

ABSTRACT
Bacteria use multidrug efflux pumps to export drugs and toxic compounds out of the cell. One of the most important efflux pumps in Escherichia coli is the AcrAB-TolC system. Small regulatory RNAs (sRNAs) are known to be major posttranscriptional regulators that can enhance or repress translation by binding to the 5′ untranslated region (UTR) of mRNA targets with the help of a chaperone protein, Hfq. In this study, we investigated the expression of acrA, acrB, and tolC translational fusions using 27 Hfq-dependent sRNAs overexpressed from plasmids. No significant sRNA regulation of acrA or acrB was detected. SdsR (also known as RyeB), an abundant and well-conserved stationary-phase sRNA, was found to repress the expression of tolC, the gene encoding the outer membrane protein of many multidrug resistance efflux pumps. This repression was shown to be by direct base pairing occurring upstream from the ribosomal binding site. SdsR overexpression and its regulation of tolC were found to reduce resistance to novobiocin and crystal violet. Our results suggest that additional targets for SdsR exist that contribute to increased antibiotic sensitivity and reduced biofilm formation. In an effort to identify phenotypes associated with single-copy SdsR and its regulation of tolC, the effect of a deletion of sdsR or mutations in tolC that should block SdsR pairing were investigated using a Biolog phenotypic microarray. However, no significant phenotypes were identified. Therefore, SdsR appears to modulate rather than act as a major regulator of its targets.
IMPORTANCE AcrAB-TolC is a major efflux pump present in E. coli and Gram-negative bacteria used to export toxic compounds; the pump confers resistance to many antibiotics of unrelated classes. In this study, we found that SdsR, a small RNA expressed in stationary phase, repressed the expression of tolC, resulting in increased sensitivity to some antibiotics. This extends the findings of previous studies showing that sRNAs contribute to the regulation of many outer membrane proteins; manipulating or enhancing their action might help in sensitizing bacteria to antibiotics.
INTRODUCTION
One of the major characteristics of bacterial pathogens is their ability to rapidly acquire antibiotic resistance. Antibiotic resistance continues to be a global issue due to the abuse and misuse of current antibiotics. While horizontal transfer of genes encoding resistance to specific antibiotics is a major factor in the spread of resistance, the efflux pumps that export both antibiotics and other compounds that accumulate in the cell under normal growth conditions play a significant role in allowing bacteria to survive antibiotic treatment even in the absence of specific resistance mechanisms. These pumps play an important role in survival when bacteria encounter toxic substances, and this is easily demonstrated by the increased sensitivity of bacterial mutants lacking efflux pump genes (1).
Multidrug efflux pumps in Escherichia coli and related bacteria consist of an outer membrane protein, TolC, an inner membrane protein, and a periplasmic fusion protein to link and stabilize protein complexes in the inner and outer membranes. The major efflux pump in E. coli, the AcrAB-TolC system, belongs to the resistance-nodulation-cell division (RND) superfamily of efflux pumps, and these complexes are abundant in the cell (2–4). AcrAB-TolC has a broad spectrum of substrates that include high- and low-molecular-weight drugs, chemotherapeutic compounds, most lipophilic antibiotics, cationic dyes, detergents, and bile acids (3, 5, 6). The AcrAB-TolC efflux pump serves as a housekeeping pump in Gram-negative bacteria, such as E. coli, which are more resistant to chemicals and antibiotics than Gram-positive bacteria (7). TolC is also a component of eight additional efflux transport systems (7, 8). The acrAB genes are encoded within an operon; tolC is located separately on the E. coli chromosome and is part of an operon that also includes ygiB and ygiC (9). The function of ygiBC-encoded proteins is unknown, but at least one phenotype of a tolC mutant, impaired growth on glucose minimal medium, was exacerbated by the deletion of ygiBC and their homologs, yjfM and yjfC (9, 10). Both the acrAB and tolC promoters are members of the marA, soxS, and rob transcriptional regulon and are upregulated during superoxide stress and exposure to heavy metals, bile salts, and antibiotics (11, 12), consistent with the role of the efflux pump in exporting these molecules from the cell.
In this study, the role of small RNAs in the regulation of the AcrAB-TolC drug efflux pump was examined. Small regulatory RNAs (sRNAs) are known to regulate genes at the posttranscriptional level (13). The expression of sRNAs is tightly controlled at the transcriptional level and is usually regulated in response to a stress signal sensed by the cell (13). In E. coli, many sRNAs have been identified that posttranscriptionally regulate target genes by direct base pairing to the 5′ untranslated region; these sRNAs frequently play a role in the modulation of translation and mRNA stability (14, 15). Regulation by sRNAs is dependent on the protein chaperone Hfq, which stabilizes the sRNA and aids in the binding to target mRNAs (16). Hfq is related to the Sm and Sm-like proteins in eukaryotes associated with mRNA splicing (17). Hfq-dependent sRNAs can enhance the translation of mRNA targets, usually by binding target mRNA transcripts, preventing the formation of hairpin structures that inhibit translation. A classic example is the positive regulation of rpoS, encoding the stationary-phase sigma factor. The sRNAs DsrA, RprA, and ArcZ bind to the mRNA of rpoS and open up the inhibitory hairpin structure (13, 18, 19). sRNAs can negatively regulate by repressing translation by binding to target mRNAs, blocking translation, and/or signaling for RNase E degradation. Most of the Hfq-dependent sRNAs are trans-acting (are encoded far from their target genes) and negatively regulate targets (14, 15). In particular, many sRNAs have been reported to negatively regulate the translation of outer membrane proteins (OMPs), and these OMPs are upregulated in Gram-negative pathogens lacking Hfq (20–25). The global regulator Hfq is known to contribute to virulence in pathogenic bacteria and has been associated with intrinsic drug resistance (8, 21, 26).
In E. coli, hfq mutants have increased drug susceptibility and increased accumulation of various compounds that are substrates for the AcrAB-TolC efflux pump; no effect on either the acrAB or tolC promoters was found (8). In addition, phenotypic data from a chemical biology screen of all E. coli mutants showed a high correlation in the phenotypes of hfq mutants with those of the acrA, acrB, and tolC drug efflux mutants (27). These results suggest that sRNAs might be important regulators of drug resistance in E. coli (8, 27), possibly regulating components of the efflux pump. Because hfq mutants are more sensitive to various drugs, our initial model was that positive sRNA regulators of the efflux pump would play a role in promoting drug resistance and might thus serve as potential targets for the treatment of Gram-negative bacterial infections. In contrast, sRNAs that negatively regulate the pump would be expected to reduce efflux and thus enhance the effects of antibiotic treatment.
The purpose of this study was to examine the posttranscriptional regulation of acrAB-tolC by sRNAs. We found a strong negative regulation of tolC, the gene encoding the common component of E. coli efflux pumps, by a stationary-phase-expressed sRNA, RyeB, named SdsR in Salmonella to reflect its expression during stationary phase (28). Here, we refer to this sRNA as SdsR. SdsR is one of the most abundant and highly conserved sRNAs and accumulates to high levels during stationary phase in enterobacteria (28). Although the exact functions of SdsR remain unclear, previous studies have suggested it has a role in modulating susceptibility to quinolone antibiotics. The overexpression of SdsR reduced resistance to quinolone antibiotics, such as levofloxacin, nalidixic acid, and norfloxacin, in multiple-drug-resistant strains of E. coli and Salmonella (29). In addition, SdsR was reported to contribute to an increase in the mutagenesis rate in E. coli cells treated with subinhibitory levels of ampicillin, presumably via its interaction with mutS (30). In our study, we reveal phenotypes with additional antibiotics and report findings associated with reduced biofilm formation in E. coli.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are derivatives of wild-type (WT) MG1655 and are listed in Table S1 in the supplemental material. The construction of strains is described in detail below and in Table S1 in the supplemental material. P1 transduction (31) was used to move the markers between strains. The primers and probes are listed in Table S2 in the supplemental material. Plasmids are also listed in Table S1 in the supplemental material and were introduced into strains using transformation and storage solution (TSS) transformation (32). The sRNA-expressing plasmids were derivatives of the pBR-plac vector described by Guillier and Gottesman (33).
Construction of translational fusions.
The lacZ translational reporter fusions were constructed using lambda Red recombineering into strain PM1205, as described previously (34). In these fusions, transcription is under the control of the PBAD promoter; the fusion contains the untranslated region (UTR) and the first part of the open reading frame of the gene of interest, in frame with lacZ (34). PCR primers (ASP1 and ASP2, ASP3 and ASP4, ASP5 and ASP6, and ASP7 and ASP6) (see Table S2 in the supplemental material), each carrying flanking homology to the PBAD promoter or to lacZ, were used to amplify the respective 5′ UTR using MG1655 genomic DNA from the +1 position of transcription (as annotated in EcoCyc [9]) through the 11th codon. The PCR fragments were purified using a MinElute PCR purification kit (Qiagen) and further analyzed for band sizes using DNA gel electrophoresis. For the tolC* fusion, 3-way PCR was used to introduce mutations in the 5′ UTR. The primers tolCp1.Mut4NT.UTR For and ASP6 were used in one reaction, and ASP5 and tolCp1.Mut4NT.UTR Rev were used in a separate reaction to amplify MG1655 genomic DNA. The PCR fragments were mixed in a 1:1 ratio, and the full-length fragment was generated using primers ASP5 and ASP6. The PCR products obtained after a 1- or 3-step PCR protocol (ABI) were recombined into PM1205, carrying a counterselectable sacB gene and cat gene between PBAD and lac; sucrose-resistant colonies were selected and screened for loss of cat (chloramphenicol resistance). All fusions were confirmed by DNA sequencing (ABI) using the lacIq.seq forward and DEEPlac reverse primers (Applied Biosystems BigDye Terminator version 1.1/3.1 cycle sequencing kit). The resulting translational fusions are annotated ASP6000 (acrA-lacZ), ASP6003 (tolCp3-lacZ), ASP6004 (tolCp1-lacZ), ASP6005 (acrB-lacZ), and ASP6018 (tolCp1.Mut4.0-lacZ).
Site-directed mutagenesis.
The regions in the SdsR sRNA nucleotide sequence that were predicted to pair with tolC were modified using the QuikChange Lightning site-directed mutagenesis kit (Stratagene) with pSdsR.Mut4 For forward and pSdsR.Mut4 Rev reverse primers that contained the nucleotide changes of interest (see Table S2 in the supplemental material).
Construction of specificity mutants in the chromosome.
Unmarked specificity mutants for tolC (tolC*) and sdsR (sdsR*), as used in the translational fusion and SdsR plasmid (see Table S1 in the supplemental material), were constructed in the chromosome in two steps, each in strains expressing the lambda recombination functions (35). In the first step, the native gene was replaced with cat-sacB, encoding resistance to chloramphenicol (cat) and sensitivity to sucrose (sacB) by selecting for chloramphenicol resistance. In the second step, a PCR fragment containing the desired mutations was used to replace cat-sacB, selecting for resistance to sucrose and screening for a loss of chloramphenicol resistance (34). The cat-sacB gene fusion was amplified from NC397 genomic DNA using the following primers with complementarity to the chromosomal site: forward cat-sacB.tolC.UTR and reverse cat-sacB.tolC primers for the tolC insertion and forward cat-sacB.sdsR and reverse cat-sacB.sdsR primers for the sdsR gene. The PCR fragments were purified using a MinElute PCR purification kit (Qiagen) and further analyzed for band sizes using DNA gel electrophoresis. The PCR fragments were combined into the chromosome of the parental strain E. coli MG1655 containing mini-λ::tet (NM1100) using the lambda Red recombinase protocol, as described previously, saving derivatives that also retained the mini-λ::tet (34); the resulting tolC::cat-sacB strain was named ASP7008A, and the sdsR::cat-sacB strain was named ASP7009. The forward tolC.Mut4.0 oligonucleotide primer was then used to replace cat-sacB in ASP7008A with tolC* to create ASP7010C (34). The insertion in ASP7009 was replaced with the sdsR.Mut4.0 forward primer to create strain ASP7011D (sdsR*) containing point mutations in the chromosomal copy of sdsR. The strains containing the specific mutations were confirmed by DNA sequencing.
β-Galactosidase assays.
Strains containing fusions were grown under conditions appropriate for expression of the PBAD promoter (arabinose concentrations as noted in figure legends), and β-galactosidase was assayed either in a microtiter plate (microtiter assay) or in tubes (Miller assay).
For the microtiter assays, a single colony was selected and grown for 5 to 6 h or until late stationary phase at 37°C in 5 ml of LB broth containing 100 μg/ml ampicillin, 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG), and 0.01 to 0.0001% arabinose, depending on the fusion. One hundred microliters of cells was dispensed in wells of a 96-well microtiter plate containing 50 μl of permeabilization buffer (100 mM Tris, 32 mM Na2HPO4·7H2O, 8 mM dithiothreitol [DTT], 8 mM cyclohexane diamine tetraacetic acid [CDTA], 4% Triton 100), and the plate was incubated for 15 min at room temperature (36). Fifty microliters of o-nitrophenyl-β-d-galactopyranoside (ONPG) (4 mg/ml) was added to each well, and readings at 420 nm were immediately made using the SpectraMax microtiter plate reader and then every 15 s for 4 h. The SpectraMax averages the first 30 points in the linear region of the curve to determine the Vmax. The specific β-galactosidase activities were calculated and averaged, using the formula Vmax/optical density at 600 nm (OD600), for a minimum of three independent experiments, as described previously (37). The average specific activities were normalized to the plac control vector to determine the relative fold change. The machine units are about 25 times smaller than Miller units.
The tolC translational fusions were further analyzed for activity using multicopy plasmids overexpressing SdsR after growth in flasks. A single colony was selected and grown at 37°C in LB broth containing 100 μg/ml ampicillin, 100 μM IPTG, and 0.0001% arabinose to an OD600 of 1.6 to 2.5. Two hundred microliters of bacterial culture was permeabilized with 800 μl of Z buffer, 24 μl of 0.1% SDS, and 48 μl of chloroform, vortexed for 10 s, and immediately placed on ice. The samples were incubated for 5 min in a 28°C water bath and were assayed for β-galactosidase activity, as described by Miller (38).
Mfold predicted pairing.
Mfold was used to determine possible regions of base pairing between the sRNA SdsR and tolC mRNA. The RNA sequence of tolC containing the 5′ UTR and the first 10 codons after the ATG translational start was linked with 6 to 9 Ns, followed by the RNA sequence of SdsR. The inserted sequence was entered into the mfold Web server, using the default settings (39), and the folded RNA structure was inspected to identify base-pairing regions between SdsR and tolC.
RNA extraction and Northern blot analysis.
Single colonies of strains containing the sRNA-expressing plasmids were inoculated into LB broth containing ampicillin (100 μg/ml), and the cultures were grown at 37°C with shaking in a water bath until the OD600 reached 0.5. The plasmids were then induced with 100 μM IPTG to overexpress the sRNA, and RNA samples were collected immediately before induction (0 min) and after 2, 5, 10, and 15 min. Total RNA from bacterial cell cultures was isolated using hot phenol RNA extraction, as described previously (40). Northern blot analysis of tolC mRNA levels was performed by fractionating 15 μg of RNA from each sample, and 3 μg of RNA was fractionated to analyze the SdsR RNA levels, as described previously (41). Gels of 1.2% agarose in 1× morpholinepropanesulfonic acid (MOPS) and 10% Tris-borate-EDTA (TBE)–urea (Invitrogen) were used for electrophoresis and transferred to nylon membranes (Zeta-Probe GT). Probes for tolC, SdsR, or SsrA, each labeled with biotin at the 5′ end (see Table S2 in the supplemental material), were used for detection, as described previously (41). The RNA bands were quantified using the MultiGauge software program, represented as arbitrary units (AU); the tolC mRNA levels were normalized to the SsrA loading control and were further compared to the plac vector control at time zero for the respective strains.
Western blot analysis.
Single colonies were inoculated into LB only or LB containing 100 μg/ml ampicillin for plasmid-containing strains, and cultures were grown in a shaking water bath at 37°C. Protein samples were collected at an OD600 of 0.5 (time zero), 100 μM IPTG was added to the culture to induce the plasmids, and additional samples were taken at 1, 2, and 2.5 h after induction. One milliliter of culture was removed for each sample and centrifuged at 20,800 × g and 4°C for 10 min. The supernatant was discarded, and the pellets were dissolved in 1× SDS sample buffer with 1× DTT final concentration; the samples were kept on ice. The protein samples were denatured at 95°C for 10 min in a thermomixer shaking at maximum speed (14,000 rpm) until the pellets were completely dissolved. Protein samples (6 to 10 μl) were loaded and run on a NuPAGE 12% Bis-Tris precast gel using 1× MOPS SDS buffer for electrophoresis. The proteins were transferred to a nitrocellulose membrane using an iBlot transfer system (Invitrogen), and the membrane was blocked with 5% milk in phosphate-buffered saline (PBS) for 30 min at room temperature. The primary anti-TolC antibody (gift from R. Misra) was diluted 1:2,000 in 5% PBS–milk and incubated with the membrane for 1 h. The membrane was washed once for 15 min and twice for 5 min with 0.03% PBS–Tween, incubated for 1 h with alkaline phosphatase (AP)-linked anti-rabbit secondary antibody diluted 1:1,000 in PBS, and washed again with the same cycle in 0.03% PBS–Tween. The protein bands were detected by chemiluminescence using the Lumi-Phos substrate for 3 min and exposing the membrane to light using the LAS-4000 mini luminescent image analyzer (42). Protein bands were analyzed using the MultiGauge program; the intensity of the bands was measured in arbitrary units (AU). TolC protein bands were normalized to the EF-Tu control protein and further compared to the plac control at 0 h for the respective strains to determine the relative amounts of TolC.
Antibiotic sensitivity assay.
Individual colonies from strains streaked on LB-only or LB-ampicillin plates, as appropriate, were inoculated into 8 ml of LB alone or LB containing 100 μg/ml ampicillin for strains with plasmids. The cultures were grown in a shaking water bath at 37°C until stationary phase. The cultures were diluted 1:10,000 in 25 ml of LB alone or LB containing 100 μg/ml ampicillin with 100 μM IPTG. The cultures containing plasmids were grown under the same conditions for 1 h to induce the plasmids. One hundred microliters of cell culture was dispensed into a 96-well plate, except the first well, which received 200 μl of cells. Four microliters of each antibiotic was added to the first well containing 200 µl of cells to make a final antibiotic concentration of 200 μg/ml. Two-fold serial dilutions of the antibiotic were made along the columns of the plate using a multichannel pipette. The 96-well plates were sealed with Parafilm to prevent evaporation and were incubated overnight at 37°C in a shaking microtiter plate holder. The OD600 was measured as an endpoint using a SpectraMax microtiter plate reader. Cells containing no added antibiotic were used as the positive control, and LB–100 μg/ml ampicillin–100 μM IPTG was dispensed into a single column of each plate as a negative control. Antibiotic stock solutions at a concentration of 10 mg/ml were made fresh each day using the appropriate solvent and were filter sterilized. Crystal violet and novobiocin were dissolved in ultrapure water, rifampin (95% ethanol with 3 to 4 drops of 1 N NaOH), and erythromycin (50% ethanol).
Biofilm assay.
Single colonies were inoculated into 5 ml of LB only or LB containing 100 μg/ml ampicillin for strains containing plasmids, and cultures were grown overnight at 37°C. The OD600 of the overnight culture was measured, and the cells were diluted to an OD600 of 0.05 in YESCA growth medium (Casamino Acids and yeast extract) (43) or YESCA supplemented with 100 μM IPTG and 100 μg/μl ampicillin. Two hundred microliters of diluted cells was dispensed into 96-well microtiter plates; the plates were wrapped with Parafilm and incubated without shaking at 32°C for 48 h. The OD600 was measured using a SpectraMax microtiter plate reader, and the planktonic cells were removed from the plate using a multichannel pipette without disturbing the biofilm area. Two hundred microliters of deionized water was added to each well and then gently removed in two repeated pipetting steps. Two hundred twenty microliters of 0.1% crystal violet (CV) solution was dispensed and allowed to stain for 10 min. The CV staining solution was removed, and the wells were washed with deionized water in two repeated steps, as described above. Three hundred microliters of deionized water was added and removed for the final wash, and the plate was allowed to dry in a 37°C incubator for 20 min. Two hundred thirty microliters of 20% acetone–80% ethanol solution was dispensed into each well for 10 min, and the OD570 was measured using the SpectraMax plate reader. The biofilm formation was determined as the ratio of the OD570 to the OD600, as described previously (43).
Biolog assay.
E. coli strains from freezer stocks were streaked for isolated colonies on LB agar plates and grown overnight at 37°C. Cells were removed using sterile loops and inoculated in 16 ml of IF-0 until the cells were turbid, at an OD600 of approximately 0.3. The remaining cell suspension was used to prepare the appropriate 85% T cell suspension diluted in IF-0 or IF-10 with Biolog redox dye mixture A, as described previously (44). One hundred microliters of cells was dispensed into the Biolog microarray plates PM1 to PM20 to test the strains for metabolic activity (44). All experiments were performed in duplicate using separate cultures.
Plasmid extraction.
Plasmids carrying the sRNA of interest were extracted using a QIAprep spin miniprep kit (Qiagen), PCR amplified (ABI), and DNA sequenced (ABI) using the pBR-for and pBR-rev primers.
RESULTS
tolC is subject to sRNA regulation.
Translational fusions of acrA, acrB, and tolC to lacZ were constructed using lambda Red recombination (35). The fusions included the 5′ UTR and first 11 codons of each gene, fused to lacZ, downstream from an arabinose-inducible promoter (Fig. 1A; see also Fig. S1B and C in the supplemental material). The acrA-lacZ and acrB-lacZ translational fusions were tested for activity on MacConkey lactose agar plates (see Fig. S2A in the supplemental material). The acrA-lacZ fusion was very active in the absence of arabinose and formed red colonies on the MacConkey lactose agar plates (see Fig. S2A in the supplemental material). Therefore, acrA may have a strong Shine-Dalgarno sequence that increases the efficiency of ribosomal loading and translation or an unmapped active promoter within the leader. The basal level of the acrB-lacZ fusion, expressed from the start site of a promoter within acrA (see Fig. S1A in the supplemental material), was extremely low on MacConkey lactose compared to those of the acrA and tolC fusions (see Fig. S2A in the supplemental material), possibly suggesting that ribosome loading directly on the acrB translation initiation region is not efficient and that the bicistronic acrAB operon is the source for most AcrB translation, or that this fusion leads to unstable mRNA. The number of copies per cell of AcrB is approximately 10- to 14-fold less than that of AcrA during the log phase of growth (4), further supporting the observation of significantly lower translation activity.
FIG 1.

Screen of sRNA library on the tolC-lacZ reporter fusion. (A) Schematic indicating the promoters and construction of the PBAD tolC-lacZ translational fusion (ASP6004). tolC promoters (P1 and regions of P2) were replaced with an arabinose-inducible promoter, with internal promoters (P3 and P4) remaining intact. The resulting tolC 5′ untranslated region (UTR) of 111 nucleotides in length with 11 codons was fused with lacZ. (B) The sRNA plasmid library was transformed into the tolC-lacZ reporter strain (ASP6004). Bacterial cultures were grown in LB broth containing 100 μg/ml ampicillin, 100 μM IPTG, and 0.0001% arabinose and assayed for β-galactosidase activity using the microtiter assay method. The relative β-galactosidase activity as fold change was determined by comparing the 27 individual sRNAs to the plac empty vector (β-galactosidase-specific activity, 4.2 machine units). The experiment was performed in triplicate. Error bars represent the standard deviations.
In E. coli, tolC is reported to have four promoters (45, 46). Translational fusions were designed to begin at the start sites for the first (Fig. 1A) and third promoters; P2 is 8 nucleotides downstream of P1, and P4 is 12 nucleotides downstream of P3. The leader from P1 is 111 nucleotides, and the leader of the second fusion (P3) is 53 nucleotides in length (45, 46). Similar to the acrA-lacZ fusion, the tolC-lacZ fusions starting either at the P1 or P3 start site were very active in the absence of arabinose on the MacConkey lactose agar plates (see Fig. S2A in the supplemental material). There are known internal promoters within the 5′ UTR of the longer tolC P1-lacZ fusion (Fig. 1A). However, the basal level of the shorter tolC P3-lacZ fusion was still significant without the induction of arabinose, suggesting that nothing in the longer leader (for instance, the downstream promoters) is required for significant translation activity (see Fig. S2A in the supplemental material). TolC is an abundant protein with 1,500 copies per cell (4) and thus might be expected to be translated very efficiently. In addition, the −10 region of the fourth promoter is present in the P3 fusion and may contribute to expression of the fusion.
The sRNA plasmid library containing 27 individually cloned sRNAs was transformed into the acrA-lacZ, acrB-lacZ, and tolC-lacZ reporter fusion strains, and these transformants were tested for sRNA regulation using the 96-well microtiter β-galactosidase assay. The activity of each fusion containing one member of the sRNA library was measured and compared to the plac empty vector to determine the relative activity; a change of ≥2-fold was considered significant. For the acrA-lacZ fusion, only DicF showed >2-fold increased activity (see Fig. S1B in the supplemental material), but the OD600 of the culture was considerably lower than that for the other plasmids. The overexpression of pGcvB caused a 1.9-fold increase in acrB-lacZ activity (see Fig. S1C in the supplemental material). However, the β-galactosidase-specific activity of the acrB-lacZ reporter was extremely low (10-fold lower than that of the acrA fusion, even with higher concentrations of arabinose). The effect of GcvB was not studied further, given the very poor translation of this fusion.
Multicopy SdsR repressed the tolC P1-lacZ translational fusion >3-fold (Fig. 1B) and showed similar effects on the tolC-lacZ (P3) fusion (see Fig. S2B in the supplemental material). Other effects on tolC, such as repression by ArcZ, fell below the 2-fold cutoff for significance (Fig. 1). The overexpression of pOxyS and pRybB increased the expression of tolC-lacZ, but the effects were not ≥2-fold (Fig. 1), although the high basal level of expression of this fusion may have made the detection of stimulatory effects difficult. The negative regulation of tolC by the sRNA SdsR was investigated further.
Direct pairing of SdsR to tolC.
The mfold computational program (39) predicted a region of 8 nucleotides of potential pairing between SdsR and tolC. The predicted pairing region within SdsR began at nucleotide 33 (Fig. 2A). In Salmonella, SdsR was found to repress an outer membrane protein, OmpD, and the base-pairing region with ompD is close to but separate from that predicted here for tolC (Fig. 2A) (28). The pairing region with tolC was in the 5′ UTR, located 26 to 33 nucleotides upstream from the translational start (Fig. 2A).
FIG 2.
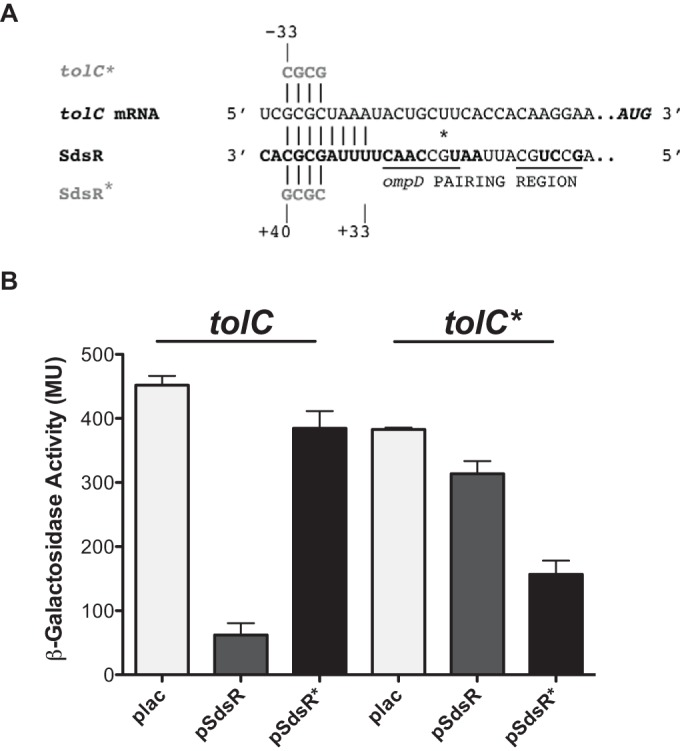
Predicted pairing and regulation of tolC with multicopy SdsR. (A) The pairing regions of tolC and SdsR are shown. Mutations in the predicted base-pairing regions were designed in SdsR (SdsR*) and the compensatory mutations in the tolC fusion (tolC*), as described in Materials and Methods. The tolC pairing region is 33 nucleotides upstream of the translational start site, and the nucleotides in SdsR in bold are highly conserved (28). Pairing in SdsR begins at position +33 downstream from the transcriptional start site and extends to position +40. The underlined region shows the full region of pairing with ompD, and the asterisk indicates the position of SdsR that was mutated in the previous work (28). (B) The plasmids plac (vector control), pSdsR, and pSdsR.Mut4.0 (SdsR*) were transformed into the WT tolC-lacZ fusion (ASP6007) and compensatory mutant tolC* (ASP6026). These strains were grown at 37°C in LB broth containing 100 μg/ml ampicillin, 100 μM IPTG, and 0.0001% arabinose to an OD600 of 1.6 to 2.5 and assayed for β-galactosidase activity using the Miller assay (38). The experiment was performed in triplicate. Error bars represent the standard deviations.
The importance of the predicted pairing for the regulation of tolC translation was tested using point mutations in the region of pairing between tolC and SdsR, which were designed to abrogate regulation (tolC*), and compensatory mutations in SdsR designed to restore regulation (SdsR*) (Fig. 2A). Four mutants in the tolC fusion (see Fig. S3A in the supplemental material) were constructed, and their basal levels of activity were compared to the activity of the WT tolC-lacZ fusion by streaking on MacConkey lactose agar plates containing 0.0005% arabinose (see Fig. S3B in the supplemental material). White or pink colonies formed after streaking tolC-lacZ mutants 4.1, 4.2, and 8.0 on the indicator plates, suggesting distinctively lower activity for these mutants than that of the WT fusion. The appearance and basal level of activity of the tolCp1.Mut4.0-lacZ mutant were similar to those of the WT fusion (see Fig. S3 in the supplemental material) and were therefore used for additional studies.
The overexpression of the WT SdsR from a plasmid in the WT-tolC fusion strain with the chromosomal sdsR deletion significantly reduced the β-galactosidase expression level compared to that of the Plac vector control, resulting in >7-fold repression, thus confirming the results seen with the sRNA library (Fig. 2B). This repression was lost with the mutant SdsR plasmid (SdsR*) that contained four mutations in the region that was predicted to pair with the 5′ UTR of tolC mRNA (Fig. 2). Therefore, this specific region in the sRNA was important in the pairing and regulation of tolC. To confirm that this specific region was required for regulation by direct base pairing, the tolC* compensatory mutations were introduced into the tolC-lacZ fusion and measured for β-galactosidase activity. In the tolC* fusion, there was no significant difference in activity between the plac vector control and the pSdsR+ plasmid, suggesting that regulation by SdsR was disrupted by the tolC* mutation (Fig. 2B). However, overexpression of the pSdsR* mutant plasmid in this strain led to a 2.4-fold reduction in tolC* activity, suggesting that the restoration of this predicted base pairing is sufficient to restore negative regulation (Fig. 2). While the fold change in the repression of tolC* was not as great as that observed in the WT tolC reporter, these data fully support the hypothesis that SdsR negatively regulates tolC posttranscriptional expression by direct base pairing.
SdsR negatively regulates tolC mRNA and TolC protein.
Given the confirmation of specific base-pairing regions of SdsR with tolC, the next question was whether this regulation leads to significant changes in the native tolC RNA and protein levels when SdsR is expressed from a multicopy plasmid. Expression of the WT SdsR plasmid and the mutant SdsR (SdsR*) was tested in both a WT strain and a strain carrying the compensatory tolC* mutation in the 5′ UTR of the native tolC gene (see Fig. 2A for mutations).
The overexpression of the WT SdsR significantly decreased the tolC mRNA containing the WT leader sequence, confirming the negative regulation observed in the tolC-lacZ fusion (Fig. 3A and B). The tolC mRNA levels were low even before the induction with IPTG (time zero) for both WT and mutant SdsR plasmids, suggesting that the WT plasmids are leaky although they have lacIq in the chromosome (Fig. 3A and B). WT SdsR did not decrease the level of the tolC* mRNA (Fig. 3C and D), confirming that the tolC* mutation is resistant to regulation by SdsR+.
FIG 3.

Expression of tolC RNA with induction of multicopy SdsR. Representative Northern blots of tolC mRNA in the presence of the WT and mutant SdsR plasmids are shown. (A) The strains bearing WT plac (ASP5056), WT pSdsR (ASP5057), and WT pSdsR* (ASP5058) were grown in LB broth containing ampicillin at 37°C and induced with IPTG, and samples were taken, as described in Materials and Methods. The sRNA was detected using the SdsR probe (top), tolC mRNA was detected using the tolC probe (middle), and SsrA, a highly abundant RNA, was used as an internal control, using the SsrA probe (bottom). (B) The tolC RNA bands were quantified using the MultiGauge program, represented in arbitrary units (AU); the tolC mRNA levels were normalized relative to time zero for the WT plac empty vector (ASP5056), which was set to 1. The quantification is based on experiments performed in duplicate. (C) The strains bearing tolC* plac (ASP5062), tolC* pSdsR (ASP5063), and tolC* pSdsR* (ASP5064) were assayed as described for panel A. (D) The tolC RNA bands were quantified using the MultiGauge program, represented in arbitrary units (AU); the tolC mRNA levels were normalized relative to time zero for the TolC* plac empty vector (ASP5062). The Northern analysis was quantified from duplicate experiments. Error bars represent the standard deviations.
The SdsR* plasmid was not able to significantly reduce levels of WT tolC (Fig. 3A and B), although for reasons that are not currently understood, the 0′ point was low in these strains. In both the vector control and the SdsR* strain, tolC mRNA levels increased with time (Fig. 3B). The overexpression of the pSdsR* plasmid in the tolC* strain, in which SdsR-tolC pairing and regulation are restored (Fig. 2B), decreased the level of tolC* mRNA significantly below that of the vector control (Fig. 3C and D), confirming that the effects on the tolC mRNA are due to direct pairing. This observation confirms that the identified pairing region of SdsR is required for full repression of the mRNA.
The TolC protein was also analyzed after expression of multicopy SdsR in the same strains shown in Fig. 3. However, induction was followed over 2.5 h to allow better detection of the changes in the stable and abundant TolC protein. As for the mRNA, a decrease in TolC protein was seen at time zero (before the induction with IPTG) in cells expressing either SdsR+ or SdsR* (Fig. 4A and B). TolC levels were lowest in the presence of the multicopy SdsR; repression was significantly less for SdsR* repression of TolC (Fig. 4A and B). However, we note that SdsR* is able to reduce TolC levels below those of the vector control.
FIG 4.

TolC protein levels in the presence of multicopy SdsR. Shown are representative Western blots of TolC with the overexpression of WT and mutant SdsR plasmids. (A) Protein samples were collected from strains bearing WT plac (ASP5056), WT pSdsR (ASP5057), and WT pSdsR*(ASP5058) at an OD600 of 0.5 (time zero), and additional samples were taken at 1, 2, and 2.5 h after induction with IPTG. (B) TolC protein bands were analyzed using the MultiGauge program; the intensity of the bands was measured in arbitrary units (AU). TolC protein bands were normalized to the control protein EF-Tu and further compared to the WT plac control at 0 h (which was set at 1) to determine the relative amounts of TolC. The quantification is based on experiments performed in duplicate. Error bars represent the standard deviations. (C) Strains ASP5062 (TolC* plac), ASP5063 (TolC* pSdsR), and ASP5064 (TolC* pSdsR*) were assayed as described for panel A. (D) TolC protein levels were first normalized to the loading control EF-Tu, and the relative amount of TolC was calculated, dividing each TolC value by the TolC* plac value before the induction with IPTG (time zero). TolC was detected using the anti-TolC primary antibody, and EF-Tu was used as an internal control probed with anti-EF-Tu antibody. Duplicate Western blotting experiments were used to quantitate TolC protein levels; however, the experiment was repeated more than three times, with consistent patterns of protein expression. Error bars represent the standard deviations.
The overexpression of either the WT SdsR plasmid or the SdsR* plasmid in strains carrying the tolC* chromosomal mutation led to lower levels of TolC than those of the plac control (Fig. 4C and D). While the reduction by the compensating SdsR* plasmid was somewhat more effective in reducing TolC protein levels, the reduction by SdsR was unexpected, given the results with both the fusion tests (Fig. 2) and mRNA (Fig. 3).
Two explanations are possible for the apparent residual regulation of tolC by SdsR* and of tolC* by SdsR, as seen in Fig. 4. Based on the tests with the fusions (Fig. 2), there is some small remaining ability of SdsR to regulate tolC* and SdsR* to regulate tolC, which might explain the effects on TolC protein levels when measured over many hours. However, we favor the idea that this regulation is in fact different from that seen in Fig. 2. Figure 4 shows measurements of the protein level from its native promoter, and we suggest that some of the additional targets of SdsR may act at the level of the tolC promoter (not present in the fusion). Over a number of hours, this may lead to the repression of TolC, independently of pairing.
The tested strains shown in Fig. 4 carried a WT copy of SdsR in the chromosome. A modest decrease in TolC levels after 2.5 h was observed for the plac vector controls in the tolC+ strain but not in the tolC* strain (Fig. 4B and D, plac). This may suggest that the decrease is dependent on the sequences within the 5′ UTR mutated in tolC* and is possibly due to repression by chromosomally encoded SdsR. We further investigated TolC protein levels by deleting sdsR and examined the expression of tolC from the chromosome. SdsR is highly expressed during stationary phase, and the expression is known to be RpoS dependent (28). This observation was confirmed in our experiments. SdsR was detectable only in stationary phase at an OD600 of ≥1.6 (see Fig. S4A in the supplemental material). TolC was detected using Western blot analysis. No significant differences in TolC protein levels between the WT and the sdsR mutant were detected (see Fig. S4B and C in the supplemental material), and the decrease in TolC in later stationary phase noted in Fig. 4B was not seen in these experiments. TolC levels were maintained in MG1655 along the growth curve (see Fig. S4C in the supplemental material). Therefore, the endogenous SdsR does not have a significant effect on TolC protein levels under these growth conditions.
SdsR repression of TolC leads to changes in antibiotic sensitivity.
One major role of TolC is in the export of antibiotics from the cell; tolC deletion mutants are significantly more sensitive to a broad set of antibiotics than are strains without the deletion (47). We tested the effects of both multicopy SdsR and the deletion of sdsR on sensitivity to three different antibiotics and one cationic dye. As for the experiments shown in Fig. 3 and 4, we also examined any observed changes in drug sensitivity for the requirement for pairing with tolC by using specificity mutations in pSdsR (pSdsR*) and tolC (tolC*), with the expectation that changes in drug sensitivity due to SdsR-mediated regulation of tolC should be seen only when pairing was present.
No significant differences in antibiotic sensitivity were observed in strains containing the tolC* mutations in the 5′ leader of tolC or with a deletion in sdsR; the strain containing a deletion of tolC was hypersensitive to the antibiotics, as expected (see Fig. S5 in the supplemental material). This is consistent with the lack of observable changes in TolC protein levels in the ΔsdsR strain (see Fig. S4 in the supplemental material).
The overexpression of SdsR or SdsR* had only mild effects on sensitivity to erythromycin and rifampin (Fig. 5C and D) and somewhat stronger effects on sensitivity to crystal violet and novobiocin (Fig. 5A and B). Strikingly, the effects for novobiocin, erythromycin, and rifampin correlate well with effects seen on the TolC protein (Fig. 4). tolC+ cells expressing high levels of SdsR were more drug sensitive, while high levels of SdsR* had little or no effect on sensitivity in tolC+ strains (Fig. 5B to D, left graphs). This is consistent with the decrease in TolC upon overexpression of SdsR but not SdsR* (Fig. 4B). However, in the tolC* host, both the SdsR+ and SdsR* plasmids increased the sensitivity and led to lower levels of the TolC protein (Fig. 4D). This pattern is not fully consistent with a simple model in which SdsR acts on drug sensitivity only by pairing with the tolC leader, and this is discussed further below.
FIG 5.

Antibiotic sensitivity using multicopy SdsR. Strains ASP5074 (WT plac), ASP5075 (WT pSdsR), ASP5076 (WT pSdsR*), ASP5077 (TolC* plac), ASP5078 (TolC* pSdsR), and ASP5079 (TolC* pSdsR*) were assayed for antibiotic sensitivity to 2-fold serially diluted crystal violet (A), novobiocin (B), rifampin (C), and erythromycin (D), as described in Materials and Methods. The average cell density measured by OD600 was determined using a SpectraMax 96-well plate reader. The experiment was performed in triplicate. Error bars represent the standard deviations.
In the tolC+ strain, both pSdsR and pSdsR* overexpression increased susceptibility to crystal violet 2- to 3-fold compared to that of the plac vector control (Fig. 5A). In the tolC* cells, only the SdsR* plasmid significantly increased sensitivity to crystal violet (Fig. 5A, right graph). This pattern is generally consistent with increased crystal violet sensitivity due to SdsR-mediated regulation of tolC. In addition, the results suggest that SdsR* may have additional targets contributing to sensitivity, as seen by the sensitivity in the tolC+ strain (Fig. 5A, left graph). We note that pSdsR* did decrease TolC protein levels (Fig. 4A and B), possibly suggesting that the other target(s) affects the synthesis of TolC or that residual direct regulation of tolC+ by SdsR* is sufficient to explain this.
SdsR and TolC are both involved in biofilm production.
The results described above and those of other studies suggest targets other than tolC for SdsR. Thus, experiments to examine other possible roles of SdsR were carried out and analyzed for dependence on tolC pairing. Native sdsR is expressed in response to the alternative sigma factor RpoS. Therefore, it seemed possible that some phenotypes known to be dependent upon RpoS would also depend upon regulation by SdsR.
One interesting phenotype of RpoS is its role in biofilm formation. rpoS deletion mutants assayed in YESCA medium had reproducibly smaller amounts of biofilm than the MG1655 (WT) control, with a decrease of >2-fold after 48 h of incubation (Fig. 6A). However, the effect of rpoS mutants on biofilm formation is independent of sdsR expression and pairing with tolC, since no changes in biofilm levels were observed in sdsR deletion mutants, the tolC* mutant strain, or strains carrying the sdsR* allele in place of the chromosomal sdsR gene (Fig. 6A). However, the deletion of tolC led to a decrease in biofilm through an unknown mechanism (Fig. 6A).
FIG 6.

Biofilm formation of chromosomal and multicopy-SdsR mutants. (A) Chromosomal deletion and specificity mutants (WT MG1655, ASP7003 [Δrpos], ASP7005 [ΔsdsR], ASP6028 [ΔtolC], ASP7011D [sdsR*], and ASP7010C [tolC*]) were assayed for biofilm formation using YESCA medium, as described in Materials and Methods. (B) Strains bearing WT tolC plac (ASP5056), WT tolC pSdsR (ASP5057), WT tolC pSdsR* (ASP5058), tolC* plac (ASP5062), tolC* pSdsR (ASP5063), and tolC* pSdsR* (ASP5064) were assayed for biofilm formation in YESCA medium supplemented with 100 μM IPTG and 100 μg/ml ampicillin. The experiments were performed in triplicate. Error bars represent the standard deviations.
WT strains and TolC specificity mutants containing multicopy SdsR plasmids were also assayed for biofilm formation in YESCA medium. The induction of pSdsR reduced biofilm formation >2-fold in the WT (tolC+) strain and >3-fold in the tolC* strain (Fig. 6B); the induction of pSdsR* had no significant effect on biofilm production. The phenotypes observed suggest that SdsR represses biofilm formation independently of pairing to tolC, but this repression is dependent on pairing with another target or targets using the region of SdsR mutated in SdsR*.
SdsR deletion mutants have no significant metabolic phenotypes.
The Biolog phenotypic microarray allows bacterial strains to be tested for nearly 2,000 phenotypes, reported by color changes in microtiter plates. The assays include the use of multiple carbon sources, nitrogen sources, other nutrients, osmolytes, and sensitivity to a variety of antibiotics (44). A set of 20 Biolog plates was used to compare the sdsR deletion mutant and the tolC* strain to the WT parent. The expectation was that if a phenotype shown by an ΔsdsR strain was due to a loss of tolC repression, the tolC* mutant would show a similar phenotype.
The strongest defect in growth in the ΔsdsR deletion strain was seen in the presence of manganese chloride (see Fig. S6A in the supplemental material, boxed on left side), which was not seen in the tolC* strain (see Fig. S6A in the supplemental material, boxed on right side). Poor growth with the addition of manganese was reproducible using growth assays and spot titer assays on LB agar plates containing manganese (data not shown). However, after transfer of the ΔsdsR mutation to a different strain background by P1 transduction, only 50% of the transductants retained the Mn sensitivity, suggesting this was due to a linked mutation. The mntP gene, encoding a manganese efflux protein and known to lead to Mn sensitivity when mutated (48), was nearby, and sequence analysis of the ΔsdsR strain revealed a missense mutation (guanine to adenine) that resulted in a glycine (GGT)-to-aspartic acid (GAT) amino acid substitution at the 25th codon of mntP. This mutation was present in PM1205 (34), the parental strain used for constructing the original ΔsdsR strain (NRD750); it is derived from DJ480 (49), so all derivatives of that may contain this mntP mutation. Our WT MG1655 strain was also tested and does not contain mutations in mntP.
The most significant growth advantage for the ΔsdsR mutant was with the antibiotics phleomycin and bleomycin (see Fig. S6B and C in the supplemental material, left side), which was mildly observed in the tolC* strain (see Fig. S6B and C in the supplemental material, right side). The ΔsdsR strain contains a zeocin antibiotic resistance cassette, providing resistance to this family of antibiotics.
Therefore, once these two phenotype changes were set aside as being unrelated to SdsR itself, the Biolog phenotypic microarray did not detect any significant growth phenotypes of a deletion of sdsR.
DISCUSSION
Bacteria have developed various mechanisms to survive harsh environments. One major strategy employed under stressful conditions is the use of efflux pumps, an important drug resistance mechanism. The major efflux pump in E. coli is AcrAB-TolC. Although the architecture, assembly, and function of AcrAB-TolC are well known (50, 51), and the transcriptional control of the genes encoding AcrAB and TolC have been examined (46, 52–56), very little has been done to study posttranscriptional regulation. We investigated the role of Hfq-dependent sRNAs in the regulation of genes encoding the efflux pump. SdsR (RyeB), an Hfq-dependent sRNA synthesized in stationary phase, was found to downregulate tolC by base pairing with its 5′ UTR, 33 nucleotides upstream of the ribosomal binding site (Fig. 2A). The overexpression of SdsR led to a loss of tolC mRNA, a decrease in TolC protein, and increased sensitivity to some antibiotics. Other phenotypes of SdsR are independent of tolC, suggesting the existence of additional targets for this sRNA (Fig. 6B).
Upstream pairing at the site of a translational enhancer.
SdsR pairs with the tolC 5′ UTR at a conserved sequence upstream of the translation initiation site (−33 to −26 relative to the ATG). This region of the 5′ UTR appears to act as a translational enhancer, since at least some mutations in this region drastically reduced the expression of a downstream tolC-lacZ fusion (see Fig. S3 in the supplemental material). The nature of this enhancer has not been determined, but it seems likely that SdsR pairing at this site decreases tolC translation and/or stability, in part by blocking enhancer function. The sRNA GcvB has also been found to pair with translational enhancers, inhibiting translation (57, 58). ArcZ negatively regulates flhDC expression by direct base pairing, upstream from the ribosomal binding site (−47 to −64), although the mechanism of repression has not been investigated (59). Furthermore, the existence of this strong translational enhancer within the tolC 5′ UTR suggests the possibility of other sRNA-independent regulation of TolC translation, and this would be consistent with significant conservation within the tolC leader.
Roles for SdsR in TolC-dependent and TolC-independent phenotypes.
The overexpression of SdsR repressed tolC reporter activity more than ∼9-fold (Fig. 2) and led to a loss of tolC mRNA (Fig. 3) and a reduction of the TolC protein (Fig. 4). TolC was also identified as one of a large number of targets of SdsR in Salmonella, using microarray analysis (K. S. Fröhlich, K. Haneke, K. Papenfort, and J. Vogel, personal communication). In another study published while the current work was being prepared for publication, the same plasmid library as that used here was screened for effects on sensitivity or resistance to a panel of antibiotics, and the overexpression of SdsR was found to increase sensitivity to multiple antibiotics, in particular, quinolones (29). Based on that observation and target prediction, the authors predicted tolC as a possible target, and a decrease in tolC mRNA was confirmed by reverse transcription-quantitative PCR (qRT-PCR) analysis after overexpression of the sRNA (29).
We also observed increased sensitivity to a number of antibiotics when SdsR was overexpressed (Fig. 5). We made use of mutations in the tolC 5′ UTR that specifically abrogate tolC regulation by SdsR (tolC*) to allow us to distinguish SdsR effects on other targets from those wholly dependent upon pairing with tolC. Thus, the overexpression of SdsR increased sensitivity to crystal violet (Fig. 5A), and the increased sensitivity was seen only when pairing between tolC and SdsR was preserved. Increased sensitivity to novobiocin appeared to depend upon either pairing with tolC (shown by SdsR* overproduction in the tolC* host [Fig. 5B, right graph]) or another SdsR target (SdsR+ overproduction in a tolC* host). The modest increase in sensitivity to rifampin and erythromycin (Fig. 5C and D) demonstrated a pattern similar to that seen for novobiocin, with SdsR effective in either the tolC+ or the tolC* host and SdsR* effective only when it could pair with tolC (tolC* host) (Fig. 5B to D, graphs on right). These results suggest that targets other than tolC, likely regulated by SdsR+ but not by SdsR* (that is, pairing in the same region of SdsR as tolC), contribute to antibiotic sensitivity. These alternative targets remain to be identified but suggest that multiple targets of SdsR may be involved in antibiotic resistance. One possibility is that the overexpression of either SdsR or SdsR* binds enough Hfq to titrate this chaperone away from other sRNAs and/or targets, thus partially mimicking the antibiotic-sensitive phenotype of an hfq deletion (8). However, we did not find SdsR (RyeB) to be a particularly effective Hfq competitor in a previous study (60).
Sublethal doses of β-lactam antibiotics have been reported to induce RpoS and thus increase levels of SdsR; in that study, the antibiotic treatment increased mutagenesis, and the deletion of sdsR restored the lower level of mutagenesis (30). This was suggested to be due to SdsR-mediated repression of mutS, although that was not directly demonstrated. We had difficulty reproducing the RpoS induction and thus were unable to test whether SdsR-mediated regulation of TolC contributed to this phenotype.
The overexpression of SdsR also reduced biofilm formation (Fig. 6B). In this case, the target of SdsR is evidently not tolC, since the SdsR* mutant is unable to reduce biofilm in the tolC* strain. However, the target or targets must pair with SdsR in the same region as tolC, since the SdsR* mutant is inactive for repressing biofilms in both the tolC+ and tolC* hosts (Fig. 6B). Biofilms formed by bacteria are known to be extremely resistant to antibiotics (62). The transcriptional regulator CsgD is known to have a major role in biofilm formation (63), and in Salmonella, the loss of SdsR led to the downregulation of csgD mRNA and protein levels, therefore decreasing the red, dry, and rough (rdar) morphotype expression; the direct target(s) of SdsR for this phenotype was not identified (64). However, this effect (positive contribution of SdsR to biofilm formation) would be in the opposite direction to what we observed here, i.e., a negative effect of SdsR overexpression and little to no effect of deletion of sdsR on E. coli biofilm formation in YESCA (Fig. 6), suggesting that the effect of SdsR on biofilms in these different organisms is likely to be complex and contingent upon specific growth conditions. Other sRNAs have been implicated in biofilm regulation, including McaS, which both represses the transcriptional regulator CsgD, a regulator of curli biogenesis, and activates flhD, the regulator of flagellar synthesis (43). In a previous study, SdsR was shown to have a modest (<2-fold) effect on motility and flhDC (59); this effect may also contribute to the decrease in biofilm. Although our results suggest that the biofilm phenotype is not associated with SdsR-mediated regulation of tolC, this is the first report of a tolC deletion mutant that significantly reduced biofilm formation in YESCA medium (Fig. 6A). TolC has been reported to be required for adhesion and biofilm formation in pathogenic enteroaggregative E. coli; deletion mutants have decreased aggregation and adhesion under growth conditions with high-glucose Dulbecco's minimal medium (65, 66), consistent with our findings.
In a broader attempt to identify roles for SdsR, we carried out a full Biolog panel of tests (carbon, nitrogen, osmolytes, pH, and multiple antibiotic conditions) comparing WT cells, cells with the sdsR deletion mutation, and cells carrying the tolC* mutation. No strong and reproducible phenotypes were identified. Overall, our results suggest no dramatic phenotypes associated with a loss of SdsR, at least under the growth conditions used in the laboratory and in Biolog assays.
Although this is the initial report of direct regulation at the posttranscriptional level, tolC is known to have complex transcriptional regulation involving four promoters and regulation of some promoters by MarA, SoxS, and Rob, increasing tolC transcription in response to some substrates (12, 46). Our results suggest that transcripts from all of these promoters should be subject to regulation by SdsR. This sRNA is possibly important in the return to equilibrium after a toxin-inducing signal has been cleared from the cell via TolC-dependent efflux. Alternatively, the overexpression of efflux pumps may also result in the loss of nutrients and metabolic intermediates necessary for growth or survival (67), which are likely to be particularly important in stationary phase. For instance, TolC protein expression levels have been reported to be lower than the maximum levels during starvation in PBS at 20°C when E. coli cells are in a viable-but-nonculturable (VBNC) state (68); this reduction in TolC expression may be due to the posttranscriptional regulation by SdsR.
Alternatively, TolC downregulation by SdsR may simply be another example of the wholesale downregulation of outer membrane porins by sRNAs (25, 61); in Salmonella, the porin OmpD is a target of SdsR (28). It is common for outer membrane proteins to decrease under stationary and stressful growth conditions via sRNAs, and changes in these surface proteins can have significant effects on the ability to obtain and retain nutrients, to interact with surfaces, and other properties (61). Therefore, the expression profiles of outer membrane proteins vary in biofilms, and this variation may be dependent on the roles they play at the onset and maturation phases of biofilm formation. One known effect of downregulating OMPs is to reduce sigmaE induction (69–71); such downregulation possibly favors RpoS-dependent transcription by reducing competition for the RNA polymerase core. The outer membrane porins OmpC and OmpF are classic examples of how the proper balance of outer membrane proteins is significant under specific growth conditions; although they are expressed differently depending on the growth conditions, the total numbers of these two porins are relatively constant (61). OmpF and OmpT had significantly repressed expression levels in biofilms, whereas OmpC expression levels increased in biofilms compared to those in stationary-phase cultures (72), emphasizing the changes in OMPs during this developmental program. The absence of strong phenotypes with a loss of SdsR possibly reflects the cooperative nature of these sRNA-based regulatory programs, with multiple sRNAs collaborating to modulate cell surface proteins.
We began this study with the expectation that we would find sRNAs that positively regulated drug efflux, based on reports of increased antibiotic sensitivity of hfq mutants (8, 27). Our results suggest that this increase in sensitivity is not likely due to the direct regulation of tolC, acrA, and/or acrB. A modest activation of acrB by GcvB was found (see Fig. S1C in the supplemental material), although acrB fusion activity was low; further work will be necessary to clarify the importance of this observation. We suggest that the increased sensitivity to these drugs may be due to more global effects of deleting hfq-dependent sRNAs, including the general increase in OMP synthesis (25). Nonetheless, continued work on the regulation of tolC may well uncover conditions under which this regulation becomes important.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Misra for donating the anti-TolC primary antibody and M. Maurizi for the EF-Tu primary antibody. We thank members of the laboratory for their comments on the manuscript. We thank K. Fröhlich, K. Papenfort, and J. Vogel for comments on the manuscript and for sharing unpublished data. We also acknowledge Yasmine Kanaan at Howard University Cancer Center, Department of Microbiology, for serving as academic advisor and contributing to the discussion of the research progress.
Funding Statement
Funding was provided primarily by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. A.P. was supported in part by Howard University during her Ph.D. study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00971-15.
REFERENCES
- 1.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. 2001. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother 45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernández L, Hancock RE. 2012. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev 25:661–681. doi: 10.1128/CMR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol 16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- 4.Tikhonova EB, Zgurskaya HI. 2004. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem 279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- 5.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. 2011. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480:565–569. [DOI] [PubMed] [Google Scholar]
- 6.Nikaido H, Takatsuka Y. 2009. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta 1794:769–781. doi: 10.1016/j.bbapap.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikaido H. 1996. Multidrug efflux pumps of Gram-negative bacteria. J Bacteriol 178:5853–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamada J, Yamasaki S, Hirakawa H, Hayashi-Nishino M, Yamaguchi A, Nishino K. 2010. Impact of the RNA chaperone Hfq on multidrug resistance in Escherichia coli. J Antimicrob Chemother 65:853–858. doi: 10.1093/jac/dkq067. [DOI] [PubMed] [Google Scholar]
- 9.Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martinez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, Latendresse M, Muniz-Rascado L, Ong Q, Paley S, Schroder I, Shearer AG, Subhraveti P, Travers M, Weerasinghe D, Weiss V, Collado-Vides J, Gunsalus RP, Paulsen I, Karp PD. 2013. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res 41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhamdhere G, Zgurskaya HI. 2010. Metabolic shutdown in Escherichia coli cells lacking the outer membrane channel TolC. Mol Microbiol 77:743–754. doi: 10.1111/j.1365-2958.2010.07245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alekshun MN, Levy SB. 1999. The mar regulon: multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol 7:410–413. doi: 10.1016/S0966-842X(99)01589-9. [DOI] [PubMed] [Google Scholar]
- 12.Rosner JL, Martin RG. 2009. An excretory function for the Escherichia coli outer membrane pore TolC: upregulation of marA and soxS transcription and Rob activity due to metabolites accumulated in tolC mutants. J Bacteriol 191:5283–5292. doi: 10.1128/JB.00507-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harbor Symp Quant Biol 71:1–11. doi: 10.1101/sqb.2006.71.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottesman S. 2005. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet 21:399–404. doi: 10.1016/j.tig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Lay N, Schu DJ, Gottesman S. 2013. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Biol Chem 288:7996–8003. doi: 10.1074/jbc.R112.441386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang A, Wassarman KM, Ortega J, Steven AC, Storz G. 2002. The Sm-like Hfq protein increases OxyS RNA interaction with target mRNAs. Mol Cell 9:11–22. doi: 10.1016/S1097-2765(01)00437-3. [DOI] [PubMed] [Google Scholar]
- 18.Majdalani N, Hernandez D, Gottesman S. 2002. Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol Microbiol 46:813–826. doi: 10.1046/j.1365-2958.2002.03203.x. [DOI] [PubMed] [Google Scholar]
- 19.Mandin P, Gottesman S. 2010. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J 29:3094–3107. doi: 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen J, Forst SA, Zhao K, Inouye M, Delihas N. 1989. The function of micF RNA. micF RNA is a major factor in the thermal regulation of OmpF protein in Escherichia coli. J Biol Chem 264:17961–17970. [PubMed] [Google Scholar]
- 21.Chao Y, Vogel J. 2010. The role of Hfq in bacterial pathogens. Curr Opin Microbiol 13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Douchin V, Bohn C, Bouloc P. 2006. Down-regulation of porins by a small RNA bypasses the essentiality of the regulated intramembrane proteolysis protease RseP in Escherichia coli. J Biol Chem 281:12253–12259. doi: 10.1074/jbc.M600819200. [DOI] [PubMed] [Google Scholar]
- 23.Mizuno T, Chou MY, Inouye M. 1984. A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (micRNA). Proc Natl Acad Sci U S A 81:1966–1970. doi: 10.1073/pnas.81.7.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Udekwu KI, Darfeuille F, Vogel J, Reimegård J, Holmqvist E, Wagner EG. 2005. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev 19:2355–2366. doi: 10.1101/gad.354405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel J, Papenfort K. 2006. Small non-coding RNAs and the bacterial outer membrane. Curr Opin Microbiol 9:605–611. doi: 10.1016/j.mib.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Schweizer HP. 2012. Understanding efflux in Gram-negative bacteria: opportunities for drug discovery. Expert Opin Drug Discov 7:633–642. doi: 10.1517/17460441.2012.688949. [DOI] [PubMed] [Google Scholar]
- 27.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fröhlich KS, Papenfort K, Berger AA, Vogel J. 2012. A conserved RpoS-dependent small RNA controls the synthesis of major porin OmpD. Nucleic Acids Res 40:3623–3640. doi: 10.1093/nar/gkr1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim T, Bak G, Lee J, Kim KS. 2015. Systematic analysis of the role of bacterial Hfq-interacting sRNAs in the response to antibiotics. J Antimicrob Chemother 70:1659–1668. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez A, Laureti L, Crussard S, Abida H, Rodriguez-Rojas A, Blazquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, Matic I. 2013. β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun 4:1610. doi: 10.1038/ncomms2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- 32.Chung CT, Niemela SL, Miller RH. 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A 86:2172–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guillier M, Gottesman S. 2006. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol 59:231–247. doi: 10.1111/j.1365-2958.2005.04929.x. [DOI] [PubMed] [Google Scholar]
- 34.Mandin P, Gottesman S. 2009. A genetic approach for finding small RNAs regulators of genes of interest identifies RybC as regulating the DpiA/DpiB two-component system. Mol Microbiol 72:551–565. doi: 10.1111/j.1365-2958.2009.06665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poteete AR. 2001. What makes the bacteriophage lambda Red system useful for genetic engineering: molecular mechanism and biological function. FEMS Microbiol Lett 201:9–14. doi: 10.1111/j.1574-6968.2001.tb10725.x. [DOI] [PubMed] [Google Scholar]
- 36.Schupp JM, Travis SE, Price LB, Shand RF, Keim P. 1995. Rapid bacterial permeabilization reagent useful for enzyme assays. Biotechniques 19:18–20. [PubMed] [Google Scholar]
- 37.Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. 2010. Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci U S A 107:9602–9607. doi: 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller JH. 1972. Experiments in bacterial genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 39.Zuker M. 2003. Mfold Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Massé E, Vanderpool CK, Gottesman S. 2005. Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol 187:6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Lay N, Gottesman S. 2009. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 191:461–476. doi: 10.1128/JB.01157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Battesti A, Tsegaye YM, Packer DG, Majdalani N, Gottesman S. 2012. H-NS regulation of IraD and IraM antiadaptors for control of RpoS degradation. J Bacteriol 194:2470–2478. doi: 10.1128/JB.00132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomason MK, Fontaine F, De Lay N, Storz G. 2012. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol 84:17–35. doi: 10.1111/j.1365-2958.2012.07965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bochner BR, Gadzinski P, Panomitros E. 2001. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res 11:1246–1255. doi: 10.1101/gr.186501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eguchi Y, Oshima T, Mori H, Aono R, Yamamoto K, Ishihama A, Utsumi R. 2003. Transcriptional regulation of drug efflux genes by EvgAS, a two-component system in Escherichia coli. Microbiology 149:2819–2828. doi: 10.1099/mic.0.26460-0. [DOI] [PubMed] [Google Scholar]
- 46.Zhang A, Rosner JL, Martin RG. 2008. Transcriptional activation by MarA, SoxS and Rob of two tolC promoters using one binding site: a complex promoter configuration for tolC in Escherichia coli. Mol Microbiol 69:1450–1455. doi: 10.1111/j.1365-2958.2008.06371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Augustus AM, Celaya T, Husain F, Humbard M, Misra R. 2004. Antibiotic-sensitive TolC mutants and their suppressors. J Bacteriol 186:1851–1860. doi: 10.1128/JB.186.6.1851-1860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waters LS, Sandoval M, Storz G. 2011. The Escherichia coli MntR miniregulon includes genes encoding a small protein and an efflux pump required for manganese homeostasis. J Bacteriol 193:5887–5897. doi: 10.1128/JB.05872-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCullen CA, Benhammou JN, Majdalani N, Gottesman S. 2010. Mechanism of positive regulation by DsrA and RprA small noncoding RNAs: pairing increases translation and protects rpoS mRNA from degradation. J Bacteriol 192:5559–5571. doi: 10.1128/JB.00464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nikaido H. 2011. Structure and mechanism of RND-type multidrug efflux pumps. Adv Enzymol Relat Areas Mol Biol 77:1–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zgurskaya HI, Krishnamoorthy G, Ntreh A, Lu S. 2011. Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front Microbiol 2:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirakawa H, Takumi-Kobayashi A, Theisen U, Hirata T, Nishino K, Yamaguchi A. 2008. AcrS/EnvR represses expression of the acrAB multidrug efflux genes in Escherichia coli. J Bacteriol 190:6276–6279. doi: 10.1128/JB.00190-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu C, Bentley WE, Rao G. 2003. Comparisons of oxidative stress response genes in aerobic Escherichia coli fermentations. Biotechnol Bioeng 83:864–870. doi: 10.1002/bit.10732. [DOI] [PubMed] [Google Scholar]
- 54.Monsieurs P, De Keersmaecker S, Navarre WW, Bader MW, De Smet F, McClelland M, Fang FC, De Moor B, Vanderleyden J, Marchal K. 2005. Comparison of the PhoPQ regulon in Escherichia coli and Salmonella Typhimurium. J Mol Evol 60:462–474. doi: 10.1007/s00239-004-0212-7. [DOI] [PubMed] [Google Scholar]
- 55.Rosenberg EY, Bertenthal D, Nilles ML, Bertrand KP, Nikaido H. 2003. Bile salts and fatty acids induce the expression of Escherichia coli AcrAB multidrug efflux pump through their interaction with Rob regulatory protein. Mol Microbiol 48:1609–1619. doi: 10.1046/j.1365-2958.2003.03531.x. [DOI] [PubMed] [Google Scholar]
- 56.Ruiz C, Levy SB. 2010. Many chromosomal genes modulate MarA-mediated multidrug resistance in Escherichia coli. Antimicrob Agents Chemother 54:2125–2134. doi: 10.1128/AAC.01420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma CM, Darfeuille F, Plantinga TH, Vogel J. 2007. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev 21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Q, Figueroa-Bossi N, Bossi L. 2014. Translation enhancing ACA motifs and their silencing by a bacterial small regulatory RNA. PLoS Genet 10:e1004026. doi: 10.1371/journal.pgen.1004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Lay N, Gottesman S. 2012. A complex network of small non-coding RNAs regulate motility in Escherichia coli. Mol Microbiol 86:524–538. doi: 10.1111/j.1365-2958.2012.08209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moon K, Gottesman S. 2011. Competition among Hfq-binding small RNAs in Escherichia coli. Mol Microbiol 82:1545–1562. doi: 10.1111/j.1365-2958.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guillier M, Gottesman S, Storz G. 2006. Modulating the outer membrane with small RNAs. Genes Dev 20:2338–2348. doi: 10.1101/gad.1457506. [DOI] [PubMed] [Google Scholar]
- 62.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 63.Zakikhany K, Harrington CR, Nimtz M, Hinton JC, Römling U. 2010. Unphosphorylated CsgD controls biofilm formation in Salmonella enterica serovar Typhimurium. Mol Microbiol 77:771–786. doi: 10.1111/j.1365-2958.2010.07247.x. [DOI] [PubMed] [Google Scholar]
- 64.Monteiro C, Papenfort K, Hentrich K, Ahmad I, Le Guyon S, Reimann R, Grantcharova N, Römling U. 2012. Hfq and Hfq-dependent small RNAs are major contributors to multicellular development in Salmonella enterica serovar Typhimurium. RNA Biol 9:489–502. doi: 10.4161/rna.19682. [DOI] [PubMed] [Google Scholar]
- 65.Imuta N, Nishi J, Tokuda K, Fujiyama R, Manago K, Iwashita M, Sarantuya J, Kawano Y. 2008. The Escherichia coli efflux pump TolC promotes aggregation of enteroaggregative E. coli 042. Infect Immun 76:1247–1256. doi: 10.1128/IAI.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soto SM. 2013. Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence 4:223–229. doi: 10.4161/viru.23724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Webber MA, Piddock LJ. 2003. The importance of efflux pumps in bacterial antibiotic resistance. J Antimicrob Chemother 51:9–11. doi: 10.1093/jac/dkg050. [DOI] [PubMed] [Google Scholar]
- 68.Muela A, Seco C, Camafeita E, Arana I, Orruño M, Lopez JA, Barcina I. 2008. Changes in Escherichia coli outer membrane subproteome under environmental conditions inducing the viable but nonculturable state. FEMS Microbiol Ecol 64:28–36. doi: 10.1111/j.1574-6941.2008.00453.x. [DOI] [PubMed] [Google Scholar]
- 69.Johansen J, Rasmussen AA, Overgaard M, Valentin-Hansen P. 2006. Conserved small non-coding RNAs that belong to the sigmaE regulon: role in down-regulation of outer membrane proteins. J Mol Biol 364:1–8. doi: 10.1016/j.jmb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 70.Mecsas J, Rouviere PE, Erickson JW, Donohue TJ, Gross CA. 1993. The activity of sigma E, an Escherichia coli heat-inducible sigma-factor, is modulated by expression of outer membrane proteins. Genes Dev 7:2618–2628. doi: 10.1101/gad.7.12b.2618. [DOI] [PubMed] [Google Scholar]
- 71.Thompson KM, Rhodius VA, Gottesman S. 2007. SigmaE regulates and is regulated by a small RNA in Escherichia coli. J Bacteriol 189:4243–4256. doi: 10.1128/JB.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schembri MA, Kjaergaard K, Klemm P. 2003. Global gene expression in Escherichia coli biofilms. Mol Microbiol 48:253–267. doi: 10.1046/j.1365-2958.2003.03432.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.