

ABSTRACT
Feo is the major ferrous iron transport system in prokaryotes. Despite having been discovered over 25 years ago and found to be widely distributed among bacteria, Feo is poorly understood, as its structure and mechanism of iron transport have not been determined. The feo operon in Vibrio cholerae is made up of three genes, encoding the FeoA, FeoB, and FeoC proteins, which are all required for Feo system function. FeoA and FeoC are both small cytoplasmic proteins, and their function remains unclear. FeoB, which is thought to function as a ferrous iron permease, is a large integral membrane protein made up of an N-terminal GTPase domain and a C-terminal membrane-spanning region. To date, structural studies of FeoB have been carried out using a truncated form of the protein encompassing only the N-terminal GTPase region. In this report, we show that full-length FeoB forms higher-order complexes when cross-linked in vivo in V. cholerae. Our analysis of these complexes revealed that FeoB can simultaneously associate with both FeoA and FeoC to form a large complex, an observation that has not been reported previously. We demonstrate that interactions between FeoB and FeoA, but not between FeoB and FeoC, are required for complex formation. Additionally, we identify amino acid residues in the GTPase region of FeoB that are required for function of the Feo system and for complex formation. These observations suggest that this large Feo complex may be the active form of Feo that is used for ferrous iron transport.
IMPORTANCE The Feo system is the major route for ferrous iron transport in bacteria. In this work, the Vibrio cholerae Feo proteins, FeoA, FeoB, and FeoC, are shown to interact to form a large inner membrane complex in vivo. This is the first report showing an interaction among all three Feo proteins. It is also determined that FeoA, but not FeoC, is required for Feo complex assembly.
INTRODUCTION
Iron is an indispensable component of enzymes involved in a wide range of biological processes and, as a result, is essential for life in almost all organisms (1). Notwithstanding the fact that iron is abundant in nature, aerobic, neutral-pH environments favor the formation of Fe(OH)3, a highly insoluble ferric iron (Fe3+) complex. In contrast, in anoxic environments, free ferrous iron (Fe2+) is more readily available (2). In order to meet their iron requirement, bacteria have multiple iron transport systems that allow use of the various forms of iron present in their environment, and Vibrio cholerae is no exception (3).
V. cholerae is a human pathogen that causes cholera, a severe diarrheal disease resulting from the ingestion of contaminated food or water (4). V. cholerae must acquire iron from different environments, such as the gut of its human host or the fresh, brackish, and ocean waters that make up its natural habitat. This need to acquire the various forms of iron is reflected in the observation that approximately 1% of its genome is devoted to the acquisition of iron (3). In V. cholerae, ferric iron is commonly acquired through its endogenous siderophore vibriobactin or through siderophores synthesized by other organisms (5–8). Vibriobactin is synthesized by the Vib system and is transported through the outer and inner membrane by the Viu and Vct systems (9–14). The Vct system is also capable of siderophore-free iron transport through an unknown mechanism (14). Ferric iron can also be transported independently of the siderophores by Fbp, an ATP-binding cassette (ABC) transporter (15). Further, V. cholerae can take up heme as a source of iron through the Hut and Has systems (16–19). Ferrous iron can be transported through the inner membrane by Feo, a major system for bacterial ferrous iron uptake (15).
The bacterial ferrous iron transport (Feo) system was discovered over 25 years ago in Escherichia coli K-12 (20, 21) and contributes to virulence in several organisms (22–24). The feo loci of E. coli and V. cholerae are made up of three genes, encoding the FeoA, FeoB, and FeoC proteins (Fig. 1A) (21, 25–27). FeoA is a small hydrophilic protein whose role is unknown, but it has been shown to be required for Feo function in several bacterial species (27–29). FeoA shows structural similarity to an Src-homology-3 (SH3) domain fold, suggesting that FeoA may mediate protein-protein interactions (26, 30–32). FeoA was shown to interact with FeoB in a bacterial two-hybrid (BACTH) assay in Salmonella enterica (29).
FIG 1.
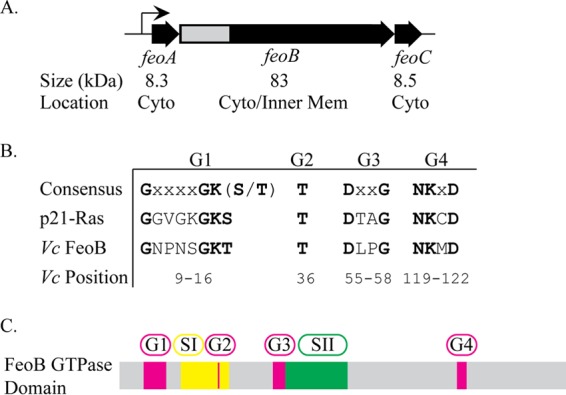
Genetic organization of V. cholerae feoABC and conserved residues of the FeoB GTPase. (A) Genetic map of V. cholerae feoABC. Below each gene are listed the predicted protein sizes and the subcellular location in the either cytoplasm (Cyto) or inner membrane (Inner Mem). The gray region indicates the section of feoB encoding the GTPase domain that is shown in more detail in panel C. (B) Consensus sequences of conserved GTPase sequence elements compared to human p21-Ras and V. cholerae FeoB, with conserved residues shown in bold. The positions of amino acid residues of each motif in V. cholerae FeoB are indicated. No G5 motif sequence has been identified in FeoB through sequence alignment. (C) Organization of the conserved GTPase sequence elements and switch regions, switch I and switch II, within the FeoB GTPase domain. The locations of switch regions I (amino acids 24 to 39) and II (amino acids 59 to 81) were predicted by sequence alignment.
FeoB is a large protein with an N-terminal, cytoplasmic GTPase domain that is thought to regulate transport (33, 34) and a C-terminal region embedded in the cytoplasmic membrane. The integral membrane domain is predicted to contain 8 to 12 membrane-spanning helices, and it is likely to form the ferrous permease. There is sequence homology between the Feo GTPase domain and eukaryotic G proteins, including the human oncogene p21-Ras, and the highly conserved signature G1, G2, G3, and G4 motifs have been identified in FeoB (Fig. 1B and C). The G5 motif is the least conserved motif and has been identified in FeoB through structural analysis but not through sequence alignments (33, 35). These G motifs are necessary for binding and hydrolysis of the guanine nucleotide (36–38). Another hallmark of GTPases found in FeoB is the presence of switch I and switch II regions that undergo large conformational changes or “switches” upon GTP association and hydrolysis (34, 39). It is believed that the GTPase domain is required for proper regulation of Feo activity, rather than providing the energy source for transport (33, 34). The energy source for transport of iron via Feo has not been determined.
FeoC is a small protein and the least conserved part of the Feo system. It appears that the feo operon lacks a feoC gene in many organisms, and the role of this protein is not understood. Structural studies of Klebsiella pneumoniae FeoC have revealed that it contains a winged-helix fold, a motif that is sometimes involved in DNA binding (26, 40). However, experimental evidence does not support the idea of a role for FeoC in DNA binding or transcription regulation (27, 41). Rather, FeoC appears to bind the N-terminal cytoplasmic domain of FeoB (N-FeoB), as shown both in BACTH assays (27, 41) and in a structural study in which N-FeoB and FeoC were crystalized in a 1:1 ratio (42).
The mechanism of iron transport via the Feo system is not understood, and the fundamental issues that remain to be elucidated include the role of the FeoB GTPase domain, the function and topology of the FeoB membrane-spanning region, and the source of energy for active transport. Although all three Feo proteins are required in V. cholerae, the structure of the active transporter is unknown. In this study, we investigated intermolecular interactions among the Feo proteins. We show that FeoB makes higher-order molecular complexes in vivo, including a large complex that contains all three Feo proteins. Further, we provide insight into the mechanism of Feo-mediated iron transport by investigating the roles of the GTPase motifs and of each of the Feo proteins in complex formation.
MATERIALS AND METHODS
Bacterial strain, plasmids, and growth conditions.
The bacterial strain and plasmids used in this study are listed in Table 1. Bacteria were grown at 37°C in Luria-Bertani (LB) medium. Ampicillin was used at 50 μg/ml. Heme was used at 10 μM.
TABLE 1.
Bacterial strain and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference |
|---|---|---|
| V. cholerae O395 EPV6 | Δvib fbp::Camr vct::Genr feo::Kanr | E. Peng, unpublished data |
| Plasmids | ||
| pWKS30 | Low-copy-no. cloning vector | 61 |
| pFeo101 | pWKS30 carrying feoABC | 15 |
| pFeo122 | pFeo101 with a BamHI site immediately downstream of the last codon of feoC | This study |
| pFeoAB-V5C | pFeo101 carrying a V5 tag on the N terminus of FeoB | This study |
| pFeoABC-V5 | pFeo122 carrying a V5 tag on the C terminus of FeoC | This study |
| pFeoAB-V5C.FeoB-K15D | pFeoAB-V5C carrying feoB with a K15D mutation | This study |
| pFeoAB-V5C.FeoB-T36K | pFeoAB-V5C carrying feoB with a T36K mutation | This study |
| pFeoAB-V5C.FeoB-D55K | pFeoAB-V5C carrying feoB with a D55K mutation | This study |
| pFeoAB-V5C.FeoB-T36K.D55K | pFeoAB-V5C carrying feoB with T36K and D55K mutations | This study |
| pFeoAB-V5C.FeoB-D72A | pFeoAB-V5C carrying feoB with a D72A mutation | This study |
| pFeoAB-V5C.FeoB-N119K | pFeoAB-V5C carrying feoB with a N119K mutation | This study |
| pFeoAB-V5C.FeoB-D122N | pFeoAB-V5C carrying feoB with a D122N mutation | This study |
| pFeoΔC | pFeo101 with an in-frame deletion in feoC | 27 |
| pFeoAB-V5 | pFeoΔC with a V5 tag on the N terminus of FeoB | This study |
| pFeoAB-V5C.FeoA-G32K | pFeoAB-V5C carrying feoA with a G32K mutation | This study |
| pFeoAB-V5C.FeoA-A45D | pFeoAB-V5C carrying feoA with a A45D mutation | This study |
| pFeoAB-V5C.FeoA-P50R | pFeoAB-V5C carrying feoA with a P50R mutation | This study |
| pFeoAB-V5C.FeoA-V72K | pFeoAB-V5C carrying feoA with a V72K mutation | This study |
Camr, chloramphenicol resistance; Genr, gentamicin resistance; Kanr, kanamycin resistance.
Plasmid construction.
Plasmid pFeoAB-V5C carries the entire V. cholerae feo operon, encoding the full-length FeoB protein N-terminally tagged with a V5 tag. For its construction, an NsiI restriction enzyme site was incorporated into pFeo101 through site-directed mutagenesis using the pFeo101.FeoB.Nsi.Fwd and pFeo101.FeoB.Nsi.Rev primers (Table 2). Complementary V5 tag primers, FeoB.V5.Fwd and FeoB.V5.Rev (Table 2), were annealed and inserted into the NsiI site of the above plasmid.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′–3′) |
|---|---|
| pFeo101.FeoB.Nsi.Fwd | TGTGGAGAGAGCACAATGATGCATCAAGTACTCACCGTAGG |
| pFeo101.FeoB.Nsi.Rev | CCTACGGTGAGTACTTGATGCATCATTGTGCTCTCTCCACA |
| FeoB.V5.Fwd | TGGCAAGCCCATCCCCAACCCCTTGCTTGGCTTGGACTCCCACCGTGCA |
| FeoB.V5.Rev | CGGTGGAGTCCAAGCCAAGCAAGGGGGTTGGGGATGGGCTTGCCATGCA |
| FeoC.V5.Fwd | GATCCGGCAAGCCCAYCCCCAACCCCYTGCTTGGCTTGGACTCCACCTGAT |
| FeoC.V5.Rev | CATGATCAGGTGGAGTCCAACCCAAGCAAGGGGTTGGGGATGGGCTTGCCG |
| pFeoAB-V5C.FeoB.K15D.Fwd | GACCATTGAATAAGGTTGTGTCTCCACTATTCGGGTTGCCTACG |
| pFeoAB-V5C.FeoB.K15D.Rev | CGTAGGCAACCCGAATAGTGGAGACACAACCTTATTCAATGGTC |
| pFeoAB-V5C.FeoB.T36K.Fwd | TACCCGTTTTTTTCTCAACTTTGACCCCCGCCCAGTTAC |
| pFeoAB-V5C.FeoB.T36K.Rev | GTAACTGGGCGGGGGTCAAAGTTGAGAAAAAAACGGGTA |
| pFeoAB-V5C.FeoB.D55K.Fwd | AAAGCATAAATTCCGGGCAGCTTGGTGAGTGAAAATTCATCGC |
| pFeoAB-V5C.FeoB.D55K.Rev | GCGATGAATTTTCACTCACCAAGCTGCCCGGAATTTATGCTTT |
| pFeoAB-V5C.FeoB.D72A.Fwd | TAATGACAGTAACAGTATTGCGGAATCGATCGCATCGCGTGC |
| pFeoAB-V5C.FeoB.D72A.Rev | GCACGCGATGCGATCGATTCCGCAATACTGTTACTGTCATTA |
| pFeoAB-V5C.FeoB.N119K.Fwd | CGCTTAAGCGCATCCATTTTCTTAAGTACGACAATCATTGG |
| pFeoAB-V5C.FeoB.N119K.Rev | CCAATGATTGTCGTACTTAAGAAAATGGATGCGCTTAAGCG |
| pFeoAB-V5C.FeoB.D122N.Fwd | TCGCGCTTAAGCGCATTCATTTTGTTAAGTACGACAATCA |
| pFeoAB-V5C.FeoB.D122N.Rev | TGATTGTCGTACTTAACAAAATGAATGCGCTTAAGCGCGA |
| pFeoAB-V5C.FeoA.G32K.Fwd | CGACTTCGGTATTGGGGAGAACTTTCATCACCATCAGTTTCTTTCTG |
| pFeoAB-V5C.FeoA.G32K.Rev | CAGAAAGAAACTGATGGTGATGAAAGTTCTCCCCAATACCGAAGTCG |
| pFeoAB-V5C.FeoA.A45D.Fwd | CGTATTAATTCGCCGAGATCCAATGGGTGATCCGC |
| pFeoAB-V5C.FeoA.A45D.Rev | GCGGATCACCCATTCGAGCTCGGCGAATT |
| pFeoAB-V5C.FeoA.P50R.Fwd | GTACTTCAACTTGAAGCCGATCACCCATTGGAGCT |
| pFeoAB-V5C.FeoA.P50R.Rev | AGCTCCAATGGGTGATCGGCTTCAAGTTGAAGTAC |
| pFeoAB-V5C.FeoA.V72K.Fwd | CATCATTGTGCTCTCTCTCCTTATCAATATTTGCCGCAATATTTTCGC |
| pFeoAB-V5C.FeoA.V72K.Rev | GCGAAAATATTGCGGCAAATATTGATAAGGAGAGAGCACAATGATG |
Plasmid pFeoABC-V5 carries the V. cholerae feo locus, where FeoC has a C-terminal V5 tag. The complementary V5 tag primers, FeoC.V5.Fwd and FeoC.V5.Rev (Table 2), were annealed and inserted into the BamHI and XbaI restriction sites of pFeo122. pFeo122 is pFeo101 modified to have a BamHI site immediately downstream of the final codon of feoC. It was constructed by deleting the BamHI fragment containing the tandem affinity purification (TAP) tag from plasmid pFeoABCtap (27).
Plasmid pFeoAB-V5 encodes only V. cholerae FeoA and FeoB, where FeoB is N-terminally tagged with a V5 tag. This plasmid was cloned as described above for the pFeoAB-V5C plasmid, except plasmid pFeoΔC (27) was used instead of pFeo101.
pFeoAB-V5C-derived plasmids containing feoA or feoB mutations were prepared through site-directed mutagenesis using the QuikChange (Agilent Technologies, Inc.) protocol. The primers used for site-directed mutagenesis are listed in Table 2.
DNA sequences of all constructs were confirmed through nucleotide sequencing.
In vivo cross-linking.
Overnight cultures were diluted 1:100 in 50 ml of LB broth with ampicillin and grown to mid-log phase at 37°C. Cells were pelleted and washed twice with 25 ml of phosphate-buffered saline (PBS). All centrifugations were done at 8,600 × g for 4 min. Cells were then resuspended in 25 ml of 0.6% (vol/vol) formaldehyde–PBS. Cells were incubated at room temperature for 6 min and then centrifuged and resuspended in 10 ml of 1.25 M glycine–PBS. Cells were centrifuged and washed in 25 ml of PBS, and the final cell pellet was frozen at −80°C until further processing.
Subcellular fractionation.
Overnight cultures were diluted 1:100 in 50 ml of LB broth with ampicillin and grown to mid-log phase at 37°C. Cells were pelleted at 8,600 × g for 4 min. In some cases, cells were cross-linked as described above. Cell pellets were frozen at −80°C until further processing. Cell pellets were thawed on ice and resuspended in 5 ml of fractionation lysis buffer (10 mM Na2HPO4 [pH 7], 5 mM MgSO4, 1 mM phenylmethylsulfonyl fluoride [PMSF]). Samples were sonicated to induce cell lysis. Cell debris was removed by centrifuging samples at 12,000 × g for 15 min. Total membrane pellets were separated from the cytoplasmic fractions at 50,000 rpm for 45 min using a TLA-100.3 rotor (Beckman Coulter). Total membrane pellets were washed with 1 ml of 1× TBS (50 mM Tris-HCl [pH 7.4], 150 mM NaCl)–1× SigmaFAST protease inhibitor (Sigma) and centrifuged at 50,000 rpm for 45 min using a TLA-100.3 rotor. Washed total membrane pellets were resuspended in 0.2 ml of 0.5% (wt/vol) Sarkosyl–1× TBS–1× SigmaFAST protease inhibitor. Membranes were incubated at room temperature for 20 min with mild shaking. Inner membrane fractions were separated from outer membrane pellets at 50,000 rpm for 45 min using a TLA-100.3 rotor. Outer membrane pellets were washed with 1 ml of 1% (wt/vol) Sarkosyl–1× TBS–1× SigmaFAST protease inhibitor and centrifuged at 50,000 rpm for 45 min using a TLA-100.3 rotor. Outer membrane pellets were resuspended in 0.2 ml of 1× TBS–1× SigmaFAST protease inhibitor. Subcellular fractionation samples were mixed with 4× SDS-PAGE loading buffer (100 mM Tris [pH 6.8], 2% SDS, 0.1% bromophenol blue, 10% glycerol, 1 mM 2-mercaptoethanol) to achieve a 1× final concentration and stored at −20°C until further processing.
Immunoprecipitation.
Frozen cross-linked cell pellets, prepared as described above, were thawed on ice. The cells were resuspended in 0.75 ml of lysis buffer (50 mM Tris-Cl [pH 7.5], 150 mM NaCl, 0.5% Triton X-100, 1% glycerol, 1 mM EDTA, 1 mM PMSF, 1 mg/ml lysozyme) and allowed to rest on ice for 20 min. Samples were briefly sonicated to shear the DNA and then centrifuged at 12,000 × g for 15 min to remove cell debris. Protein concentrations of cell lysates were determined by the use of the DC (detergent-compatible) protein assay (Bio-Rad), using bovine serum albumin (BSA) for determination of the standard curve. Cell lysate containing 1 μg of protein was mixed with 40 μl of anti-V5–agarose bead slurry (Sigma) that had been previously washed three times with 1× TBS. Cell lysates and beads were mixed overnight in the cold room. Cell lysate was removed from the anti-V5–agarose beads by brief centrifugation, and then beads were washed 10 times with wash buffer (50 mM Tris-Cl [pH 7.5], 150 mM NaCl, 1% Triton X-100, 2% glycerol, 1 mM EDTA, 1 mM PMSF). Proteins were eluted from the beads by adding 4× SDS-PAGE loading buffer and incubating at 65°C for 20 min.
BN-PAGE.
Blue native PAGE (BN-PAGE) was carried out as described by Wittig et al. (43) with the following modifications. Overnight cultures were diluted 1:100 in 50 ml of LB broth with ampicillin and grown to mid-log phase at 37°C. Cells were pelleted at 8,600 × g for 4 min. The pellet was resuspended in 1.5 ml of 1× TBS–1× SigmaFAST protease inhibitor (Sigma), and the cells were lysed by sonication. Cell debris was removed by centrifuging samples at 12,000 × g for 15 min. Membrane fractions were isolated from soluble cell lysate at 50,000 rpm for 45 min using a TLA-100.3 rotor (Beckman Coulter). Membrane pellets were washed with 1× TBS–1× SigmaFAST protease inhibitor (Sigma). A small aliquot was used to determine the protein concentration using the DC protein assay (Bio-Rad). Membrane fractions were pelleted again at 50,000 rpm for 45 min using a TLA-100.3 rotor. Membrane pellets were resuspended in a mixture containing 20 mM bis-Tris (pH 7), 500 mM aminocaproic acid (pH 7), 20 mM NaCl, and 1 mM EDTA such that the final protein concentration was 10 mg/ml. Triton X-100 (Fisher Scientific) was added to give a final concentration of 2% (vol/vol). Membranes were incubated on ice for 30 min and then centrifuged at 20,000 × g for 20 min. The supernatant was mixed with 100% (vol/vol) glycerol and a 5% (wt/vol) Coomassie blue-G250 solution to give final concentrations of 5% (vol/vol) and 0.2% (vol/vol), respectively. Samples were resolved at 4°C on bis-Tris gels. Gels were run on a standard gel electrophoresis system at 80 V using 50 mM bis-Tris (pH 7) as the anode buffer and a mixture containing 15 mM bis-Tris (pH 7), 50 mM Tricine, and 0.02% Coomassie brilliant blue G250 as the cathode buffer. When the dye front had traveled one-third of the way to the end of the gel, the cathode buffer was changed to a mixture containing 15 mM bis-Tris (pH 7), 50 mM Tricine, and 0.002% Coomassie brilliant blue G250. When the dye front had traveled two-thirds of the way to the end of the gel, cathode buffer was switched to a mixture containing 15 mM bis-Tris (pH 7) and 50 mM Tricine. Gels were stained using Gel Code Blue stain (Pierce). Native protein sizes were estimated using NativeMark unstained protein standard (Life Technologies). Immunoblot analysis was carried out as described below, except resolved proteins were transferred to an Amersham Hybond P polyvinylidene difluoride (PVDF) membrane (GE Healthcare).
Immunoblot analysis.
Cross-linked and non-cross-linked whole-cell lysates, cross-linked subcellular fractions, or immunoprecipitation elutions were resolved on Tris-acetate gradient SDS-PAGE gels, and Precision Plus protein dual-color standards (Bio-Rad) or HiMark prestained protein standard (Life Technologies) was used to estimate protein sizes. Non-cross-linked inner membrane fractions were resolved on nongradient SDS-PAGE gels, and Precision Plus protein dual-color standards were used to estimate protein size. After electrophoresis, resolved proteins were transferred to a Hybond ECL nitrocellulose membrane (GE Healthcare) and V5-tagged proteins were visualized using anti-V5 antibodies derived from mouse (Sigma) or rabbit (Santa Cruz) followed by incubation with a horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit antibody (Bio-Rad). Signal was detected by development using a Pierce ECL Western blotting substrate kit (Thermo-Fisher Scientific). Analyses were performed at least 3 times, and representative gels are shown. Quantitation of protein bands was performed using GeoQuant.NET software provided by BiochemLab Solutions.
Mass spectrometry analysis.
Immunoprecipitation elution samples were resolved on Tris-acetate gradient SDS-PAGE gels. After electrophoresis, gels were stained using Gel Code Blue stain. The desired bands were excised from the gel, and proteins were identified at The University of Texas at Austin Institute for Cellular and Molecular Biology Proteomics Facility via liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a Dionex Ultimate 3000 RSLCnano LC instrument coupled to a Thermo Orbitrap Elite spectrometer. Scaffold (version Scaffold_4.3.2; Proteome Software Inc.) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability to achieve a false-discovery rate (FDR) of less than 1.0% by the Peptide Prophet algorithm with Scaffold delta mass correction (44). Protein identifications were accepted if they could be established at greater than 99.9% probability to achieve an FDR of less than 1.0% and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (45). Proteins that contained similar peptides and that could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins were annotated with gene ontology (GO) terms from gene_association.goa_uniprot (downloaded 25 July 2013) (46).
RESULTS
V5-tagged FeoB and FeoC retain function.
To facilitate immunoprecipitation studies to determine the interactions among the Feo proteins, a V5 tag was added to the N terminus of FeoB or the C terminus of FeoC. To ensure that the tag did not significantly alter the function of the proteins, we used EPV6, a V. cholerae strain in which the feo, fbp, vib, and vct operons were mutated, rendering the strain unable to grow on LB medium unless the medium was supplemented with heme. EPV6 is able to grow in the absence of heme supplementation only when a functional iron transport system is supplied on a plasmid. EPV6 carrying a plasmid containing the feo operon with a V5 tag on the N terminus of FeoB (pFeoAB-V5C) or a V5 tag on the C terminus of FeoC (pFeoABC-V5) allowed growth at a level similar to that of EPV6 carrying a plasmid with the untagged feoABC operon, pFeo101 (Fig. 2). Except where indicated otherwise, all structural studies were performed with the plasmids carried in EPV6 grown in the absence of heme, so that structural determinations were made under conditions where Feo must be active.
FIG 2.

The V5 tag does not affect function of FeoB or FeoC. V. cholerae EPV6 bacteria carrying plasmids pWKS30 (vector), pFeo101 (FeoABC), pFeoAB-V5C (FeoAB-V5C), and pFeoABC-V5 (FeoABC-V5) were streaked on either LB agar (A) or LB agar supplemented with 10 μM heme (B).
FeoB makes higher-order complexes.
To determine whether there are intermolecular interactions among the Feo proteins in V. cholerae, we used in vivo formaldehyde cross-linking of EPV6 carrying pFeoAB-V5C. Mid-log-phase cells were treated with formaldehyde, and proteins from the whole-cell lysate were resolved by SDS-PAGE and analyzed by immunoblotting using an anti-V5 antibody. Results show that FeoB-V5, with a monomeric molecular mass of 84.6 kDa, is found in several higher-order complexes, indicating that FeoB is involved in intermolecular interactions (Fig. 3A). Although cross-linking may interfere with determining accurate sizes of protein complexes, comparison to a HiMark prestained protein standard (Life Technologies) suggested that these complexes were approximately 250 kDa, 500 kDa, and larger than 500 kDa. The size of the largest complex could not be determined, since there are no commercial ladders that provide molecular standards with masses greater than 460 kDa under denaturing conditions. To determine the intracellular location of these complexes, we carried out membrane fractionation of the V. cholerae lysate (Fig. 3B). Immunoblot analysis of the cytoplasm, inner membrane, and outer membrane fractions showed that the FeoB complexes were located in the inner membrane fraction. This is the expected location for FeoB (21).
FIG 3.

V. cholerae FeoB makes complexes in vivo that are located in the inner membrane. (A) A culture of EPV6/pFeoAB-V5C was cross-linked using formaldehyde (CH2O), and the proteins were separated by SDS-PAGE and analyzed by immunoblotting using an anti-V5 antibody. (B) The lysate from cross-linked cells was fractionated to separate the cytoplasm (Cyto), inner membrane (IM), and outer membrane (OM). Proteins were resolved by SDS-PAGE and analyzed by immunoblotting using an anti-V5 antibody. The monomeric form of FeoB is shown with an asterisk.
Complex formation was corroborated through the use of BN-PAGE, a technique that is commonly used to determine native membrane protein masses and oligomeric states (47). BN-PAGE results (Fig. 4) showed that FeoB formed two major complexes, one of approximately 250 kDa and another one of more than 720 kDa. Native protein masses were estimated using a NativeMark unstained protein standard (Life Technologies). Overall, the results described above indicate that full-length FeoB forms complexes in vivo.
FIG 4.
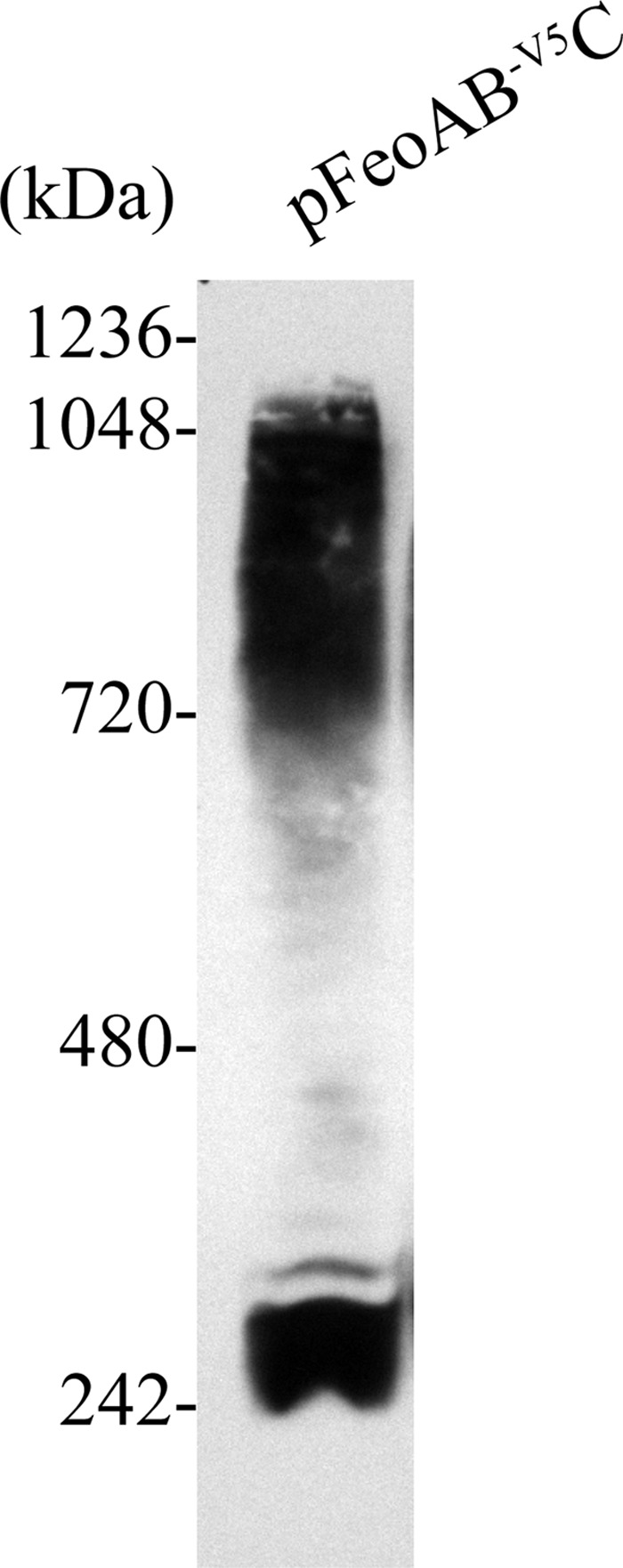
V. cholerae FeoB makes higher-order native complexes as shown by BN-PAGE analysis. EPV6/pFeoAB-V5C total membrane fraction proteins were separated on a bis-Tris native gel and immunoblotted using an anti-V5 antibody.
All three Feo proteins interact to form a complex.
To determine the composition of the complexes, we employed immunoprecipitation followed by LC-MS/MS analysis. Cultures of EPV6/pFeoAB-V5C were grown to mid-log phase and cross-linked in vivo, and FeoB complexes were immunoprecipitated from the cell lysate using an anti-V5 antibody. Immunoprecipitated proteins were then separated by SDS-PAGE and stained with Gel Code Blue (Fig. 5A), and the presence and mobility of the V5-tagged proteins were verified by immunoblotting using an anti-V5 antibody (Fig. 5B). The indicated bands (Fig. 5A) were excised, subjected to in-gel digestion using trypsin, and analyzed by LC-MS/MS (Table 3). FeoA and FeoB were detected during analysis of the largest band (marked as complex 1 in Fig. 5A). FeoC was not detected at the specified confidence level. To determine whether FeoC is present in complex 1, we immunoprecipitated cross-linked V5-tagged FeoC complexes (Fig. 5A) and verified the presence of FeoC through immunoblotting (Fig. 5C). The largest band, which comigrated with complex 1 when electrophoresed on the same gel, was named complex 1° (Fig. 5A) to differentiate it from complex 1 immunoprecipitated with V5-tagged FeoB. Complexes 1 and 1° are likely the same, but this cannot be unequivocally stated at this time. Analysis of complex 1° showed the presence of FeoA and FeoB, demonstrating that FeoC is in a complex with both FeoA and FeoB. FeoC was not detected by LC-MS/MS at the specified confidence level, even though FeoC was the V5-tagged component of the immunoprecipitated complex. Lack of detection of FeoC by LC-MS/MS may have been due to the presence of a low concentration of FeoC in the complex. Overall, these results indicate that FeoA, FeoB, and FeoC interact to form one complex. To our knowledge, this is the first demonstration that all three Feo proteins can form a single complex.
FIG 5.

Immunoprecipitation of in vivo cross-linked Feo complexes using V5-tagged FeoB or FeoC. (A) Proteins were formaldehyde cross-linked in vivo and immunoprecipitated using a mouse anti-V5 antibody. Cultures of EPV6 containing the empty vector (pWKS30), a plasmid encoding the Feo proteins with V5-tagged FeoB (pFeoAB-V5C), or a plasmid encoding the Feo proteins with V5-tagged FeoC (pFeoABC-V5) were grown to mid-log phase and cross-linked with formaldehyde. Proteins were separated by SDS-PAGE and visualized using Gel Code Blue. Numbered arrowheads indicate Feo complexes that were excised and analyzed by mass spectrometry. The asterisk denotes the monomeric FeoB. (B and C) V5-labeled complexes immunoprecipitated from either cross-linked FeoB-V5 (B) or cross-linked FeoC-V5 (C) were visualized with a rabbit-derived anti-V5 antibody.
TABLE 3.
Immunoprecipitated Feo proteins detected by mass spectrometry
| Protein used for immunoprecipitation | Complexa | Protein | No. of peptide spectral matches |
|---|---|---|---|
| FeoB | 1 | FeoB | 97 |
| FeoA | 7 | ||
| 2 | FeoB | 113 | |
| 3 | FeoB | 188 | |
| 4 | FeoB | 355 | |
| FeoA | 23 | ||
| FeoC | 4 | ||
| FeoC | 1° | FeoB | 150 |
| FeoA | 9 | ||
| 5 | FeoB | 54 | |
| FeoC | 4 |
FeoC was below the limit of detection in the complex 1 and complex 1° samples. However, FeoC was detected in complex 1° by Western blotting.
Only FeoB was detected in two of the smaller complexes that were immunoprecipitated using cross-linked V5-tagged FeoB (marked as complexes 2 and 3 in Fig. 5A). Neither FeoA nor FeoC was detected in either of these complexes, and no complexes of similar size were observed in the V5-tagged FeoC immunoprecipitation eluate (Fig. 5A). Complex 4 (Fig. 5A), located near the FeoB monomer, contained FeoB, FeoA, and FeoC (Table 3). A similarly sized cross-linked complex (marked as complex 5 in Fig. 5A) was immunoprecipitated using V5-tagged FeoC, and it contained detectable FeoB and FeoC but not FeoA. Since FeoA and FeoC are both approximately 8 kDa, it is possible that the band marked as complex 4 actually represents two separate cross-linked complexes consisting of FeoB cross-linked to either FeoA or FeoC that are migrating to the same position on the gel. This hypothesis is supported by the fact that V5-tagged FeoC complex 5 immunoprecipitated only FeoB and not FeoA, suggesting that the FeoB monomer may interact with either FeoA or FeoC but not with both at the same time. Complexes 2, 3, 4, and 5 may represent intermediates in the assembly of the larger complex 1 or breakdown/disassembly products. Alternatively, the components of complex 1 may not be fully cross-linked by this procedure, and cross-links may form more easily within the trimers than between trimers. This would result in the appearance of the intermediate-sized complexes upon electrophoresis.
Attempts to perform immunoprecipitation using either an N-terminally or C-terminally V5-tagged FeoA were unsuccessful. The tags on FeoA did not affect the function of the Feo system, but we were unable to detect a level of FeoA protein sufficient for protein analysis (data not shown).
Residues in the FeoB GTPase domain are required for complex formation and iron transport.
In order to determine whether or not the GTPase domain plays a role in FeoB complex formation, we constructed mutations in each of the four conserved sequence elements, G1 to G4, and in the regulatory switch I and switch II regions of FeoB (Fig. 1B). A FeoB G1 element mutation, K15D, was introduced into pFeoAB-V5C. The equivalent lysine residue in p21-Ras contacts the β- and γ-phosphates of GTP (48–50). The K15D mutation in FeoB abolished the function of the Feo system, as EPV6 carrying this FeoB mutant on a plasmid was unable to grow without heme supplementation (Table 4). Cross-linking analysis of the K15D mutant showed no complex formation; FeoB remained in the monomeric form (Fig. 6B). Since the equivalent K15 FeoB residue in p21-Ras is known to contact GTP, it is possible that bound nucleotide is required for complex formation to occur. Densitometry analysis of the immunoblot revealed that the level of the K15D FeoB mutant protein was approximately 40% of the wild-type (WT) level (Fig. 6A), indicating that this FeoB mutant may be less stable or less efficiently localized in the inner membrane than the wild type. However, the amount of FeoB detected in the cross-linked sample (Fig. 6B) should have been sufficient for detection of complexes, if they were formed.
TABLE 4.
Effect of FeoB mutations on growth of EPV6 and on Feo complex formation
| Protein or mutation(s)a | Mutation location | Feo functionb | Feo complex formation |
|---|---|---|---|
| WT FeoB-V5 | NAc | + | + |
| FeoB K15D | FeoB G1 | − | − |
| FeoB T36K | FeoB switch I/G2 | − | + |
| FeoB D55K | FeoB G3 | − | + |
| FeoB T36K and D55K | FeoB switch I/G2/G3 | − | + |
| FeoB D72A | FeoB switch II | − | − |
| FeoB N119K | FeoB G4 | + | NDe |
| FeoB D122N | FeoB G4 | + | ND |
| FeoB D93K | FeoB potential G4 | + | ND |
| ΔCd | NA | − | + |
| FeoA G32K | FeoA | − | − |
| FeoA A45D | FeoA | − | − |
| FeoA P50R | FeoA | − | − |
| FeoA V72K | FeoA | − | − |
All the indicated mutations were made in the plasmid pFeoAB-V5C.
Feo function was assessed by comparing growth of EPV6 carrying a mutated pFeoAB-V5C plasmid to that of EPV6/pFeoAB-V5C on LB agar without heme.
NA, not applicable.
ΔC indicates the pFeoAB-V5 plasmid.
ND, not determined.
FIG 6.

Effect of mutations in the FeoB GTPase G motifs and switch regions on FeoB subcellular localization and complex formation. (A) Immunoblot analysis of equal protein amounts of inner membrane fractions of EPV6 carrying pFeoAB-V5C or of pFeoAB-V5C carrying the indicated FeoB mutations. (B) Immunoblot analysis of in vivo formaldehyde (CH2O) cross-linked EPV6/pFeoAB-V5C and EPV6/pFeoAB-V5C with FeoB K15D or D72A mutations. These mutations are in the GTPase G1 and switch II regions, respectively. (C) Immunoblot analysis of in vivo formaldehyde (CH2O) cross-linked EPV6/pFeoAB-V5C with FeoB carrying no mutation or the T36K, D55K, or T36K/D55K mutations. The T36K mutation is in the GTPase switch I and G2 region, while the D55K mutation is in the GTPase G3 region. All immunoblot analyses were performed using an anti-V5 antibody. An asterisk denotes monomeric FeoB.
To look at the roles of the switch I region and the G2 and G3 elements, the T36 and D55 residues were mutated. T36 is equivalent to a critical p21-Ras threonine residue located in the G2 element and switch I region and coordinates the Mg2+ ion that is needed for GTP hydrolysis (36). D55, located in the G3 element, plays a role in coordination of the catalytic Mg2+ ion through a water molecule, as shown in crystal structures of p21-Ras and Methanococcus jannaschii FeoB (50, 51). Both the T36K and D55K mutations in pFeoAB-V5C resulted in loss of Feo function when the plasmids were transformed into EPV6 (Table 4). However, loss of these residues did not affect complex formation, membrane localization, or protein stability (Fig. 5A and C). Since it is expected that T36 interacts directly and D55 interacts indirectly with the catalytic Mg2+ ion, we also determined the effect on complex formation when both residues were mutated. The plasmid encoding the FeoB T36K and D55K double mutant was transformed into EPV6, and, as expected, no Feo function was observed (Table 4); however, inner membrane localization of FeoB (Fig. 6A) and complex formation (Fig. 6C) were similar to those seen with the wild type. These data suggest that GTP hydrolysis is required for ferrous iron transport but not for complex formation.
The results of mutation of the predicted G4 element were unexpected. Alignment of V. cholerae FeoB with p21-Ras suggests that the G4 motif, which provides guanine base specificity (35), is positioned at amino acids 119 to 122 (Fig. 1B). However, the versions of FeoB with mutations at two conserved positions, N119K and D122N, were both fully active, as plasmids carrying these mutations allowed wild-type growth of EPV6 without heme (Table 4). Cross-linking analysis was not performed for either of these mutants, because Feo system function was not affected. A second possible G4 motif at positions 90 to 93 was identified, but mutation of the conserved Asp residue (D93K) did not alter the activity of the transporter. These data suggest that a noncanonical G4 region that has not yet been identified fulfills this role in V. cholerae FeoB.
An additional mutation was made to test the role of the switch II region of FeoB. The switch II region of GTPases undergoes a conformational change upon GTP binding, and in Legionella pneumophila, mutations in this region of FeoB cause a decrease in FeoB nucleotide binding (35, 50). The V. cholerae FeoB D72A mutation was unable to promote growth of EPV6 without heme, indicating that the D72A mutation resulted in loss of function. Cellular fractionation analysis showed that FeoB D72A was located in the inner membrane and was stable, since the protein levels were comparable to those of unmutated FeoB (Fig. 6A). Cross-linking analysis showed that FeoB D72A remained in the monomeric form and was not able to form complexes (Fig. 6B). These data suggest that the switch II region, and possibly nucleotide binding, is required for Feo complex formation.
FeoC is required for WT FeoB protein levels, while FeoA is involved in complex formation.
In addition to FeoB, FeoA and FeoC are both found in the large Feo complex; thus, we wanted to determine whether they are required for complex formation. To determine the possible role of FeoC, we transformed EPV6 with a plasmid containing feoA and N-terminal V5-tagged feoB. This plasmid did not support growth of EPV6 in the absence of heme (Table 4), consistent with our previous finding that FeoC is required for the activity of V. cholerae Feo (27). Membrane fractionation demonstrated that FeoB was still located in the inner membrane in the absence of FeoC (Fig. 7A); however, the level of FeoB protein was somewhat decreased. Densitometry indicated that the amount of FeoB in the absence of FeoC was approximately 75% of the amount of FeoB expressed when the feo operon was intact (Fig. 7A, top). Coomassie blue staining confirmed that the total amounts of protein in the samples were similar (Fig. 7A, bottom). Although the amount of FeoB was reduced, the amount present was sufficient to observe FeoB in cross-linked complexes similar to those observed with the intact feo operon (Fig. 7B). Thus, FeoC is not required for complex formation.
FIG 7.

FeoC is required for wild-type FeoB levels but not for complex formation. (A) Immunoblot and SDS-PAGE analysis of EPV6/pFeoAB-V5C or EPV6/pFeoAB-V5 inner membrane fractions. (B) Immunoblot and SDS-PAGE analysis of whole-cell lysate of in vivo formaldehyde (CH2O) cross-linked EPV6/pFeoAB-V5C or EPV6/pFeoAB-V5. Immunoblot analyses were performed using an anti-V5 mouse antibody. Coomassie-stained gels are shown as a loading control. An asterisk denotes monomeric FeoB.
In initial attempts to determine the role of FeoA in complex formation, we employed a plasmid containing the feo operon with an in-frame deletion of feoA. When this plasmid was transformed into EPV6, the amount of FeoB was reduced to nearly undetectable levels, such that we were unable to ascertain whether complexes were formed (data not shown). When feoA was supplied on a separate plasmid, activity was restored but the amount of FeoB protein did not increase, indicating that the decrease in FeoB levels was due to a polar effect of the deletion, rather than to the absence of the FeoA protein (data not shown). Since we were not able to delete the feoA gene without severely affecting the level of FeoB, we created feoA point mutations that abolished Feo activity. FeoA residues conserved across several bacterial species were mutated within the context of the pFeoAB-V5C plasmid, and EPV6 carrying the plasmids was tested for the ability to grow without heme supplementation. Four FeoA amino acid substitutions that eliminated Feo function were identified: G32K, A45D, P50R, and V72K. Membrane fractionation showed that FeoB was expressed from each of these mutant plasmids and that it localized to the inner membrane, albeit at reduced levels for the G32K (61% of wild type) and V72K (64% of wild type) mutants (Fig. 8A). However, FeoB expressed from plasmids containing the FeoA mutations failed to form complexes and remained in monomeric form (Fig. 8B), indicating that FeoA is required for complex formation.
FIG 8.
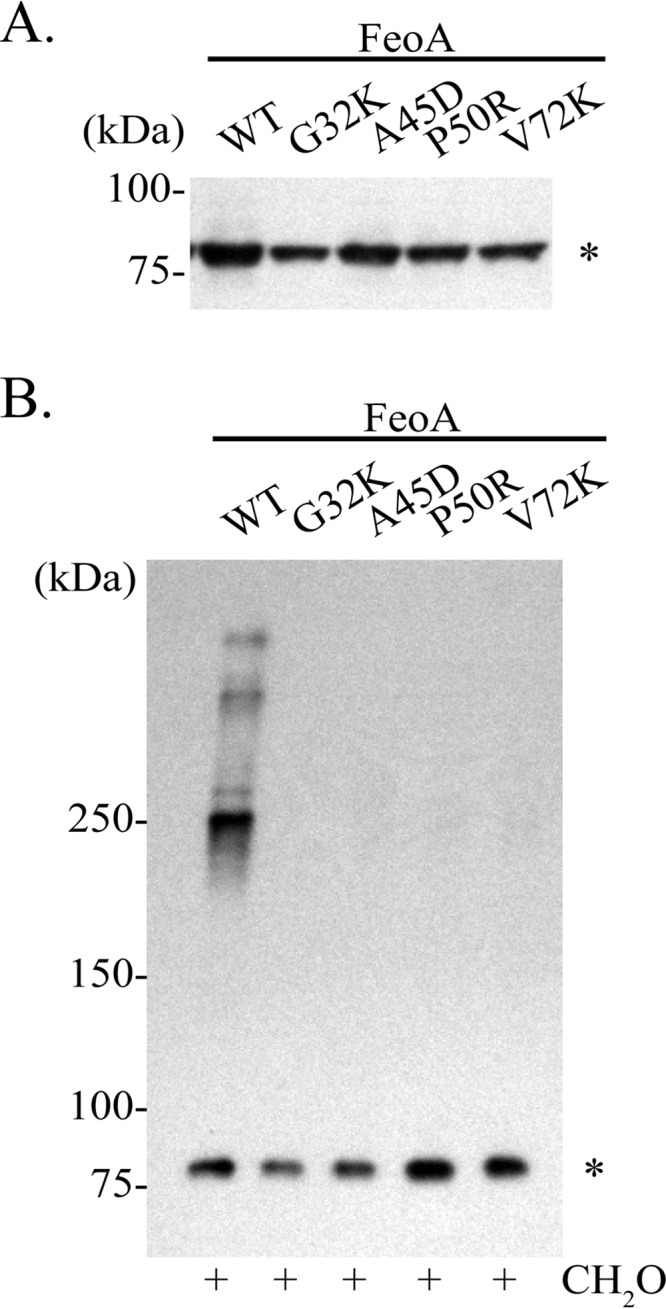
Mutations in FeoA abolish FeoB complex formation. (A) Immunoblot analysis of equal protein amounts of inner membrane fractions of EPV6 carrying pFeoAB-V5C (WT) or plasmids encoding the indicated FeoA mutations. (B) Immunoblot analysis of in vivo formaldehyde cross-linked EPV6 carrying pFeoAB-V5C (WT) or plasmids encoding the indicated FeoA mutations. Immunoblot analyses were performed using an anti-V5 antibody. An asterisk denotes monomeric FeoB.
DISCUSSION
The V. cholerae Feo system is made up of three proteins, FeoA, FeoB, and FeoC, and all three Feo proteins are required for its function (27). Prior to our study, several lines of evidence suggested that the Feo proteins may interact to form a functional Feo structure. Interactions among FeoB monomers have been suggested by crystallography studies (51–54). An interaction between FeoC and FeoB was observed in a BACTH assay for both the V. cholerae (27) and S. enterica (41) proteins. Similarly, K. pneumoniae FeoB and FeoC were found to cocrystallize (42). Further, in S. enterica, FeoA was shown to interact with FeoB in a BACTH assay (29).
Crystallography studies of the large membrane protein, FeoB, have provided important insights into its structure; however, these structural studies have been limited to the N-terminal domain of FeoB (N-FeoB), since it is readily soluble and has measurable enzymatic (GTPase) activity. Further, significant limitations to studying Feo structure by crystallography have been observed, since a consensus on the oligomeric state of FeoB has never been reached. For example, FeoB N-terminal domains from several organisms have crystallized as monomers (35, 53, 55–57) whereas the FeoB N-terminal domains from other organisms have crystallized as dimers or trimers (51–54). Since those studies used a truncated FeoB protein, interactions involving the membrane-spanning regions or C terminus of FeoB were not detected. Further, crystallization of FeoB in the presence of both FeoA and FeoC has not been reported, in spite of evidence indicating that both FeoA and FeoC are required for Feo activity and that each can interact with FeoB (27, 29).
In our study, we undertook in vivo structural analysis using the more functionally relevant full-length FeoB protein. In vivo cross-linking showed that FeoB forms several large complexes that are approximately 250 kDa (complex 3), 500 kDa (complex 2), and larger than 500 kDa (complex 1). This observation was validated by BN-PAGE analysis, which showed that FeoB was able to form two large native complexes, 250 kDa and around 720 kDa or larger, which are consistent with the sizes of complex 3 and 1 seen through in vivo cross-linking. Complex 2, the 500-kDa cross-linked complex, was not detected through BN-PAGE, suggesting that this complex may be a FeoB transition state of the larger complex, complex 1. Overall, these observations demonstrate, for the first time, that full-length FeoB is found in complexes in vivo.
Although FeoA and FeoC were both determined to be in the large complex (complex 1°), neither of these proteins was found in complex 2 or 3. Since both FeoA and FeoC are required for Feo system function, the large complex may be the active form of the Feo system. The smaller complexes may represent intermediates in the assembly or may reflect incomplete cross-linking of proteins within the large complex. In E. coli, N-FeoB was found to crystallize as a trimer, and it was proposed that this trimer forms an intracellular pore for ferrous iron transport (52). Consistent with that study, complex 3, made up of FeoB, was observed to have an approximate size of 250 kDa; this complex may represent a trimer of full-length FeoB proteins, since the tagged monomeric FeoB protein has a size of 84.6 kDa. Taking into consideration the sizes of the other two FeoB complexes, complex 1 and 2, it is possible that 500-kDa complex 2 is a dimer of FeoB trimers and that 720-kDa complex 1 is a trimer of trimers. FeoB may form trimers in the absence of the other Feo proteins and assemble with FeoA and FeoC to form the large complex, complex 1°, that is likely the functional complex. While we were not able to determine the exact stoichiometry of this complex, we propose that this complex is a trimer of FeoB trimers that interacts with one or more molecules of FeoA and FeoC to transport ferrous iron into V. cholerae (Fig. 9).
FIG 9.

Model of the active form of the Feo system. Complex 1/1° is most likely made up of a trimer of FeoB trimers, as indicated by shading, in addition to FeoA and FeoC. They may come together to form a pore in the inner membrane (IM) through which ferrous iron is transported into the cytoplasm (Cyto). The ratio of FeoB to FeoA or FeoC is not clear at this time.
To gain more insight into the assembly of the functional complex, we sought to determine the role of FeoC in complex formation. Deletion of feoC did not affect FeoB complex formation, although the quantity of FeoB, and therefore the amount of the complexes, was decreased. This suggests that FeoC may be needed to protect FeoB from proteolysis in V. cholerae such as was found in S. enterica (41). Nonetheless, we believe that this may not be the only function of FeoC in V. cholerae, since the activity of the Feo system was completely eliminated by deletion of the feoC gene. If FeoC were required only for protecting FeoB from proteolysis, it follows that at least some Feo system activity should be retained, since there was still a significant level of FeoB protein, as well as FeoB complexes, in the absence of FeoC. FeoC proteins are poorly conserved, and it would not be surprising to find that they have different functions in different organisms. For example, S. enterica FeoC, as well as FeoC proteins from other species, features a conserved cysteine motif that is involved in iron-sulfur (Fe-S) cluster formation and that may be used as an oxygen sensor (26, 58, 59). V. cholerae FeoC, on the other hand, does not contain any cysteine residues and therefore would not be able to bind an iron-sulfur cluster, indicating that it may function in a different manner.
The role of FeoA is not well defined, even though it is present in the majority of organisms that contain a Feo system (31). We investigated the role of FeoA in FeoB complex formation by mutational analysis. Mutation of several conserved FeoA residues that resulted in loss of Feo system function also resulted in loss of FeoB complexes, indicating that FeoA is required for complex formation and is an integral part of the Feo transport system.
Genetic analysis showed that mutations within certain regions of the N-terminal GTPase domain of FeoB resulted in loss of complex formation. FeoB was unable to form complexes when residues in the G1 and switch II regions, known to be closely associated with nucleotide binding, were mutated. These results suggest that nucleotide binding may be essential for formation or maintenance of the Feo complex. Further, these findings are consistent with the observation that a residue in the switch II region of p21-Ras is important for p21-Ras complex formation (60). We observed that mutations in conserved residues in the G2 and G3 elements of FeoB, which are predicted to be involved in the coordination of the catalytic Mg2+ ion and were shown to be required for FeoB GTP hydrolysis in Streptococcus thermophilus (49), did not alter complex formation. This suggests that although nucleotide binding may be required for FeoB complex formation, GTP hydrolysis is not. However, GTP hydrolysis is required for transport of iron into the cell, since mutations in the G2 and G3 elements abolished Feo function in V. cholerae.
Our results are consistent with FeoA, FeoB, and FeoC forming a large complex in the inner membrane of V. cholerae. Mutational analysis showed a correlation between function and complex formation. The large complex, complex 1/1°, was always present when Feo activity was detected. Mutants that failed to form this complex were inactive. However, complex formation was not sufficient for activity, since some mutants that lacked Feo function were still able to form complexes that appeared to be the same size as wild-type complexes. How FeoA and the GTPase region of FeoB participate in complex formation and the mechanism of ferrous iron transport through the complex remain to be determined.
ACKNOWLEDGMENTS
National Institutes of Health grant AI091957 supported this work.
We are thankful to Eric Peng for his generous gift of V. cholerae EPV6. We thank Alexandra Mey for reading of the manuscript and helpful comments. We gratefully acknowledge Maria Person for excellent assistance with mass spectrometry experiments.
REFERENCES
- 1.Cairo G, Bernuzzi F, Recalcati S. 2006. A precious metal: iron, an essential nutrient for all cells. Genes Nutr 1:25–39. doi: 10.1007/BF02829934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neilands JB. 1991. A brief history of iron metabolism. Biol Met 4:1–6. doi: 10.1007/BF01135550. [DOI] [PubMed] [Google Scholar]
- 3.Wyckoff EE, Mey AR, Payne SM. 2007. Iron acquisition in Vibrio cholerae. Biometals 20:405–416. doi: 10.1007/s10534-006-9073-4. [DOI] [PubMed] [Google Scholar]
- 4.Sack DA, Sack RB, Nair GB, Siddique AK. 2004. Cholera. Lancet 363:223–233. doi: 10.1016/S0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths GL, Sigel SP, Payne SM, Neilands JB. 1984. Vibriobactin, a siderophore from Vibrio cholerae. J Biol Chem 259:383–385. [PubMed] [Google Scholar]
- 6.Rogers MB, Sexton JA, DeCastro GJ, Calderwood SB. 2000. Identification of an operon required for ferrichrome iron utilization in Vibrio cholerae. J Bacteriol 182:2350–2353. doi: 10.1128/JB.182.8.2350-2353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mey AR, Wyckoff EE, Oglesby AG, Rab E, Taylor RK, Payne SM. 2002. Identification of the Vibrio cholerae enterobactin receptors VctA and IrgA: IrgA is not required for virulence. Infect Immun 70:3419–3426. doi: 10.1128/IAI.70.7.3419-3426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wyckoff EE, Allred BE, Raymond KN, Payne SM. 2015. Catechol siderophore transport by Vibrio cholerae. J Bacteriol 197:2840–2849. doi: 10.1128/JB.00417-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keating TA, Marshall CG, Walsh CT. 2000. Vibriobactin biosynthesis in Vibrio cholerae: VibH is an amide synthase homologous to nonribosomal peptide synthetase condensation domains. Biochemistry 39:15513–15521. doi: 10.1021/bi001651a. [DOI] [PubMed] [Google Scholar]
- 10.Butterton JR, Stoebner JA, Payne SM, Calderwood SB. 1992. Cloning, sequencing, and transcriptional regulation of viuA, the gene encoding the ferric vibriobactin receptor of Vibrio cholerae. J Bacteriol 174:3729–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butterton JR, Calderwood SB. 1994. Identification, cloning, and sequencing of a gene required for ferric vibriobactin utilization by Vibrio cholerae. J Bacteriol 176:5631–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wyckoff EE, Valle A-M, Smith SL, Payne SM. 1999. A multifunctional ATP-binding cassette transporter system from Vibrio cholerae transports vibriobactin and enterobactin. J Bacteriol 181:7588–7596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wyckoff EE, Stoebner JA, Reed KE, Payne SM. 1997. Cloning of a Vibrio cholerae vibriobactin gene cluster: identification of genes required for early steps in siderophore biosynthesis. J Bacteriol 179:7055–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wyckoff EE, Payne SM. 2011. The Vibrio cholerae VctPDGC system transports catechol siderophores and a siderophore-free iron ligand. Mol Microbiol 81:1446–1458. doi: 10.1111/j.1365-2958.2011.07775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wyckoff EE, Mey AR, Leimbach A, Fisher CF, Payne SM. 2006. Characterization of ferric and ferrous iron transport systems in Vibrio cholerae. J Bacteriol 188:6515–6523. doi: 10.1128/JB.00626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoebner JA, Payne SM. 1988. Iron-regulated hemolysin production and utilization of heme and hemoglobin by Vibrio cholerae. Infect Immun 56:2891–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson DP, Payne SM. 1994. Characterization of the Vibrio cholerae outer membrane heme transport protein HutA: sequence of the gene, regulation of expression, and homology to the family of TonB-dependent proteins. J Bacteriol 176:3269–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Occhino DA, Wyckoff EE, Henderson DP, Wrona TJ, Payne SM. 1998. Vibrio cholerae iron transport: haem transport genes are linked to one of two sets of tonB, exbB, exbD genes. Mol Microbiol 29:1493–1507. doi: 10.1046/j.1365-2958.1998.01034.x. [DOI] [PubMed] [Google Scholar]
- 19.Mey AR, Payne SM. 2001. Haem utilization in Vibrio cholerae involves multiple TonB-dependent haem receptors. Mol Microbiol 42:835–849. [DOI] [PubMed] [Google Scholar]
- 20.Hantke K. 1987. Ferrous iron transport mutants in Escherichia coli K12. FEMS Microbiol Lett 44:53–57. doi: 10.1111/j.1574-6968.1987.tb02241.x. [DOI] [Google Scholar]
- 21.Kammler M, Schön C, Hantke K. 1993. Characterization of the ferrous iron uptake system of Escherichia coli. J Bacteriol 175:6212–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velayudhan J, Hughes NJ, McColm AA, Bagshaw J, Clayton CL, Andrews SC, Kelly DJ. 2000. Iron acquisition and virulence in Helicobacter pylori: a major role for FeoB, a high-affinity ferrous iron transporter. Mol Microbiol 37:274–286. doi: 10.1046/j.1365-2958.2000.01987.x. [DOI] [PubMed] [Google Scholar]
- 23.Naikare H, Palyada K, Panciera R, Marlow D, Stintzi A. 2006. Major role for FeoB in Campylobacter jejuni ferrous iron acquisition, gut colonization, and intracellular survival. Infect Immun 74:5433–5444. doi: 10.1128/IAI.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cianciotto NP. 2007. Iron acquisition by Legionella pneumophila. Biometals 20:323–331. doi: 10.1007/s10534-006-9057-4. [DOI] [PubMed] [Google Scholar]
- 25.Hantke K. 2003. Is the bacterial ferrous iron transporter FeoB a living fossil? Trends Microbiol 11:192–195. doi: 10.1016/S0966-842X(03)00100-8. [DOI] [PubMed] [Google Scholar]
- 26.Cartron ML, Maddocks S, Gillingham P, Craven CJ, Andrews SC. 2006. Feo—transport of ferrous iron into bacteria. Biometals 19:143–157. doi: 10.1007/s10534-006-0003-2. [DOI] [PubMed] [Google Scholar]
- 27.Weaver EA, Wyckoff EE, Mey AR, Morrison R, Payne SM. 2013. FeoA and FeoC are essential components of the Vibrio cholerae ferrous iron uptake system, and FeoC interacts with FeoB. J Bacteriol 195:4826–4835. doi: 10.1128/JB.00738-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry RD, Mier I, Fetherston JD. 2007. Roles of the Yfe and Feo transporters of Yersinia pestis in iron uptake and intracellular growth. Biometals 20:699–703. doi: 10.1007/s10534-006-9051-x. [DOI] [PubMed] [Google Scholar]
- 29.Kim H, Lee H, Shin D. 2012. The FeoA protein is necessary for the FeoB transporter to import ferrous iron. Biochem Biophys Res Commun 423:733–738. doi: 10.1016/j.bbrc.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Pawson T, Gish GD. 1992. SH2 and SH3 domains: from structure to function. Cell 71:359–362. doi: 10.1016/0092-8674(92)90504-6. [DOI] [PubMed] [Google Scholar]
- 31.Lau CKY, Ishida H, Liu Z, Vogel HJ. 2013. Solution structure of Escherichia coli FeoA and its potential role in bacterial ferrous iron transport. J Bacteriol 195:46–55. doi: 10.1128/JB.01121-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su YC, Chin KH, Hung HC, Shen GH, Wang AHJ, Chou SH. 2010. Structure of Stenotrophomonas maltophilia FeoA complexed with zinc: a unique prokaryotic SH3-domain protein that possibly acts as a bacterial ferrous iron-transport activating factor. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:636–642. doi: 10.1107/S1744309110013941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marlovits TC, Haase W, Herrmann C, Aller SG, Unger VM. 2002. The membrane protein FeoB contains an intramolecular G protein essential for Fe(II) uptake in bacteria. Proc Natl Acad Sci U S A 99:16243–16248. doi: 10.1073/pnas.242338299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eng ET, Jalilian AR, Spasov KA, Unger VM. 2008. Characterization of a novel prokaryotic GDP dissociation inhibitor domain from the G protein coupled membrane protein FeoB. J Mol Biol 375:1086–1097. doi: 10.1016/j.jmb.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petermann N, Hansen G, Schmidt CL, Hilgenfeld R. 2010. Structure of the GTPase and GDI domains of FeoB, the ferrous iron transporter of Legionella pneumophila. FEBS Lett 584:733–738. doi: 10.1016/j.febslet.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 36.Bourne HR, Sanders DA, McCormick F. 1991. The GTPase superfamily: conserved structure and molecular mechanism. Nature 349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 37.Leipe DD, Wolf YI, Koonin EV, Aravind L. 2002. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol 317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 38.Rudack T, Xia F, Schlitter J, Kötting C, Gerwert K. 2012. The role of magnesium for geometry and charge in GTP hydrolysis, revealed by quantum mechanics/molecular mechanics simulations. Biophys J 103:293–302. doi: 10.1016/j.bpj.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vetter IR, Wittinghofer A. 2001. The guanine nucleotide-binding switch in three dimensions. Science 294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 40.Hung K-W, Juan T-H, Hsu Y-L, Huang TH. 2012. NMR structure note: the ferrous iron transport protein C (FeoC) from Klebsiella pneumoniae. J Biomol NMR 53:161–165. doi: 10.1007/s10858-012-9633-6. [DOI] [PubMed] [Google Scholar]
- 41.Kim H, Lee H, Shin D. 2013. The FeoC protein leads to high cellular levels of the Fe(II) transporter FeoB by preventing FtsH protease regulation of FeoB in Salmonella enterica. J Bacteriol 195:3364–3370. doi: 10.1128/JB.00343-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hung K-W, Tsai J-Y, Juan T-H, Hsu Y-L, Hsiao C-D, Huang T-H. 2012. Crystal structure of the Klebsiella pneumoniae NFeoB/FeoC complex and roles of FeoC in regulation of Fe2+ transport by the bacterial Feo system. J Bacteriol 194:6518–6526. doi: 10.1128/JB.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wittig I, Braun H-P, Schägger H. 2006. Blue native PAGE. Nat Protoc 1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 44.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 45.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schägger H, Cramer WA, von Jagow G. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 217:220–230. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- 48.Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A. 1989. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 341:209–214. doi: 10.1038/341209a0. [DOI] [PubMed] [Google Scholar]
- 49.Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, Kim SH. 1990. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 50.Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A. 1990. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J 9:2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Köster S, Wehner M, Herrmann C, Kühlbrandt W, Yildiz O. 2009. Structure and function of the FeoB G-domain from Methanococcus jannaschii. J Mol Biol 392:405–419. doi: 10.1016/j.jmb.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 52.Guilfoyle A, Maher MJ, Rapp M, Clarke R, Harrop S, Jormakka M. 2009. Structural basis of GDP release and gating in G protein coupled Fe2+ transport. EMBO J 28:2677–2685. doi: 10.1038/emboj.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hung K-W, Chang Y-W, Eng ET, Chen J-H, Chen Y-C, Sun Y-J, Hsiao C-D, Dong G, Spasov KA, Unger VM, Huang T-H. 2010. Structural fold, conservation and Fe(II) binding of the intracellular domain of prokaryote FeoB. J Struct Biol 170:501–512. doi: 10.1016/j.jsb.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deshpande CN, McGrath AP, Font J, Guilfoyle AP, Maher MJ, Jormakka M. 2013. Structure of an atypical FeoB G-domain reveals a putative domain-swapped dimer. Acta Crystallogr Sect F Struct Biol Cryst Commun 69:399–404. doi: 10.1107/S1744309113005939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ash M-R, Guilfoyle A, Clarke RJ, Guss JM, Maher MJ, Jormakka M. 2010. Potassium-activated GTPase reaction in the G protein-coupled ferrous iron transporter B. J Biol Chem 285:14594–14602. doi: 10.1074/jbc.M110.111914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hattori M, Jin Y, Nishimasu H, Tanaka Y, Mochizuki M, Uchiumi T, Ishitani R, Ito K, Nureki O. 2009. Structural basis of novel interactions between the small-GTPase and GDI-like domains in prokaryotic FeoB iron transporter. Structure 17:1345–1355. doi: 10.1016/j.str.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 57.Ash M-R, Maher MJ, Guss JM, Jormakka M. 2011. The initiation of GTP hydrolysis by the G-domain of FeoB: insights from a transition-state complex structure. PLoS One 6:e23355. doi: 10.1371/journal.pone.0023355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsueh K-L, Yu L-K, Chen Y-H, Cheng Y-H, Hsieh Y-C, Ke S, Hung K-W, Chen C-J, Huang T. 2013. FeoC from Klebsiella pneumoniae contains a [4Fe-4S] cluster. J Bacteriol 195:4726–4734. doi: 10.1128/JB.00687-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim H, Lee H, Shin D. 2015. Lon-mediated proteolysis of the FeoC protein prevents Salmonella enterica from accumulating the Fe(II) transporter FeoB under high-oxygen conditions. J Bacteriol 197:92–98. doi: 10.1128/JB.01826-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin W-C, Iversen L, Tu H-L, Rhodes C, Christensen SM, Iwig JS, Hansen SD, Huang WYC, Groves JT. 2014. H-Ras forms dimers on membrane surfaces via a protein–protein interface. Proc Natl Acad Sci U S A 111:2996–3001. doi: 10.1073/pnas.1321155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]