

Abstract
Bone fractures constitute a major cause of morbidity and mortality especially in the elderly. Complications associated with osteoporosis drugs and the age of the patient slow bone turnover and render such fractures difficult to heal. Increasing the speed of fracture repair by administration of a fracture-targeted bone anabolic agent could find considerable application.
Aspartic acid oligopeptides are negatively charged molecules at physiological pH that adsorb to hydroxyapatite, the mineral portion of bone. This general adsorption is the strongest where bone turnover is highest or where hydroxyapatite is freshly exposed. Importantly, both of these conditions are prominent at fracture sites.
GSK3β inhibitors are potent anabolic agents that can promote tissue repair when concentrated in a damaged tissue. Unfortunately, they can also cause significant toxicity when administered systemically and are furthermore difficult to deliver due to their strong hydrophobicity. In this paper, we solve both problems by conjugating the hydrophobic GSK3β inhibitor to a hydrophilic aspartic acid octapeptide using a hydrolyzable bond, thereby generating a bone fracture-targeted water-soluble form of the drug. The resulting amphiphile is shown to assemble into micelles, extending its circulation time while maintaining its fracture-targeting abilities. For measurement of pharmacokinetics, an 125I was introduced at the location of the bromine in the GSK3β inhibitor to minimize any structural differences. Biodistribution studies demonstrate a greater than 4-fold increase in fracture accumulation over healthy bone.
Keywords: Bone fracture, GSK3β inhibitor, 6-Bromoindirubin-3’-Oxime, Biodistribution, Micelles, Octaaspartic acid
INTRODUCTION
Maintaining healthy bone requires a delicate balance of resorption and anabolic processes. Resorption is controlled primarily by osteoclasts, monocyte lineage cells that due to Receptor Activator of Nuclear Factor κ B Ligand (RANKL) to Receptor Activator of Nuclear Factor κ B (RANK) interactions have differentiated into bone resorptive cells.4,5 Anabolic processes begin as mesenchymal stem cells (MSCs) are stimulated to become osteoblasts by the BMP-2/Runx2 and Wnt/β-catenin pathways. Matured osteoblasts then deposit osteoid, a component of the bone matrix primarily composed of type I collagen, which mineralizes and becomes new bone. 6,7 Alteration of this balance results in diseased bone and fractures. Osteoporosis occurs when catabolism surpasses anabolism wherein a two-standard deviation decrease in bone density from healthy bone is observed. In the US, approximately 44 million people have low bone density, and 10 million people suffer from osteoporosis. By 2020, an estimated 61 million are projected to have osteoporosis.8 In general, osteoporosis can be treated with a regimen of bisphosphonates, which inhibits osteoclasts thereby slowing catabolism as well as healthy bone turnover. This becomes problematic when bone fractures occur and proper bone turnover is retarded by bisphosphonates. These complications include crippling vertebral and hip fractures with estimated costs between in $13.7 billion and $20.3 billion in 2005.9
Clinical treatment of these fractures generally does not include site-specific anabolic drugs. In fact, the only drugs approved for clinical use on fractures are BMP-2 and BMP-7, which are applied locally for use in open long bone fractures and spinal fusions.10 However, the need for broader application of anabolic drugs to treat bone maladies such as osteoporotic fractures is evident when one considers that 85% of the use of anabolics are off-label.11 Still, the FDA judiciously continues to limit approved use of locally administered drugs to fractures that are already open and at risk of infection. This limitation necessitates a clinically relevant approach to treating these fractures: a fracture treatment drug that is administered systemically yet targets the fracture site.
Efficacy in targeting fractures in humans has been demonstrated for quite some time as radiolabeled methylene diphosphate and hydroxymethylene diphosphate are used to diagnose hairline fractures. The use of bisphosphonates as fracture-targeting ligands inhibits both osteoclasts and angiogenesis, returning to the original problem of retarding bone turnover critical for fracture healing.12-14 Nature has a more biocompatible answer. Sialoprotein is one of several naturally-occuring proteins that displays a strong affinity for hydroxyapatite (HAp), a mineral found in bone. Several strings of acidic amino acids are responsible for sialoprotein’s bone affinity. Modeled after sialoprotein, a biocompatible targeting ligand can be produced by combining a string of eight aspartic acids.15 The aspartic acid octapeptide prefers the crystalline state of HAp, which is exposed at the time of a fracture.3,16 Our previous studies involving aspartic acid octapeptide targeted doxorubicin containing micelles for the treatment of osteosarcoma demonstrated that after conjugation to a drug and self-assembly into a micelle, the aspartic acid retains its HAp binding abilities.17
Having a bone-targeted peptide that targets fractures locally means that it is important to select a drug which acts locally to stimulate bone growth. Of particular interest in this area are GSK3β inhibitors. GSK3β is an enzyme that can promote degradation of beta-catenin and thus impact the Wnt/catenin pathway. Inhibition of GSK3β, therefore, promotes a robust osteoblast population and aggressive bone anabolism, as several groups have shown.18 Chen et al. demonstrated that levels of β-catenin in fractures far exceed those of healthy bone. By using a β-catenin knockout transgenic mouse model, fracture healing is essentially stopped with the removal of β-catenin. More importantly, they demonstrated that the addition of lithium chloride, a basic GSK3β inhibitor, to the mouse diet increased the bone density of the fracture callus. 19 Sisask et al. reported that a much more potent GSK3β inhibitor administered orally increased load failure of fractured femurs by 270% compared to the vehicle alone.20 Toxicity profiles of GSK3β inhibitors may permit short term administration, however, because the targets of GSK3β, Wnt and Hedgehog signaling are found in many cell types, one must be cautious of both short-term and long-term side effects. Nevertheless, the potent anabolic effects of GSK3β inhibitors render them excellent candidates for a targeted treatment of bone fractures. We have selected 6-bromoindirubin-3’-oxime (6Bio), a potent GSK3β inhibitor with an enzymatic IC50 of 5 nM, because of its osteoinductive nature.18,21-24
Conjugating the targeting ligand aspartic acid oligopeptide to 6BIO via a hydrolyzable oxime ester bond would yield a simple drug that would likely improve drug accumulation in bone. However, with slight modifications it is possible to target the site of inflammation in addition to the bone.25,26 6BIO is a drug that is hydrophobic, while the aspartic acid oligopeptide-targeting moiety is very hydrophilic. Upon addition of aliphatic hydrocarbon chain between 6BIO and the aspartic acid chain a micelle unimer is formed (Figure 1). The micelle increases the overall size of the drug carrier, thereby increasing uptake in the inflamed tissue. Unlike traditional micelles, however, each unimer contains a targeting ligand as well as a drug, ensuring high drug loading. Additionally, should the micelle destabilize in the blood stream, the drug will remain targeted to the fracture.
Figure 1.

Fracture-targeting micelles from amphiphilic unimer consisting of aspartic acid octapeptide, miniPEG spacer, and hydrophobic tail based on 11-aminoundecanoic acid conjugated to a GSK3β inhibitor by copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC).
EXPERIMENTAL SECTION
Materials
Solvents, dimethylformamide (DMF), dichloromethane (DCM), methanol (MeOH), dimethyl sulfoxide (DMSO), ethyl acetate, ether, and acetonitrile (ACN) were purchased from VWR, Fisher Scientific, or Sigma-Aldrich and were reagent grade or better. Piperidine, diisopropyl ethylamine (DIPEA), trifluoroacetic acid (TFA), triisopropylsilane (TIS), ethylenebis(diphenylphosphine) (DPPE), lithium hydroxide, bis(benzonitrile)palladium(II) chloride, copper(I) bromide, Chloramine T trihydrate, lithium diisopropyl amide (LDA), propargyl chloride, and sodium carbonate (Na2CO3), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) were purchased from Sigma-Aldrich. Sodium sulfate was purchased from Macron Chemicals. Fmoc-11-aminoundecanoic acid (Fmoc-AUA), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]-pyridinium 3-oxide hexafluorophosphate (HATU), 2-chlorotrityl resin, and Fmoc-L-Asp(OtBu)-OH) were purchased from AKSci. N-2-N-6-Bis(9-fluorenyl-methyloxycarbonyl)-L-lysine (Fmoc-Lys(Fmoc)-OH) was purchased from Aapptec, and 9-fluorenylmethoxycarbonyl-8-amino-3,6-dioxaoctanoic acid (Fmoc-miniPEG) was purchased from BioBlocks. Sephadex LH20 beads were purchased from Amersham Pharmacia Biotech AM. Fmoc–Azidohomoalanine (Fmoc-Dab(N3)-OH), 3-acetoxylindole, and Bromostatin were purchased from Chem-Impex International.
Synthesis of 6’-bromoindirubin (I)
6’-Bromoindirubin was synthesized as previously described 27 with modifications. Briefly, 615 mL of anhydrous methanol was set stirring under nitrogen in a 2 L round bottom flask. 6-Bromoisatin 3.37 g (15 mmol) and 3-acetoxylindole 2.63 g (15 mmol) were then added and the solution was bubbled with nitrogen for 45 min. Anhydrous sodium carbonate 3.98 g (37.5 mmol) was added and the reaction was stirred for 5 h in the dark. The resultant dark purple precipitate was isolated by centrifugation, the solid decanted with methanol and re-suspended in ethyl acetate. The 6’-bromoindirubin was washed 3x with brine. The organic layer was dried with anhydrous sodium sulfate and the solvent removed under reduced pressure to yield a dark purple solid. Yield: >90%, mass: 338.94, 340.93 (the two masses are due to the two common isotopes of bromine).
Synthesis of 6’-bromoindirubin-3’-oxime (II, 6BIO)
6’-bromoindirubin-3’-oxime (6BIO) was synthesized as described in Polychronopoulos et al.27 6-Bromoindirubin 3.06 g (9 mmol) and hydroxylamine hydrochloride 6.25g (90 mmol) were combined in a pressure reaction tube. Ninety mL of pyridine was added and purged under argon for 30 min. The reaction tube was sealed and stirred at 115-120° C for 2.5 h. The pyridine was removed under reduced pressure and the dark red product solid was washed with hexanes. The product was suspended in ethyl acetate and washed 2x with water followed by a wash with brine. The organic phase was dried with sodium sulfate and solvent removed under reduced pressure yielding a dark red solid (See Supporting Information). Yield >90% mass: 353.96, 355.96.
Synthesis of 2,2-dimethylpent-4-ynoic acid
Briefly, a round bottom flask was dried and purged with nitrogen and cooled to -78°C. LDA 2M 6.1 mL (13 mmol) was added and a stream of nitrogen was used to bubble the reaction. Methyl isobutyrate 1.6 mL (14 mmol) was added dropwise as the reaction was stirred. Propargyl chloride 891 μL (10 mmol) was also added dropwise as the reaction was stirred vigorously. The solution was allowed to warm to room temperature and was stirred over night. The reaction was then quenched with concentrated NH4Cl and extracted with ethyl acetate. The organic layer was dried with sodium sulfate and solvent removed under reduced pressure until a yellow oil remained.
Lithium hydroxide 480 mg (20 mmol) and MeOH were added to the product and stirred overnight, removing the methyl ester. Water was added to the reaction and acidified to pH 2 prior to extracting twice with ethyl acetate. The organic layer was dried with sodium sulfate and the solvent was removed under reduced pressure. Yield was 7% (2,2-dimethylpent-4-ynoic acid).
Synthesis of (2Z,3E)-6’-bromo-3-(((2,2-dimethylpent-4-ynoyl)oxy)imino)-[2,3’-biindolinylidene]-2’-one (III)
6BIO was conjugated to 2,2-dimethylpent-4-ynoic acid using standard DCC-assisted ester coupling. 6BIO 18 mg (0.05 mmol), 2,2-dimethylpent-4-ynoic acid (7.5 mg; 0.06 mmol), and DCC 12.4 mg (0.06 mmol) were added to 3 mL of DMF under nitrogen. DIPEA 26 μL (0.15 mmol) was then added and the reaction was stirred under nitrogen for 4 h. The product was filtered and dissolved in ethyl acetate. The solution was then washed 3 times with water and dried with sodium sulfate. The solvent was removed under reduced pressure onto celite (diatomite) and purified using flash chromatography (hexanes / ethyl acetate) (see Supporting Information). Yield: 80%.
Synthesis of amphiphilic unimers flanked with C-terminal azide
Unimers were synthesized as previously described with slight modifications.17 Briefly, in a small glass vial, 2-chlorotrityl resin (1.11 mmol/g) was loaded at 0.4 mmol/g with Fmoc-Dab(N3)-OH overnight in DCM and DIPEA. The resin was transferred to a SPPS vessel capable of bubbling nitrogen. The resin was then capped with four 5 mL washes of DCM:MeOH:DIPEA (17:2:1) followed by three washes of DCM and DMF, respectively. Following each amino acid coupling reaction, Fmoc-groups were removed by two 20-min incubations with 20% (v/v) piperidine in DMF. The resin was then washed three times with DMF prior to the next amino acid being added. Fmoc-AUA was added twice in a five-fold excess using HATU/DIPEA. Following addition of Fmoc-AUA, branched unimer (N3-A2-K-D4) was prepared by adding Fmoc-Lys(Fmoc)-OH followed by Fmoc-miniPEG and 4 Fmoc-L-Asp(OtBu)-OHs. Linear unimer (N3-A2-D8) was created by adding Fmoc-miniPEG followed by 8 Fmoc-L-Asp(OtBu)-OHs to AUA. Both unimers were capped with acetic acid, HATU, and DIPEA. The final resin-bound products were washed with DMF, DCM, MeOH (3 x each) and dried under vaccum. The products were cleaved from resin using TFA:TIS:H2O (90:5:5) and precipitated in diethyl ether prior to HPLC purification.
Click conjugation of (2Z, 3E)-6’-bromo-3-(((2,2-dimethylpent-4-ynoyl)oxy)imino)-[2,3’-biindolinylidene]-2’-one to unimers
N3-A2-D8 and N3-A2-K-D4 (both 0.01 mmol) were dissolved in DMF and purged with nitrogen for 20 min. Additional DMF was purged with nitrogen and was used to dissolve a mixture of (2Z,3E)-6’-bromo-3-(((2,2-dimethylpent-4-ynoyl)oxy)imino)-[2,3’-biindolinylidene]-2’-one (0.03 mmol), copper (I) bromide (0.01 mmol), and TBTA (0.01 mmol). The two solutions were combined and stirred under nitrogen for 20 min. Ascorbic acid (0.01 mmol) and a drop of water were then added. The reaction proceeded for 40 min after which it was diluted in MeOH and purified on LH20 column. The fractionated peaks were dried under reduced pressure. Yields for BIO-N3-A2-D8 and BIO-N3-A2-K-D8 were 65% and 48%, respectively (see Supporting Information).
Dynamic light scattering measurements of micelles
The hydrodynamic radius of the micelles were determined using Malvern Zetasizer Nano ZS. Measurements of both unimers were taken using 2.4 nmol/mL unimer at 25°C. Malvern software was used to determine hydrodynamic diameter.
Release kinetics of 6BIO from micelles
Release kinetics was measured for each of the micelles. Unimer (0.1 mg) was dissolved in 1 mL of human plasma. Samples for each time point (0, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h) were made in triplicate. Each sample was incubated in a water bath at 37°C for the allotted length of time. Each sample was then measured at λ=450 nm on an Agilent 1200 (0.01 M NH4HCO3, ACN). Area under the curve was measured using MestReNova software and the percent of the drug peak released was calculated as follows:
Stannylation of 6BIO
Stannylation of 6BIO was performed as previously described for other indols.28 To a pressure tube, 6BIO (1 mmol) was dissolved in 20 mL dioxane and purged with argon for 30 min. PdCl2(PhCN)2 (0.05 mmol), DPPE (0.1 mmol), and tributyl tin (3 mmol) were then added. The tube was purged with argon for an additional 10 min and sealed. The reaction was stirred vigorously at 110°C for 12 h. The product was dried on celite and purified using flash chromatography (hexanes 1% TEA/ EtOAc). The solvent was removed under reduced pressure resulting in a brown red solid (2Z,3E)-3-(hydroxyimino)-6’-(tributylstannyl)-[2,3’-biindolinylidene]-2’-one (see Supporting Information).
O-Alkylation of stannylated BIO ((2Z,3E)-3-(hydroxyimino)-6’-(tributylstannyl)-[2,3’-biindolinylidene]-2’-one)
DMF was added to a mixture of (2Z,3E)-3-(hydroxyimino)-6’-(tributylstannyl)-[2,3’-biindolinylidene]-2’-one (56 mg; 0.1 mmol), propargyl chloride (7.2 μL; 0.1 mmol), and cesium carbonate 38 mg (0.1 mmol) and stirred vigorously.29 The reaction was monitored every half hour throughout the reaction by LC/MS. At 1.5 h a small amount of dialkylated side-product was formed, and the reaction was stopped by adding water. The product was then extracted into EtOAc and washed 3 times with water and dried on sodium sulfate. Celite was added and the solvent was removed by reduced pressure. The product was purified by reverse phase chromatography (H2O-1% DIPEA; ACN-1% DIPEA). The product, (2Z,3E)-3-((prop-2-yn-1-yloxy)imino)-6’-(tributylstannyl)-[2,3’-biindolinylidene]-2’-one, was brownish purple (see Supporting Information).
Conjugation of alkylated stannylated 6BIO to unimers
Each unimer (N3-A2-D8 or N3-A2-K-D4) (0.01 mmol) was dissolved in DMF and purged with nitrogen for 20 min. To a separate vial of DMF purged with nitrogen (2Z,3E)-3-((prop-2-yn-1-yloxy)imino)-6’-(tributylstannyl)-[2,3’-biindolinylidene]-2’-one (0.03 mmol), copper(I)bromide (0.01 mmol), DIPEA (0.1 mmol), and TBTA (0.01 mmol) were added. The two solutions were combined and stirred under nitrogen for 40 min. Upon completion the reaction was diluted in MeOH and purified on LH20 column (MeOH containing 1% DIPEA). The fractionated peaks were dried under reduced pressure yielding a bluish purple solid.
Radioiodination of micelles and 6BIO
The stannylated unimers and stannylated 6BIO (0.3–0.5 μmol) were dissolved in 100 μL DMF. The solutions were transferred to Wheaton V-vials containing a solution of [125I]NaI (5.0 mCi) in 100 μL DMF phosphate-buffered saline (PBS) (pH 7.4). Chloramine-T was added, and the reaction mixture stirred for 15 min at ambient temperature. Purification of the iodinated micelles was performed using a LH20 column eluted with MeOH. Iodinated 6BIO was dissolved in EtOAc and was washed twice with water. The solvent of all three reactions was evaporated using a stream of nitrogen, and the residue was dissolved with PBS (pH 7.4) to obtain solutions with an activity concentration of 370 kBq/100 μL. The overall radiochemical yield was ca. 10% (see Supporting Information).
Murine fracture induction
All animal studies were reviewed and approved by Purdue’s animal care and use committee protocol and were performed as described in the literature. CD4 Swiss mice (30-35 g) acquired from Harlan laboratories were used for these experiments. A stabilized femoral fracture was performed under aseptic conditions with isoflurane anesthesia. Skin around the knee was shaved and cleaned with an alcohol pad first, then with Betadine solution. The skin incision was made medial parapatellar. The patella was then dislocated and an incision was made under the patella. A 25 gauge needle was used to ream the intramedullary canal. A 22 guage locking nail (where both ends are flattened to produce rotational stability), was then inserted. The wound was sutured and the bone was then fractured using a three point bending device that has a built-in stop to prevent excess injury. Subcutaneous Buprenorphine (0.05 – 0.1 mg/kg) was administered at the time of surgery, followed by a dose every 12 h for 3-7 days post operation.
Injection and dissection techniques/counting for 24 h biodistribution
From the iodinated products described previously 1 μCi/mL solutions were reconstituted in sterile PBS and equilibrated at room temperature for 1 h. The study was performed on 3 groups of mice with 5 mice per group. The injections were performed two weeks following fracture induction, ensuring blood flow had returned to the area. Each mouse received a 0.1 μCi (0.1 mL) dose of the radio-iodinated linear micelles, branched micelles, or free drug by i.v. tail-vein injection. Animals were then placed back in their cages for 24 h. At 24 h they were sacrificed using CO2 excess. Blood was drawn from a cardiopuncture and all other organs measured were removed (lungs, liver, spleen, kidneys, fractured femur, healthy femur). The activity was counted using Packard Cobra Auto-Gamma and was normalized by dividing the count rate by the weights of each organ.
Injection and techniques for imaging
From the iodinated products described previously, 1 mCi/mL solutions reconstituted in sterile PBS and equilibrated at room temperature for 1 h. The study was performed on 3 groups of mice with 5 mice per group. Two weeks following fracture induction each mouse received 0.1 mCi (0.1 mL) of dose by i.v. tail-vein injection. Animals were then returned to their cages until euthanasia by CO2 overdose at the desired time point (1 h, 4 h, 24 h). Animals were imaged using MiLabs U-SPECT-II/CT. 3D reconstructions were performed using Image J software.
RESULTS AND DISCUSSION
Design and characterization of micelles
We set out to develop a fracture-targeted anabolic-loaded micelle by utilizing the unique microenvironment available in bone fractures. That is, freshly exposed HAp due to a fracture, additional HAp exposed by osteoclasts during resorption, newly exposed calcified bone, and the local inflammatory response.
HAp can be targeted using any one of several calcium adsorbing ligands including bisphosphonates, tetracycline, and acidic oligopeptides.30-33 Both bisphosphonates and tetracycline display various biocompatibility and toxicity issues which may inhibit healthy bone turnover in fractures.34-38 In order to maintain bone homeostasis, the more biocompatible option is acidic oligopeptides. Aspartic acid octapeptide, as evidence suggests, has sufficient number of aspartic acids to ensure rapid binding.39 Evidence also suggests that aspartic acid octapeptide has sufficient HAp affinity to target poor quality bone as found in osteoporosis.40 Unlike bisphosphonates and tetracyclines, aspartic acid octapeptide has a low toxicity even in very high concentrations.17,41
Targeting a drug to a fracture site via aspartic acid octapeptides by itself does not fully exploit the microenvironment of a fracture. In fractured bones, the extent of bone accumulation is expected to be far more dramatic when using large molecules. Yuan et al. proposed a mechanism by which inflamed tissue leads to leaky vasculature and subsequent inflammatory cell-mediated sequestration (ELVIS). This theory essentially states that large molecules will readily be extravasated from the leaky vasculature of the inflamed fracture callus. Then, inflammatory cells actively phagocytose and sequester any large molecules in the area.42,43 Bone fracture calluses certainly fall under the category of inflamed tissue, and as such, higher drug accumulation could hypothetically be achieved by increasing the size of a drug delivery system.
The proposed micellar design is a simplified way of increasing the size of the drug delivery system to improve accumulation in inflamed tissue. By using the hydrophilic targeting peptide aspartic acid octapeptide covalently bound by a hydrolyzable linker to a hydrophobic drug a micelle-forming unimer was created. The designed unimer was based on our previous studied bone-targeted micelles and was largely synthesized similarly using solid-phase peptide synthesis (SPPS) methodology.17 Synthesis began with an azide-containing amino acid (azidohomoalanine) that provided a clickable site for attachment of a drug/linker. To the azide-containing amino acid, two 11-aminoundecanoic acids (AUAs) were attached to increase the overall hydrophobicity and therefore the stability of the micelle. A miniPEG spacer (8-amino-2,6-dioxaoctanoic acid) was added for flexibility followed by aspartic acid octapeptide for targeting. Finally, the unimer was capped with acetic acid, preventing the terminal primary amine from initiating premature hydrolysis of the amine sensitive drug linker yielding the finalized linear unimer, BIO-A2-D8 (Figure 2). BIO-A2-D8 indicates that it is a 6BIO (BIO)-containing unimer with two aminoundecanoic acids (A2) and eight aspartic acids in a linear fashion (D8).
Figure 2.
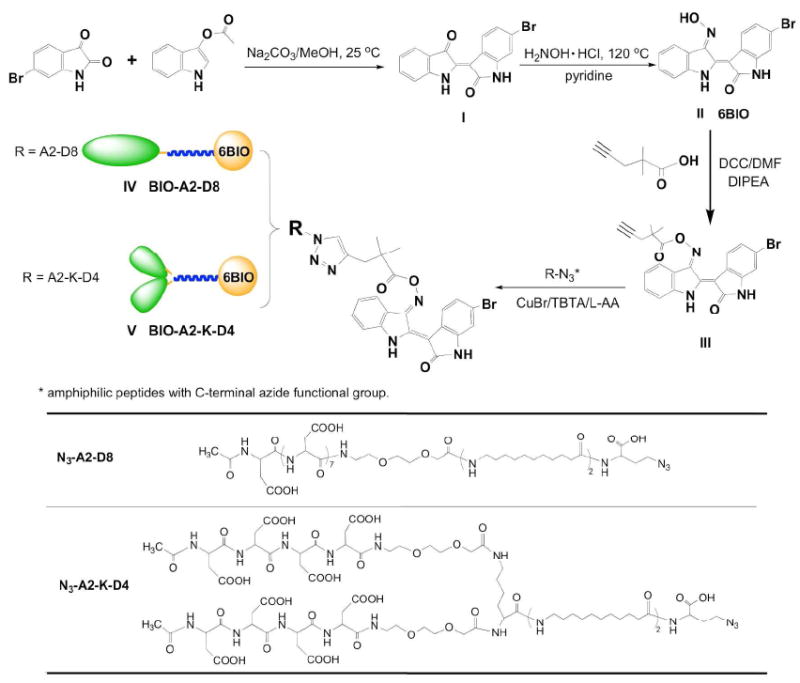
Synthesis of a clickable 6BIO and conjugation of 6BIO and linker to micelle unimers.
The stability of the micelle can be tailored by the number of AUAs. However, increasing the number of AUAs in a linear micelle by too much can result in aggregates and non-micellar structures. To increase the number of AUAs and avoid the formation of non-micellar structures, we also studied a branched aspartic acid head group. Building from the original azide-aminoundacanoic acid base, lysine was added to the second aminoundecanoic acid. Both primary amines on the lysine were deprotected and miniPEG was added to both, followed by four aspartic acids each and capping with acetic acid. The 6BIO (BIO) containing unimer contains two aminoundecanoic acids (A2) is branched at the lysine (K) and ends in two branches of four aspartic acids (D4) yielding BIO-A2-K-D4. This branched conformation increased the head group size and therefore the conicity of the unimer. By increasing the head group size, far greater hydrophobic portions could be incorporated before the conicity of the unimer was lost, and a bilayer, aggregate, or other conformation was formed.17
The designed micelles, in addition to increasing the fracture accumulation by increasing the size, have several unique features. By building the unimers by SPPS, the unimers were monodisperse, which aided in characterization. Furthermore, each unimer featured a targeting ligand and a drug. This means that in the event of the micelle rupturing in the blood stream, the drug still could accumulate in the fracture site by aspartic acid octapeptide-HAp interactions.44 In addition, whereas micelles often vary in drug loading depending on the formulation method used, the linear and branched micelles contained a consistent 17% or 15% drug loading by weight, respectively.45
The drug being attached to the unimers was a GSK3β inhibitor, 6BIO. It has been used for increasing osteoblast activity and has an enzymatic IC50 of 5 nM.18,21,46 6BIO by itself, however, is only sparingly soluble in water. By including 6BIO in the unimer design, the unimer solubilized the drug while the drug stabilized the micelle. The oxime of 6BIO was first conjugated to the carboxylic group of a 4-pentynoic acid derivative and then clicked to the azide-containing unimer by the alkyne on the 4-pentynoic acid derivative. Upon completion of the click reaction, the unimer was purified, dried, and formed micelles upon reconstitution in PBS.
Size determination by dynamic light scattering
Dynamic light scattering was used to determine the hydrodynamic diameter of the micelles. The micelles self-assembled at the concentrations theorized to be sufficient for injection for fracture healing. BIO-A2-D8 and BIO-A2-K-D4 were 28.8 nm and 11.2 nm respectively (Figure 3). Sizes were slightly smaller that the micelles with similar structures in our previous work with doxorubicin loaded micelles.17
Figure 3.

Dynamic light scattering of the linear micelle BIO-A2-D8 and the branched micelle BIO-A2-K-D4 in PBS at 25°C.
Micellar drug release
Drug-linker selection plays an important role in maintaining targeting ligand to drug integrity until the drug has reached the target tissue. This micelle features an oxime linkage which is base sensitive and hydrolytically cleavable at the pH ranges found in a bone fractures (6.9-7.6).47 Simple 4-pentynoic oxime esters degrade within hours (see Supporting Information) and would likely release 6BIO prior to fracture accumulation. To improve the hydrolytic stability of the conjugate, germinal dimethyl substituents were introduced into the structure (Figure 2). This increased steric hindrance to the 107° Bürgi-Dunitz angle required for a Bac2 reaction to release the drug and completely eliminate the possibility of an E1cb reaction. The stabilized oxime ester linkage slowed the drug release, reaching 54% and 63% released drug after 6 h for the linear and branched micelles respectively (Figure 4). Though the linker is not specific to the fracture microenvironment, the slow degradation would reduce the amount of drug released in the blood stream prior to fracture accumulation and reduces the frequency of necessary dosing for treatment.
Figure 4.

Time dependent 6BIO release from the linear micelle BIO-A2-D8 and from the branched micelle BIO-A2-K-D4 in plasma at 37°C
Biodistribution
Organ biodistribution at 24 h
BIO-A2-D8 and BIO-A2-K-D4 have hydrolyzable oxime ester linkages designed to release the 6BIO for treatment of bone fractures. Although a cleavable linker is necessary for therapy studies, it may pose a problem with biodistribution studies. At 24 h of an in vivo study, 90-95% of the drug may be released. In a biodistribution study, premature release would not give accurate information on how well the targeting ligand is able to remain in the fracture site. A non-degradable oxime ether linker replaced the oxime-ester linker in the micelles for biodistribution. Radiolabeling BIO-A2-D8, BIO-A2-K-D4 and 6BIO was done by substituting the bromine on 6BIO with 125I. These minor modifications allowed the majority of the molecule to remain unmodified while giving information on targeting and free drug clearance (Figure 5).
Figure 5.
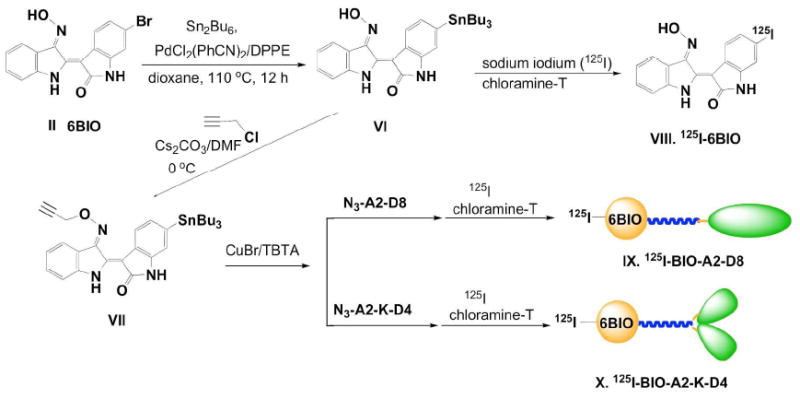
Synthesis of radioiodinated micelles and 6BIO.
In bone, HAp increases in crystallinity over time. It is this higher crystalline state to which acidic oligopeptides preferentially bind.3,16 In bone fracture patients, highly active osteoclasts produce extensive resorption surfaces by exposing highly crystalline HAp surfaces to which acidic oligopeptides are able to target.48 This specificity to highly crystalline HAp may additionally reduce non-specific binding to the majority of the non-fractured bone. An organ biodistribution was performed to elucidate this and other questions about the fate of the micelles in vivo.
Several interesting observations emerged from the biodistribution. 6BIO is a hydrophobic drug and its high accumulation in the lungs, liver, and spleen was likely due to the drug aggregating. By turning the drug into an amphiphile, the particle size was controlled, and the liver, spleen, and lung accumulation all decreased significantly (p<0.001) (Figure 6). However, kidney uptake dramatically increased with both micelles (p<0.001). The kidney accumulation may be due to the kidney’s ability to recycle peptides such as the aspartic acid octapeptide back into the blood stream.49,50 As long as the micelle is not internalized into renal cells, the 6BIO should eventually hydrolyze, be cleared, and not inhibit GSK3β activity.
Figure 6.
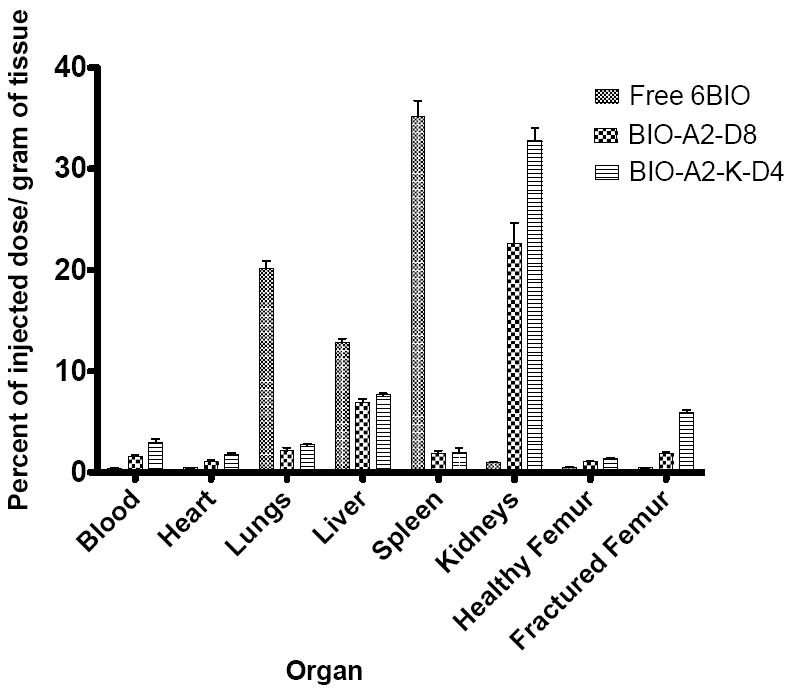
Organ biodistribution of free 6BIO, the linear micelle BIO-A2-D8, and the branched micelle BIO-A2-K-D4 measured in percent of injected dose/gram of tissue. Of particular interest the drug accumulation was significantly higher in the fractured femur over the healthy femur in both of our targeted micelles (p<0.001). The accumulation in fractured femurs was also significantly higher in the targeted groups over the free drug (p<0.001).
The primary purposes of this study were to elucidate the bone and fracture targeting potential of targeted micelles over free drug, as well as the differences between branched and linear designs. Naturally, accumulation in healthy femurs was higher in both the linear (1.0±0.27% injected dose/g; p<0.01) and the branched (1.4±0.14% injected dose/g; p<0.001) micelles than free 6BIO (0.5% injected dose/g ± 0.10). This is also the first evidence of the branched micelle having a higher accumulation than the linear micelle in bone (p<0.05). These trends were amplified when comparing the fractured femurs. The fractured femurs of the free 6BIO, linear, and branched micelles were each statistically different from each other (p<0.001) with percent of injected dose per gram of 0.4±0.05, 1.9±0.32, and 6.0±0.51, respectively. Both unimers demonstrated an important increase of accumulation in the fractured femurs over the free drug control.
The micelles were able to show fracture-specific targeting compared to healthy bone. While there was no statistical difference in accumulation of the free drug between the healthy and the fractured femurs, both the linear and the branched micelles showed significantly (p<0.001) higher drug uptake in the fractured femur by a 1.8 and a 4.3 fold increase, respectively.
SPECT/CT biodistribution
In order to validate the organ biodistribution and study the clearance of the micelles, SPECT/CT imaging was performed at 1 h, 4 h, and 24 h (Figure 7) after administration of micelles. In general, fracture-specific uptake was more evident at later time points than earlier ones. At 1 h, a small amount of fracture accumulation was visible in both micelles; however, high signal intensities in both the lungs and kidneys made them difficult to discern. On the other hand, at one hour the free drug was primarily found in the stool. At 4 h, free drug was still primarily contained in the bowel; however, spleen accumulation began to be more evident. Again this bowel clearance was likely due to aggregation and clearance though the liver of free 6BIO, an observation in accordance with the lungs, liver, and spleen accumulation seen in the 24 h organ biodistribution. Meanwhile, the linear micelle had cleared somewhat from the lungs, was still strong in the kidneys, but much more visible in the fracture. The branched unimer showed superior targeting at 4 h with the majority of accumulation in the femur and kidneys. At 24 h, free drug appears to be cleared; all signals were background. Both micelles had their highest fracture accumulation intensities at 24 h relative to other organs. Liver to kidney accumulation was lower in the branched unimer, which correlates with the 24-h organ biodistribution data (Figure 7-24 h compared to Figure 6). In both micelles throughout the experiment there was no apparent accumulation in the bowels, leading us to believe that most was excreted through urination. Most importantly, there was no noticeable uptake in the healthy bone while the fractured bone had high accumulation.
Figure 7.

SPECT/CT of mice at 1 h, 4 h, and 24 h. The upper frames are a 3D reconstruction of the whole mouse. The lower frames are a cross section of femurs with the fractured femur on the right. Organs are labeled as follows: A: fracture, B: kidneys, C: Liver, D: Lungs, E: Spleen, F: Bowels.
Between the SPECT data and the organ biodistribution data there were a few discrepancies. Primarily, the high ratio of renal accumulation to fracture accumulation in the organ biodistribution is not as pronounced in the SPECT data. There are several explanations for this. First, the kidneys contain proteins for the reabsorption of peptides from the urine.49,50 It is plausible that peptide kidney transporters were saturated in the SPECT study due to a 100-fold increase in drug dose compared to the organ biodistribution. Bone fracture targeting is not limited by a finite number of receptors as in the kidney; rather, it is limited by the surface area of the bone and may not have been similarly saturated.
In addition, the organ biodistribution is measured in injected dose/gram and does not account for differences in tissue type or location of accumulation within an organ. Bone is far more dense (1900 kg/m3) than soft tissue organs such as kidneys or liver (1030-1060 kg/m3).51 In the organ biodistribution, this discrepancy in density of tissues will dilute the signal per gram tissue in bone to nearly half of what would be observed in soft tissue such as a kidney with an equal amount of 125I signal. While no hotspots are found in the kidney due to the accumulation being distributed throughout the entire kidney, the signal in fractured femur is further muted by the entire bone being weighed for the measurement rather than the fracture alone. The combination of these factors better explains why in the SPECT study, accumulation per volume is much higher in the fracture compared to the kidney.
CONCLUSIONS
In this study we created two fracture-targeted micelles (branched or linear) designed to increase the rate of healing in bone fractures. These micelles are built on the concept that the micellar corona can function as both a moiety that gives amphiphilicity, as well as being a low-toxicity targeting ligand. Likewise, the 6BIO drug functions both as a pharmaceutical as well as giving stability to the micelle core. The micelles also feature a hydrolyzable oxime ester bond to the drug that releases the drug unmodified over several days.
In vivo, both the branched and linear micelle designs demonstrated excellent uptake in fractured bone versus healthy bone. The branched unimer demonstrated both a higher ratio of fracture to kidney uptake as well as fracture to healthy bone accumulation over the linear unimer. Future treatment studies will elucidate fracture-healing capabilities of these micelles and include histology of the kidneys and liver to assess any acute toxicities associated with the treatment.
Fracture treatment in the elderly is a clear unsolved problem. The lifetime incidence of hip fractures is estimated to be 8.1% and 19.5% in males and females respectively.52 The frequency of those fractures increasing exponentially after the age of 65.52 Using current therapies 24% of elderly with hip fractures will die within the first year and 50% will not regain the ability to walk.53 The problem needs to be approached in a two-fold manner. First, more basic research into understanding biochemical pathways in the fracture that might be employed in developing anabolic agents and understanding the bone microenvironment in order to better tune the release mechanism. Second, applied research needs to be conducted determining the in vivo stability, potential immunogenicity, controlling the deposition, and release of anabolic agents. This research answers some of the questions. With additional studies, a nonsurgical treatment for delayed union and nonunion fractures may be possible.
Supplementary Material
Acknowledgments
We thank Drs. Ananda K Kanduluru, Skarapalayam Mahalingam, and Pengcheng Lu for assisting with animal work. We also thank Dr. Aaron B. Taylor and Jennifer Lu for their advising on instrumentation. This research was supported in part by NIH grant RO1 GM69847 (to J.K.) and Purdue University.
Footnotes
SUPPORTING INFORMATION
Pysiochemical characterization of compounds, unimers, and micelles. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.Shea JE, Miller SC. Adv Drug Deliv Rev. 2005;57(7):945–957. doi: 10.1016/j.addr.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 2.Posner AS, Betts F. Acc Chem Res. 1975;8(8):273–281. [Google Scholar]
- 3.Wang D, Miller SC, Shlyakhtenko LS, Portillo AM, Liu X-M, Papangkorn K, Kopečková P, Lyubchenko Y, Higuchi WI, Kopeček J. Bioconjug Chem. 2007;18(5):1375–1378. doi: 10.1021/bc7002132. [DOI] [PubMed] [Google Scholar]
- 4.Lacey D, Timms E, Tan HL, Kelley M, Dunstan C, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Cell. 1998;93(2):165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 5.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Proc Natl Acad Sci. 1998;95(7):3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Väänänen H, Zhao H, Mulari M, Halleen JM. J Cell Sci. 2000;113(3):377–381. doi: 10.1242/jcs.113.3.377. [DOI] [PubMed] [Google Scholar]
- 7.Harada S. Nature. 2003;423(6937):349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 8.Bartl R, Frisch B, Bartl C. Osteoporosis: Diagnosis, prevention, therapy. Springer; 2009. [Google Scholar]
- 9.Dempster DW. Am J Manag Care. 2011;17:S164–S169. [PubMed] [Google Scholar]
- 10.Bishop GB, Einhorn TA. Int Orthop. 2007;31(6):721–727. doi: 10.1007/s00264-007-0424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ong KL, Villarraga ML, Lau E, Carreon LY, Kurtz SM, Glassman SD. Spine. 2010;35(19):1794–1800. doi: 10.1097/BRS.0b013e3181ecf6e4. [DOI] [PubMed] [Google Scholar]
- 12.Fournier P, Boissier S, Filleur S, Guglielmi J, Cabon F, Colombel M, Clézardin P. Cancer Res. 2002;62(22):6538–6544. [PubMed] [Google Scholar]
- 13.Giraudo E, Inoue M, Hanahan D. J Clin Invest. 2004;114(5):623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebart T, Pabst A, Klein MO, Kämmerer P, Gauss L, Brüllmann D, Al-Nawas B, Walter C. Clin Oral Investig. 2009;15(1):105–111. doi: 10.1007/s00784-009-0365-2. [DOI] [PubMed] [Google Scholar]
- 15.Ishizaki J. J Bone Miner Metab. 2009;27(1):1–8. doi: 10.1007/s00774-008-0004-z. [DOI] [PubMed] [Google Scholar]
- 16.Miller S, Pan H, Wang D, Bowman B, Kopečková P, Kopeček J. Pharm Res. 2008;25(12):2889–2895. doi: 10.1007/s11095-008-9706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Low SA, Yang J, Kopeček J. Bioconjug Chem. 2014;25(11):2012–2020. doi: 10.1021/bc500392x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baron R, Rawadi G. Endocrinology. 2007;148(6):2635–2643. doi: 10.1210/en.2007-0270. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R, Alman BA. PLoS Med. 20074(7):1216–1229. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sisask G, Marsell R, Sundgren-Andersson A, Larsson S, Nilsson O, Ljunggren Ö, Jonsson KB. Bone. 2013;54(1):126–132. doi: 10.1016/j.bone.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 21.Piters E, Boudin E, Van Hul W. Arch Biochem Biophys. 2008;473(2):112–116. doi: 10.1016/j.abb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Wang F-S, Ko J-Y, Weng L-H, Yeh D-W, Ke H-J, Wu S-L. Life Sci. 2009;85(19-20):685–692. doi: 10.1016/j.lfs.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Georgiou KR, King TJ, Scherer MA, Zhou H, Foster BK, Xian CJ. Bone. 2012;50(6):1223–1233. doi: 10.1016/j.bone.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda T, Kokabu S, Ohte S, Sasanuma H, Kanomata K, Yoneyama K, Kato H, Akita M, Oda H, Katagiri T. Differentiation. 2010;80(1):46–52. doi: 10.1016/j.diff.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Metselaar JM, van den Berg WB, Holthuysen AEM, Wauben MHM, Storm G, van Lent PLEM. Ann Rheum Dis. 2004;63(4):348–353. doi: 10.1136/ard.2003.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quan L, Zhang Y, Crielaard BJ, Dusad A, Lele SM, Rijcken CJF, Metselaar JM, Kostková H, Etrych T, Ulbrich K, Kiessling F, Mikuls TR, Hennink WE, Storm G, Lammers T, Wang D. ACS Nano. 2014;8(1):458–466. doi: 10.1021/nn4048205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polychronopoulos P, Magiatis P, Skaltsounis A-L, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M, Greengard P, Meijer L. J Med Chem. 2004;47(4):935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 28.Corcoran EB, Williams AB, Hanson RN. Org Lett. 2012;14(17):4630–4633. doi: 10.1021/ol302076d. [DOI] [PubMed] [Google Scholar]
- 29.McCarroll AJ, Walton JC. J Chem Soc Perkin Trans 2. 2000;2(9):1868–1875. [Google Scholar]
- 30.Rogers MJ, Gordon S, Benford HL, Coxon FP, Luckman SP, Monkkonen J, Frith JC. Cancer. 2000;88(S12):2961–2978. doi: 10.1002/1097-0142(20000615)88:12+<2961::aid-cncr12>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 31.Myers HM. Am J Phys Anthropol. 1968;29(2):179–182. doi: 10.1002/ajpa.1330290211. [DOI] [PubMed] [Google Scholar]
- 32.Wang D, Miller S, Sima M, Kopecková P, Kopeček J. Bioconjug Chem. 2003;14(5):853–859. doi: 10.1021/bc034090j. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Miller SC, Kopečková P, Kopeček J. Adv Drug Deliv Rev. 2005;57(7):1049–1076. doi: 10.1016/j.addr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Marx RE, Sawatari Y, Fortin M, Broumand V. J Oral Maxillofac Surg. 2005;63(11):1567–1575. doi: 10.1016/j.joms.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 35.O Hoff A, Toth BB, Altundag K, Johnson MM, Warneke CL, Hu M, Nooka A, Sayegh G, Guarneri V, Desrouleaux K, Cui J, Adamus A, Gagel RF, Hortobagyi GN. J Bone Miner Res. 2008;23(6):826–836. doi: 10.1359/JBMR.080205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madison JF. Arch Dermatol. 1963;88:58–59. doi: 10.1001/archderm.1963.01590190064008. [DOI] [PubMed] [Google Scholar]
- 37.Vernillo AT, Rifkin BR. Adv Dent Res. 1998;12(1):56–62. doi: 10.1177/08959374980120012101. [DOI] [PubMed] [Google Scholar]
- 38.Solomon DH, Hochberg MC, Mogun H, Schneeweiss S. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. 2009;20(6):895–901. doi: 10.1007/s00198-008-0759-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Low SA, Kopeček J. Adv Drug Deliv Rev. 2012;64(12):1189–1204. doi: 10.1016/j.addr.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan H, Sima M, Miller SC, Kopečková P, Yang J, Kopeček J. Biomaterials. 2013;34(27):6528–6538. doi: 10.1016/j.biomaterials.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sekido T, Sakura N, Higashi Y, Miya K, Nitta Y, Nomura M, Sawanishi H, Morito K, Masamune Y, Kasugai S, Yokogawa K, Miyamoto K. J Drug Target. 2001;9(2):111–121. doi: 10.3109/10611860108997922. [DOI] [PubMed] [Google Scholar]
- 42.Yuan F, Quan L, Cui L, Goldring SR, Wang D. Adv Drug Deliv Rev. 2012;64(12):1205–1219. doi: 10.1016/j.addr.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren K, Purdue PE, Burton L, Quan L-D, Fehringer EV, Thiele GM, Goldring SR, Wang D. Mol Pharm. 2011;8(4):1043–1051. doi: 10.1021/mp2000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Sima M, Mosley RL, Davda JP, Tietze N, Miller SC, Gwilt PR, Kopečková P, Kopeček J. Mol Pharm. 2006;3(6):717–725. doi: 10.1021/mp0600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jia Z, Zhang Y, Chen YH, Dusad A, Yuan H, Ren K, Li F, Fehringer EV, Purdue PE, Goldring SR, Daluiski A, Wang D. J Controlled Release. 2015;200:23–34. doi: 10.1016/j.jconrel.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krause U, Harris S, Green A, Ylostalo J, Zeitouni S, Lee N, Gregory CA. Proc Natl Acad Sci USA. 2010;107(9):4147–4152. doi: 10.1073/pnas.0914360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swenson O. J Bone Joint Surg Am. 1946;28:288–293. [PubMed] [Google Scholar]
- 48.Schindeler A, McDonald MM, Bokko P, Little DG. Semin Cell Dev Biol. 2008;19(5):459–466. doi: 10.1016/j.semcdb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 49.Kanai Y, Hediger MA. Nature. 1992;360(6403):467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 50.Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. Biochem Biophys Res Commun. 1998;246(2):470–475. doi: 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- 51.Newman J. Physics of the life sciences. Springer Science & Business Media; 2008. [Google Scholar]
- 52.Oden A, Dawson A, Dere W, Johnell O, Jonsson B, Kanis JA. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. 1998;8(6):599–603. doi: 10.1007/s001980050105. [DOI] [PubMed] [Google Scholar]
- 53.Looker AC, Melton LJ, Harris TB, Borrud LG, Shepherd JA. J Bone Miner Res. 2010;25(1):64–71. doi: 10.1359/jbmr.090706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.