

Abstract
Wnt pathway-driven proliferation and renewal of the intestinal epithelium must be tightly controlled to prevent development of cancer and barrier dysfunction. Although type I interferons (IFN) produced in the gut under the influence of microbiota are known for their antiproliferative effects, the role of these cytokines in regulating intestinal epithelial cell renewal is largely unknown. Here we report a novel role for IFN in the context of intestinal knockout of casein kinase 1α (CK1α), which controls the ubiquitination and degradation of both β-catenin and the IFNAR1 chain of the IFN receptor. Ablation of CK1α leads to the activation of both β-catenin and IFN pathways and prevents the unlimited proliferation of intestinal epithelial cells despite constitutive β-catenin activity. IFN signaling contributes to the activation of the p53 pathway and the appearance of apoptotic and senescence markers in the CK1α-deficient gut. Concurrent genetic ablation of CK1α and IFNAR1 leads to intestinal hyperplasia, robust attenuation of apoptosis, and rapid and lethal loss of barrier function. These data indicate that IFN play an important role in controlling the proliferation and function of the intestinal epithelium in the context of β-catenin activation.
INTRODUCTION
The mature mammalian gut epithelial lining requires continuous cell proliferation that enables the replacement of lost cells and almost weekly tissue renewal (1). This renewal driven by the Wnt–β-catenin pathway must be tightly controlled to ensure the proper differentiation of intestinal epithelial cells (IECs). IEC differentiation is required to maintain normal intestinal functions and to prevent the development of colorectal cancers (2, 3). Development of these cancers often depends on the interaction of the host with commensal microbiota and elicited tissue inflammation (4, 5). Inflammatory cytokines regulate the innate immune responses shaped by the microbiota and affect the rate of intestinal epithelial cell proliferation (6–9). Among these cytokines are type I interferons (IFN) that activate a cognate cell surface receptor (consisting of the IFNAR1 and IFNAR2 chains) and signal to induce the transcription of IFN-stimulated genes (ISGs), some of which are known for their antiproliferative properties (10). Despite the known suppressive effects of IFN on cell proliferation and constitutive induction of IFN in the gut (11, 12), the role of these cytokines in regulating intestinal epithelium proliferation and function remains poorly understood.
Adequate expression of the IFNAR1 chain of the IFN receptor is required for all IFN effects (13–15). Genetic studies using the Ifnar1 knockout in mice have yet to establish the role of IFN in regulating intestinal epithelial cell proliferation. Ablation of Ifnar1, specifically in IECs (Ifnar1ΔIEC), resulted in only a modest increase in bromodeoxyuridine (BrdU) labeling, and this increase was dependent on the changes in the commensal microbiota (16). However, neither alteration in the microbiome profile (17) nor increased IEC proliferation (17, 18) was observed in mice lacking Ifnar1 in all tissues compared to wild-type mice. These data suggest either that IFN do not play an important role in regulating intestinal epithelium proliferation or that genetic differences between Ifnar1+/+ and Ifnar1−/− animals are masked by additional factors.
Indeed, high levels of cell surface IFNAR1 are specifically required for the antiproliferative effects of IFN as opposed to their ability to elicit an antiviral state (14, 19). These levels of IFNAR1 are tightly regulated by its phosphorylation-dependent ubiquitination and subsequent endocytosis and lysosomal degradation (20–22). The rate-limiting event in these processes is the phosphorylation of serine residues within the IFNAR1 degron that enables the recruitment of beta-transducin repeat containing protein ( βTrcp) E3 ubiquitin ligase and IFNAR1 ubiquitination (21, 23). Importantly, while this phosphorylation can be induced by IFN via the activation of protein kinase D2 (24, 25), there is also a ligand-independent pathway that removes IFNAR1 from the surface of cells that are yet to encounter IFN (23, 26). This pathway was shown to be activated by inflammatory cytokines (27) and may contribute to the lack of phenotypic differences between wild-type and Ifnar1 knockout mice under inflammatory conditions (28). We previously purified and characterized casein kinase 1α (CK1α) as a major ligand-independent kinase that is capable of phosphorylating IFNAR1 in vitro (29).
Importantly, CK1α is also a critical mediator of β-catenin ubiquitination and degradation (2). Priming phosphorylation of β-catenin by CK1α greatly increases the phosphorylation of the β-catenin degron by glycogen synthase kinase 3β (GSK3β) (30, 31) that is required for its recognition by the βTrcp E3 ubiquitin ligases and subsequent ubiquitination and proteasomal degradation (32–36). Our recent studies demonstrated that gut-specific knockout of the Csnk1a1 gene that encodes CK1α leads to robust stabilization of β-catenin and activation of Wnt target genes (37, 38). Intriguingly, ablation of Csnk1a1 alone did not lead to either epithelial cell hyperproliferation or tumorigenesis. Instead, inactivation of CK1α induced the DNA damage response (DDR) and p53/p21-dependent senescence. These events appear to prevent tumorigenesis driven by hyperactive β-catenin because the concurrent ablation of CK1α with either p53 or p21 resulted in hyperproliferation and a rapid development of aggressive and invasive intestinal tumors (37, 38).
Here we determined the role of CK1α in the regulation of IFNAR1 ubiquitination and levels in vivo. We found that despite the accumulation of IFNAR1 protein (but not mRNA), the ubiquitination of IFNAR1 was decreased in CK1α-deficient intestinal tissues. In addition, the expression of IFN-stimulated genes was increased in the gut upon Csnk1a1 ablation. As the lack of CK1α stabilized both β-catenin and IFNAR1, the phenotype associated with the loss of Csnk1a1 highlighted the contribution of IFN signaling to control IEC proliferation and function. Intriguingly, IFN signaling was required for the effective activation of p53 and p21 and induction of senescence and apoptosis in the CK1α-deficient intestinal epithelium. Furthermore, the concurrent ablation of CK1α with Ifnar1 led to unrestricted IEC proliferation to an extent that caused profound aberrations of gut barrier function and rapid animal death. These results demonstrate that IFN play an important role in restricting intestinal epithelial cell proliferation elicited by the activated β-catenin pathway.
MATERIALS AND METHODS
Animals.
All experiments with animals were carried out under protocol 803995 approved by the IACUC of the University of Pennsylvania. All mice were on the C57BL/6 background, had water ad libitum, and were fed regular chow. Ifnar1−/− mice (a kind gift of Dong-Er Zhang, UCSD) were crossed with Csnk1a1lox/lox mice, which bear floxed Csnk1a1 and Vil1-Cre-ERT2 (37), to generate Csnk1a1lox/lox; Ifnar1+/+ or Csnk1a1lox/lox; Ifnar1−/− mice bearing Vil1-Cre-ERT2 (Vil1-Cre-ERT2 in genotype identification is omitted for simplicity). Genotyping was performed on the tail of 4-week-old pups according to standard protocols using previously described primers (37).
Tamoxifen (Sigma) was dissolved in corn oil (Sigma), and mice were injected intraperitoneally (120 mg/kg of body weight) on two consecutive days. On day 5 after the last injection, mice were euthanized. The jejunum, the ileum, and the entire large intestine were flushed with ice-cold phosphate-buffered saline (PBS); cut open longitudinally; and subjected to fixation in 4% formaldehyde and paraffin embedding. Small pieces of the jejunum were embedded in Tissue-Tek OCT compound (Sakura) and frozen at −80°C. IECs were isolated from the middle part of the small intestine as described previously (39) but with the following slight modifications: intestinal cells were separated into single cells in Hanks' balanced salt solution containing 5 mM EDTA at 4°C for 30 min.
For antibiotic treatment, Csnk1a1Δgut; Ifnar1−/− mice were gavaged with 100 mg streptomycin (Sigma), and the drinking water was immediately replaced with filter-sterilized water containing ampicillin (1 g/liter; American Bioanalytical), vancomycin (0.5 g/liter; MP Biomedicals), neomycin (1 g/liter; Sigma), metronidazole (1 g/liter; Sigma), and 1% sucrose (Fisher). Antibiotic-containing water was replaced at least once a week during the course of the experiment. For all experimental groups, either mice were cohoused or their feces were swapped daily between cages to minimize potential differences in the gut microbiota.
Histology and immunotechniques.
Sections (5 μm) were cut for hematoxylin and eosin (H&E) staining and immunohistochemistry analysis. For immunohistochemistry, sections were incubated with antibodies to detect CKIα (C-19 [1:1,000]; Santa Cruz Biotechnology), β-catenin (1:200; Cell Signaling), cyclin D1 (SP4 [1:100]; Thermo Scientific), Ki67 (2.5 mg/ml; BioLegend), cleaved caspase-3 (1:100; Cell Signaling Technology), and p53 (1:500; NovoCasta). Secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-rabbit, anti-goat, and anti-rat antibodies (Millipore, Cell Signaling Technology). 3,3′-Diaminobenzidine (DAB) chromogen (Lab Vision) was used for detection. For whole-tissue immunofluorescence, pieces of small intestine harvested from mice were frozen in Tissue-Tek OCT compound, cryosectioned by using Leica CM3050 S cryostats, fixed in acetone, washed, and blocked with PBS containing 5% goat serum. The sections were incubated for 1 h with primary antibodies to detect IFNAR1 (2 μg/ml; Sino Biological), γH2AX (1:100; Millipore), E-cadherin (1:500; Millipore), or TJP1/ZO-1 (1:100; Thermo Fisher). The sections were then washed, incubated with the corresponding secondary antibodies labeled with Alexa Fluor 488 or 594 (Invitrogen) for 1 h, washed again, and mounted onto coverslips by using mounting solution with 4′,6-diamidino-2-phenylindole (DAPI) (Prolong Gold). For senescence-associated β-galactosidase (SA-βGal) staining (described in detail in reference 40), 10-μm sections were cut from OCT-embedded frozen tissue and allowed to adhere to coated slides at 25°C for 1 min before fixation for 15 min. Staining was performed according to the instruction provided with the senescence β-galactosidase staining kit (catalog number 9860S; Cell Signaling). After staining, sections were counterstained with nuclear fast red, dehydrated, and mounted. Periodic acid-Schiff (PAS) staining for goblet cell determination was performed according to standard protocols. Numbers of γH2AX-, IFNAR1-, and Ki67-positive foci per crypt/villus axis were determined by counting foci in 40 or 20 low-power fields (magnification of ×200 or ×400). The number of positive cells per crypt (Ki67, cleaved caspase-3, and SA-βGal) was determined by counting foci in 20 low-power fields (magnification of ×200).
Western blotting.
Proteins were extracted from intestinal epithelial cell pellets in protein lysis buffer containing protease and phosphatase inhibitors according to whole-cell extract protocols. IFNAR1 was immunoprecipitated from whole-cell lysates by using MAR1-5A3 (Leinco Technologies, Inc.), as previously described (28). Membranes were incubated with antibodies to detect IFNAR1 (2 μg/ml; Sino Biological), ubiquitin (Ub) (P4D1 [1:1,000]; Santa Cruz Biotechnology), CKIα (C-19 [1:1,000]; Santa Cruz Biotechnology), p21CIP1/WAF1 (ab7960 [1:1,000]; Abcam), interferon regulatory factor 7 (IRF7) (ab62505 [1:1,000]; Abcam), ZO-1 (1:100; Thermo Fisher), and β-actin (AC-74 [1:5,000]; Sigma). Secondary antibodies conjugated to horseradish peroxidase were purchased from Millipore Bioscience Research Reagents. Blots were processed as previously described (23) and developed by using ECL (GE Healthcare).
RNA analysis.
Total RNA was extracted from cell pellets by using TRIzol reagent and phenol-chloroform methods. RNA (1 μg) was subjected to reverse transcription using a first-strand cDNA synthesis kit (Thermo Scientific), and mRNA expression levels were measured by quantitative real-time PCR using an Applied Biosystems 7500 Fast real-time PCR system. Relative quantities of gene transcripts were normalized to β-actin transcript levels. Sequences of PCR primers are as follows: 5′-TAGGCGGAATGAAGATGGAC (forward primer for Axin2), 5′-CTGGTCACCCAACAAGGAGT (reverse primer for Axin2), 5′-CAGTATCTCCCGGACTGAGG (forward primer for Cd44), 5′-GCCAACTTCATTTGGTCCAT (reverse primer for Cd44), 5′-GGTGCGGAAGATCGGATCT (forward primer for Csnk1a1), 5′-TTCACTGCCACTTCCTCGC (reverse primer for Csnk1a1), 5′-TTGACTGCCGAGAAGTTGTG (forward primer for cyclin D1), 5′-CCACTTGAGCTTGTTCACCA (reverse primer for cyclin D1), 5′-CTGCTGCCTGGGCTTCATAG (forward primer for Ifitm3), 5′-GGATGCTGAGGACCAAGGTG (reverse primer for Ifitm3), 5′-TCCACAGCGATATCCAGACA (forward primer for Cdkn1a), 5′-AGACAACGGCACACTTTGCT (reverse primer for Cdkn1a), 5′-GGAGCTCAGCAAGACTCTGG (forward primer for Sox9), 5′-TGTAATCGGGGTGGTCTTTCT (reverse primer for Sox9), 5′-GCCTACTCGTCGGAGGAA (forward primer for Ascl2), 5′-CCAACTGGAAAAGTCAAGCA (reverse primer for Ascl2), 5′-CCCTGTGAAGGAAGTGGCTA (forward primer for Oas2), 5′-CTGTTGGAAGCAGTCCATGA (reverse primer for Oas2), 5′-GTCAGAGTGGAAATCCTAAG (forward primer for Ifnβ), 5′-ACAGCATCTGCTGGTTGAAG (reverse primer for Ifnβ); 5′-CGACCAAGTGTGAATTCTCTTTAC (forward primer for Ifnar1), 5′-ATCAACCTCATTCCACGAAGAT (reverse primer for Ifnar1), 5′-ACCCGAAACTGATGCTGTGGATAG (forward primer for Tjp1), 5′-AAATGGCCGGGCAGAGACTTGTGTA (reverse primer for Tjp1), 5′-TGAAACGCCGACCTATCCTTA (forward primer for Trp53), 5′-GGCACAAACACGAACCTCAAA (reverse primer for Trp53), 5′-ATATTAACCGGCGCTACGAC (forward primer for Bak1), 5′-AGGCGATCTTGGTGAAGAGT (reverse primer for Bak1), 5′-ATGCTGTGGATCTGGGCTGTCCT (forward primer for Fas), 5′-GCATAATGGTTCTTGTCCATG (reverse primer for Fas), 5′-CCTCAAGTTTTGCCCTTTA (forward primer for Casp1), 5′-CCTTCTTAATGCCATCATCTT (reverse primer for Casp1), 5′-AGAGGGAAATCGTGCGTGAC (forward primer for β-actin), and 5′-CAATAGTGATGACCTGGCCGT (reverse primer for β-actin).
Isolation and culture of intestinal crypts.
Crypt culture was performed as previously described (41). After intestinal crypt isolation, a total of 500 crypts were mixed with 50 μl of Matrigel (BD Biosciences) and plated into 24-well plates. After polymerization of Matrigel, 500 μl of crypt culture medium (Advanced Dulbecco's modified Eagle's medium [DMEM]–F-12 medium containing 50 ng/ml epidermal growth factor [EGF] [Invitrogen], 1 μg/ml R-spondin [Peprotech], and 100 ng/ml Noggin [Peprotech]) was added. Organoids were treated with 10 μM the CK1 inhibitor D4476 (Sigma), 10 μg/ml IFN-β neutralizing antibody (Leinco Technologies), and the corresponding vehicle or isotype controls. After 4 days of culture at 37°C, the numbers of live organoids were quantified.
Microarray.
Microarray analyses were performed with an Illumina whole-genome array. Total RNA was isolated from intestinal epithelial cells of Csnk1a1Δgut; Ifnar1+/+ (single knockout [SKO]) and Csnk1a1Δgut;Ifnar1−/− (double knockout [DKO]) mice at day 5 after the last tamoxifen treatment by using an miRNeasy minikit (Qiagen). Biotin-labeled cRNA samples were prepared by using a TargetAmp-Nano labeling kit (Epicentre) as recommended by the manufacturer. Thereafter, 0.75 μg cRNA was hybridized to Illumina Sentrix Mouse-6 v.1 BeadChips, which were scanned with an Illumina BeadStation 500 instrument (both from Applied Biosystems-Life Technologies, Inc.). Data were collected with Illumina BeadStudio 3.1.1.0 software, and statistical analyses were conducted with IlluminaGUI R-package15,70.
Intestinal permeability assay.
Barrier function was evaluated by measuring in vivo paracellular permeability to fluorescence-labeled dextran. Mice were fasted for 4.5 h and then gavage fed fluorescein isothiocyanate (FITC)-labeled 4.4-kDa dextran (FD4; Sigma). Plasma was obtained 5 h after gavage administration by terminal cardiac puncture after CO2 anesthesia. The plasma FD4 concentration was calculated by comparing samples with serial dilutions of known standards by using a Varioskan Flush fluorimeter (Thermo Scientific) with excitation at 485 nm and emission at 530 nm.
Statistical analyses.
Data are presented as averages ± standard errors of the means (SEM). Statistical analysis was performed by using Microsoft Excel. Statistical significance was calculated by using a two-tailed Student t test. A P value of <0.05 was considered significant.
Microarray data accession number.
Raw data were deposited in the GEO database under accession number GSE76512.
RESULTS
Activation of IFN signaling in response to CK1α ablation.
To determine the role of CK1α in the regulation of IFNAR1 levels and signaling in the intestinal epithelium, we crossed Csnk1a1f/f mice with animals expressing Vil1-Cre-ERT2 (37). Both strains harbored a double-wild-type allele for Ifnar1 (Ifnar1+/+). Treatment of the progeny from these crosses with tamoxifen resulted in mice lacking CK1α exclusively in the intestinal epithelium (Csnk1a1Δgut; Ifnar1+/+ [SKO]). The decrease in CK1α expression in the intestinal epithelium was monitored by assessing the levels of Csnk1a1 mRNA (Fig. 1A) and protein (Fig. 1B and C). We next crossed these mice to mice lacking Ifnar1 and observed a similar extent of CK1α ablation in these DKO mice (Fig. 1A to C).
FIG 1.

Activation of IFN signaling in response to CK1α ablation. (A) Relative levels of Csnk1a1 and Ifnar1 mRNAs in intestinal tissues from mice of the indicated genotypes. Levels of mRNA in intestinal tissues from untreated Csnk1a1lox/lox; Ifnar1+/+ mice were taken as 1.0. Data are shown as averages ± SEM; asterisks above Csnk1a1Δgut (SKO) indicate a significant difference between wild-type (WT) and SKO mice, and asterisks above Csnk1a1Δgut; Ifnar1−/− (DKO) indicate a significant difference between SKO and DKO mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Immunohistochemistry analysis of CK1α levels in intestinal tissues from mice of the indicated genotypes. For all immunochemistry, brown indicates specific immunostaining and purple indicates nuclear hematoxylin staining. Bars, 100 μm. (C) Western blotting of intestinal epithelial cells from mice of the indicated genotypes. IRF7 protein levels serve as an IFN-induced gene product control. Levels of β-actin were used as a loading control. (D) Immunofluorescence analysis of IFNAR1 expression in intestinal tissues from mice of the indicated genotypes. Bars, 100 μm. (E) Relative mRNA levels of the indicated IFN-stimulated genes in IECs from mice of the indicated genotypes assessed by quantitative PCR (levels in untreated Csnk1a1lox/lox; Ifnar1+/+ mice are taken as 1.0). (F) Heat map showing upregulation of IFN-induced genes in Csnk1a1Δgut/+ (wild-type) versus Csnk1a1Δgut (SKO) IECs at day 5 after CK1α ablation. Total RNA samples from three mice per genotype were pooled for analysis. The IFN gene signature is the Bosco interferon-induced antiviral module.
CK1α can phosphorylate IFNAR1 within its degron in vitro (29). Phosphorylation of this degron enables ubiquitination of IFNAR1 (21, 22). Here we aimed to determine the role of CK1α in the regulation of IFNAR1 ubiquitination and levels in the intestinal tissues. We detected IFNAR1 protein and its ubiquitination in intestinal epithelial tissue lysates from Csnk1a1Δgut; Ifnar1+/+ mice (Fig. 1C). Importantly, deletion of Csnk1a1 resulted in decreased IFNAR1 ubiquitination concurrent with a dramatic increase in the overall level of IFNAR1 protein (Fig. 1C and D). Given that Ifnar1 mRNA levels were not affected by the ablation of Csnk1a1 (Fig. 1A), these data are consistent with the hypothesis that CK1α is a major regulator of IFNAR1 ubiquitination and proteolytic degradation in vivo.
Importantly, the IFN-inducible IRF7 protein was upregulated in CK1α-deficient mice (Fig. 1C). Furthermore, CK1α ablation induced the mRNA levels of Ifnb along with a number of other IFN-stimulated genes (Fig. 1E and F). Induction of mRNA of the ligand IFN-β and stimulation of Oas2 and Ifitm3 expression were likely dependent on intact IFN signaling because these increases were not seen in tissues from Csnk1a1Δgut; Ifnar1−/− mice (Fig. 1E). Collectively, these data suggest that the ablation of CK1α leads to the accumulation of IFNAR1, induction of IFN expression, and activation of expression of IFN-stimulated genes in the intestinal epithelium.
Inhibition of IFN signaling attenuates DNA damage responses in CK1α-deficient mouse intestines.
Our previous work demonstrated that ablation of CK1α in the gut resulted in the stabilization and accumulation of β-catenin, activation of Wnt target genes, and induction of DDR signaling (37). Moreover, DNA damage was shown to both induce IFN production (42, 43) and be further stimulated by IFN signaling (44). Thus, we sought to investigate the role of IFN signaling in the DDR by comparing the consequences of CK1α ablation in Ifnar1+/+ and Ifnar1−/− mice.
Regardless of the status of Ifnar1, we observed that CK1α deletion leads to a robust accumulation of β-catenin (Fig. 2A). Consistent with this result, similar levels of induction of cyclin D1 protein was observed in Csnk1a1Δgut; Ifnar1+/+ and Csnk1a1Δgut;Ifnar1−/− mice (Fig. 2B). Furthermore, animals of both genotypes exhibited comparable increases in the levels of several Wnt target genes, including Sox9, Ccnd1, Axin2, Ascl2, and Cd44 (Fig. 2C). These results indicate that IFN signaling is dispensable for β-catenin stabilization and transcriptional activation in the absence of CK1α.
FIG 2.

Knockout of the gene encoding CK1α induces β-catenin stabilization and Wnt hyperactivation in murine intestines from Ifnar1−/− mice. (A) Immunohistochemistry analysis of β-catenin expression in intestinal tissues from mice of the indicated genotypes. Bars, 100 μm. (B) Immunohistochemistry analysis of cyclin D1 expression in intestinal tissues from mice of the indicated genotypes. Bars, 100 μm. (C) Relative mRNA levels of the indicated Wnt target genes in intestinal epithelial cells from mice of the indicated genotypes as assessed by quantitative PCR (levels in untreated Csnk1a1lox/lox; Ifnar1+/+ mice are taken as 1.0). Data are shown as averages ± SEM; an asterisk above Csnk1a1Δgut (SKO) indicates a significant difference between wild-type (WT) and SKO mice. *, P < 0.05.
Intriguingly, the concurrent ablation of CK1α and Ifnar1 noticeably decreased the extent of DDR signaling as seen from the number of phosphorylated histone H2AX foci in CK1α-deficient tissues (Fig. 3A and B). Our previously reported results showed that IFN-β induced in response to DNA damage can further augment the extent of this damage. Furthermore, DDR-stimulated IFN was shown to play an important role in the induction of the p53 tumor suppressor protein transcriptional target gene Cdkn1a, which encodes the p21CIP1/WAF1 protein (43). The accumulation of p53 protein seen upon the deletion of Csnk1a1 was not affected by the status of Ifnar1 (Fig. 3C). However, inactivation of Ifnar1 led to noticeable decreases in the levels of p21CIP1/WAF1 protein (Fig. 1C) and mRNA (Fig. 3D) in CK1α-deficient tissues. Given that Ifnar1 ablation also attenuated the induction of other p53-dependent genes (Fig. 3D and E), these results strongly suggest an important role for IFN signaling in the activation of DDR and p53 activities in CK1α-deficient intestinal tissues.
FIG 3.

Inhibition of IFN signaling suppresses DNA damage responses in CK1α-deficient mouse intestines. (A) Immunofluorescence analysis of γH2AX expression in intestinal tissues from mice of the indicated genotypes. Bar, 100 μm. (B) Quantitation of the number of γH2AX-positive foci per basal crypt in small intestines of three mice (30 to 50 crypts/villi were analyzed for each mouse). Data are shown as averages ± SEM; asterisks above Csnk1a1Δgut (SKO) indicate a significant difference between wild-type (WT) and SKO mice, and asterisks above Csnk1a1Δgut; Ifnar1−/− (DKO) indicate a significant difference between SKO and DKO mice. *, P < 0.05; ***, P < 0.001. (C) Immunohistochemical analysis of p53 levels in intestinal tissues from mice of the indicated genotypes. Magnification, ×10. (D) Relative mRNA levels of the indicated p53 target genes in IECs from mice of the indicated genotypes as assessed by quantitative PCR (levels in untreated Csnk1a1lox/lox; Ifnar1+/+ mice are taken as 1.0). (E) GSEA of the transcriptome profiles showing a significant enrichment of p53 target gene signatures in Csnk1a1Δgut (SKO) versus Csnk1a1Δgut; Ifnar1−/− (DKO) IECs at day 5 after CK1α ablation. NES, normalized enrichment score; FDR, false discovery rate.
Endogenous IFN restrict proliferation of intestinal epithelial cells and elicit their apoptosis.
We previously postulated that Csnk1a1 ablation simultaneously elicits the proproliferative activity of canonical Wnt pathway target genes through β-catenin stabilization along with another yet-to-be-understood antiproliferative pathway that activates p53-p21, restricts IEC proliferation, and prevents tumorigenesis in spite of constitutive β-catenin activity (37). We therefore investigated the role of IFN in inhibiting epithelial cell proliferation. Intriguingly, immunofluorescence staining demonstrates the mutual exclusion of the IFNAR1 protein with the marker of cell replication Ki67 in normal intestine (Fig. 4A and B). Whereas Ki67 staining in Ifnar1+/+ mice was elevated upon Csnk1a1 ablation, a further significant increase was seen in DKO mice (Fig. 4C and D). Consistent with this finding, the number of senescence-associated β-galactosidase (SA-βgal)-positive cells was also dramatically lower in intestinal tissues of Csnk1a1Δgut; Ifnar1−/− double-knockout mice (Fig. 4E and F). Finally, gene set enrichment analysis (GSEA) revealed a significant increase in the expression of genes associated with cell proliferation in Csnk1a1Δgut; Ifnar1−/− double-knockout mice versus mice with the Csnk1a1Δgut knockout alone (Fig. 4G). Collectively, these results indicate that IFN signaling plays an important role in restricting IEC proliferation within the context of β-catenin stabilization caused by the inactivation of CK1α.
FIG 4.

Endogenous IFN restrict proliferation of intestinal epithelial cells and elicit their apoptosis. (A) Double-immunofluorescence analysis of IFNAR1 and Ki67 expression in intestinal tissues from mice of the indicated genotypes. Bar, 100 μm. (B) Quantitation of the number of IFNAR1-positive, Ki67-positive, or double-positive cells per crypt/villus axis in the small intestines of three mice (30 to 50 crypts/villi were analyzed for each mouse). Data are shown as averages ± SEM; asterisks above Csnk1a1Δgut (SKO) indicate a significant difference between wild-type (WT) and SKO mice, and asterisks above Csnk1a1Δgut; Ifnar1−/− (DKO) indicate a significant difference between SKO and DKO mice. ***, P < 0.001. (C) Immunohistochemistry analysis of Ki67 levels in intestinal tissues from mice of the indicated genotypes. Bars, 100 μm. (D) Quantitation of the number of Ki67-positive cells per basal crypt in the small intestines of three mice (30 to 50 crypts were analyzed for each mouse). ***, P < 0.001 for differences between SKO and WT and between SKO and DKO mice. (E) Analysis of SA-βGal-positive cells in intestinal tissues (counterstained with nuclear fast red) from mice of the indicated genotypes. (F) Quantitation of the number of SA-βGal-positive cells per basal crypt in the small intestines of three mice (30 to 50 crypts were analyzed for each mouse). (G) GSEA of transcriptome profiles showing a significant enrichment of proliferation-associated gene signatures in Csnk1a1Δgut; Ifnar1−/− (DKO) versus Csnk1a1Δgut (SKO) intestinal epithelial cells at day 5 after CK1α ablation. NES, normalized enrichment score; FDR, false discovery rate. (H) GSEA of transcriptome profiles showing a significant enrichment of apoptosis-associated gene signatures in Csnk1a1Δgut (SKO) versus Csnk1a1Δgut; Ifnar1−/− (DKO) IECs at day 5 after CK1α ablation. (I) Immunohistochemistry analysis of cleaved caspase-3 levels in intestinal tissues from mice of the indicated genotypes. (J) Quantitation of the numbers of cleaved caspase-3-positive cells per basal crypt in the small intestines of three mice (30 to 50 crypts were analyzed for each mouse).
Persistent activation of the Wnt pathway by stabilized β-catenin was shown to increase the number of apoptotic cells in the intestine (37, 45). Importantly, the concurrent deletion of Ifnar1 and Csnk1a1 decreased the enrichment of the apoptosis-associated gene signature (Fig. 4H) and decreased the number of cleaved caspase-3-positive cells (Fig. 4I and J). Taken together with the lower level of induction of p53-dependent proapoptotic genes such as Casp1, Fas, and Bak1 (Fig. 3D), these results implicate IFN signaling in apoptosis that occurs upon constitutive β-catenin stabilization.
Alterations in CK1α and IFNAR1 levels can affect intestinal cell growth and survival directly or/and indirectly. To assess the direct contribution of these regulators, we assessed the proliferation and viability of IECs grown ex vivo as organoid cultures. Treatment of mouse intestinal crypt cultures with the CK1 inhibitor D4476 resulted in decreased overall organoid size and viability (Fig. 5). Importantly, organoids from Ifnar1 knockout mice were less sensitive to the effects of D4476 (Fig. 5A and B). Furthermore, the inhibition of organoid growth by D4476 was noticeably attenuated by the addition of neutralizing antibodies against mouse IFN-β (Fig. 5C and D). These results suggest that IFN mediate cell-autonomous growth restriction upon CK1 inactivation.
FIG 5.
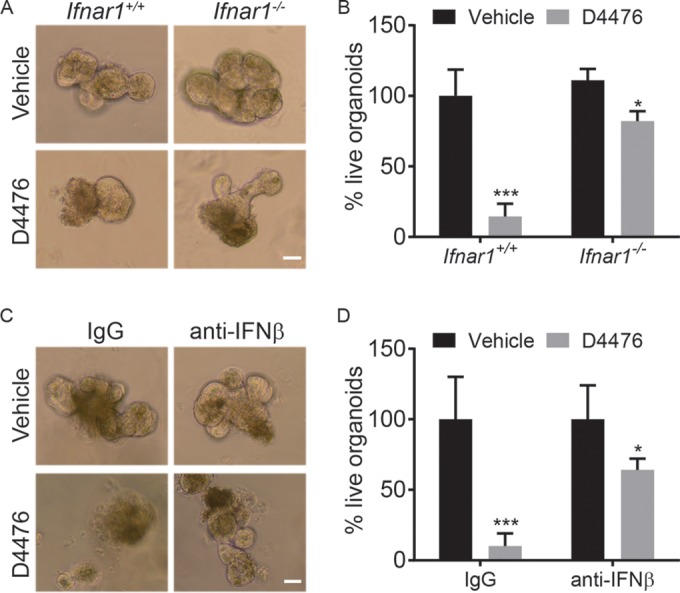
Alterations in CK1α and IFNAR1 levels affect intestinal organoid growth and survival. (A) Representative bright-field images of wild-type (WT) and Ifnar1−/− intestinal organoids treated with 10 μM the CK1α inhibitor D4476 at day 4. (B) Quantitation of live organoids in wild-type and Ifnar1−/− cultures compared with their vehicle-treated counterparts at day 4 (at least 10 random fields were analyzed for each culture). Data are shown as averages ± SEM. *, P < 0.05; ***, P < 0.001. (C) Representative bright-field images of WT organoids treated with 10 μM the CK1α inhibitor D4476 in the presence or absence of 10 μg/ml IFN-β neutralizing antibodies at day 4. (D) Quantitation of live organoids in WT cultures compared with vehicle- and IgG-treated counterparts at day 4 (at least 10 random fields were analyzed for each culture).
CK1α and IFN pathways regulate intestinal barrier function.
Loss of the tumor suppressor and β-catenin destruction complex component Apc in the small and large intestines leads to dramatic increases in proliferation, loss of differentiated goblet cells, and rapid animal death, probably due to an inability to absorb water and electrolytes (46). While the inactivation of both CK1α and adenomatous polyposis coli (APC) in the gut led to the stabilization of β-catenin and activation of the Wnt-inducible genes, a markedly different gene expression profile was seen in Csnk1a1Δgut; Ifnar1+/+ mice (37) and in ApcΔgut; Ifnar1+/+ mice (46). Intriguingly, a significant enrichment for the genes altered in the latter mice was seen upon the concurrent ablation of CK1α and IFNAR1 (Fig. 6A). The mild hyperplasia of the small intestine (Fig. 6B) and colonic epithelium (Fig. 6C) as well as the progressive loss of goblet cells (Fig. 6D and E) observed in Csnk1a1Δgut; Ifnar1−/− mice were indeed somewhat reminiscent of the phenotype in ApcΔgut; Ifnar1+/+ animals.
FIG 6.

Inhibition of IFN signaling increases proliferation and decreases numbers of goblet cells in intestinal tissues from Csnk1a1-deleted mice. (A) GSEA of transcriptome profiles showing a significant enrichment of genes upregulated upon APC deletion in Csnk1a1Δgut; Ifnar1−/− (DKO) versus Csnk1a1Δgut (SKO) intestinal epithelial cells at day 5 after CK1α ablation. NES, normalized enrichment score; FDR, false discovery rate. (B) H&E staining of small intestinal tissues from mice of the indicated genotypes. Bars, 100 μm. (C) H&E staining of colonic tissues from mice of the indicated genotypes. Bars, 100 μm. (D) PAS staining of small intestinal tissues from mice of the indicated genotypes. (E) Quantitation of the number of PAS-positive goblet cells per crypt/villus axis in the small intestines of three mice (30 to 50 crypts/villi were analyzed for each mouse). Data are shown as averages ± SEM; asterisks above SKO indicate a significant difference between wild-type (WT) and SKO mice, and asterisks above DKO indicate a significant difference between SKO and DKO mice. ***, P < 0.001.
Furthermore, Csnk1a1Δgut; Ifnar1−/− mice rapidly became moribund within a week after tamoxifen treatment was completed (Fig. 7A). Their life span could be extended by intraperitoneal injections of buffered saline solution, suggesting that an imbalance of water and electrolytes due to a differentiation block and potential loss of barrier function contributed to this early lethality. To test barrier function, we administered fluorescent dextran by gavage into the gastrointestinal tract of Csnk1a1/Ifnar1-deficient and control animals. Csnk1a1Δgut; Ifnar1−/− double-knockout mice exhibited markedly increased translocation of dextran into the bloodstream, consistent with compromised barrier function (Fig. 7B). While the levels of E-cadherin in Csnk1a1Δgut mice were not affected by the status of Ifnar1, the double-knockout mice displayed decreased levels of the ZO-1 tight junction protein (Fig. 1C and 7C) and its mRNA (Fig. 7D). These results implicate the IFN pathway in regulating the barrier function of the gut.
FIG 7.

CK1α and IFN pathways regulate intestinal barrier function. (A) Kaplan-Meier survival analysis of mice of the indicated genotypes after induction of intestinal CK1α ablation. Csnk1a1Δgut; Ifnar1−/− mice received intraperitoneal injections of 1 ml saline every day starting on day 1 after intestinal CK1α ablation (DKO+Saline) or were administered antibiotics (Ab) in drinking water. ***, P < 0.001. Asterisks next to the DKO line indicate comparisons between SKO and DKO mice, asterisks next to the DKO+Saline indicate comparisons between DKO and DKO+Saline mice, and asterisks next to the DKO+Ab line indicate comparisons between DKO and DKO+Ab mice. Neither wild-type nor Ifnar1−/− mice died during the course of the experiment. (B) Levels of FITC-dextran (FD4) in plasma samples from mice of the indicated genotypes 5 h after FD4 gavage (n > 3 for each group). Data are shown as averages ± SEM; an asterisk above Csnk1a1Δgut (SKO) indicates a significant difference between wild-type (WT) and SKO mice, and an asterisk above Csnk1a1Δgut; Ifnar1−/− (DKO) indicates a significant difference between SKO and DKO mice. *, P < 0.05. (C) Immunofluorescence analysis of E-cadherin (top) and tight junction protein 1 (TJP1) (zona occludens 1 [ZO-1]) (bottom) expression in intestinal tissues from mice of the indicated genotypes. Bar, 100 μm. (D) Relative mRNA levels of Tjp1 in intestinal epithelial cells from mice of the indicated genotypes as assessed by quantitative PCR (levels in untreated Csnk1a1lox/lox; Ifnar1+/+ mice are taken as 1.0). (E) H&E staining of intestinal tissues from mice of the indicated genotypes at day 30 after tamoxifen treatment (DKO mice were treated with saline). Bar, 200 μm.
Importantly, Csnk1a1/Ifnar1-deficient mice eventually became moribund despite the administration of buffered saline solution (Fig. 7A), suggesting additional mechanisms by which the IFN pathway contributes to intestinal homeostasis. A number of studies suggested an important role of microbiota in IFN induction and effects of IFN on innate immunity responses to commensal bacteria (reviewed in reference 47). Consistent with this role of IFN, the stromal elements of the gut in Csnk1a1Δgut; Ifnar1−/− mice displayed signs of injury and leukocyte infiltration and contained enlarged and inflamed lymph nodes. A robust inflammatory response was seen near the basement membrane, including leukocyte infiltration, combined with mucosal injury and increased epithelium permeability (Fig. 7E). Observed alterations were indicative of an increased microbiota-induced ability to penetrate the barrier and induce inflammation. Indeed, treatment of Csnk1a1/Ifnar1-deficient mice with antibiotics efficiently prevented their death (Fig. 7A). These data collectively suggest that IFN signaling plays an important role in maintaining intestinal barrier functions and homeostasis of the host interaction with the microbiota.
DISCUSSION
Our data presented here demonstrate that induction of IFN signaling appears to contribute to the activation of the DNA damage responses and apoptotic pathways as well as the suppression of intestinal epithelium proliferation that occurs upon the inactivation of CK1α. The latter event stabilizes both β-catenin and IFNAR1, thereby highlighting the conditions that determine the role of IFN signaling in restricting IEC proliferation. In addition, IFN contributes to the vitally important function of maintenance of intestinal barrier function.
CK1α is capable of phosphorylating numerous proteins and affecting a multitude of signaling pathways and transcriptional activities toward specific genes (reviewed in reference 48). Whereas CK1α was capable of phosphorylating the IFNAR1 degron in vitro (29), the role of other kinases in stimulating the recruitment of βTrcp to IFNAR1 and promoting its ubiquitination, endocytosis, and degradation was also demonstrated (24, 25). Our current data clearly characterize CK1α as a major regulator of IFNAR1 ubiquitination and stability in vivo (Fig. 1). Furthermore, these results implicate this kinase in the negative regulation of the IFN pathway in intestinal tissues and underscore the importance of CK1α function for the proliferation of the intestinal epithelium and its permeability.
Furthermore, our data suggest that the role of CK1α in regulating intestinal homeostasis is at least in part mediated by its effects on the stability and levels of IFNAR1 and the ensuing alterations in IFN signaling. Although microbiota-supported constitutive tonic IFN signaling has been described in the gut (11, 12), the role of this signaling in regulating gut renewal and function was a challenge to evaluate due to the potential phenotypic similarity of mice lacking the Ifnar1 gene and wild-type mice exhibiting rapid IFNAR1 degradation under inflammatory conditions (28). The concurrent stabilization of IFNAR1 and β-catenin upon CK1α inactivation enabled us to determine that IFN plays an important role in restricting the proliferation and viability of the intestinal epithelium (Fig. 4 and 5) and contributes to the maintenance of the equilibrium of the host-microbiota interaction and barrier function (Fig. 7).
Our data further indicate that the IFN pathway contributes to the expression of p53-driven genes (including proapoptotic genes and Cdkn1a) stimulated in the absence of CK1α (Fig. 3). These data are consistent with our recently reported data demonstrating that IFN can amplify DNA damage responses (43) as well as with data from previous reports that exogenous IFN can trigger an increase in p53 activities (44, 49). Importantly, these effects of IFN may provide an additional mechanism for the activation of the p53 pathway in the CK1α-deficient gut in addition to the previously reported downregulation of MDMX and the ensuing stabilization of the p53 protein (37).
Intriguingly, activation of the p53 pathway was not observed in the Apc-deficient gut (37, 46). Given that the ablation of either CK1α or APC results in the stabilization of β-catenin and stimulation of the Wnt pathway, the difference between these phenotypes and underlying gene profile signatures can be explained by at least two diverse reasons. First, APC possesses important functions that do not depend on the timely degradation of β-catenin. For example, APC can bind RNA and regulate the microtubule scaffold (50). Furthermore, the loss of Apc leads to the upregulation of Musashi proteins, which are pleiotropic translational regulators that affect numerous important signaling cascades, including the mTorc1 pathway (51, 52). Second, characteristic for the deletion of Cnsk1a1 (but not Apc) induction of the DNA damage response, activation of the p53-driven genes, and cell senescence are likely augmented by IFN signaling activated by CK1α deletion. Indeed, massive hyperproliferation of the intestinal epithelium leading to a defective barrier function and rapid animal death that was seen in Cnsk1a1/Ifnar1-deficient mice is reminiscent of the phenotype observed upon intestinal deletion of APC (46) and is characterized by a similar gene expression signature (Fig. 6).
The restriction of proliferation of Csnk1a1-deficient IECs could be lifted by the concurrent inactivation of either p53, p21CIP1/WAF1 (37, 38), or, to a lesser extent, IFNAR1 (this work). Importantly, ablation of either p53 or p21CIP1/WAF1 in CK1α-deficient animals led to the development of malignant tumors (37, 38). However, we did not observe these tumors in any groups of CK1α/IFNAR1-deficient mice that developed a lethal disruption of intestinal barrier function (Fig. 7), suggesting a contribution of IFN-independent pathways to p53 function as a tumor suppressor in CK1α-null intestines.
A few overlapping and non-mutually exclusive mechanisms by which IFN contributes to the maintenance of the barrier function of the gut can be proposed. First, IFN-mediated restriction of the rate of cell proliferation should enable better differentiation, maturation, and establishment of the cohesive sheet of enterocytes and colonocytes. Second, IFN can contribute to immune defenses against conditionally pathogenic microbiota and intestinal inflammation. Katakura and coauthors previously reported a greater sensitivity of Ifnar1-null mice to intestinal inflammation caused by dextran sodium sulfate (53). Those authors also mentioned a positive effect of IFN on the protection of barrier properties of the epithelial cell sheet in vitro. Importantly, our results implicate IFN in regulating the expression of the ZO-1 protein involved in the formation of tight junctions that separate the basolateral epithelial space from the microbiota. Potential immune-related effects of IFN on the ability to withstand the pathogenic effects of microbiota are illustrated by the rescue of Cnsk1a1/Ifnar1-deficient mice upon administration of antibiotics (Fig. 7).
Genetic alterations in the IFNAR1 gene in humans were linked with the susceptibility locus for inflammatory bowel disease (54), and IFN-based pharmaceutical formulations have been used to treat patients with these disorders albeit with variable results (reviewed in reference 55). As evident from the literature and our current results, the effects of IFN on intestinal homeostasis are pleiotropic. Roles of IFN in antigen recognition and immune function, cell differentiation and proliferation, and gut barrier function are likely to contribute to the complexity of patient responses to IFN. Although some of the long-term IFN effects, such as suppression of tissue-regenerative abilities in the gastrointestinal tract, could be genuinely detrimental (17, 28), it is also plausible that rapid degradation of IFNAR1 may constitute an additional challenge for the efficacy of IFN-based therapies (reviewed in reference 13). Thus, future studies may determine the potential application of poorly bioavailable gut-restricted CK1α inhibitors to stabilize IFNAR1 and improve intestinal barrier function.
ACKNOWLEDGMENTS
We declare no conflict of interest.
This work was supported by NIH/NCI grants RO1 CA092900 and PO1 CA165997 (to S.Y.F.), including help from the Scientific Cell/Tissue Morphology Core and its principal investigator, Qian-Chun Yu.
We thank D. E. Zhang for reagents and A. Ortiz for help with manuscript preparation.
REFERENCES
- 1.Heath JP. 1996. Epithelial cell migration in the intestine. Cell Biol Int 20:139–146. doi: 10.1006/cbir.1996.0018. [DOI] [PubMed] [Google Scholar]
- 2.Clevers H. 2006. Wnt/beta-catenin signaling in development and disease. Cell 127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Pinto D, Clevers H. 2005. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res 306:357–363. doi: 10.1016/j.yexcr.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terzic J, Grivennikov S, Karin E, Karin M. 2010. Inflammation and colon cancer. Gastroenterology 138:2101–2114. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 6.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, Artis D. 2012. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37:158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAleer JP, Kolls JK. 2012. Maintaining poise: commensal microbiota calibrate interferon responses. Immunity 37:10–12. doi: 10.1016/j.immuni.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Santaolalla R, Abreu MT. 2012. Innate immunity in the small intestine. Curr Opin Gastroenterol 28:124–129. doi: 10.1097/MOG.0b013e3283506559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santos RL, Raffatellu M, Bevins CL, Adams LG, Tukel C, Tsolis RM, Baumler AJ. 2009. Life in the inflamed intestine, Salmonella style. Trends Microbiol 17:498–506. doi: 10.1016/j.tim.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 11.Kole A, He J, Rivollier A, Silveira DD, Kitamura K, Maloy KJ, Kelsall BL. 2013. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J Immunol 191:2771–2779. doi: 10.4049/jimmunol.1301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, Staeheli P, Weiss S. 2009. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol 183:3229–3236. doi: 10.4049/jimmunol.0804277. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs SY. 2013. Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J Interferon Cytokine Res 33:211–225. doi: 10.1089/jir.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piehler J, Thomas C, Garcia KC, Schreiber G. 2012. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev 250:317–334. doi: 10.1111/imr.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uze G, Schreiber G, Piehler J, Pellegrini S. 2007. The receptor of the type I interferon family. Curr Top Microbiol Immunol 316:71–95. [DOI] [PubMed] [Google Scholar]
- 16.Tschurtschenthaler M, Wang J, Fricke C, Fritz TM, Niederreiter L, Adolph TE, Sarcevic E, Kunzel S, Offner FA, Kalinke U, Baines JF, Tilg H, Kaser A. 2014. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 63:1921–1931. doi: 10.1136/gutjnl-2013-305863. [DOI] [PubMed] [Google Scholar]
- 17.Rauch I, Hainzl E, Rosebrock F, Heider S, Schwab C, Berry D, Stoiber D, Wagner M, Schleper C, Loy A, Urich T, Muller M, Strobl B, Kenner L, Decker T. 2014. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur J Immunol 44:2749–2760. doi: 10.1002/eji.201344401. [DOI] [PubMed] [Google Scholar]
- 18.Sun L, Miyoshi H, Origanti S, Nice TJ, Barger AC, Manieri NA, Fogel LA, French AR, Piwnica-Worms D, Piwnica-Worms H, Virgin HW, Lenschow DJ, Stappenbeck TS. 2015. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 17:85–97. doi: 10.1016/j.chom.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levin D, Harari D, Schreiber G. 2011. Stochastic receptor expression determines cell fate upon interferon treatment. Mol Cell Biol 31:3252–3266. doi: 10.1128/MCB.05251-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY. 2007. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol 179:935–950. doi: 10.1083/jcb.200706034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar KG, Krolewski JJ, Fuchs SY. 2004. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem 279:46614–46620. doi: 10.1074/jbc.M407082200. [DOI] [PubMed] [Google Scholar]
- 22.Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY. 2003. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J 22:5480–5490. doi: 10.1093/emboj/cdg524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Plotnikov A, Banerjee A, Suresh Kumar KG, Ragimbeau J, Marijanovic Z, Baker DP, Pellegrini S, Fuchs SY. 2008. Ligand-independent pathway that controls stability of interferon alpha receptor. Biochem Biophys Res Commun 367:388–393. doi: 10.1016/j.bbrc.2007.12.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng H, Qian J, Baker DP, Fuchs SY. 2011. Tyrosine phosphorylation of protein kinase D2 mediates ligand-inducible elimination of the type 1 interferon receptor. J Biol Chem 286:35733–35741. doi: 10.1074/jbc.M111.263608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng H, Qian J, Varghese B, Baker DP, Fuchs S. 2011. Ligand-stimulated downregulation of the alpha interferon receptor: role of protein kinase D2. Mol Cell Biol 31:710–720. doi: 10.1128/MCB.01154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, HuangFu WC, Kumar KG, Qian J, Casey JP, Hamanaka RB, Grigoriadou C, Aldabe R, Diehl JA, Fuchs SY. 2009. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5:72–83. doi: 10.1016/j.chom.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huangfu WC, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, Fuchs SY. 2012. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 31:161–172. doi: 10.1038/onc.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, Yu Q, Zheng H, Carbone CJ, Katlinskaya YV, Leu NA, McCorkell KA, Srinivasan S, Girondo M, Rui H, May MJ, Avadhani NG, Rustgi AK, Fuchs SY. 2014. Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol Med 6:384–397. doi: 10.1002/emmm.201303236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Carvalho LP, Bhattacharya S, Carbone CJ, Kumar KG, Leu NA, Yau PM, Donald RG, Weiss MJ, Baker DP, McLaughlin KJ, Scott P, Fuchs SY. 2009. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol 29:6401–6412. doi: 10.1128/MCB.00478-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. 2002. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16:1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. 2002. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837–847. doi: 10.1016/S0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 32.Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z. 1999. HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene 18:2039–2046. doi: 10.1038/sj.onc.1202760. [DOI] [PubMed] [Google Scholar]
- 33.Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R, Polakis P. 1999. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol 9:207–210. doi: 10.1016/S0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 34.Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, Hattori K, Nakamichi I, Kikuchi A, Nakayama K, Nakayama K. 1999. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J 18:2401–2410. doi: 10.1093/emboj/18.9.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latres E, Chiaur DS, Pagano M. 1999. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 18:849–854. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- 36.Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. 1999. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev 13:270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, Snir-Alkalay I, Burstain I, Haffner-Krausz R, Jung S, Wiener Z, Alitalo K, Oren M, Pikarsky E, Ben-Neriah Y. 2011. CKIalpha ablation highlights a critical role for p53 in invasiveness control. Nature 470:409–413. doi: 10.1038/nature09673. [DOI] [PubMed] [Google Scholar]
- 38.Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, Burstain I, Morgenstern Y, Brachya G, Billauer H, Biton S, Snir-Alkalay I, Vucic D, Schlereth K, Mernberger M, Stiewe T, Oren M, Alitalo K, Pikarsky E, Ben-Neriah Y. 2013. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24:242–256. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. 2004. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Katlinskaya YV, Carbone CJ, Yu Q, Fuchs SY. 2015. Type 1 interferons contribute to the clearance of senescent cell. Cancer Biol Ther 16:1214–1219. doi: 10.1080/15384047.2015.1056419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 42.Yu Q, Carbone CJ, Katlinskaya YV, Zheng H, Zheng K, Luo M, Wang PJ, Greenberg RA, Fuchs SY. 2015. Type I interferon controls propagation of long interspersed element-1. J Biol Chem 290:10191–10199. doi: 10.1074/jbc.M114.612374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, Lengner CJ, Greenberg RA, Johnson FB, Fuchs SY. 2015. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep 11:785–797. doi: 10.1016/j.celrep.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. 2006. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell 17:1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong MH, Rubinfeld B, Gordon JI. 1998. Effects of forced expression of an NH2-terminal truncated beta-catenin on mouse intestinal epithelial homeostasis. J Cell Biol 141:765–777. doi: 10.1083/jcb.141.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, Clarke AR, Winton DJ. 2004. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev 18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mangan NE, Fung KY. 2012. Type I interferons in regulation of mucosal immunity. Immunol Cell Biol 90:510–519. doi: 10.1038/icb.2012.13. [DOI] [PubMed] [Google Scholar]
- 48.Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. 2005. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal 17:675–689. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 50.Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young-Pearse TL, Flanagan JG. 2014. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158:368–382. doi: 10.1016/j.cell.2014.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valvezan AJ, Huang J, Lengner CJ, Pack M, Klein PS. 2014. Oncogenic mutations in adenomatous polyposis coli (Apc) activate mechanistic target of rapamycin complex 1 (mTORC1) in mice and zebrafish. Dis Model Mech 7:63–71. doi: 10.1242/dmm.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang S, Li N, Yousefi M, Nakauka-Ddamba A, Li F, Parada K, Rao S, Minuesa G, Katz Y, Gregory BD, Kharas MG, Yu Z, Lengner CJ. 2015. Transformation of the intestinal epithelium by the MSI2 RNA-binding protein. Nat Commun 6:6517. doi: 10.1038/ncomms7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. 2005. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest 115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D'Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, et al. 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]