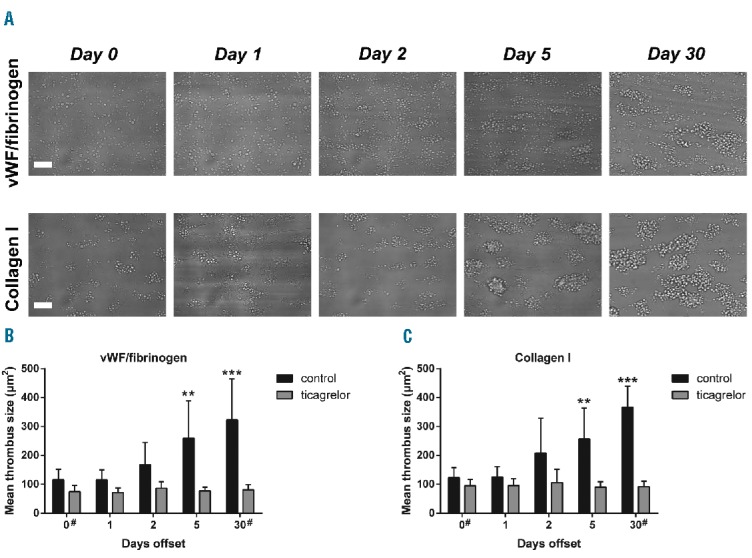
Restored thrombus formation under flow upon prasugrel offset. Whole blood samples from patients (at indicated days after stopping prasugrel intake) were co-perfused with 0.1 μM 2MeS-ADP over microspots containing vWF/fibrinogen or collagen type I, at a shear rate of 1600 s−1 for 4 min. Vehicle (control) or 10 mM ticagrelor was added to the blood. (A) Brightfield images of thrombi from a representative patient formed on the two microspots. Bars = 25 mm. Assessment of mean thrombus size for surfaces with vWF/fibrinogen (B) or collagen I (C). Means ± SD (n=16), **P<0.01, ***P<0.001 versus day 0 (ANOVA with Bonferroni correction); #n=15.
Staining of thrombi with AF647-labeled fibrinogen allowed assessment of integrin αIIbβ3 activation. On both the vWF/fibrinogen and collagen microspots, a marked increase in fibrinogen binding to the aggregated platelets was detected for the later blood samples (Online Supplementary Figure S6A and B). Addition of ticagrelor to the blood resulted in a nearly complete abolition of platelet aggregation at all offset days, but did not block platelet adhesion. With blood samples from a limited number of patients, staining with DiOC6 made it possible to record z-stacks by confocal microscopy for 3D visualization of the thrombi. This illustrated the gradual increase in thrombus size from day 0 to day 5 and 30 (Online Supplementary Figure S6C). Taken together, these results indicate that the accumulation of highly P2Y12 -responsive, juvenile platelets upon prasugrel offset led to a gradual increase in formation of large-size thrombi.
Discussion
In this paper, we confirm earlier findings7,9,15,16,37 that when treatment with an irreversible P2Y12 antagonist is stopped, platelet aggregation in response to ADP gradually recovers in time, when measured by light transmission aggregometry. Near maximal aggregation after stopping prasugrel intake was already reached at day 5, which is in line with results from earlier trials, such as Recovery showing recuperation of this platelet response after 7–9 days of prasugrel cessation.9 However, we also found that other platelet function tests, such as ADP-induced whole blood aggregation and integrin αIIbβ3 activation, were incompletely recovered at day 5 in comparison to day 30. In vitro addition of ticagrelor completely antagonized the time-dependent increase in platelet responses, thereby proving that the recuperation was due to regained P2Y12 signaling.
Detailed flow cytometric analysis indicated that the functional recovery during prasugrel offset was caused by the appearance of a population of juvenile platelets that was increasingly responsive towards ADP. Separation of newly formed and older platelets with two mRNA probes, thiazole orange and a new Cy5-conjugated oligo-dT probe, revealed increased responsiveness to ADP of the positively stained platelet population in terms of integrin αIIbβ3 activation and fibrinogen binding. However, both probes also gave unexpected results. First, we observed a marked increase in ADP responsiveness of the juvenile platelet population after only two days of offset, and for the older platelet population after five days. This suggested that the majority of juvenile platelets formed during the first days still had inhibited P2Y12 receptors, taking into account the presence of the prasugrel active metabolite in the circulation for 7–8 h post prasugrel administration.38 Second, we found a steady rise in the responsiveness of juvenile platelets up to days 12 and 30 of offset. This also points towards residual ADP receptor inhibition of new platelets, likely at the megakaryocytic level, for more than five days. In agreement with this hypothesis, experimental animal models have shown that prasugrel is present in the bone marrow.39 An alternative explanation for this phenomenon might be a long-term increase in autocrine platelet-stimulating effects due to the larger population of P2Y12-responsive platelets.3,40 However, flow cytometric analysis did not point to a higher extent of integrin αIIbβ3 activation of the ADP-responsive platelet population as a whole.
To determine how the increased reactivity of juvenile platelets translates into hemostasis, we studied thrombus formation under flow on two different adhesive surfaces using whole blood. Platelet deposition and aggregate formation on the vWF/fibrinogen and the collagen surfaces restored during the offset and was only maximal at day 30. The regained P2Y12 activity was most apparent from thrombus size, with larger thrombi towards the end phase of the offset. This is in agreement with earlier work showing that signaling via P2Y12 is crucial for thrombus formation and stabilization.1,36 Others have also found a relative preponderance of thiazole orange-stained platelets in a thrombus.41
In recent years, it has been debated whether the termination of clopidogrel or prasugrel intake leads to a rebound effect of recurrent cardiovascular events, perhaps related to platelet hyper-reactivity. Several research groups did report a rebound effect within 90 days after cessation of clopidogrel,13–17 while other researchers could not confirm this.37,42–44 This discussion has led to clinical trials investigating the effect of tapering clopidogrel medication with the idea to prevent platelet hyper-reactivity, but with no beneficial effect so far.42,43,45 The present work may explain this ambiguity in clinical offset effects. On the one hand, cessation of clopidogrel or prasugrel medication will lead to the appearance of the newly formed platelets with uninhibited P2Y12 receptors, which preferentially partake in thrombus formation. On the other hand, as shown in this study, at least during the first few days these juvenile platelets do not appear to be hyperactive, possibly due to residual receptor blockage at the megakaryocyte level. When tapering the medication, the prolonged time interval between two consecutive dosages will result in the alternative formation of uninhibited and inhibited platelets.
The present study has potential limitations, as we have investigated a relatively small number of patients. Further, in our initial ex vivo studies we used the active metabolite of clopidogrel. Although prasugrel is a more potent P2Y12 antagonist in comparison to clopidogrel,5 we added the active metabolite of clopidogrel at concentrations high enough for maximal inhibition.
Patients on dual antiplatelet therapy who require surgery are recommended to stop prasugrel intake seven days beforehand.9 Our findings that prasugrel can still affect the reactivity of juvenile platelets during several days after treatment cessation does not plea for a shortening of this period. The compromised reactivity of juvenile platelets during the initial days of offset can contribute to a risk of bleeding upon surgery. When urgent surgery is required, or when bleeding has to be controlled, platelet transfusions have shown to be effective in restoring hemostasis at 6 h after a loading dose of prasugrel.46 Altogether, the present study provides clinically relevant detailed insights into the mechanisms of prasugrel offset, and thereby provides better insight into the optimal treatment regimen of P2Y12 inhibitors.
Acknowledgments
The authors thank S. Lelieveld and the central diagnostic laboratory for technical support and R. Dennert for assisting in patient recruitment.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Cosemans JM, Munnix IC, Heller R, Jackson SP, Heemskerk JW. Continuous signaling via PI3K isoforms b and g is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood. 2006;108(9):3045–3052. [DOI] [PubMed] [Google Scholar]
- 2.Cattaneo M. The platelet P2Y12 receptor for adenosine diphosphate: congenital and drug-induced defects. Blood. 2011;117(7):2102–2112. [DOI] [PubMed] [Google Scholar]
- 3.Swieringa F, Kuijpers MJ, Heemskerk JW, van der Meijden PE. Targeting platelet receptor function in thrombus formation: the risk of bleeding. Blood Rev. 2014;28(1):9–21. [DOI] [PubMed] [Google Scholar]
- 4.Steg PG, James SK, Atar D, et al. ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33(20):2569–2619. [DOI] [PubMed] [Google Scholar]
- 5.Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–2015. [DOI] [PubMed] [Google Scholar]
- 6.Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. [DOI] [PubMed] [Google Scholar]
- 7.Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the onset and offset of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the onset/offset study. Circulation. 2009;120(25):2577–2585. [DOI] [PubMed] [Google Scholar]
- 8.Cannon CP, Harrington RA, James S, et al. Comparison of ticagrelor with clopidogrel in patients with a planned invasive strategy for acute coronary syndromes (PLATO): a randomised double-blind study. Lancet. 2010;375:283–293. [DOI] [PubMed] [Google Scholar]
- 9.Price MJ, Walder JS, Baker BA, et al. Recovery of platelet function after discontinuation of prasugrel or clopidogrel maintenance dosing in aspirin-treated patients with stable coronary disease. J Am Coll Cardiol. 2012;59(25):2338–2343. [DOI] [PubMed] [Google Scholar]
- 10.Floyd CN, Passacquale G, Ferro A. Comparative pharmacokinetics and pharmacodynamics of platelet adenosine diphosphate receptor antagonists and their clinical implications. Clin Pharmacokinet. 2012;51(7):429–442. [DOI] [PubMed] [Google Scholar]
- 11.Van Giezen JJJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Sem Thromb Hemost. 2005;31(2):195–204. [DOI] [PubMed] [Google Scholar]
- 12.Van Giezen JJJ, Nilsson L, Berntsson P, et al. Ticagrelor binds to human P2Y12 independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. J Thromb Haemost. 2009;7(9):1556–1565. [DOI] [PubMed] [Google Scholar]
- 13.Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA. 2008;299(5):532–539. [DOI] [PubMed] [Google Scholar]
- 14.Ho PM, Tsai TT, Wang TY, et al. Adverse events after stopping clopidogrel in post-acute coronary syndrome patients: insights from a large integrated healthcare delivery system. Circ Cardiovasc Qual Outcomes. 2010;3(3):303–308. [DOI] [PubMed] [Google Scholar]
- 15.Mylotte D, Peace AJ, Tedesco AT, et al. Clopidogrel discontinuation and platelet reactivity following coronary stenting. J Thromb Haemost. 2011;9(1):24–32. [DOI] [PubMed] [Google Scholar]
- 16.Charlot M, Nielsen LH, Lindhardsen J, et al. Clopidogrel discontinuation after myocardial infarction and risk of thrombosis: a nationwide cohort study. Eur Heart J. 2012;33(20):2527–2534. [DOI] [PubMed] [Google Scholar]
- 17.Mauri L, Kereiakes DJ, Yeh RW, et al. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med. 2014;371(23):2155–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuijpers MJ, Megens RT, Nikookhesal E, et al. Role of newly formed platelets in thrombus formation in rat after clopidogrel treatment: comparison to the reversible binding of P2Y12 antagonist ticagrelor. Thromb Haemost. 2011;106(6):1179–1188. [DOI] [PubMed] [Google Scholar]
- 19.Ault KA, Knowles C. In vivo biotinylation demonstrates that reticulated platelets are the youngest platelets in circulation. Exp Hematol. 1995;23(9):996–1001. [PubMed] [Google Scholar]
- 20.Harrison P, Goodall AH. “Message in the platelet”- more than just vestigial mRNA! Platelets. 2008;19(6):395–404. [DOI] [PubMed] [Google Scholar]
- 21.Roth GJ, Hickey MJ, Chung DW, Hickstein DD. Circulating human blood platelets retain appreciable amounts of poly (A)+ RNA. Biochem Biophys Res Commun. 1989;160(2):705–710 [DOI] [PubMed] [Google Scholar]
- 22.Robinson M, Machin S, Mackie I, Harrison P. In vivo biotylation studies: specificity of labelling of reticulated platelets by thiazole orange and mepacrine. Br J Haematol. 2000;108(4):859–864. [DOI] [PubMed] [Google Scholar]
- 23.Rinder HM, Tracey JB, Recht M, et al. Differences in platelet a-granule release between normals and immune thrombocytopenic patients and between young and old platelets. Thromb Haemost. 1998;80(3):457–462. [PubMed] [Google Scholar]
- 24.Karpatkin S. The heterogeneity of human platelets. Functional evidence suggestive of young and old platelets. J Clin Invest. 1969;48(6):1083–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guthikonda S, Alviar CL, Vaduganathan M, et al. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol. 2008;52(9):743–749. [DOI] [PubMed] [Google Scholar]
- 26.Perl L, Lerman-Shivek H, Rechavia E, et al. Response to prasugrel and levels of circulating reticulated platelets in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2014;63(6):513–517. [DOI] [PubMed] [Google Scholar]
- 27.Ibrahim H, Nadipalli S, DeLao T, Guthikonda S, Kleiman NS. Immature platelet fraction (IPF) determined with an automated method predicts clopidogrel hyporesponsiveness. J Thromb Thrombolysis. 2012;33(2):137–142. [DOI] [PubMed] [Google Scholar]
- 28.Van Gorp RM, Feijge MA, Vuist WM, Rook MB, Heemskerk JW. Irregular spiking in free calcium concentration in human platelets. Regulation by modulation of the inositol trisphosphate receptors. Eur J Biochem. 2002;269(5):1543–1552. [DOI] [PubMed] [Google Scholar]
- 29.Schols SE, Feijge MA, Lance MD, et al. Effects of plasma dilution on tissue factor-induced thrombin generation and thromboelastography: partly compensating role of platelets. Transfusion. 2008;48(11):2384–2394. [DOI] [PubMed] [Google Scholar]
- 30.Ninivaggi M, Feijge MA, Baaten CC, et al. Additive roles of platelets and fibrinogen in whole-blood fibrin clot formation upon dilution as assessed by thromboelastometry. Thromb Haemost. 2014;111(3):447–457. [DOI] [PubMed] [Google Scholar]
- 31.Van der Meijden PE, Bouman AC, Feijge MA, et al. Platelet dysfunction in thrombosis patients treated with vitamin K antagonists and recurrent bleeding. Plos One. 2013;8(5):e64112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velik-Salchner C, Maier S, Innerhofer P, et al. Point-of-care whole blood impedance aggregometry versus classical light transmission aggregometry for detecting aspirin and clopidogrel: the results of a pilot study. Anesth Analg. 2008;107(6):1798–1806. [DOI] [PubMed] [Google Scholar]
- 33.Alexopoulos D, Galati A, Xanthopoulou I, et al. Ticagrelor versus prasugrel in acute coronary syndrome patients with high onclopidogrel platelet reactivity following percutaneous coronary intervention: a pharmacodynamic study. J Am Coll Cardiol. 2012;60(3):193–199. [DOI] [PubMed] [Google Scholar]
- 34.McCabe DJ, Harrison P, Sidhu PS, Brown MM, Machin SJ. Circulating reticulated platelets in the early and late phases after ischaemic stroke and transient ischaemic attack. Br J Haematol. 2004;126(6):861–869. [DOI] [PubMed] [Google Scholar]
- 35.De Witt SM, Swieringa F, Cavill R, et al. Identification of platelet function defects by multiparameter assessment of thrombus formation. Nat Commun. 2014;5:4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nergiz-Unal R, Cosemans JM, Feijge MA, et al. Stabilizing role of platelet P2Y12 receptors in shear dependent thrombus formation on ruptured plaques. Plos One. 2010;5(4):e10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frelinger AL, Barnard MR, Fox ML, Michelson AD. The platelet activity after clopidogrel termination (PACT) study. Circulation. 2010;3(5):442–449. [DOI] [PubMed] [Google Scholar]
- 38.Farid NA, Kurihara A, Wrighton SA. Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel and prasugrel in humans. J Clin Pharmacol. 2010;50(2):126–142. [DOI] [PubMed] [Google Scholar]
- 39.Hagihara K, Kurihara A, Kawai K, et al. Absorption, distribution and excretion of the new thienopyridine agent prasugrel in rats. Xenobiotica. 2007;37(7):788–801. [DOI] [PubMed] [Google Scholar]
- 40.Cosemans JM, van Kruchten R, Olieslagers S, et al. Potentiating role of Gas6 and Tyro3, Axl and Mer (TAM) receptors in human and murine platelet activation and thrombus stabilization. J Thromb Haemost. 2010;8(8):1797–1808. [DOI] [PubMed] [Google Scholar]
- 41.McBane RD, Gonzalez C, Hodge DO, Wysokinski WE. Propensity for young reticulated platelet recruitment into arterial thrombi. J Thromb Thrombolysis. 2014;37(2):148–154. [DOI] [PubMed] [Google Scholar]
- 42.Sibbing D, Stegherr J, Braun S, et al. A double-blind, randomized study on prevention and existence of a rebound phenomenon of platelets after cessation of clopidogrel treatment. J Am Coll Cardiol. 2010;55(6):558–565. [DOI] [PubMed] [Google Scholar]
- 43.Fiedler KA, Mehilli J, Kufner S, et al. Randomized, double-blind trial on the value of tapered discontinuation of clopidogrel maintenance therapy after drug-eluting stent implantation. Intracoronary stenting and antithrombotic regimen: caution in discontinuing clopidogrel therapy. Thromb Haemost. 2014;111(6):1041–1049. [DOI] [PubMed] [Google Scholar]
- 44.Jakubowski JA, Li YG, Payne CD, Small DS, Winters KJ. Absence of "rebound" platelet hyperreactivity following cessation of prasugrel. Thromb Haemost. 2011;106(1):174–176. [DOI] [PubMed] [Google Scholar]
- 45.Yedidya I, Netzer A, Vaduganathan M, Solodky A, Kornowski R, Lev EI. Clopidogrel tapering as a strategy to attenuate platelet rebound phenomenon in patients with bare-metal stents. J Thromb Thrombolysis. 2012;33(1):16–21. [DOI] [PubMed] [Google Scholar]
- 46.Zafar MU, Santos-Gallego DA, Vorchheimer DA, et al. Platelet function normalization after a prasugrel loading dose: time-dependent effect of platelet supplementation. J Thromb Haemost. 2013;11(1):100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]