

Abstract
Mutations in Cx26 gene are found in most cases of human genetic deafness. Some mutations produce syndromic deafness associated with skin disorders, like Keratitis Ichthyosis Deafness syndrome (KID). Because in the human skin Cx26 is co-expressed with other connexins, like Cx43 and Cx30, and since KID syndrome is inherited as autosomal dominant condition, it is possible that KID mutations change the way Cx26 interacts with other co-expressed connexins. Indeed, some Cx26 syndromic mutations showed gap junction dominant negative effect when co-expressed with wild type connexins, including Cx26 and Cx43. The nature of these interactions and the consequences on hemichannels and gap junction channels functions remain unknown. In this study we demonstrate that syndromic mutations at the N-terminus segment of Cx26, change connexin oligomerization compatibility, allowing aberrant interactions with Cx43. Strikingly, heteromeric oligomer formed by Cx43/Cx26 (syndromic mutants) show exacerbated hemichannel activity, but nonfunctional gap junction channels; this also occurs for those Cx26 KID mutants that do not show functional homomeric hemichannels. Heterologous expression of these hyperactive heteromeric hemichannels increases cell membrane permeability, favoring ATP release and Ca2+ overload. The functional paradox produced by oligomerization of Cx43 and Cx26 KID mutants could underlie the severe syndromic phenotype in human skin.
INTRODUCTION
Gap junction channels (GJCs) allow metabolic and electrical coupling between adjacent cells and are formed by the oligomerization of connexin (Cx) protein subunits. Cxs oligomerize to form hexamers called hemichannels (HCs), which reach the appositional plasma membrane and dock with other complementary HCs provided by an adjacent cell to form gap junction plaques (Segretain and Falk, 2004). In non-appositional plasma membrane, HCs connect the cytoplasm with the external milieu, allowing paracrine and autocrine signaling mediated by ATP and Ca2+, among others (Saez et al., 2005; Stout et al., 2004). HCs can be homomeric, if all subunits are the same Cxs or heteromeric if they are formed by two or more different Cxs; however, some Cxs are incompatible to form heteromeric channels, like Cx26 and Cx43 (Gemel et al., 2004; Jara et al., 2012; Martinez et al., 2011). Indeed, heteromeric channels have different functional properties than homomeric channels; therefore, regulation of Cx-Cx interaction could be important in controlling intercellular communication (Jara et al., 2012; Martinez et al., 2002; Martinez et al., 2011).
Mutations in the human Cx26 gene accounts for about 50% of genetic deafness. To date, more than 100 Cx26 mutations have been identified, but only 16 of them, mainly located at the N-terminus and the transition between the first transmembrane segment and the extracellular loop segment, cause the syndromic phenotype (Martinez et al., 2009). Although syndromic Cx26 mutations are sparse, they have an autosomal dominant inheritance pattern that affects proliferation and differentiation of skin keratinocytes, and produces progressive corneal neovascularization and hair and nail dystrophies like in Keratitis Ichthyosis Deafness (KID) syndrome (Arita et al., 2006; Caceres-Rios et al., 1996; Kelsell et al., 2000). It is hypothesized that syndromic mutations produced gain of function HCs (Gerido et al., 2007; Lee et al., 2009; Levit et al., 2012; Sanchez et al., 2010). However, some syndromic mutations, like Cx26S17F, lead to non-functional HCs, but they still developed a severe KID syndrome phenotype (Lee et al., 2009; Mazereeuw-Hautier et al., 2007; Richard et al., 2002). Most of these studies have been performed in homomeric HCs, which may not reproduce what occurs in the disease. In human skin, Cx26 is co-expressed with Cx43, Cx30 and Cx31 (Salomon et al., 1994); since KID syndrome is inherited as autosomal dominant condition, it is possible that KID mutations change the interaction and functional properties of Cx26 with other Cxs. Thus, to better understand the disease mechanisms, it is necessary to investigate the effect of Cx26 KID mutations when co-expressed with other Cxs found in the human skin.
Here, we functionally analyzed several N-terminus Cx26 mutants that were co-expressed, or not, with wild type Cx26 and Cx43. We focused on N14Y and S17F syndromic mutations, as well as two mutations located at residue 12 that can cause syndromic (G12R) or non-syndromic (G12V) deafness. The residue G12 is located inside the pore at the plasma membrane level (Kwon et al., 2011; Maeda et al., 2009), while residues N14 and S17 face the cytoplasm in the hinge region of the Cx26 N-terminus (Figure 1, a1). The effect of two non-syndromic mutations, V37I and A40G, positioned at the TM1/ECL1 transition, was also assessed to compare interactions with wild type Cx26 and Cx43.
Figure 1. Syndromic mutations reduce or eliminate GJC formation.
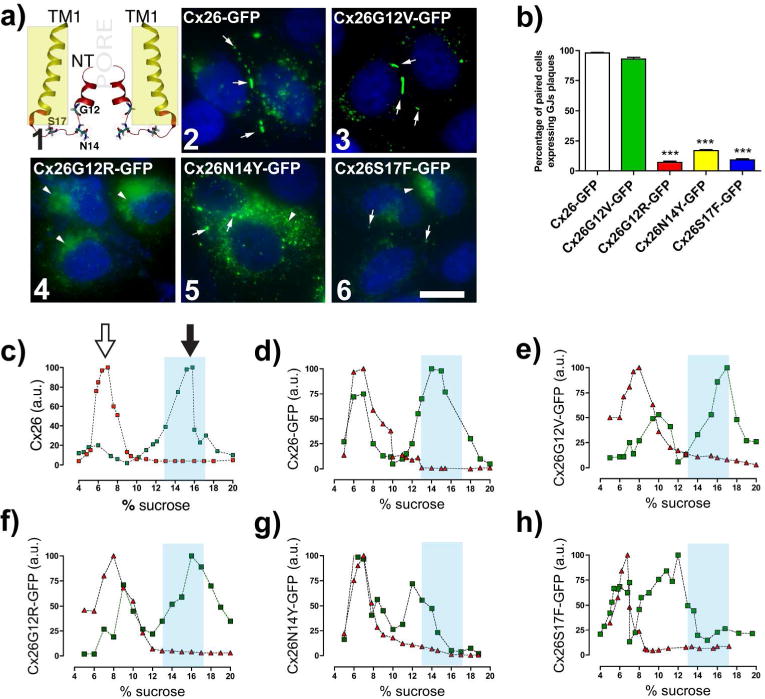
(a1) TM1 and NT of two Cx26 subunits with highlighted residues at the NT that are linked to deafness mutations. Yellowish rectangle indicates plasma membrane region. (a2–6) Confocal images show the respective subcellular localization of cells expressing Cx26 wild type or mutants. White arrows indicate GJ plaques. (a2 and 6) Arrowheads indicate perinuclear labeling. Bar=15 μm. (b) Percentage of paired cells expressing GJ plaques per condition. (***p<0.001; n=5, 250 paired cells for condition). (c–h) Syndromic mutations, N14Y and S17F, affect oligomerization. Graphs represent the sedimentation profile of monomer and oligomers (in arbitrary units of Cx amount, a.u.) of WT or mutants Cx26. SDS treated fractions (red triangles). Black-filled arrows in (c) show the hexameric peak, and the unfilled arrows show monomeric peaks. Light blue columns indicate where WT Cx26 hexamers sedimented (n=4).
RESULTS
Non-syndromic and syndromic mutations located at the N-terminus segment of Cx26 reduce or eliminate GJC formation and/or function
To assess the effect of Cx26 mutations on the traffic, oligomerization and functional state of GJCs, HeLa cells were transfected with wild type Cx26 (Cx26 hereinafter) or deafness mutants attached to GFP to visualize their subcellular localization. All transfections were performed in a HeLa parental cell line that either does not express Cx26 and Cx43 (Figure S1a) or does not show GJCs currents (Table S1), confirming that this line is Cx-expression deficient. In contrast, we observed large GJ plaques in HeLa cells expressing Cx43, Cx26 (Figure S1b), Cx26-GFP or the non-syndromic Cx26G12V-GFP mutant (Figure 1, a2,3) similar to that previously reported for non-syndromic mutants Cx26V37I and Cx26A40G (Jara et al., 2012). Conversely, syndromic mutants form few GJ plaques, suggesting intracellular trapping or preferential localization in non-appositional plasma membrane (Figure 1, a4–6 and b).
We next evaluated the functional state of GJCs formed by homomeric mutations at the N-terminus. Dye-transfer and transjunctional conductance (gj), between cells expressing syndromic or non-syndromic mutants were indistinguishable from those obtained in untransfected cells (Table S1). Thus, these mutations completely abolished the formation of functional GJCs.
Syndromic N-terminus mutations affect Cx26 oligomerization
Diffuse cytoplasmic and/or perinuclear localizations (Figure 1, a4–6) identified for syndromic mutants may reflect partial intracellular trapping due to defective protein oligomerization. This possibility was analyzed by velocity sedimentation assays in sucrose gradients. We found that the oligomeric and monomeric fractions of Cx26-GFP, Cx26G12V-GFP and Cx26G12R-GFP present similar sedimentation patterns to Cx26 (Figure 1, c–f), which was consistent with those reported previously (Gemel et al., 2004; Jara et al., 2012). However, for syndromic mutants Cx26N14Y-GFP and Cx26S17F-GFP (Figure 1, g–h), their oligomeric fractions shifted to lower sucrose densities, suggesting the formation of intermediate arrangements with fewer hexamers.
To identify a potential dominant negative effect of Cx26 mutants on Cx26 GJC function, we co-expressed the GFP-tagged mutants with Cx26 containing a hemagglutinin tag (Cx26-HA). In these cells, all mutants co-localize with Cx26-HA and formed abundant and large GJ plaques between pair of co-expressing cells (Figure S2, a–f), suggesting that Cx26 rescues the traffic and delivery of syndromic mutants to appositional areas. Interestingly, all N-terminus mutants, either syndromic or non-syndromic, were not dominant negative of Cx26 GJCs. Nevertheless, we cannot rule out subtle changes in the channel permeability, since we found that GJCs formed by Cx26 and either syndromic mutant Cx26N14Y or Cx26S17F showed reduced coupling to Lucifer yellow (−2), but normal coupling to the smaller tracer neurobiotin (+1) (Table S1), suggesting that these channels are less permeable to negatively charged molecules and/or they have a reduce pore size.
Syndromic mutations in the N-terminus change the oligomerization compatibility of Cx26
Although Cx26 is co-expressed with Cx43 in cochlear cells (Forge et al., 2003) and skin keratinocytes (Arita et al., 2006; Beyer et al., 2001; Richard, 2000; Richard et al., 2002; Salomon et al., 1994) they are incompatible to form heteromeric channels (Gemel et al., 2004). Hence we aim to study whether oligomerization of these incompatible Cxs result in compatible interactions as consequence of the syndromic mutations located at the N-terminus of Cx26. HeLa cells expressing Cx43 were transiently transfected with Cx26-GFP or mutants. We found that Cx43 and Cx26-GFP localize at the same plaques, but segregate in different regions (Figure 2, a1–4), which is consistent with their oligomerization incompatibility (Gemel et al., 2004). No co-localization was observed between Cx43 and the non-syndromic mutants Cx26G12V-GFP (Figure 2a, 5–8), Cx26V37I-GFP or Cx26A40G-GFP (Figure S3). However, all syndromic mutants perfect co-localized with Cx43 in intracellular compartments and in GJ plaques (Figure 2, a9–20), suggesting the formation of heteromeric channels. Consistently, Cx sedimentation analysis showed that only co-expression of Cx43 with any syndromic mutant (Figure 2, e–g) promoted a shift in the sedimentation profiles of both proteins to overlap in a new oligomeric peak, which varied depending on the protein pair analyzed, confirming the formation of heteromeric channels between Cx43 and syndromic mutants. However, these heteromeric pairs produced non-functional GJCs, suggesting a total transdominant-negative effect of the syndromic mutants on Cx43 GJCs (Table S1). To rule out that the null GJC observed under previous conditions were not consequences of mutant Cx26 overexpression with respect to Cx43, we did transient transfection using different ratios of plasmid cDNAs carrying Cx43-RFP and Cx26-GFP or its mutant variants (1:1, 1:0.3 ratios, respectively) and then quantified the fluorescence for each fluorescent protein to demonstrate the relative reduction in the expression levels of each mutant Cx26 (Figure S4). Regardless of the ratio, we did not observed any junctional coupling in cell co-expressing Cx43-RFP with any Cx26 syndromic mutant (Table S3). Although we cannot estimate the molecular stoichiometry, our experiments suggest that the dominant negative effect of syndromic mutant over Cx43 GJC is strong. In addition, a previous report by Rouan and colleagues also showed that Cx26 with a syndromic mutation in TM1/EL1 is a negative dominant of Cx43 GJCs (Rouan et al., 2001), suggesting that interaction with Cx43 would be a general feature of syndromic Cx26 mutants.
Figure 2. Syndromic mutations change the oligomerization compatibility of Cx26.
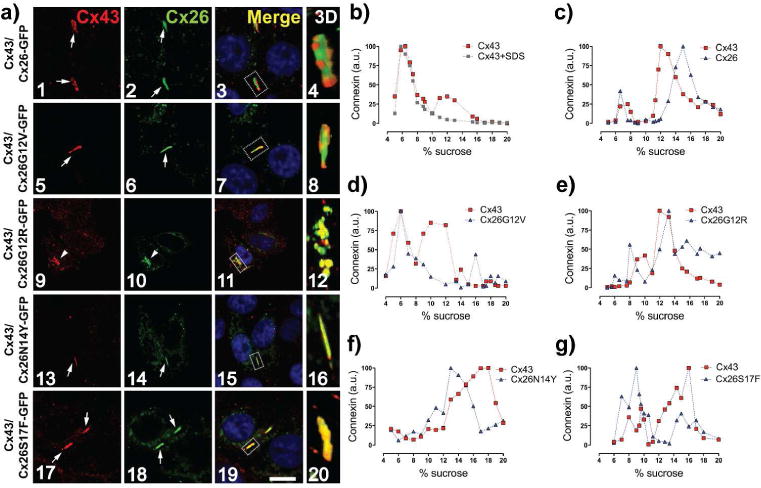
(a) Confocal images of HeLa cells co-expressing Cx43 (red labeling) with WT or mutants Cx26. Arrows point GJ plaques. Arrowheads show perinuclear staining. Nuclei were staining with DAPI. Dashed rectangles in the merged panels show regions of interest for 3D image projections. Bar=10 μm. (b,c) Cx43 and Cx26-GFP do not co-sediment in the same oligomeric fractions. Graphs represent the levels of Cx43 and Cx26GFP in each sucrose fractions in samples from HeLa cells that were transfected with Cx43 (b) or co-transfected with Cx43 and Cx26-GFP (c). Samples treated with SDS (red square). (d–g) Syndromic mutants co-sediment with Cx43 in new oligomeric fractions. Graphs show levels of Cx43 and mutant Cx26 in sucrose gradient fractions from cells co-transfected with Cx43 and the respective mutant. (n=4).
Syndromic mutations G12R and N14Y, but not S17F, produce homomeric HCs with gain of function
Because syndromic mutations are linked to gain in HCs function, we performed YO-PRO uptake assays to address HCs function. Under physiological extracellular Ca2+ concentration, HCs are mostly closed; however, cells that expressed the syndromic mutants Cx26G12R-GFP or Cx26N14Y-GFP showed uptake rates that were ~10 and ~4 times greater than what was observed in cells expressing Cx26-GFP, respectively, suggesting that both mutants increased the activity of homomeric HCs enhancing permeation to positively (+2) charged molecules (Figure 3, b–c). This result is consistent with previous reports indicating that Cx26G12R mutation produces high HC activity in the presence of physiological extracellular Ca2+ concentrations (Lee et al., 2009). To further examine the presence of HCs in the cell membrane, dye-uptake was evaluated in divalent cation-free Hanks’ balanced salt solution (DCF-HBSS), which enhances HC activity or in the presence of the lanthanum, a widely used HCs blocker. In this condition, YO-PRO uptake was significantly increased in cells that expressed Cx26 or all the mutants, except for Cx26S17F-GFP (Figure 3, a–d). Conversely, YO-PRO uptake was partially or completely reduced in the presence of lanthanum in wild type and mutant HCs. The variation in YO-PRO uptake was not related to differences in expression levels (Figure S1c) neither depends on the expression levels of the mutants as revealed by the correlation analysis of the relative-uptake rate/GFP-expression ratio (Figure S5a).
Figure 3. All syndromic mutants form hyperactive heteromeric HCs with Cx43.
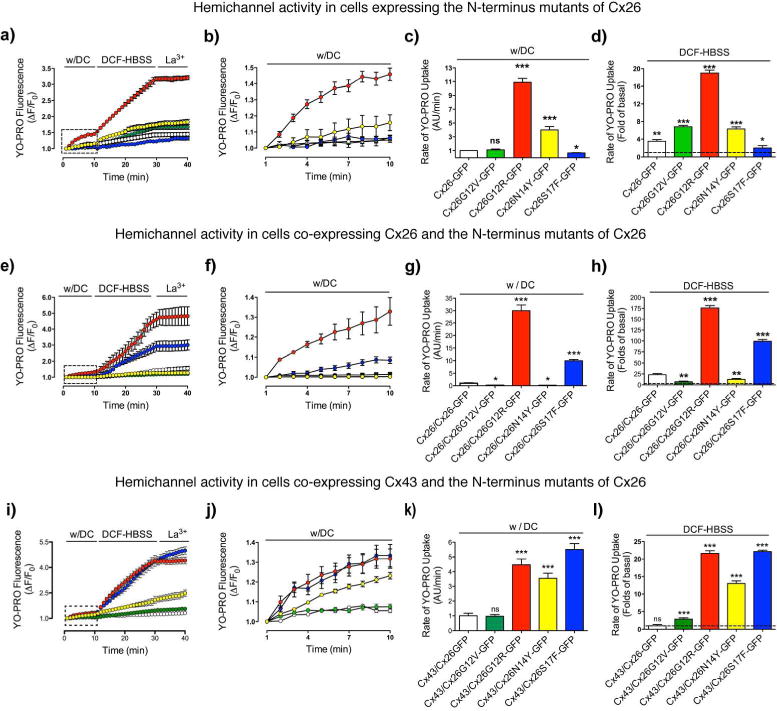
The functional state of HCs was determined in time-lapse experiments of YO-PRO uptake by HeLa cells expressing Cx26-GFP (open symbols), Cx26G12V-GFP (light blue), Cx26G12R-GFP (red), Cx26N14Y-GFP (yellow) or Cx26S17F-GFP (blue) (a–d) or by cells co-expressing these constructs with WT Cx26 (e–h) or Cx43 (i–l). a, e, i) Uptake under physiological extracellular divalent cation concentrations (w/DC) for 10 min, followed by a 20 min bath in Ca2+/Mg2+ free (DCF-HBSS). Then, 100 μM La3+ was added to block HCs. (b, f, j) Magnification of the area indicated by the rectangle in the graph (a,e,i). (c,d; g,h; k,l) The rate of uptake was determined calculating the uptake slope in cells bathed with physiological (c, g, k) or DCF-HBSS (d, h, l) solution. Data is presented as average ± SEM (n= 7). t-Test for unpaired data. *** p<0.001; **p<0.01; *p<0.05
A significant increase in YO-PRO uptake under normal extracellular Ca2+ concentration was recorded in cells that co-expressed Cx26 with Cx26G12R-GFP or Cx26S17F-GFP. Such increase was several times greater when compared to their respective homomeric counterparts or cells co-expressing Cx26 and Cx26GFP (Figure 3, e–h). Nevertheless, HCs composed by Cx26 and Cx26G12V-GFP or syndromic Cx26N14Y-GFP are less active than their respective homomeric conformations and Cx26/Cx26-GFP HCs, which would reflect the formation of inactive HCs. Correlation analysis indicates that levels of uptake observed were not related to differences in expression levels (Figure S5b).
Syndromic mutants form hyperactive heteromeric HCs with Cx43
We found no significant difference in YO-PRO uptake in cells co-expressing Cx43 with Cx26-GFP or with non-syndromic mutants Cx26G12V-GFP (Figure 3, j–k), Cx26V37I-GFP or Cx26A40G-GFP (Figure S3). In contrast, uptake mediated by heteromeric HC formed by all syndromic mutants with Cx43 showed higher uptake, even in the presence of divalent cations (Figure 3, j–k), which was boosted after the removal of extracellular Ca2+ (Figure 3, l), suggesting that these heteromeric HCs are still sensitive to Ca2+. The effects described here were not a consequence of different expression levels of Cx26 mutants, because they were observed even in cells that express low levels of syndromic mutant Cxs (Figure S4, S5c).
To unequivocally assess those Cx26 mutations that increase HCs activity when forming homomeric and heteromeric HCs, we performed electrophysiological studies using the two-electrode voltage clamp technique. Macroscopic currents were elicited by depolarizing pulses in Xenopus laevis oocytes expressing homomeric and heteromeric HCs, to examine the current-voltage relationship. Consistent with previous reports (Hansen et al., 2014; White et al., 1999), oocytes expressing human Cx43 did not exhibit detectable HCs macroscopic currents (Figure 4, a). The lack of HCs currents was also observed in oocytes expressing Cx26G12V, Cx26N14Y or Cx26S17F alone (Figure S6). To evaluate heteromeric HCs currents, equal amounts of cRNA were co-injected in oocytes to obtain 1:1 protein expression ratios. Co-expression of Cx43 with the non-syndromic G12V mutant did not elicited HCs currents (Figure 4; c,g), supporting our findings than these proteins do not interact. Conversely, co-expression of Cx43 with the syndromic Cx26G12R and Cx26S17F mutants promotes larger HCs currents (Figure 4; d,f). Lower HCs currents were detected in oocytes co-expressing Cx43 and the Cx26N14Y mutant (Figure 4, e), yet they were significantly larger at high positive potentials (>50 mV) than those observed for oocytes expressing Cx43 or Cx26N14Y alone. Co-expression of Cx26 and Cx43 showed HCs currents with similar kinetic to those previously described for Cx26 alone (Lopez et al., 2013a; Lopez et al., 2013b).
Figure 4. Heteromeric HCs composed by Cx43 and the syndromic mutants produce large macroscopic currents.
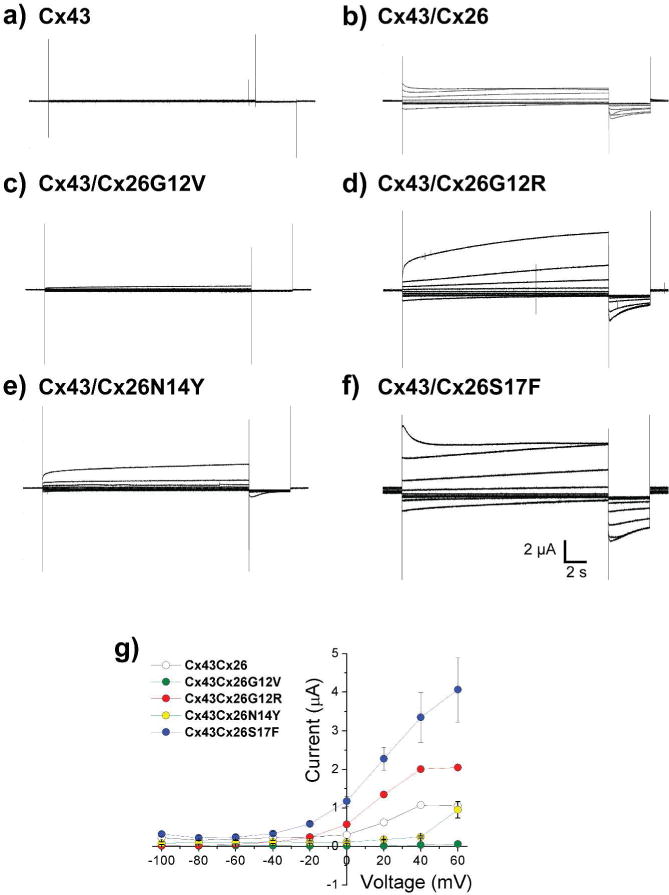
Membrane currents from oocytes expressing Cx43 alone or Cx43 with Cx26 mutants were recorded using the two-electrode voltage clamp technique. HC currents were activated in response to depolarizing voltage steps from a holding potential of −10 mV, and stepped in 20 mV increments from −100 mV to + 60 mV. Representative currents traces are showed for hCx43 alone (a) or for hCx43 coexpressed with wild type Cx26 (b), hCx26G12V (c), hCx26G12R (d), hCx26N14Y (e) or Cx26S17F (f). (g) Graphic depicted the current voltage relationship of macroscopic heteromeric HCs. The data represent mean ± SEM of at least five independent experiments.
Expression of heteromeric channels formed by Cx26 syndromic mutants and Cx43 promotes intracellular Ca2+ overload and ATP release
Compelling evidence has indicated the existence of a strong relationship between HC activity and increments in intracellular Ca2+ concentration and ATP release (Fiori et al., 2012; Saez et al., 2010; Stout et al., 2002), both are important for normal functioning of the cochlea and skin (Anselmi et al., 2008; Djalilian et al., 2006). To determinate changes in basal cytosolic Ca2+ levels from HeLa cells expressing homomeric and heteromeric HCs, cells loaded with the non-ratiometric probe Fura-Red were incubated in normal extracellular solution. Only cells that co-expressed Cx43 with syndromic mutants showed higher intracellular Ca2+ signal, which was almost saturated as indicated by treating these cells with 10 μM ionomycin that only produced a minor increase in the Ca2+ signal (Figure 5; a,b). Because Fura-Red has very limited sensitivity to Ca2+ concentration (above 1 μM), we also used Indo-1AM to more accurately estimate changes in the intracellular Ca2+ concentration among different mutants. HeLa cells expressing homomeric HCs, formed by Cx26G12R or Cx26N14Y mutants, but not Cx26S17F, exhibit higher levels of the intracellular Ca2+ when compared to HeLa cells expressing homomeric wild type Cx26 (Figure S7). Consistently with the Ca2+ overload reported by the Fura-Red experiments, cells co-expressing Cx43 with any syndromic mutants showed elevated intracellular Ca2+ concentration. In particular, cell co-expressing Cx43 and Cx26S17F display intracellular Ca2+ levels almost two folds than those measured in cell co-expressing the wild type counterparts (Figure 5, c).
Figure 5. Heteromeric hemichannels formed by syndromic mutants and Cx43 promote cellular Ca2+ overload and ATP release.
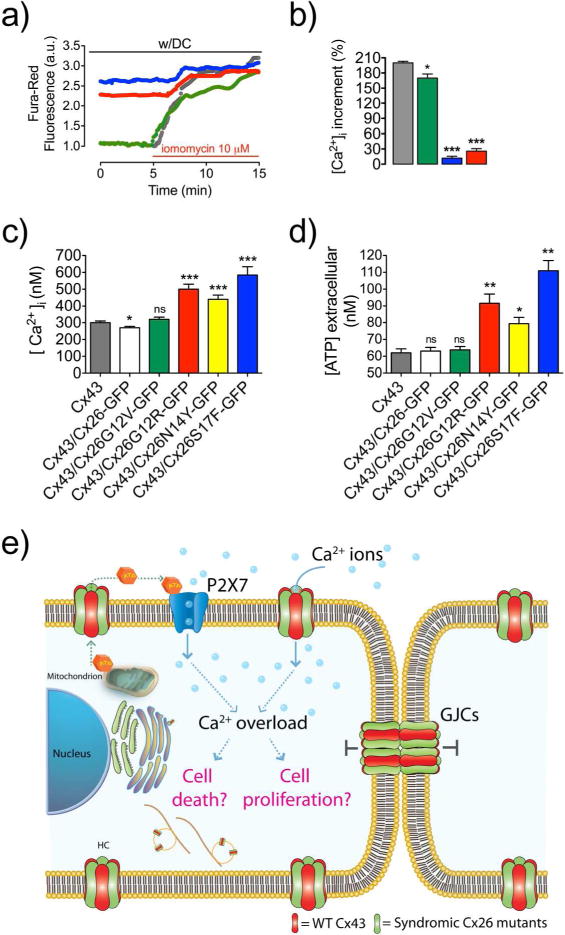
(a) Time course of Ca2+ signal obtained with probe Fura-Red in cells expressing Cx43 (gray), or co-expressing Cx43 with non-syndromic Cx26G12V-GFP (green), or syndromic Cx26G12R-GFP (red) and Cx26S17F-GFP (blue) before and after application of the Ca2+ ionophore ionomycin (100 μM). (b) Percentage of the Ca2+ signal increments induced by ionomycin. (c) Intracellular Ca2+ concentration in HeLa cells or in cells expressing Cx43, or co-expressing Cx43 with Cx26-GFP or Cx26S17F-GFP. (d) Measurement of ATP release during 10 min of cellular incubation of HeLa cells co-expressing Cx43 with the N-terminus mutants. Data is presented as average ± SEM (n= 20). t-Test for unpaired data. *** p<0.001; **p<0.01; *p<0.05 (e) Model of cell signaling induced by expression of aberrant heteromeric HCs but nonfunctional GJCs in KID syndrome.
Because extracellular ATP release correlates with HC activity (Stout et al., 2002), we explore HCs basal ATP release. As expected for hyperactive HCs, we found that ATP release augmented in cells expressing heteromeric HCs formed by Cx43 with any syndromic Cx26 mutants (Figure 5, d).
DISCUSSION
In the present work, we propose a possible molecular mechanism that might underlie the severity of KID syndrome by mutations at the N-terminus segment of Cx26. We demonstrate that Cx26 syndromic mutations produce aberrant Cx-Cx interactions leading to the formation of heteromeric HCs and GJCs with Cx43. These heteromeric HCs, but not GJCs, present exacerbate activity (gain in function) that may lead to dysfunction of paracrine signaling and Ca2+ overload (Figure 5, c; Table S4). Although KID syndromic mutations have been previously associate to hyperactive HCs, some syndromic mutations formed inactive homomeric HCs contradicting the previous mechanistic explanation for the disease. Because both, Cx26 and Cx43, are highly expressed in the human skin (Salomon et al., 1994), we propose that all syndromic mutations at the N-terminus could produce the functional paradox of hyperactive heteromeric HCs (Cx43/Cx26 mutants), but non-functional GJCs, which can enhanced severe skin damage in patients carrying these mutations (Mazereeuw-Hautier et al., 2007).
At the molecular level, our data demonstrate a critical role of the N-terminus in controlling Cx oligomerization. We found that mutations facing the cytoplasmic environment (i.e., Cx26N14Y and Cx26S17F) disrupt the proper formation of hexamers, but still allow the formation of intermediate Cx oligomers (Figure 1; g,h). The latter is consistent with the formation of Cx26 dimers that depend on TM1 (Jara et al., 2012) as the first step into the oligomerization process. However, mutations in the N-terminus might affect subsequent steps in hexamer formation, like the check point for oligomerization compatibility between Cxs. Our results support the idea that such check point is located in the Cx26 N-terminus, as it has been previously suggested for this Cx (Jara et al., 2012; Lagree et al., 2003; Martinez et al., 2009) and for acetylcholine receptors (Verrall and Hall, 1992). These results are consistent with the notion that an intact N-terminus is critical for the formation (Kyle et al., 2008) and proper functioning of GJCs, since this segment is part of the transjunctional voltage sensor (Verselis et al., 1994) and gating mechanism (Bargiello et al., 2012; Barrio et al., 1991; Maeda et al., 2009).
Consistent with previous studies (Lee et al., 2009) and with the YO-PRO uptake experiments, we observed that Xenopus oocytes expressing homomeric Cx26S17F mutants do not display HCs currents upon membrane depolarization. Suggesting that this mutation drastically decrease open probability and/or ionic conductance in homomeric HCs. Similarly, Cx43 when expressed in Xenopus oocytes does not show macroscopic HCs currents, but it does allow uptake of ethidium bromide (Hansen et al., 2014). Only few single Cx43 HCs with very low open probability have been detected in the whole-cell configuration from cells expressing high levels of Cx43 proteins at the plasma membrane (Contreras et al., 2003), yet significant dye uptake is observed over larger periods of time-video recording (Saez et al., 2005; Saez et al., 2010). Remarkably, we showed that the co-expression of Cx43 and Cx26S17F results in large HCs currents (Figure 4, f), which can only be explained by formation of heteromeric HCs between these two Cxs, as supported by co-localization and velocity sedimentation assays. Similar results were found when Cx26N14Y and Cx26G12R were co-expressed with Cx43. Conversely, the non-syndromic mutant Cx26G12V did not show significant levels of HCs currents or dye uptake when co-expressed with Cx43, consistently with the biochemical data showing that this mutation does not lead to the formation of heteromeric HCs with Cx43, but it does interacts with wild type Cx26, without the formation of hyperactive functional HCs. Similar results were observed for non-syndromic mutants Cx26V37I and Cx26A40G.
In KID syndrome several tissues are affected but the most extensive damage is observed at the epidermis, a tissue were Cx26 and Cx43 are the main Cxs expressed (Goliger and Paul, 1994; Salomon et al., 1994; Wiszniewski et al., 2000) playing a critical role in keratinocytes homeostasis maintenance and migration from the basal to cornified layer (Risek et al., 1992). Overexpression and ectopic expression of Cx26 are associated with psoriatic plaques and ATP release, delayed epidermal barrier recovery and inflammatory responses (Djalilian et al., 2006). In addition, a KID patient carrying mutation N14Y present also ectopic skin expression of Cx26 that co-localized with Cx43 (Arita et al., 2006). Additionally, P2X purinergic receptors are expressed in the skin and ATP signaling is important for differentiation and proliferative processes in keratinocytes as well as hyperplasia induced after skin barrier disruption (Denda et al., 2002). Our findings show that the expression of syndromic heteromeric HCs and GJC increases basal concentrations of intracellular Ca2+ and ATP release, are consistent with the notion that both signaling agents permeate through HCs (Fiori et al., 2012; Stout et al., 2002). Our results support the notion that KID syndrome involves keratinocyte homeostasis misregulation as consequences of leaky heteromeric HCs but non-functional GJC, which might promote ATP release, intracellular Ca2+ overload and cell death and/or proliferation (Figure 5, e). Hence this study supports the idea to use HC blockers as possible therapeutic agents for this disease.
MATERIALS AND METHODS
Molecular cloning
Wild type rat Cx26 cDNA (accession: NM_001004099) was cloned in the pcDNA3.1/CT-GFP-TOPO vector (Invitrogen). Site-directed mutagenesis was generated using the Quick-Change II kit (Agilent Technologies). For some experiments Cx26 or its variants contain hemagglutinin epitope tag appended to their C-terminus. Primers sequences are detailed in supplementary Table S2. DNA was sequenced by Macrogen Inc (Seoul, Korea), and analyzed using the “Vector NTI Advance” software (Invitrogen).
Immunofluorescence
Parental HeLa cells transfection and immunofluorescence were performed as previously described (Jara et al., 2012). Antibodies against Cx43, Cx26, and anti-HA (hemagglutinin) were from Invitrogen (Invitrogen). Images acquisition was performed with a Nikon Eclipse C1-Plus confocal microscope, Image processing and 3D reconstruction, were using the EZ-C1 and the NIS-Elements Viewer 4.0 software, respectively (Nikon Instruments Inc).
Velocity of sedimentation in sucrose gradients and immunoblotting
Transfected HeLa cells were harvested and lysed as indicated previously (Jara et al., 2012). Soluble fractions were subjected to sedimentation velocity through 5–20% (w/v) linear sucrose gradients, as previously described (Berthoud et al., 2001; Jara et al., 2012). To determine the percentage of sucrose at which Cx monomers sediment, Triton X-100 soluble fractions were treated with SDS to disaggregate the oligomers. After centrifugation for 22 h at 100,000g, 250 μL-fractions were collected and analyzed by immunoblotting as previously described (Jara et al., 2012; Martinez et al., 2002).
Measurement of GJC activity
Intercellular dye-coupling was performed as previously described (Jara et al., 2012; Martinez et al., 2002), and visualized with a Nikon TE-2000U inverted microscope (Nikon ACT-2U, Tokyo, Japan). The results are reported as incidence of coupling, that is, the percentage of microinjections that resulted in tracer diffusion to one or more adjacent cells. Electrophysiological recordings in pairs of HeLa cells were done with double whole-cell patch clamp essentially as described in (Jara et al., 2012). Total gap junctional conductance (gj) was calculated by using the formula (gj=Ij/[Vj cell 1 – Vj cell 2]).
Assessment of hemichannel activity
Cells plated on glass coverslips were bathed in physiological extracellular solution containing 200 nM of YO-PRO-1 uptake (375 Dalton/+2) and subjected to dye uptake time-lapse imaging as described previously (Contreras et al., 2002; Jara et al., 2012). The bath solution was replaced by Ca2+-Mg2+-free solution (DCF-HBSS, Invitrogen) containing 200 nM YO-PRO-1, and tracer uptake was recorded for 20 min, followed by the addition of 100 μM LaCl3 (Sigma-Aldrich) for an additional 10 min. Regions of interest were obtained with a 40X objective in a NIKON TE-2000U inverted microscope, and captured with a Nikon DS-2WBc fast-cooled monochromatic digital camera (8-bit) every 60 s. Image analysis and quantification of fluorescence intensity were performed with Image J (http://rsbweb.nih.gov/ij/).
Molecular cloning and HCs electrophysiology
Wild-type human Cx26 (hCx26) cDNA was purchased from OriGene and was subcloned in the pGEM-HA vector (Promega) for in vitro translation. Mutations of hCx26 were produced with Quick-Change II Site-Directed Mutagenesis kits (Agilent Technologies). DNA sequencing performed at the New Jersey Medical School Molecular Resource Facility confirmed the amino acid substitutions. Nhe1-linearized hCx26 wild type, Cx43 and mutant DNAs were transcribed in vitro to cRNAs using the T7 Ultra mMessage Machine kit (Ambion). Electrophysiological data were collected using the two-electrode voltage-clamp technique as in our previous studies (Lopez et al., 2013a).
Ca2+ imaging
Ionomycin-induced Ca2+ overload was measured by imaging of Fura-red acetoxymethylester (Invitrogen). Briefly, HeLa cells growing in coverslips were loaded with 15 μM Fura-red for 20 min at room temperature. Each coverslip was placed in a perfusion chamber mounted on an IX81 inverted epifluorescence microscope (Olympus) equipped with a 40x/1.4 NA oil-immersion objective. Images analysis was performed using the Excellent Pro software. The relative fluorescence ratio was calculated by dividing fluorescence (F) at each time point by the initial fluorescence value (F0).
Intracellular Ca2+ concentration was measured using Indo-1AM (Molecular Probes, Invitrogen) based on the standard ratio method (Grynkiewicz et al., 1985; Jacquemond, 1997). Briefly, HeLa cells growing in 25 mm coverslips were loaded with 5 μM Indo-1AM (dissolved in 20% pluronic acid in DMSO) for 30 min at 37°C in normal extracellular solution. Each coverslip was placed in a perfusion chamber mounted on the stage of a microscope (Diaphot 200-Nikon, Japan) equipped with a photomultiplier (Micron System, MS878). Indo-1 signals were amplified with an EPC 10-USB amplifier (HeKa Electronics). Analogue/digital signal conversion was performed with a DigiData 1200 Series card (Axon Instruments, Inc). The Patch Master program (HeKa Electronics) was used for data acquisition, and data analysis was carried out with the Igor6 software.
Extracellular ATP measurements
ATP release was assessed with the luciferin/luciferase based ATP Determination Kit (Molecular Probes, Invitrogen). Briefly, for each condition, 50,000 cells/well were seeded in a 24-well plaque. Twenty-four hours later, the culture medium was washed out and replaced with HBSS containing 1.8 mM Ca2+, and cells were incubated for 10 min at 37°C. Subsequently, 5 μL samples of extracellular milieu were mixed with 45 μL ATP-mix solution in a 96-well plaque. Accumulated ATP was determined using an Appliskan Luminometer (Thermo Electro Corporation) based on a calibration curve range from 1 nM to 1 μM ATP. Data was collected with the SkanIT software (Thermo Electro Corporation).
Supplementary Material
Acknowledgments
We thank Dr. José Ignacio García-Palacios for substantial comments on the manuscript, and Victoria Devia for technical support in confocal microscopy. This work was supported by the Fondecyt-1130855 to ADM, the Anillo ACT-1104 grant to CG and ADM, Fondecyt-1120802 to CG, Fondecyt-3150634 to IEG, CONICYT fellow and Beca Apoyo a la realización de Tesis Doctoral AT24121123 fellowship grant to IEG; Fondecyt-11100047 to PO, Fondecyt-1130652 and PFB16 Fundación Ciencia y Vida to TPA. The National Institutes of Health/National Institute of General Medical Sciences (grant RO1-GM099490) to J.E.C. The Centro Interdisciplinario de Neurociencias de Valparaíso is a Chilean Millennium Institute (P09-022-F).
Footnotes
AUTHOR CONTRIBUTIONS: I.E.G and A.D.M conceived the present study; I.E.G and A.D.M designed the research; I.E.G, J.M, O.J, R.C, P.O, A.P.M, J.R, performed research; T.P.A contributed to paper discussion and tool analysis; I.E.G, J.C.S, JEC and A.D.M analyzed data; I.E.G, J.C.S, C.G, JEC and A.D.M, wrote the paper.
CONFLICT OF INTEREST
Authors declare no conflict of interest
References
- Anselmi F, Hernandez VH, Crispino G, et al. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18770–5. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K, Akiyama M, Aizawa T, et al. A novel N14Y mutation in Connexin26 in keratitis-ichthyosis-deafness syndrome: analyses of altered gap junctional communication and molecular structure of N terminus of mutated Connexin26. The American journal of pathology. 2006;169:416–23. doi: 10.2353/ajpath.2006.051242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiello TA, Tang Q, Oh S, et al. Voltage-dependent conformational changes in connexin channels. Biochimica et biophysica acta. 2012;1818:1807–22. doi: 10.1016/j.bbamem.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio LC, Suchyna T, Bargiello T, et al. Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:8410–4. doi: 10.1073/pnas.88.19.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud VM, Montegna EA, Atal N, et al. Heteromeric connexons formed by the lens connexins, connexin43 and connexin56. European journal of cell biology. 2001;80:11–9. doi: 10.1078/0171-9335-00132. [DOI] [PubMed] [Google Scholar]
- Beyer EC, Gemel J, Martinez A, et al. Heteromeric mixing of connexins: compatibility of partners and functional consequences. Cell communication & adhesion. 2001;8:199–204. doi: 10.3109/15419060109080723. [DOI] [PubMed] [Google Scholar]
- Caceres-Rios H, Tamayo-Sanchez L, Duran-Mckinster C, et al. Keratitis, ichthyosis, and deafness (KID syndrome): review of the literature and proposal of a new terminology. Pediatr Dermatol. 1996;13:105–13. doi: 10.1111/j.1525-1470.1996.tb01414.x. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Saez JC, Bukauskas FF, et al. Gating and regulation of connexin 43 (Cx43) hemichannels. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11388–93. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras JE, Sanchez HA, Eugenin EA, et al. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci U S A. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denda M, Inoue K, Fuziwara S, et al. P2X purinergic receptor antagonist accelerates skin barrier repair and prevents epidermal hyperplasia induced by skin barrier disruption. The Journal of investigative dermatology. 2002;119:1034–40. doi: 10.1046/j.1523-1747.2002.19505.x. [DOI] [PubMed] [Google Scholar]
- Djalilian AR, McGaughey D, Patel S, et al. Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. The Journal of clinical investigation. 2006;116:1243–53. doi: 10.1172/JCI27186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori MC, Figueroa V, Zoghbi ME, et al. Permeation of calcium through purified connexin 26 hemichannels. The Journal of biological chemistry. 2012;287:40826–34. doi: 10.1074/jbc.M112.383281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge A, Becker D, Casalotti S, et al. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. The Journal of comparative neurology. 2003;467:207–31. doi: 10.1002/cne.10916. [DOI] [PubMed] [Google Scholar]
- Gemel J, Valiunas V, Brink PR, et al. Connexin43 and connexin26 form gap junctions, but not heteromeric channels in co-expressing cells. Journal of cell science. 2004;117:2469–80. doi: 10.1242/jcs.01084. [DOI] [PubMed] [Google Scholar]
- Gerido DA, DeRosa AM, Richard G, et al. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. American journal of physiology Cell physiology. 2007;293:C337–45. doi: 10.1152/ajpcell.00626.2006. [DOI] [PubMed] [Google Scholar]
- Goliger JA, Paul DL. Expression of gap junction proteins Cx26, Cx31.1, Cx37, and Cx43 in developing and mature rat epidermis. Dev Dyn. 1994;200:1–13. doi: 10.1002/aja.1002000102. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. The Journal of biological chemistry. 1985;260:3440–50. [PubMed] [Google Scholar]
- Hansen DB, Braunstein TH, Nielsen MS, et al. Distinct permeation profiles of the connexin 30 and 43 hemichannels. FEBS letters. 2014;588:1446–57. doi: 10.1016/j.febslet.2014.01.036. [DOI] [PubMed] [Google Scholar]
- Jacquemond V. Indo-1 fluorescence signals elicited by membrane depolarization in enzymatically isolated mouse skeletal muscle fibers. Biophysical journal. 1997;73:920–8. doi: 10.1016/S0006-3495(97)78124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara O, Acuna R, Garcia IE, et al. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Molecular biology of the cell. 2012;23:3299–311. doi: 10.1091/mbc.E11-12-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell DP, Wilgoss AL, Richard G, et al. Connexin mutations associated with palmoplantar keratoderma and profound deafness in a single family. European journal of human genetics: EJHG. 2000;8:141–4. doi: 10.1038/sj.ejhg.5200407. [DOI] [PubMed] [Google Scholar]
- Kwon T, Harris AL, Rossi A, et al. Molecular dynamics simulations of the Cx26 hemichannel: evaluation of structural models with Brownian dynamics. The Journal of general physiology. 2011;138:475–93. doi: 10.1085/jgp.201110679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle JW, Minogue PJ, Thomas BC, et al. An intact connexin N-terminus is required for function but not gap junction formation. J Cell Sci. 2008;121:2744–50. doi: 10.1242/jcs.032482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagree V, Brunschwig K, Lopez P, et al. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. Journal of cell science. 2003;116:3189–201. doi: 10.1242/jcs.00604. [DOI] [PubMed] [Google Scholar]
- Lee JR, Derosa AM, White TW. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. The Journal of investigative dermatology. 2009;129:870–8. doi: 10.1038/jid.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levit NA, Mese G, Basaly MG, et al. Pathological hemichannels associated with human Cx26 mutations causing Keratitis-Ichthyosis-Deafness syndrome. Biochimica et biophysica acta. 2012;1818:2014–9. doi: 10.1016/j.bbamem.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez W, Gonzalez J, Liu Y, et al. Insights on the mechanisms of Ca(2+) regulation of connexin26 hemichannels revealed by human pathogenic mutations (D50N/Y) The Journal of general physiology. 2013a;142:23–35. doi: 10.1085/jgp.201210893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez W, Liu Y, Harris AL, et al. Divalent regulation and intersubunit interactions of human Connexin26 (Cx26) hemichannels. Channels. 2013b;8 doi: 10.4161/chan.26789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, et al. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Martinez AD, Acuna R, Figueroa V, et al. Gap-junction channels dysfunction in deafness and hearing loss. Antioxidants & redox signaling. 2009;11:309–22. doi: 10.1089/ars.2008.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez AD, Hayrapetyan V, Moreno AP, et al. Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circulation research. 2002;90:1100–7. doi: 10.1161/01.res.0000019580.64013.31. [DOI] [PubMed] [Google Scholar]
- Martinez AD, Maripillan J, Acuna R, et al. Different domains are critical for oligomerization compatibility of different connexins. Biochem J. 2011;436:35–43. doi: 10.1042/BJ20110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, et al. Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. The British journal of dermatology. 2007;156:1015–9. doi: 10.1111/j.1365-2133.2007.07806.x. [DOI] [PubMed] [Google Scholar]
- Richard G. Connexins: a connection with the skin. Exp Dermatol. 2000;9:77–96. doi: 10.1034/j.1600-0625.2000.009002077.x. [DOI] [PubMed] [Google Scholar]
- Richard G, Rouan F, Willoughby CE, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. American journal of human genetics. 2002;70:1341–8. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risek B, Klier FG, Gilula NB. Multiple gap junction genes are utilized during rat skin and hair development. Development. 1992;116:639–51. doi: 10.1242/dev.116.3.639. [DOI] [PubMed] [Google Scholar]
- Rouan F, White TW, Brown N, et al. trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. Journal of cell science. 2001;114:2105–13. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- Saez JC, Retamal MA, Basilio D, et al. Connexin-based gap junction hemichannels: gating mechanisms. Biochimica et biophysica acta. 2005;1711:215–24. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JC, Schalper KA, Retamal MA, et al. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res. 2010;316:2377–89. doi: 10.1016/j.yexcr.2010.05.026. [DOI] [PubMed] [Google Scholar]
- Salomon D, Masgrau E, Vischer S, et al. Topography of mammalian connexins in human skin. The Journal of investigative dermatology. 1994;103:240–7. doi: 10.1111/1523-1747.ep12393218. [DOI] [PubMed] [Google Scholar]
- Sanchez HA, Mese G, Srinivas M, et al. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. The Journal of general physiology. 2010;136:47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segretain D, Falk MM. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochimica et biophysica acta. 2004;1662:3–21. doi: 10.1016/j.bbamem.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Stout C, Goodenough DA, Paul DL. Connexins: functions without junctions. Curr Opin Cell Biol. 2004;16:507–12. doi: 10.1016/j.ceb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, et al. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. The Journal of biological chemistry. 2002;277:10482–8. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Verrall S, Hall ZW. The N-terminal domains of acetylcholine receptor subunits contain recognition signals for the initial steps of receptor assembly. Cell. 1992;68:23–31. doi: 10.1016/0092-8674(92)90203-o. [DOI] [PubMed] [Google Scholar]
- Verselis VK, Ginter CS, Bargiello TA. Opposite voltage gating polarities of two closely related connexins. Nature. 1994;368:348–51. doi: 10.1038/368348a0. [DOI] [PubMed] [Google Scholar]
- White TW, Deans MR, O’Brien J, et al. Functional characteristics of skate connexin35, a member of the gamma subfamily of connexins expressed in the vertebrate retina. The European journal of neuroscience. 1999;11:1883–90. doi: 10.1046/j.1460-9568.1999.00607.x. [DOI] [PubMed] [Google Scholar]
- Wiszniewski L, Limat A, Saurat JH, et al. Differential expression of connexins during stratification of human keratinocytes. The Journal of investigative dermatology. 2000;115:278–85. doi: 10.1046/j.1523-1747.2000.00043.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.