

Introduction
Both arrhythmogenic right ventricular cardiomyopathy (ARVC) and Brugada syndrome (BS) are inherited conditions which distinctively affect the right ventricle and predispose to arrhythmic sudden cardiac death (SCD)1–2. While ARVC is a heart muscle disorder characterized by loss of right ventricular myocardium with fibrofatty substitution which constitute a part of the substrate for malignant ventricular arrhrythmias3–5, BS is a primary electrical heart disease, and the ventricular electrical instability is not directly related to widespread, major structural myocardial changes6–8.
Since the first description of BS in 1982, problems regarding nosographic definition, pathogenetic relationship, and differential diagnosis with ARVC have emerged on the basis of common phenotypic manifestations mostly related to the predominant RV involvement with right precordial ECG repolarization abnormalities, RV (outflow tract) conduction disturbances and RV ventricular arrhythmias degenerating into ventricular fibrillation9–10. Moreover, due to the “concealed” nature of structural changes, which may fall under the resolution power of routine imaging techniques, early/minor variants of ARVC may simulate a channelopathy-like primarily electrical disorder mimicking BS11.
Whether ARVC and BS actually have a common pathogenetic denominator remained unsolved by past clinical and pathologic studies. However, recent experimental studies have renewed the interest in identifying the mechanisms responsible of the overlapping disease phenotype by demonstrating a subcellular interrelationship due to a crosstalk between desmosomal and sodium channel proteins12–21.
This article reviews the available clinical, imaging, electrophysiologic and pathologic evidence of the overlapping phenotype between ARVC and BS and reweaves the scientific discussion about the possible pathogenetic link between these two conditions on the basis of significant insights coming from recent studies of molecular biology. The purpose is to put into perspective the debate on this controversial issue, without claiming to draw definite conclusions.
ARVC versus Brugada syndrome
According to traditional diagnostic criteria ARVC and BS are distinct clinical entities. ARVC demonstrates substantial differences from Brugada syndrome with respect to involved genes, underlying cardiomyopathic changes, autonomic and antiarrhythmic drug modulation of ECG abnormalities, circumstances and mechanisms of arrhythmias and outcome (Table 1).
Table 1.
ARVC versus Brugada syndrome
| ARVC | Brugada Syndrome | |
|---|---|---|
| Age of presentation (yrs) | 15–30 | 30–40 |
| Gender | M>F (3:1) | M>F (8:1) |
| Distribution | World-wide (Italy) | World-wide (Southeast Asia) |
| Inheritance | AD (AR) | AD |
| Predominant pathogenetic genes | Desmosomal genes | SCN5A gene |
| Typical symptoms | Palpitations, syncope, cardiac arrest | Syncope, cardiac arrest |
| Imaging | Structural RV (and LV) abnormalities | Normal |
| Biopsy | Fibrofatty replacement | Normal |
| ECG: repolarization | Right precordial TWI | Right precordial high take-off ST elevation and TWI |
| ECG depolarization | Right precordial QRS prolongation, ε waves | RBBB/LAD |
| AV conduction times | Normal | Prolonged PR/HV interval |
| ECG changes | Fixed | Dynamic |
| Ventricular arrhythmias | Monomorphic VT, VF | Polymorphic VT, VF |
| Typical mechanism of VT | Scar- related reentry | Phase 2 reentry |
| Natural history | Sudden death, heart failure | Sudden death |
AD = autosomal dominant; AR = autosomal recessive; AV = atrioventricular; LAD = left axis deviation; LV = left ventricle, RBBB = right bundle branch block; RV = right ventricle; TWI = T-waves inversion; VF = ventricular fibrillation; VT = ventricular tachycardia
At variance with ARVC that is a genetic heart muscle disorder of intercellular junctions resulting from mutations of desmosomal genes, BS is a channelopathy caused by defects in genes coding for sodium channel, calcium channel, potassium channel, or channel-interacting proteins. Mutations in SCN5A, encoding the cardiac predominant sodium channel α-subunit, are found in up to 30% of patients with BS, while other mutant genes are very uncommon22.
Patients with BS typically have no signs of overt structural heart disease detectable by imaging techniques such as echocardiography, angiography and magnetic resonance imaging;6–7 in contrast, ARVC patients characteristically show right ventricular morpho-functional changes such as global dilatation, bulgings/aneurysms, and wall motion abnormalities23–24.
The ECG pattern of BS is characterized by a high take-off and downsloping ST-segment elevation (“coved-type morphology) followed by T-wave inversion in V1 and V2/V3, while right precordial T-wave inversion in ARVC is usually preceded by no or mild upward ST-segment6 (Figure 1).
Figure 1.
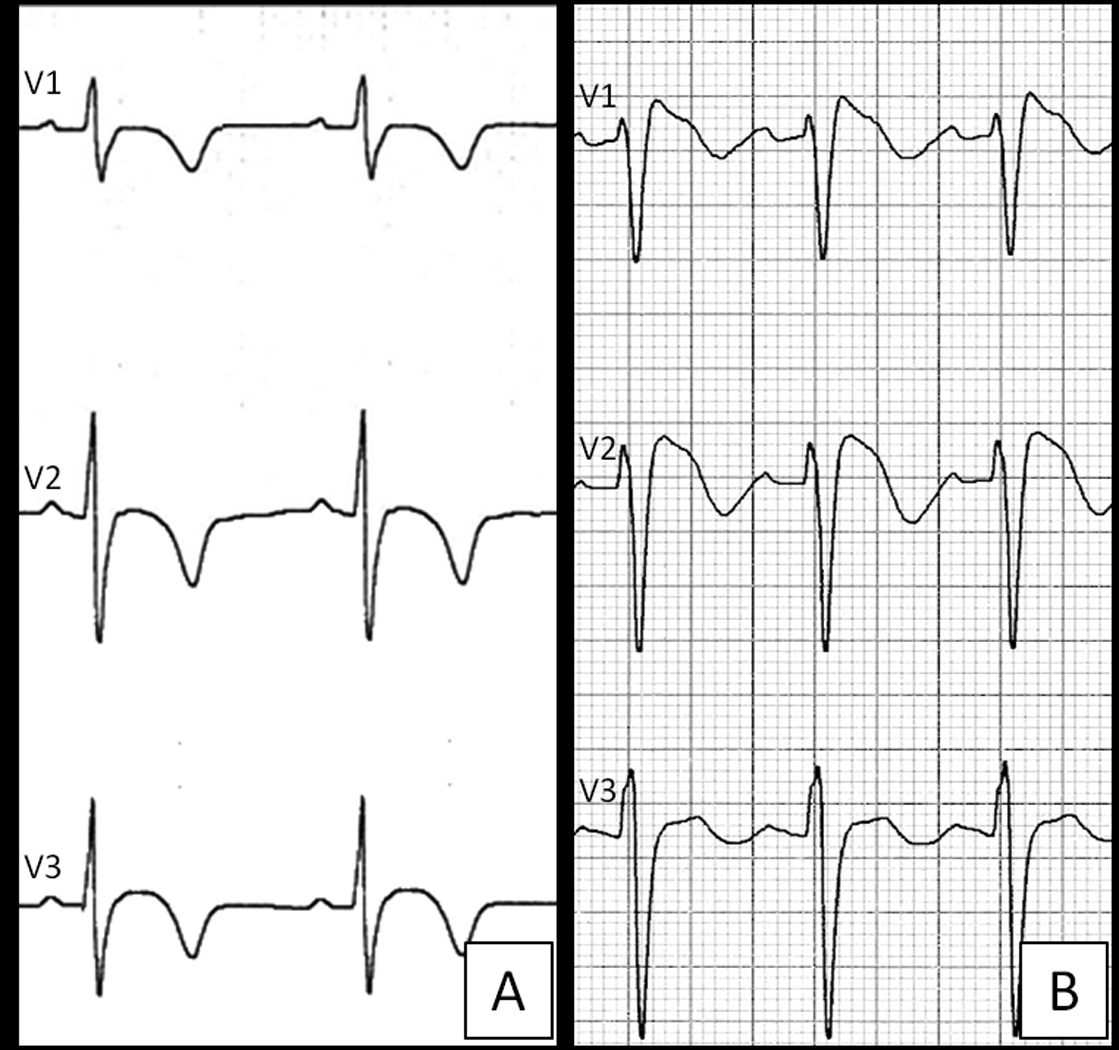
Right precordial ECG repolarization abnormalities in ARVC versus BS. Although both conditions are characterized by T-wave inversion in V1–V2/V3, ARVC shows a baseline J-point/ST-segment, while BS is characterized by a high take-off ST-segment elevation featuring a “coved type” morphology.
The most common arrhythmic event in patients with ARVC is a monomorphic ventricular tachycardia (VT) with a left bundle branch block pattern, most likely resulting from a macro-reentry around fibrofatty tissue4–5. Fibro-fatty myocardial replacement accounts for a right intraventricular conduction defect and predisposes to a scar-related VT similar to that observed in the postmyocardial infarction setting. In contrast, in patients with BS, a genetically-induced sodium channel loss of function has been proposed to lead to loss of the epicardial action potential dome and transmural/epicardial dispersion of repolarization, which, in turn, could induce elevation of the ST-segment and predispose to “phase 2 reentry (or ‘local re-excitation’) leading to rapid polymorphic ventricular tachycardia, which can degenerate into ventricular fibrillation (“repolarization theory)7–8 (Figure 2). On the other hand, impaired conduction in the right ventricular outflow tract in BS may in itself provoke or contribute to inhomogeneous repolarization to trigger malignant arrhythmic events (“depolarization” theory)25.
Figure 2.

Mechanisms of ventricular arrhythmias in ARVC versus BS. In ARVC the predominant ventricular arrhythmia is a monomorphic ventricular tachycardia due to a scar-related macro-reentry circuit (A). The typical ventricular arrhythmia of BS is a rapid polymorphic ventricular tachycardia which results from a phase-2 reentry mechanism and may degenerate into ventricular fibrillation (see the text for further explanation)
Ventricular arrhythmias in patients with ARVC are facilitated by catecholamines and mostly occur during or immediately after exercise; accordingly, participation in competitive sports has been associated with an increased risk for SCD26. Instead, in Brugada patients ST segment elevation and arrhythmias are characteristically enhanced by vagotonic agents or beta-adrenergic blockers, and cardiac arrest has been documented to occur usually at rest or during sleep7–8. Unlike ARVC, the ECG abnormalities in patients with BS can vary considerably from time to time, until complete transient normalization, mostly because of variable influences of autonomic nervous system over the time27. Administration of sodium channel blockers such as flecainide, ajmaline, and procainamide can accentuate or unmask ST segment elevation in Brugada patients, whereas it usually does not affect ventricular repolarization in patients with ARVC6, 28.
Finally, the clinical course of both conditions may be complicated by VF leading to sudden cardiac death mostly occurring in young adults, whereas RV or biventricular heart failure distinctively occur in ARVC patients as a consequence of progression of the underlying structural myocardial abnormalities over time1, 3–4, 7, 27, 29.
Phenotypic overlap between ARVC and Brugada syndrome
Review of the literature demonstrates scientific evidence that features of both ARVC and BS may occur in some patients.
Clinical features
Previous clinical studies demonstrated a phenotypic overlap between the ARVC and BS. In 1986, Martini et al30 described six patients who experienced ventricular fibrillation and had clinical evidence of underlying structural abnormalities of the right ventricle; three of them exhibited a Brugada-like ECG pattern. Tada et al. reported right ventricular morpho-functional abnormalities and/or histologic abnormalities consistent with ARVC in 5 out of 6 Japanese men with a clinical and ECG diagnosis of BS31. Corrado et al. reported an Italian family with Brugada-like ST-segment elevation, RV cardiomyopathic changes at echocardiography and diagnostic morphologic features of ARVC at histopathologic investigation of the heart specimen of the proband with SCD32.
Imaging features
Cardiac magnetic resonance studies led to identify in Brugada patients some structural and arrhythmic features consistent with ARVC such as a larger right ventricular outflow tract area and occasional high intramyocardial T1 signal suggestive of fatty infiltration or right ventricular wall motion abnormalities and increased RV end-systolic and inflow tract diameters33–34. Of interest, morpho-functional structural abnormalities on MRI were more evident in patients with spontaneous than in those with a drug-induced Brugada ECG35. Finally, RV microaneurysms on angiograpy were found in 38% of patients (7 of 18) undergoing cardiac catheterization for RV endocardial biopsy36.
Endomyocardial biopsy features
The histopathologic finding of fatty infiltration of the myocardium (non-diagnostic for ARVC) was observed by Ohkubo et al.37 in 20% (5 of 25) and by Zumhagen et al.38 in 19% (4 of 21) of Brugada patients undergoing RV endomyocardial biopsy. However, a lower prevalence of typical fibro-fatty myocardial replacement suggestive of ARVC was reported both in the series of Frustaci et al.36 and Zumhagen et al.38.
Electrophysiologic features
In patients with transient right precordial ST segment elevation and vagal-induced ventricular fibrillation, Kasanuki et al.39 by using body-surface mapping demonstrated conduction abnormalities which were predominantly localized between the anterior RV wall and the outflow tract, and advanced the hypothesis that this clinical condition may reflect an early subclinical stage of ARVC, characterized by a very localized lesion in the right ventricular outflow tract.
Letsas et al.40 evaluated the presence of ARVC “ventricular depolarization” criteria in patients with BS, in the absence of sodium channel blockers. While epsilon waves were uncommon (6 of 47 Brugada patients), other conduction abnormalities such as a terminal QRS activation delay >55 ms were detected in 40% of cases. Moreover, late potential on signal-averaged-ECG were identified by Ikeda et al.41 in the majority of Brugada patients despite using more restrictive cut-off values than those proposed by the revised Task Force diagnostic criteria for ARVC. Of interest, the presence of late-potential was an independent predictor of life-threatening events with most significant correlation on multivariable analysis. Morita et al.42 recorded a fragmented QRS characterized by multiple spikes within the QRS complex in 43% of basal ECG of Brugada patients. This ECG pattern reflected a local epicardial activation delay and was significantly more often found in the patient subgroup that experienced VF (VF subgroup 85%, syncope subgroup 50%, and asymptomatic subgroup 34%, P<0.01). The QRS fragmentation was confirmed to be an independent risk indicator (HR of 4.94; 95% CI 1.54–15.8; p= 0.007) in a large cohort of patients prospectively investigated in the PRELUDE study43.
Intracardiac electrophysiologic studies using epicardial and endocardial mapping have consistently confirmed signs of ventricular conduction delay, mostly involving the RVOT in the form of fragmented electrograms and late potentials25, 39, 42. These electrogram recordings are typically observed in patients with ARVC as an expression of a discontinuous conduction through the diseased RV myocardium. According to some authors, these findings support the “depolarization” theory that explains ECG abnormalities and life-threatening ventricular arrhythmias of BS as the result, at least in part, of an intraventricular conduction defect in the right ventricular outflow tract. Such a delayed conduction is presumably caused by relatively mild structural abnormalities, such as histologic myocardial fibrosis, which may not become evident using conventional imaging techniques. Most compelling evidence in favour of this substrate derives from electrophysiologic mapping and ablation procedures in Brugada patients, showing that elimination of fragmented electrograms and late potentials in the RVOT resulted in normalization of ST-segment elevation and non inducibility of VT/VF44.
Clinico-pathologic features
The largest clinico-pathologic study on SCD victims with an ECG pattern of right precordial ST-segment elevation was obtained by reviewing a series of 273 young (≤35 years) SCD victims of who were enrolled in the Registry of the Veneto Region of Italy from 1979 to 199845. Among 96 SCD with an available ECG, 13 (14%; 12 males, 1 female; mean age 24 years) showed ST-segment elevation in leads V1 to V2/V3, either isolated (9 cases) or associated with RBBB (4 cases). At autopsy, all these victims had pathologic features consistent with ARVC (92%) except one, who had no evidence of structural heart disease. Compared with the 19 SCD victims with ARVC and no ST-segment elevation from the same series, those with ARVC and right precordial ST-segment elevation significantly more often died suddenly at rest or during sleep, showed serial ECG changes over time (Figure 3A), and had polymorphic ventricular tachycardia. These findings provide compelling evidence that ARVC may present with phenotypic features typically observed in BS.
Figure 3.

Electrocardiographic and histopathologic findings in two sudden death victims with overlapping phenotype of ARVC and BS. Top: A 35 year-old man who died suddenly at rest. Baseline 12-lead ECG showing first degree AV block and a coved-type Brugada ECG (A). Note the serial changes over years of right precordial repolarization abnormalities with transient near-normalization (B). Panoramic histological view of RV free wall showing transmural myocardial loss with fatty replacement, mostly involving subepicardial and midmural layers (C). At higher magnification, histological examination of RV myocardium shows tiny interstitial fibrosis in the setting of myocardial atrophy and fatty tissue (D). Bottom: A 27 year-old who died suddenly while sleeping. Baseline 12-lead ECG showing a coved-type Brugada ECG (A). Day-to-day changes of right precordial ST-segment pattern, which exhibits maximum displacement upward on March 22, 1996 (B). Panoramic histological view of RV myocardium disclosing full-thickness fibrofatty myocardial replacement (C).
Adapted from Corrado et al.45
Pathogenetic hypotheses
The clinical, imaging, histopathologic, electrophysiologic and pathologic evidence of a possible overlap between ARVC and BS raises the question on which pathogenetic mechanisms underlie such an interaction.
In the past, the “phenocopy theory” was proposed based on post-mortem histopathologic findings of patients with a mixed phenotype. According to this theory, the Brugada- like ECG and Brugada arrhythmias found in some ARVC patients are due to pathologic changes which give rise to a structural “epicardial-endocardial heterogeneity” of repolarization in the right ventricular wall, which in turn may account for a voltage gradient-dependent ST-segment elevation and arrhythmias due to local re-excitation, electrical mechanisms similar to those responsible of ECG repolarization changes and arrhythmias of BS (Figure 3B)45.
The overlapping phenotype has been alternatively explained by a “genetic theory” of double genetic defects leading to the coexistence of both ARVC and Brugada phenotypes and/or genetically defective cardiac sodium channel secondarily inducing myocyte death and leading to structural cardiomyopathic changes over time that resemble those observed in ARVC patients46.
More recently, new insights from molecular biology and genetic studies demonstrated molecular and cellular mechanisms of interaction between desmosomes and sodium channel at the intercalated disc, which support the concept that ARVC and BS are not two separate entities.
Experimental cell and animal studies
It is noteworthy that intercalated disc is the host of multiple molecular complexes capable of interacting with each other. Electron microscopy images show the proximity at the intercalated disc of mechanical junctions (DS proteins and adherens junctions/area composita), gap junctions and sodium-channel complexes, suggesting the likelihood of mutual interactions between constituting proteins such as PKP2, Cx43, and NaV1.5 (i.e., the major subunit of the cardiac sodium channel), respectively. These molecules populate adhesion/excitability nodes (“mini-nodes of Ranvier”) holding the “connexome”, a protein-interacting network where multiple molecules work together to coordinate excitability, cell coupling, and cell adhesion in the heart (Figure 4)14.
Figure 4.

Diagramatic representation of the relationship between desmosomes, gap junctions, and sodium channels at the intercalated discs. The “connexome” is a protein-interacting network where these molecules work together to coordinate excitability, cell coupling, and cell adhesion in the heart. Loss of expression of desmosomal proteins may induce electrical ventricular instability by a concomitant sodium channel dysfunction with current reduction, as a consequence of the cross-talk between these molecules at the intercalated discs. Adapted from Sato et al16.
Experimental studies have provided solid scientific data that a molecular crosstalk exists between desmosomes and both voltage gated sodium-channels and gap junction proteins at intercalated discs. Particularly, cell and animal models studies have shown that loss of expression of DS proteins may affect the integrity of the voltage gated sodium-channels resident in the cardiac intercalated disc, leading to an alteration of the amplitude and kinetics of sodium current. Using a combination of conventional biochemistry, patch clamp, and optical-mapping experiments, Sato et al.15 showed for the first time that PKP2 associates with NaV1.5 and that knockdown of PKP2 expression alters the properties of the sodium current and the velocity of action potential propagation in cultured cardiomyocytes. Biochemical analysis demonstrated that PKP2 coimmunoprecipitates not only with Cx43 but also with NaV1.5. Voltage clamp study revealed that loss of PKP2 expression also leads to a decrease in amplitude and a shift in voltage-gating kinetics of the sodium current in adult cardiac myocytes. Optical mapping showed that PKP2 knockdown associates with a significant decrease in conduction velocity in cardiac cell monolayers and an increased propensity to reentrant arrhythmias, likely resulting from the combination of decreased electric coupling and impaired sodium current density15.
A subsequent study showed that the cytoskeletal adaptor protein Ankyrin-G (AnkG) may play a key role in allowing for the interaction between three molecular components previously considered independent: the desmosome, the gap junction, and the sodium channel complex16. A mouse model with PKP2 haplo-insufficiency (PKP2-Hz), which mimics the clinical situation of patients harboring truncating mutations (with an expected <50% of PKP2 availability) was used to investigate “in vivo” the modulation of sodium current amplitude by defective DS proteins in the murine heart17. The mouse model did not show signs of structural cardiomyopathy. NaV1.5 protein abundance was not altered and yet, the amplitude of sodium current in isolated ventricular cardiomyocytes was significantly decreased. Moreover, there was a shift in gating and sodium current kinetics when compared to wild-type cardiomyocytes. These findings indicate that genetically mediated partial loss of PKP2 was able to affect sodium current amplitude, similarly to what was demonstrated in cells after total loss of PKP2 expression.
To investigate the predisposition of hearts deficient in PKP2 to drug-induced arrhythmic events, the PKP2-Hz mouse model was challenged with flecainide (i.e., a class 1C sodium channel-blocker antiarrhythmic drug)17. All treated animals showed an increased sensitivity to flecainide-induced atrial and ventricular conduction prolongation, with marked increased P wave, PR and QRS interval duration and increased conduction velocity in Langendorff-perfused isolated hearts. Of importance, flecainide injection in vivo caused ventricular arrhythmias and some cases of SCD in PKP2-Hz animals but not in the wild-types. These results demonstrated that PKP2 haplo-insufficiency reduces sodium current in murine myocardium and may render the heart susceptible to sodium channel-dependent life-threatening arrhythmias, similarly to those which precipitate SCD in patients with BS.
The relation between DS integrity, and the structure and/or function of the sodium channel complex has been confirmed by other studies from different laboratories. Gomes et al. reported that patients with ARVC harboring DSP mutations showed regional conduction delay and heterogeneous Nav1.5 distribution18. In a collaborative immunohistochemistry study on heart samples from patients with ARVC, Noorman et al. showed that in most cases, Nav1.5 was reduced at the intercalated disc, even if the distribution of the N-cadherin signal remained normal. Reduced sodium current amplitude has been observed in PKP2-deficient HL1 cells and in induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from a patient with PKP2 deficiency19.
Rizzo et al. studied transgenic mice with cardiac overexpression of mutant DSG-2-N271S (Tg-NS/L) before and after the onset of cell death and replacement fibrosis20. Before the onset of myocyte necrosis, epicardial mapping in Langendorff-perfused hearts showed prolonged ventricular activation time, reduced longitudinal and transversal conduction velocity, and reduced action potential upstroke velocity due to a lower sodium current density in mice over-expressing a mutation in DSG-2. Of interest, spontaneous ventricular arrhythmias, including short run of ventricular tachycardia, occurred in mice aged ≥6 weeks that had histopathologic evidence of myocyte necrosis; instead, younger mice showed an increased arrhythmia inducibility by extrastimuli or burst pacing, but no spontaneous arrhythmias.
Recently, Asimaki et al confirmed that Zebrafish ventricular myocytes expressing genetically-defective plakoglobin showed a 70 to 80% reductions in sodium current density47.
It is noteworthy that serious cardiomyopathies with severe structural abnormalities may develop secondarily a non-specific sodium channel dysfunction with current reduction. The experimental evidence overwhelmingly supports the notion that modifications in desmosomal proteins can affect the sodium current even in the absence of a structural disease, or of a cellular environment compatible with that of a fibrotic heart. The concept is further supported by the observation that the majority of patients with overlapping phenotype reported in the literature were affected by an early or minor variant of ARVC not fulfilling International Task Force criteria for diagnosis of definitive ARVC and, thus, unlikely to induce a secondary sodium channel dysfunction.
Molecular genetic studies in humans
Among 38 Dutch BS patients in whom SCN5A were previously excluded, Koopmann et al were unable to demonstrate mutations in a variety of other candidate genes including DS genes plakoglobin and plakophilin-2. The authors concluded that the studied candidate genes are unlikely to be major causal genes of BS48. Studying a larger patient population, Cerrone at al.21 discovered five single amino acid substitutions in five unrelated patients by direct sequencing the PKP2-gene in a cohort of 200 patients with clinical diagnosis of BS and no mutations on the most prevalent genes. This missense variant in PKP2 was proved to affect the cardiac sodium current, by using an HL-1 cell line, stably silenced for the endogenous PKP2; in the absence of PKP2, these cells showed a decrease in the native sodium current (Figure 5). Moreover, cells transiently transfected with the PKP2 mutants associated with the Brugada phenotype showed significantly decrease of sodium current, when compared with cells transfected with wild type PKP2. Similar results were obtained using a line of human iPSC-derived cardiomyocytes from a patient lacking PKP2 at the cell membrane. In these cells, sodium current increased upon transfection with wild type PKP2, while transfection with one of the PKP2 mutants associated with BS was not able to restore normal sodium current. These genotype-phenotype correlation data demonstrated that missense mutations in PKP2 can induce a Brugada phenotype by a decreased cardiac sodium current.
Figure 5.

PKP2 mutations associate with Brugada syndrome and with reduced sodium current. (Top left) Representative ECG showing ST-segment elevation, diagnostic for Brugada Syndrome, in one of the five patients carriers of missense mutations on the PKP-2 gene, and correspondent electropherograms showing the specific aminoacid substitution R635Q. (Bottom left) Sodium current amplitude recorded inHL1 cells silenced for PKP-2 (PKP2-KD) transfected with the mutant (red), anempty vector (blue), or wild-ype PKP-2 (black). (Right) pedigree of the family showing co-segregation between the PKP-2 mutation and the clinical Phenotype. Hx = history; SD=sudden death. Adapted from Cerrone et al21.
Clinical implications
The clinical impact of the interaction between DS-gene mutations and sodium current is an emerging area of interest that offers arguments of discussion and has the potential to generate further scientific investigations. One clinical implication is that screening of DS-gene mutation should be considered as part of the algorithm of molecular genetic testing of Brugada patients, when the genotype is negative for other predominant genes associated with BS21.
Clinical manifestations of ARVC usually develop during adolescence and young adulthood and are preceded by a long preclinical phase (“concealed ARVC”), during which SCD has been reported to occur unexpectedly as the first manifestation of the disease. It has been suggested that fatal events occurring prior to overt structural myocardial changes may be caused by a primarily electrical mechanism, as a consequence of the crosstalk of genetically defective DS-proteins with the voltage gated sodium-channel complex, leading to reduced sodium current and arrhythmogenic mechanisms similar to those in BS. Previous experimental studies consistently showed that pure electrical abnormalities induced by genetically defective DS-proteins created a predisposing arrhythmogenic myocardial “milieu”, with induction of arrhythmias exclusively in the presence of additional triggers such as drug challenge or electrical pacing in the experimental setting17–18. These data suggest that sodium channel dysfunction secondary to defective DS proteins is unlikely to be sufficient to generate spontaneous arrhythmias; rather, it may contribute to the arrhythmogenesis in the early stage of a developing ARVC structural phenotype (Figure 6). In this regard, it has been recently reported that phenotypic expression was a prerequisite for malignant ventricular arrhythmias and SCD in a cohort of ARVC DS-gene mutation carriers prospectively investigated49. Arrhythmic events during a long-term follow-up (8.5 years) occurred in DS-gene mutation carriers who fulfilled morpho-functional ITF diagnostic criteria and showed major risk factors. DS-gene mutation carriers without a ‘definite’ ARVC phenotypic expression had an uneventful clinical course, with the exception of a 15-year-old DSP-gene mutation carrier with previously normal ECG and echocardiographic findings who died suddenly two years later while sleeping. Post-mortem evaluation in this SCD victim demonstrated the presence of an epicardial scar in the infero-lateral LV region. This finding suggests that lethal ventricular arrhythmias during the ‘concealed phase’ of ARVC, other than expression of a subcellular arrhythmogenic mechanism, may be the result of the low sensitivity of routine clinical tests such as ECG and echocardiography for detection of early/minor arrhythmogenic phenotypic substrates, such as an isolated epicardial scar of the left ventricle, for which detection requires more sophisticated imaging technology such as contrast-enhanced cardiac magnetic resonance. These data were recently confirmed by the study of Te Riele et al. showing that in patients with familial ARVC, development of structural abnormalities always preceded the occurrence of ventricular arrhythmias50.
Figure 6.

Schematic representation of the ARVC/ course from desmosomal-gene mutation to phenotypic expression and complex interaction of arrhythmogenic mechanisms leading to cardiac arrest due to ventricular fibrillation. ARVC is a heart muscle disease caused by genetically defective DS proteins. The typical phenotype is characterized by fibro-fatty myocardial replacement predisposing to scar-related ventricular arrhythmias which may lead to SCD due to ventricular fibrillation. Experimental data support the concept that life-threatening ventricular arrhythmias may occur in the absence of structural changes due to a purely electrical mechanism: the loss of expression of DS-proteins may induce electrical ventricular instability and SCD by a concomitant sodium channel dysfunction with current reduction (similarly to the electrogenesis of ventricular fibrillation in patients with Brugada syndrome), as a consequence of the cross-talk between these molecules at the intercalated discs.
A better understanding of the role of sodium current in desmosome disease is crucial to guide antiarrhythmic therapy with sodium channel blockers in patients affected with ARVC. The knowledge that ARVC and BS may share mechanisms of ventricular arrhythmias mediated by a Na+ channel dysfunction may have important pharmacogenetic implications regarding the response to sodium channel blockers of ARVC patients, in terms of either therapeutic efficacy or adverse effects. The complex molecular crosstalk between desmosomes and sodium channel invites to great caution in using “class IC” antiarrhythmic agents to treat symptomatic ventricular arrhythmias in ARVC patients, because of the inherent pro-arrhythmic risk of these drugs to enhance conduction disturbances and repolarization heterogeneity by further reduction of the Na+ current. Whether an aggressive antiarrhythmic approach with implantable defibrillator represents a more appropriate prevention strategy in the subset of patients with ARVC and Brugada-like right precordial ST-segment elevation and/or polymorphic ventricular tachycardia needs to be assessed by future prospective studies.
Sodium channel blockers (including flecainide acetate in Europe) are also currently used as a diagnostic tool in patients with suspect of BS, with the aim to provoke the occurrence of the diagnostic ECG changes by further stressing the sodium channel function28. Krishan and Antzelevitch8 showed a synergism between intramyocardial conduction defect and heterogeneous repolarization in giving rise to arrhythmic activity in canine ventricle. In some ARVC patients, the reduction in Na+ current leading to dispersion of repolarization may contribute importantly to the induction and maintenance of reentrant activity in association with delayed intraventricular conduction. As a corollary, flecainide would be expected to aid in the evaluation of an increased arrhythmia risk or progression to cardiomyopathy in DS-gene mutation carriers by unmasking a concomitant Na+ channel dysfunction with current reduction secondary to DS-gene mutation51. However, anecdotal experience of using flecainide challenge in patients with DS-gene related ARVC warns about the risk of potentially lethal arrhythmic complications (Figure 7). The exaggerated response of ARVC patients to flecainide is in keeping with the observation that Na+ current deficiency induced by DS-gene defects increases the susceptibility to flecainide-induced arrhythmias in experimental animals.
Figure 7.

Effects of flecainide test in a 16-year old patient with compound DSG-2 gene mutations. Pharmacologic sodium channel block test was interrupted after infusion of 50 mg of flecainide acetate due to the occurrence of enormous QRS widening/J wave (A) followed by AV block (B), and ventricular fibrillation (C). The patients survived thanks to an ECMO device that was promptly applied after unsuccessful cardiopulmonary resuscitation and repeated DC-shocks due to VF relapses leading to electromechanical dissociation. Subsequent genotyping showed an ARVC-causing compound DSG-2 gene mutations (1253_1257insATGA, E418fsX419; and 2983_2987delGG, G995fsX1014). The patients developed a definite ARVC phenotype with T-wave inversion in right precordial leads (D), RV dilatation/dysfunction (E) with biventricular LGE at post-contrast sequences on cardiac magnetic resonance (non shown).
Conclusions
This article has proposed the controversial topic on the relationship between ARVC and BS for scientific discussion in light of the results of recent studies of biology and genetics that provide new insights on the existence of a pathogenetic link at molecular level. Accordingly, the overlapping phenotype may be explained by the emerging theory that ARVC and BS are not completely different conditions, but the ends of a spectrum of structural myocardial abnormalities and sodium current deficiency that share a common origin as diseases of the connexome.
Future experimental and clinical studies are needed to better define the molecular basis and to assess the clinical impact of overlapping phenotypes in terms of prevalence, arrhythmic risk stratification and therapeutic approaches for prevention of SCD.
Acknowledgments
Funding Sources: This study was funded by the University of Padua Research Grant TRANSAC, Padova; NIH grants (RO1-GM57691 and HL106632M); a grant from the Children Cardiomyopathy Foundation; and a Scientist Development Grant from the American Heart Association (AHA SDG14SDG18580014).
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 2.Brugada P, Brugada J. Right bundle branch block, persistent st segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 3.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol. 1997;30:1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 4.Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 2011;97:530–539. doi: 10.1136/hrt.2010.193276. [DOI] [PubMed] [Google Scholar]
- 5.Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, Tandri H, Paul M, Schmied C, Pelliccia A, Duru F, Protonotarios N, Estes NM, 3rd, McKenna WJ, Thiene G, Marcus FI, Calkins H. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: An international task force consensus statement. Circulation. 2015;132:441–453. doi: 10.1161/CIRCULATIONAHA.115.017944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA. Proposed diagnostic criteria for the brugada syndrome: Consensus report. Circulation. 2002;106:2514–2519. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 7.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: Report of the second consensus conference: Endorsed by the heart rhythm society and the european heart rhythm association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 8.Antzelevitch C, Yan GX. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. 2015;12:1852–1866. doi: 10.1016/j.hrthm.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corrado D, Buja G, Basso C, Nava A, Thiene G. What is the brugada syndrome? Cardiol Rev. 1999;7:191–195. doi: 10.1097/00045415-199907000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Scheinman MM. Is the brugada syndrome a distinct clinical entity? J Cardiovasc Electrophysiol. 1997;8:332–336. doi: 10.1111/j.1540-8167.1997.tb00797.x. [DOI] [PubMed] [Google Scholar]
- 11.Corrado D, Basso C, Leoni L, Tokajuk B, Turrini P, Bauce B, Migliore F, Pavei A, Tarantini G, Napodano M, Ramondo A, Buja G, Iliceto S, Thiene G. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2008;51:731–739. doi: 10.1016/j.jacc.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 12.Delmar M. The intercalated disk as a single functional unit. Heart Rhythm. 2004;1:12–13. doi: 10.1016/j.hrthm.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Delmar M. Desmosome-ion channel interactions and their possible role in arrhythmogenic cardiomyopathy. Pediatr Cardiol. 2012;33:975–979. doi: 10.1007/s00246-012-0257-0. [DOI] [PubMed] [Google Scholar]
- 14.Cerrone M, Delmar M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc Med. 2014;24:184–190. doi: 10.1016/j.tcm.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-g, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, van Rijen HV, Delmar M. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012;95:460–468. doi: 10.1093/cvr/cvs218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, McKenna WJ, Tinker A, Lambiase PD. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-a combined murine and human study. Eur Heart J. 2012;33:1942–1953. doi: 10.1093/eurheartj/ehr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–419. doi: 10.1016/j.hrthm.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418. doi: 10.1093/cvr/cvs219. [DOI] [PubMed] [Google Scholar]
- 21.Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe H, Minamino T. Genetics of brugada syndrome. J Hum Genet. 2015 Jul 30; doi: 10.1038/jhg.2015.97. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 23.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task force of the working group myocardial and pericardial disease of the european society of cardiology and of the scientific council on cardiomyopathies of the international society and federation of cardiology. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilde AA, Postema PG, Di Diego JM, Viskin S, Morita H, Fish JM, Antzelevitch C. The pathophysiological mechanism underlying brugada syndrome: Depolarization versus repolarization. J Mol Cell Cardiol. 2010;49:543–553. doi: 10.1016/j.yjmcc.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruwald AC, Marcus F, Estes NA, 3rd, Link M, McNitt S, Polonsky B, Calkins H, Towbin JA, Moss AJ, Zareba W. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: Results from the north american multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36:1735–1743. doi: 10.1093/eurheartj/ehv110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adler A, Rosso R, Chorin E, Havakuk O, Antzelevitch C, Viskin S. Risk stratification in brugada syndrome: Clinical characteristics, electrocardiographic parameters and auxiliary testing. Heart Rhythm. 2015 Sep 1; doi: 10.1016/j.hrthm.2015.08.038. pii: S1547-5271(15)01128-5. [DOI] [PubMed] [Google Scholar]
- 28.Zorzi A, Migliore F, Marras E, Marinelli A, Baritussio A, Allocca G, Leoni L, Perazzolo Marra M, Basso C, Buja G, Thiene G, Iliceto S, Delise P, Corrado D. Should all individuals with a nondiagnostic brugada-electrocardiogram undergo sodium-channel blocker test? Heart Rhythm. 2012;9:909–916. doi: 10.1016/j.hrthm.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Basso C, Corrado D, Bauce B, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5:1233–1246. doi: 10.1161/CIRCEP.111.962035. [DOI] [PubMed] [Google Scholar]
- 30.Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, Daliento L, Dalla Volta S. Ventricular fibrillation without apparent heart disease: Description of six cases. Am Heart J. 1989;118:1203–1209. doi: 10.1016/0002-8703(89)90011-2. [DOI] [PubMed] [Google Scholar]
- 31.Tada H, Aihara N, Ohe T, Yutani C, Hamada S, Miyanuma H, Takamiya M, Kamakura S. Arrhythmogenic right ventricular cardiomyopathy underlies syndrome of right bundle branch block, st-segment elevation, and sudden death. Am J Cardiol. 1998;81:519–522. doi: 10.1016/s0002-9149(97)00942-9. [DOI] [PubMed] [Google Scholar]
- 32.Corrado D, Nava A, Buja G, Martini B, Fasoli G, Oselladore L, Turrini P, Thiene G. Familial cardiomyopathy underlies syndrome of right bundle branch block, st segment elevation and sudden death. J Am Coll Cardiol. 1996;27:443–448. doi: 10.1016/0735-1097(95)00485-8. [DOI] [PubMed] [Google Scholar]
- 33.Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M, Cobelli F, Baiardi P, Nastoli J, Bloise R, Monteforte N, Napolitano C, Priori SG. Magnetic resonance investigations in brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J. 2009;30:2241–2248. doi: 10.1093/eurheartj/ehp252. [DOI] [PubMed] [Google Scholar]
- 34.Papavassiliu T, Wolpert C, Fluchter S, Schimpf R, Neff W, Haase KK, Duber C, Borggrefe M. Magnetic resonance imaging findings in patients with brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:1133–1138. doi: 10.1046/j.1540-8167.2004.03681.x. [DOI] [PubMed] [Google Scholar]
- 35.Papavassiliu T, Veltmann C, Doesch C, Haghi D, Germans T, Schoenberg SO, van Rossum AC, Schimpf R, Brade J, Wolpert C, Borggrefe M. Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in brugada syndrome. Heart Rhythm. 2010;7:1790–1796. doi: 10.1016/j.hrthm.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I, Sanna T, Bellocci F, Russo MA. Cardiac histological substrate in patients with clinical phenotype of brugada syndrome. Circulation. 2005;112:3680–3687. doi: 10.1161/CIRCULATIONAHA.105.520999. [DOI] [PubMed] [Google Scholar]
- 37.Ohkubo K, Watanabe I, Okumura Y, Takagi Y, Ashino S, Kofune M, Sugimura H, Nakai T, Kasamaki Y, Hirayama A, Morimoto S. Right ventricular histological substrate and conduction delay in patients with brugada syndrome. Int Heart J. 2010;51:17–23. doi: 10.1536/ihj.51.17. [DOI] [PubMed] [Google Scholar]
- 38.Zumhagen S, Spieker T, Rolinck J, Baba HA, Breithardt G, Bocker W, Eckardt L, Paul M, Wichter T, Schulze-Bahr E. Absence of pathognomonic or inflammatory patterns in cardiac biopsies from patients with brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2:16–23. doi: 10.1161/CIRCEP.107.737882. [DOI] [PubMed] [Google Scholar]
- 39.Kasanuki H, Ohnishi S, Ohtuka M, Matsuda N, Nirei T, Isogai R, Shoda M, Toyoshima Y, Hosoda S. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation. 1997;95:2277–2285. doi: 10.1161/01.cir.95.9.2277. [DOI] [PubMed] [Google Scholar]
- 40.Letsas KP, Efremidis M, Weber R, Korantzopoulos P, Protonotarios N, Prappa E, Kounas SP, Evagelidou EN, Xydonas S, Kalusche D, Sideris A, Arentz T. Epsilon-like waves and ventricular conduction abnormalities in subjects with type 1 ecg pattern of brugada syndrome. Heart Rhythm. 2011;8:874–878. doi: 10.1016/j.hrthm.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda T, Sakurada H, Sakabe K, Sakata T, Takami M, Tezuka N, Nakae T, Noro M, Enjoji Y, Tejima T, Sugi K, Yamaguchi T. Assessment of noninvasive markers in identifying patients at risk in the brugada syndrome: Insight into risk stratification. J Am Coll Cardiol. 2001;37:1628–1634. doi: 10.1016/s0735-1097(01)01197-4. [DOI] [PubMed] [Google Scholar]
- 42.Morita H, Kusano KF, Miura D, Nagase S, Nakamura K, Morita ST, Ohe T, Zipes DP, Wu J. Fragmented qrs as a marker of conduction abnormality and a predictor of prognosis of brugada syndrome. Circulation. 2008;118:1697–1704. doi: 10.1161/CIRCULATIONAHA.108.770917. [DOI] [PubMed] [Google Scholar]
- 43.Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, Giordano U, Pappone C, Mascioli G, Rossetti G, De Nardis R, Colombo M. Risk stratification in brugada syndrome: Results of the prelude (programmed electrical stimulation predictive value) registry. J Am Coll Cardiol. 2012;59:37–45. doi: 10.1016/j.jacc.2011.08.064. [DOI] [PubMed] [Google Scholar]
- 44.Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, Likittanasombat K, Bhuripanyo K, Ngarmukos T. Prevention of ventricular fibrillation episodes in brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123:1270–1279. doi: 10.1161/CIRCULATIONAHA.110.972612. [DOI] [PubMed] [Google Scholar]
- 45.Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation. 2001;103:710–717. doi: 10.1161/01.cir.103.5.710. [DOI] [PubMed] [Google Scholar]
- 46.Corrado D, Basso C, Buja G, Nava A, Thiene G. Brugada sindrome: Relationship to other arrhythmogenic sindromes. In: Antzelevitch C, editor. The brugada syndrome. From bench to bedside. Blackwell Futura: 2004. pp. 111–118. [Google Scholar]
- 47.Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, Wu JC, MacRae CA, Kléber AG, Saffitz JE. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:261–266. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koopmann TT, Beekman L, Alders M, Meregalli PG, Mannens MM, Moorman AF, Wilde AA, Bezzina CR. Exclusion of multiple candidate genes and large genomic rearrangements in scn5a in a dutch brugada syndrome cohort. Heart Rhythm. 2007;4:752–755. doi: 10.1016/j.hrthm.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 49.Zorzi A, Rigato I, Pilichou K, Perazzolo Marra M, Migliore F, Mazzotti E, Gregori D, Thiene G, Daliento L, Iliceto S, Rampazzo A, Basso C, Bauce B, Corrado D. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace. 2015 Jul 2; doi: 10.1093/europace/euv205. pii: euv205. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 50.Te Riele AS, James CA, Groeneweg JA, Sawant AC, Kammers K, Murray B, Tichnell C, van der Heijden JF, Judge DP, Dooijes D, van Tintelen JP, Hauer RN, Calkins H, Tandri H. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur Heart J. 2015 Aug 27; doi: 10.1093/eurheartj/ehv387. pii: ehv387. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 51.Peters S. Arrhythmogenic right ventricular dysplasia-cardiomyopathy and provocable coved-type st-segment elevation in right precordial leads: Clues from long-term follow-up. Europace. 2008;10:816–820. doi: 10.1093/europace/eun030. [DOI] [PubMed] [Google Scholar]