

Abstract
Objectives:
To evaluate the nutritional status, to screen for the presence of malnutrition, and to study the possible risk factors associated with malnutrition in patients with cystic fibrosis (CF).
Methods:
A retrospective cross-sectional review of medical records of all diagnosed CF patients in the Pediatric Department, Salmaniya Medical Complex, Manama, Kingdom of Bahrain, between January 1984 and May 2015 was conducted. Demographic and anthropometric data were collected from records of last visit to CF clinic. Nutritional status and risk factors of malnutrition were assessed.
Results:
All records of 109 CF patients were reviewed. Forty-seven pediatric patients were included in the study. All included patients were on pancreatic enzyme replacement and 42 (89%) received high-calorie supplementation. Growth failure was noted in 34 (72%) patients, 19 (56%) were wasted and stunted, 8 (23.5%) were wasted only, and 7 (20.5%) were stunted. Low birth weight (p=0.032), and the presence of gastroesophageal reflux disease (GERD) (p=0.039) were the significant risk factors for malnutrition.
Conclusion:
Most CF patients in Bahrain (72%) are malnourished. Low birth weight and the presence of GERD are risk factors.
Cystic fibrosis (CF) is a multisystem progressive disorder characterized by chronic lung disease, excessive sweat electrolytes losses, exocrine pancreatic insufficiency and malnutrition.1,2 Cystic fibrosis is an autosomal recessive disease commonly seen in western countries.1 Cystic fibrosis patients have great energy demand and expenditure, which are usually associated with suboptimal caloric intake leading to malnutrition.3 Nutritional assessment, early identification of malnutrition, and prompt initiation of supportive treatments are essential parts of CF patients’ care as nutritional status can affect the patients’ response to treatment.1,4 Weight gain and normal muscle mass are linked to normal growth and good pulmonary functions.3 There are no data on the diagnostic value of the different anthropometric measure of failure to thrive (FTT) or on the sensitivity and specificity of the probable cut off points.5 According to the 2012 health statistic on growth indicators for children under 5 years in Bahrain, the percentage of total wasted children based on weight for length/height measurement is 2.1% (wasted 1.8% and severely wasted 0.3%) and the total stunted children based on length/height for age are 2.4% (stunted 1.9% and severely stunted 0.4%).6 Between 1981 and 1995, the age specific mortality among CF patients in Bahrain had fallen dramatically from 80% to 9%.7 This could be attributed to the improvement in living standards and the advances in medical care, particularly the introduction of pancreatic enzyme replacement therapy (PERT).8 The aim of this study is to evaluate the nutritional status of patients with CF via the use of anthropometric parameters, to screen for the presence of malnutrition, and to study the possible risk factors associated with malnutrition in this group of patients.
Methods
Original and review articles related to pediatric and adult CF patients were identified. We used PubMed, Ovid, Medline, Google, and the following cited references to retrieve full papers that report data on the incidence, diagnosis, risk factors, and management of malnutrition in CF patients. A retrospective cross-sectional review of medical records of all patients diagnosed with CF in the Pediatric Department, Salmaniya Medical Complex, Manama, Kingdom of Bahrain, between January 1984 and May 2015 was conducted. The diagnosis of CF was confirmed by the presence of characteristic clinical symptoms, or a family history of CF along with a positive sweat test, and/or 2 disease-causing gene mutations. Any CF patient (≤18 years) with available growth parameters was included in the study while patients with associated gastrointestinal diseases that affect digestion, or absorption such as celiac disease, inflammatory bowel diseases, or on medications affecting appetite or growth were excluded. Cystic fibrosis patients who are pancreatic sufficient were also excluded. Data regarding gestational age, type of delivery, gender, nationality, initial clinical presentation, age at presentation, age at diagnosis, consanguinity, and complications were gathered. The diagnosis of CF was confirmed using sweat chloride test (ELITechGroup, Logan, USA), or CF trans-membrane regulator (CFTR) gene mutation testing (Saudi Diagnostic Laboratory, Riyadh, Saudi Arabia). Investigations such as liver function tests, stool fat globules and tryptic activity, deep tracheal aspiration, throat swab, and sputum culture were collected in addition to data on treatment with pancreatic enzyme replacement, fat soluble vitamin replacement, high calorie supplementation, bronchodilator use, and antimicrobial therapy.
Anthropometric parameters of the last visit to the pediatric clinic were collected. Based on 2006 World Health Organization (WHO) Child Growth Standards,9 AnthroPlus (WHO, Geneva, Switzerland) anthropometric software program was used to calculate the different growth parameters. These parameters were presented as a standard deviation (SD) from age and gender-specific reference means. The patient’s weight for age (WA) in kilograms, weight for age percentile (WAP) and z score (WAZ), height for age (HA) in centimetres, height for age percentile (HAP) and z score (HAZ), body mass index (BMI; weight/height2 [kg/m2]), BMI percentile (BMIP) and z score (BMIZ), ideal body weight (IBW), percentage IBW (%IBW), weight for height percentile (WHP) and weight for height z score (WHZ) were calculated. Patients were considered to have nutritional failure if their WA or HA are <5th or BMI <10th percentile for age.5,6 If the WA was between 5th and 10th percentile, or BMI is between the 10th and 24th percentile, the patient was considered at risk of malnutrition. If the weight was >10th, HA is >5th and BMI was between 25th and 49th percentile, the patient was considered to have acceptable nutritional status. The nutritional status is considered optimal if the WA was >10th, HA >5th, and BMI ≥50th percentile.6 Patients were considered wasted if the WA was <5th percentile and stunted if the HA was <5th percentile. The results of growth parameters were compared with the reference population. To determine risk factor for FTT, we studied the statistical differences between malnourished and normal growth CF patients in regards to gender, gestational age, type of delivery, birth weight, weight at presentation, age at diagnosis (<3 versus ≥3 years), age groups (<5 versus ≥5 years), family history of CF, associated gastroesophageal reflux disease (GERD), recurrent chest infections, fat soluble vitamins intake, high calorie supplementation intake, and CFTR gene mutations.
This study was in accordance with the principles of Helsinki Declaration and was ethically approved by the secondary care medical research subcommittee, Salmaniya Medical Complex, Ministry of Health, Kingdom of Bahrain.
Statistical analysis
The patients’ data were analyzed using Statistical Package for Social Sciences (IBM SPSS Statistics, Armonk, NY) version 21. The frequencies and percentages were calculated for categorical variables. Chi-square and Fisher exact tests were used to compare categorical variables. The continuous variables were tested for normal distribution using the Kolmogorov-Smirnov test and the Shapiro-Wilk W test. Grouped data are presented as mean ± SD for normally distributed variables or median, and range for non-normally distributed variables. Independent student t-test was used to compare group means for continuous variable; while the Wilcoxon signed rank test was used to compare the non-normally distributed variables. The missing data were excluded from the analysis. Exact 2-sided p-values <0.05 were considered statistically significant.
Results
Of the 109 patients attending the Bahrain CF clinic, those >18 years (n=45) were excluded from the study. In addition, those with no available growth data (n=16), and one patient with pancreatic sufficiency were also excluded. Data of the remaining 47 CF patients were analyzed. Their clinical characteristics are presented in Table 1.
Table 1.
Clinical characteristics of 47 pediatric patients with cystic fibrosis.
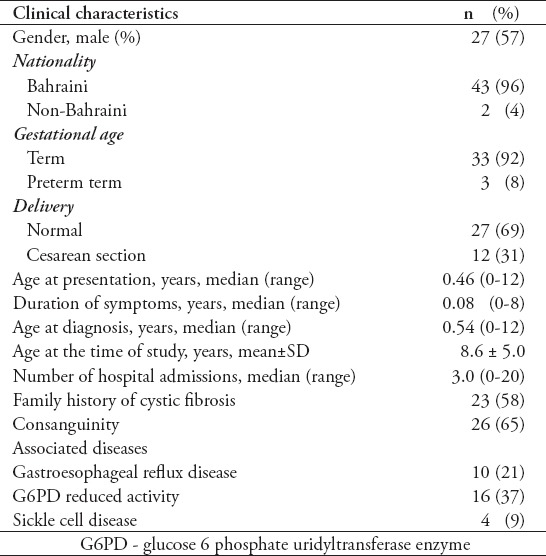
Twenty-seven (57%) were males and 20 (43%) were females. One patient presented late at the age of 12 years after a serious chest infection with swine flu (influenza A H1N1 virus) and pneumothorax. Thirty (64%) patients were diagnosed below 3 years of age and 17 (36%) at >3 years. The mean age at growth measurement was 7 years (± 4.9 SD). Nineteen patients (40%) were <5 years, 13 (28%) were between 5-9.9 years, 11 (23%) between 10-14.9 years, and 4 (9%) were >15 years. Forty-four (94%) patients were alive at the time of the study and 3 (6%) were dead (one male and 2 females). The age of death was 6, 17, and 20 years (the last patient had her last growth parameters at the age of 16.6 years). Initial clinical presentations are showed in Table 2.
Table 2.
Initial clinical presentation of 47 pediatric patients with cystic fibrosis.
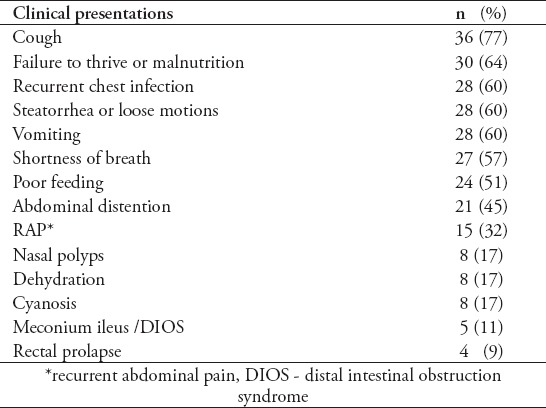
The most frequent complications were digital clubbing in 20 (43%), admission to intensive care unit in 14 (30%), and surgeries in 12 (26%). The surgeries included 3 ileostomies, 2 appendectomies, one intussusception surgery, one rectal prolapse repair, and 5 other different surgeries (tonsillectomy in 2 patients and adenoidectomy, nasal polypectomy, intercostal drainage insertion and upper gastrointestinal endoscopy each in one patient). Other complications such as pneumonia, tonsillitis, and diabetes mellitus were seen each in 2 patients, while chronic sinusitis, anemia requiring blood transfusion, developmental delay, bone fractures, pneumothorax, and biliary cirrhosis were seen each in one patient.
Laboratory features at presentation revealed a positive sweat chloride test in 36 (97%) out of 37 patients with available data. The median sweat chloride was 110 mmol/l (range 51-129 mmol/l, normal range: <60 mmol/l). Fecal microscopy was positive for fat globules in 26 (77%). Abnormal fecal tryptic activity in 12 (43%) (normal: 1/100 dilution).
Genetic testing was performed in 25 patients. Sixteen (64%) patients were positive for CFTR gene mutation while 9 (36%) had undetected gene mutations. The most common mutation was 3120+1G>A in 8 patients (50%) (3 as homozygous and 4 as heterozygous, 3120+1G>A/c.1397C>G in one, 3120+1G>A/c.1733_1734delTA in one, 3120+1G>A/other unknown mutation in 2 patients) followed by homozygous N1303K mutation in 4 (25%) patients followed by 2043del1G (2 as homozygous and one as heterozygous, 2043del1G/3120+1G>A). Other mutations were also found such as homozygous c.1418delG, c.1397C>G/c.2988+IG, c.1733_1734delTA/c.2988+IG>A, and ∆F508/other unknown mutation each in one patient (6%). Abnormal LFTs was found in 13 (30%) patients. Hypoalbuminemia was present in 25 (63%); mean serum albumin 32 ±6.7 SD (normal range: 35 to 50 g/l). Hypocalcemia werer found in 9 (27%); mean serum calcium 2.2 ±0.2 SD (normal range: 2.13-2.63 mmol/l). Four had low 25 hydroxy-vitamin D (57%) with a mean serum 25-hydroxyvitamin D level of 49±25 SD (normal level: >50 nmol/l). Positive sputum culture was found in 27 (55%) patients. The most common organisms were Pseudomonas aerogenosa in 18 (41%), Staphylococcus aureus in 11 (25%), and Klebseilla in 6 patients (14%). Other organisms such as Escherichia coli, Yeast, Candida albicans were seen each in 3 patients (7%), Hemophilius influenza and Serratia marcescens each was seen in 2 (5%), Citrobacter, Enterobacter, Achromobacter, and Aspergillus flavus were seen each in one patient (2.3%).
All patients were on pancreatic enzyme replacement therapy (Creon 10.000 capsules of 10000 IU of lipase per kilogram per day with a maximum of 15 capsules per day) (Table 3). No patient was on nasogastric or gastrostomy tube feeding.
Table 3.
Management in 47 patients with cystic fibrosis.

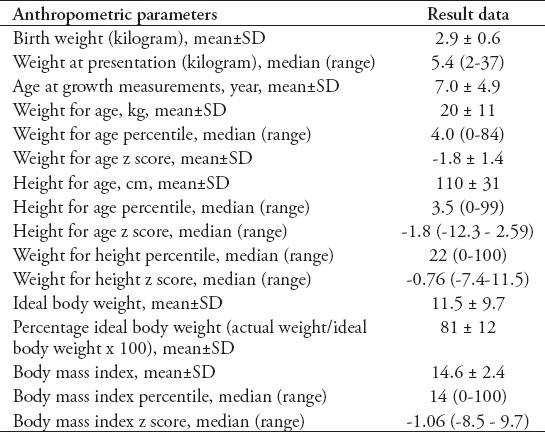
The detailed data of the anthropometric parameters are shown in Table 4. Failure to thrive was found in 34 (72%) patients. Nineteen (56%) of the malnourished CF patients were both wasted and stunted, 8 (23.5%) were only wasted, and 7 (20.5%) were only stunted. Although, 13 (28%) patients had normal growth, 4 of them (31%) were at risk of malnutrition, 5 (38%) had acceptable growth parameters, and only 4 (31%) patients had an appropriate weight/height and height/age.
Table 4.
Data of anthropometric parameters of 47 pediatric patients with cystic fibrosis.
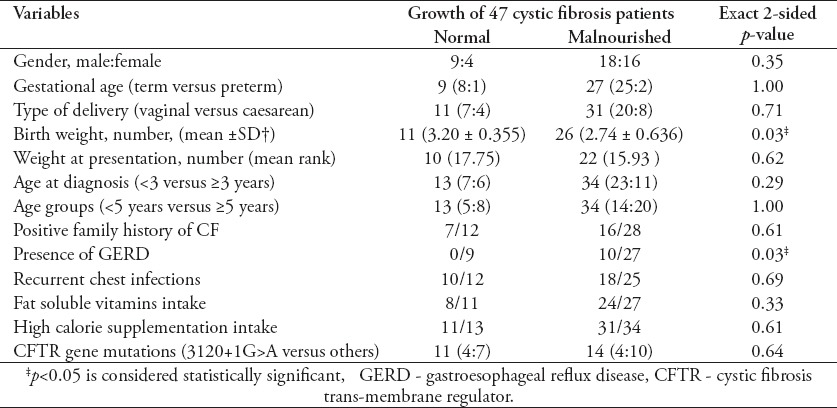
The risk factors associated with malnutrition in CF patients are shown in Table 5. Low birth weight and the presence of GERD were found to be statistically significant risk factors for abnormal growth in CF patients in this study.
Table 5.
Risk factors of malnutrition in 47 pediatric patients with cystic fibrosis (CF).
Discussion
The survival and growth of CF patients had improved over the last decades due to better understanding of the disease, early diagnosis, proper nutritional management and PERT, improved antibiotics regimen, and lung clearance therapy.1,10 Survival rate in CF patients was better among higher socioeconomic class and among males.8 In this study, the socioeconomic class was not measured, but survival rate was better in males.
Diagnosis of CF is based on pulmonary and or gastrointestinal symptoms along with FTT, which may delay the diagnosis.5 Several countries have introduced newborn screening program for CF to overcome this problem.5 However, up to date, Bahrain has no neonatal screening program for CF. Despite that, the average age at diagnosis is 6 months. It was suggested that in the absence of newborn screening, the best screening procedure for CF is proper growth monitoring, detailed medical history and physical examination,5 which are routinely carried out for the children in Bahrain as a part of their routine medical checkup and vaccination visit in the primary health care, which could explain the early diagnosis of the cases.
Testing for CF should be carried out in the work up of children with FTT using either sweat chloride testing,5,11 (a sweat chloride test >60 mmol/L suggests the diagnosis of CF5), or CFTR gene mutation analysis.12 In the European population, the most common CFTR mutation is class 2 ∆F508 mutation;12,13 however, in this study the most frequent CFTR mutation was 3120+1G>A. Patients with CF who had undetected gene mutation varies between countries across Europe ranging from 1% in Denmark to 46.9% in Hungary.13 In this study, the rate of undetected gene was 36%. Gene mutation is linked to the pancreatic exocrine dysfunction, but it is less manifested in the gastrointestinal and pulmonary disease.12
The nutritional problems in CF patients are multifactorial.14 Inadequate dietary intakes due to anorexia together with inadequate digestion due to pancreatic insufficiency in addition to increased energy demand and severe lung disease, all contributes to malnutrition.3,14 Cystic fibrosis-related diabetes also contribute to the poor nutritional status.14 This study showed a significant association between GERD and malnutrition in CF patients. Gastroesophageal reflux disease is commonly found in patients with CF.12 Gastroesophageal reflux disease may contribute to nutritional failure as it can lead to esophagitis, structuring disease and hypoproteinemia.12
Monitoring of growth and nutritional status is crucial for early intervention, effective treatment, and rehabilitation of CF patients.3,15 Nutritional status of patients cannot be identified by a single measure.16 In this study, several anthropometric measures have been used to assess nutritional status of CF patients. Growth and maturation in CF patients is slower than in healthy peers.2 Body mass index standard deviation score (SDS) is considered a useful tool for detection of CF.5 The mean highest median WAP achieved by pediatric patients with CF is at the 42nd percentile and is reached by the age of 3 years.17 A French study2 found that despite the improvement in nutritional management and survival in CF patient, they had impaired peak height velocity during adolescence.
Early treatment of suboptimal growth in CF is recommended.17 Maintenance of normal nutritional status is an essential task for the multidisciplinary team managing CF patients. Leonard et al14 found a significant improvement in nutritional outcomes by 19% in just 15 months after using standardized nutritional assessment and treatment. However, maintaining optimal nutrition is a real challenge in CF patient as they have a higher energy expenditure compared with unaffected individuals.14 High energy diet with no restrictions on fat intake, 35-40% calories from fat,15 high-calorie supplementations, and enzyme replacement therapy are the recommended management. Pancreatic enzyme replacement therapy is one of the most significant contributions in maintaining adequate nutritional status.15 Aggressive nutritional support with adequate PERT early in life can lead to preservation of lung functions and normal growth.3 When oral intake alone is not adequate to maintain normal growth, aggressive nutritional care via enteral tube feeding may be necessary.15 Probiotics was found to improve quality of life of CF patients and reduce number of pulmonary exacerbations.18
This study is limited by its retrospective nature where some data are missed or cannot be retrieved. Data on some anthropometric measures such as mid-arm circumference and triceps skinfold were also not available. In addition, some tests to confirm pancreatic insufficiency were not preformed and genetic mutation testing was limited to the Middle East panel although some other mutation might have been present. However, this study is important being the first study to tackle the growth parameters of CF patients in Bahrain, which can form a foundation for the future studies dealing with a chronic serious disease such as CF.
In conclusion, despite the rapid improvements in medical care and the significant reduction in mortality rate, most of CF children in Bahrain (72%) are failing to thrive compared to the reference population. The low birth weight and the presence of GERD were the significant risk factors for malnutrition in CF patients. A proper nutritional assessment and management of malnutrition are highly recommended. Further studies evaluating nutritional interventions will be required to achieve optimal growth in CF patients.
Acknowledgment
The authors gratefully acknowledge all those who participated in this study. Specifically to the members of the multidisciplinary team who provide care for patients with cystic fibrosis in the Kingdom of Bahrain including Prof. Fadheela Al-Mahroos, Pediatric Gastroenterologist; Dr. Hasan Zainaldeen, Pediatric Gastroenterologist; Dr. Ali Ebrahim, Pulmonology Consultant; Dr. Osama Abdulkarim, Pulmonology Consultant; Ms. Farah and Ms. Dana A. Aziz, Clinical Dietitians; Ms. Ashwaq Hasani and Mrs. Khadija Al-Khayat, Physiotherapists; and Ms. Awatif Abukhudhra, Pediatric Respiratory Nurse, Pediatric Department, Salmaniya Medical Complex, Manama, Kingdom of Bahrain.
Footnotes
References
- 1.Gaskin KJ. Nutritional care in children with cystic fibrosis: are our patients becoming better? Eur J Clin Nutr. 2013;67:558–564. doi: 10.1038/ejcn.2013.20. [DOI] [PubMed] [Google Scholar]
- 2.Bournez M, Bellis G, Huet F. Growth during puberty in cystic fibrosis: a retrospective evaluation of a French cohort. Arch Dis Child. 2012;97:714–720. doi: 10.1136/archdischild-2011-301069. [DOI] [PubMed] [Google Scholar]
- 3.Matel JL, Milla CE. Nutrition in cystic fibrosis. Semin Respir Crit Care Med. 2009;30:579–586. doi: 10.1055/s-0029-1238916. [DOI] [PubMed] [Google Scholar]
- 4.Woestenenk JW, Ent CK, Houwen RH. Pancreatic enzyme replacement therapy and coefficient of fat absorption in children and adolescents with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2015;61:355–360. doi: 10.1097/MPG.0000000000000784. [DOI] [PubMed] [Google Scholar]
- 5.Dommelen PV, Grote FK, Oostdijk W, Keizer-Scharma S, Bouquet J, Hendrix J, et al. Growth monitoring to detect children with cystic fibrosis. Horm Res. 2009;72:218–224. doi: 10.1159/000236083. [DOI] [PubMed] [Google Scholar]
- 6.Health Information Directorate Ministry of Health, Bahrain. Health Statistics 2012. [Updated 2012; Accessed 2015 Aug 13]. Available from URL: http://www.moh.gov.bh/AR/aboutMOH/Information/Statistics.aspx .
- 7.Al-Mahroos F. Cystic fibrosis in Bahrain: incidence, phenotype and outcome. J Trop Pediatr. 1998;44:35–39. doi: 10.1093/tropej/44.1.35. [DOI] [PubMed] [Google Scholar]
- 8.Barr HL, Britton J, Smyth AR, Fogarty AW. Association between socioeconomic status, sex, and age at death from cystic fibrosis in England and Wales (1959 to 2008): cross sectional study. BMJ. 2011;343:d4662. doi: 10.1136/bmj.d4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization. WHO child growth standards: length/height-for-age, weight-for-age, weight-for-length, weight-for-height and body mass index-for-age: methods and development. Geneva (CH): World Health Organization; 2006. [Google Scholar]
- 10.Tluczek A, Laxova A, Grieve A, Heun A, Brown RL, Rock MJ, et al. Long-term follow-up of cystic fibrosis newborn screening: Psychosocial functioning of adolescents and young adults. J Cyst Fibros. 2014;13:227–234. doi: 10.1016/j.jcf.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Accurso FJ, Goor FV, Zha J, Stone AJ, Dong Q, Ordonez CL, et al. Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data. J Cyst Fibros. 2014;13:139–147. doi: 10.1016/j.jcf.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haller W, Ledder O, Lewindon PJ, Couper R, Gaskin KJ, Oliver M. Cystic fibrosis: an update for clinician. Part1: nutrition and gastrointestinal complications. J Gasrtoenterol Hepatol. 2014;29:1344–1355. doi: 10.1111/jgh.12546. [DOI] [PubMed] [Google Scholar]
- 13.Boeck K, Zolin A, Cuppens H, Olesen HV, Viviani L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros. 2014;13:403–409. doi: 10.1016/j.jcf.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Leonard A, Davis E, Rosenstein BJ, Zeitlin PL, Paranjape SM, Peeler D, et al. Description of a standardized nutrition classification plan and its relation to nutritional outcomes in children with cystic fibrosis. J Pediatr Psychol. 2010;35:6–13. doi: 10.1093/jpepsy/jsp029. [DOI] [PubMed] [Google Scholar]
- 15.Kalnins D, Wilschanski M. Maintenance of nutritional status in patients with cystic fibrosis: new and emerging therapies. Drug Des Devel Ther. 2012;6:151–161. doi: 10.2147/DDDT.S9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiskin A, Johnson MJ, Leaf AA, Wootton SA, Beattie RM. How to use: nutritional assessment in children. Arch Dis Child Educ Pract Ed. 2015;100:204–209. doi: 10.1136/archdischild-2014-306516. [DOI] [PubMed] [Google Scholar]
- 17.Stark LJ, Quittner AL, Powers SW, Opopari-Arrigan L, Bean JA, Duggan C, et al. Randomized clinical trial of behavioral intervention and nutrition education to improve calori intake and weight in children with cystic fibrosis. Arch Pediatr Adolesc Med. 2009;163:915–921. doi: 10.1001/archpediatrics.2009.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jafari A, Mehdizadeh-Hakkak A, Kianifar H, Hebrani P, Ahanchian H, Abbasnejad E. Effects of probiotics on quality of life in children with cystic fibrosis;a randomized control trial. Iran J Pediatr. 2013;23:669–674. [PMC free article] [PubMed] [Google Scholar]