

ABSTRACT
The inhibition of STAT3 may exert cell-autonomous cytotoxic and cytostatic effects, yet may also stimulate anticancer immunosurveillance through the neutralization of immunosuppressive circuitries. In addition, STAT3 inhibition in cancer cells may stimulate the type 1 interferon response elicited by anthracyclines. This pathway results in an enhanced chemotherapy-associated anticancer immune response with improved therapeutic efficacy. Hence, combination therapies that include immunogenic cell death (ICD) inducers and STAT3 inhibitors can be envisaged.
A certain number of successful chemotherapeutic agents have the property to induce ICD. ICD is characterized by at least four hallmarks: (i) the exposure of calreticulin on the plasma membrane following the translocation of this protein from the lumen of the stressed endoplasmic reticulum to the cell surface; (ii) the release of ATP from the cells to the extracellular space, a process that can be favored by premortem autophagy; (iii) the induction of type 1 interferons that act in an autocrine or paracrine fashion on the common interferon α/β receptor (IFNAR) to stimulate a type 1 interferon response; and (iv) the postmortem release of HMGB1 from the nuclei of dead cells.1-3 These four hallmarks determine specific interactions with immune cells, at multiple levels, namely (i) the transfer of tumor antigens due to the interaction of the ‘eat-me’ signal calreticulin with suitable receptors on antigen-presenting cells; (ii) the action of extracellular ATP on purinergic receptors such as P2Y2 an P2RX7 to facilitate the recruitment of myeloid cells into the proximity of dying cells (via P2Y2) and to activate the inflammasome in dendritic cells (via P2RX7); (iii) the production of type 1-interferon-induced chemokines including CXCL9 and CXCL10 that act on CXCR3 to attract T lymphocytes into the tumor bed; and (iv) the ligation of toll-like receptor 4 (TLR4) expressed by immature dendritic cells by HMGB1, stimulating their capacity of tumor antigen presentation.3-5 Although it is possible that the aforementioned list of ICD hallmarks of ICD is still non-exhaustive, it does allow for the identification of ICD inducers in compound libraries by virtue of screening program designed to measure calreticulin exposure, ATP secretion and HMGB1 release.1,6
Driven by the consideration that STAT3 inhibition might be useful for anticancer immunotherapy:7-9, we recently explored the possibility that STAT3 inhibition might be combined with ICD inducers (such as anthracyclines) to improve the outcome of chemotherapy. We found that combination of the anthracycline mitoxantrone with the STAT3 inhibitor Stattic indeed provided a synergistic anticancer effect and that tumor growth reduction by the combination regimen (mitoxantrone plus Stattic) entirely depended on the contribution of T lymphocytes, meaning that its activity was lost when it was used to treat tumor-bearing nu/nu mice (which lack thymus-dependent T cells). In a subsequent round of experiments, we used MCA205 cancer cells from which we deleted the Stat3 gene by CRISPR/Cas9 technology, driven by the consideration that this would constitute a way to specifically suppress STAT3 in cancer cells without interfering with its function in other cell types including immune cells. Stat3−/− MCA205 cancers again responded better to chemotherapy with mitoxantrone than wild type (WT) control tumors, supporting the idea that it was indeed STAT3 contained in the tumor cells that was the target of Stattic. Moreover, retransfection of Stat3−/− MCA205 cells with Stat3 (but not with a mutant, inactive form of Stat3) reversed their chemosensitivity.10 Of note, after mitoxantrone treatment, Stat3−/− MCA205 cancers induced a much stronger anticancer immune response than their WT counterparts. Thus, the chemotherapy-induced infiltration of the tumors by CD11c+CD86+ dendritic cells and by CD3+CD8+ T lymphocytes was much more pronounced in Stat3−/− than in WT tumors.10 Moreover, the frequency of γ/δ T cells, CD4+ α/β T cells and CD8+ α/β T cells among tumor-infiltrating leukocytes was only enhanced by the dual experimental manipulation (Stat3 knockout plus doxorubicin injection), which also caused an increase in capacity of tumor-infiltrating CD4+ and CD8+ α/β T lymphocytes to produce IFNγ and that of γ/δ T cells to produce IL-17 (Fig. 1).
Figure 1.
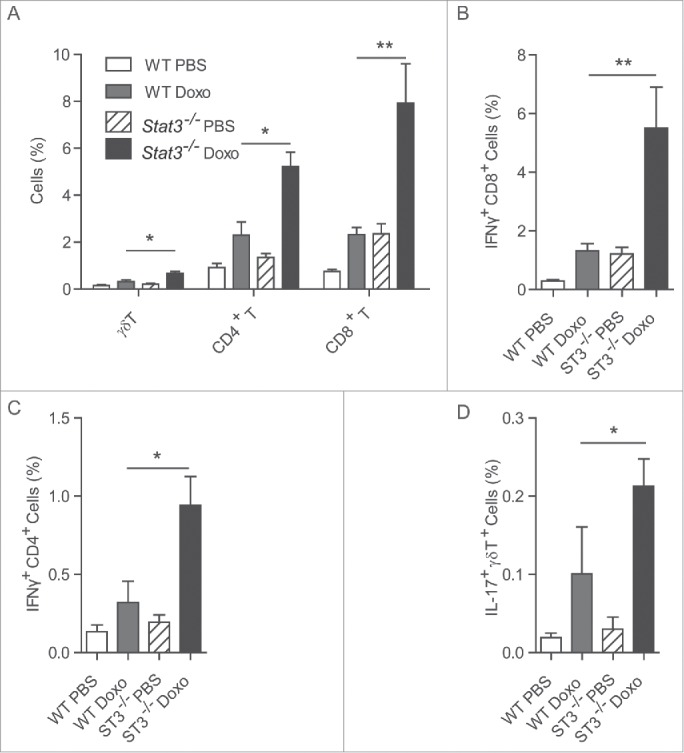
Cytofluorometric analysis of tumor-infiltrating T lymphocytes. MCA205 fibrosarcoma cells that were either wild type (WT) or Stat3−/− were inoculated into histocompatible C57Bl/6 mice. Once palpable the tumors were either injected with doxorubicin or PBS as a vehicle control. Seven days after intratumoral injection of chemotherapy, the fibrosarcomas were retrieved and digested with 0.4 Wünsch units/mL Liberase TL (Roche) and 200 U/mL DNase I (Calbiochem), cultured for 4 h in the presence of phorbol 12-myristate 13-acetate (20 ng/mL) + ionomycin (1 µg/mL) + brefeldin A (3µg/mL) and then stained to determine the expression of CD3, CD4+, CD8+, γ/δ T cell receptor on the cell surface, as well as that of interferon-γ (IFNγ) and interleukin-17 (IL-17) within the cytoplasm. (A). Proportion of γ/δ T cells, CD4+ α/β T cells and CD8+ α/β T cells among viable (Vivid yellow−) cells. (B). Proportion of IFNγ-producing CD8+ α/β T cells. (C). Proportion of IFNγ-producing CD4+ α/β T cells. (D). Proportion of IL-17-producing T cells among γ/δ T cells. Values are means ± standard error of the mean (n = 5 per group). Asterisks mark significant differences (*, p < 0.05, **, p < 0.01,) between groups, as determined by the Student t test.
We also explored whether the deletion of Stat3 might affect any among the hallmarks of ICD. To our surprise, Stat3 deletion failed to sensitize the tumor cells to cell death induction by anthracyclines in vitro, reduced calreticulin exposure and did not affect ATP secretion or HMGB1 release. Stat3 deletion only stimulated one of the hallmarks of ICD, namely the production of type 1 interferons and that of multiple type 1 interferon-inducible genes including CXCL9 and CXCL10. Accordingly, neutralization of IFNAR or CXCR3 with suitable antibodies reversed the chemosensitivity of Stat3−/− tumors in vivo.10 This finding establishes the cause–effect relationship between STAT3 inhibition, stimulation of a type 1 interferon response and improved outcome of cancer chemotherapy.
STAT3 inhibition has been proposed to mediate anticancer effects by multiple mechanisms including cell-autonomous effects (knowing that STAT3 is a potent oncogene and that many cancers are ‘addicted’ to STAT3 and hence require this transcription factor for their survival and proliferation), as well as immunological effects (knowing that STAT3 is expressed by immunosuppressive cell types including myeloid-derived suppressor cells). Our observations suggest that STAT3 inhibition may also trigger the immunostimulatory induction of the type 1 interferon response, thus reinforcing one of the hallmarks of ICD. It has been shown that breast cancer that fail to mount a type 1 interferon response have a poor prognosis.3 Hence, it may be interesting to explore whether such tumors exhibit signs of STAT3 activation and then to explore the possibility of treating them with a combination of chemotherapy and STAT3 inhibitors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
Heng Yang is supported by PACRI. Yuting Ma received fundings from the “Chinese National Thousand Young Talents Program” and Chinese Academy of Medical Sciences (2015RC310003). Guido Kroemer and Laurence Zitvogel are supported by the Ligue contre le Cancer (équipes labelisées); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); the Swiss Bridge Foundation, ISREC and the Paris Alliance of Cancer Research Institutes (PACRI).
References
- 1.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N et al.. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3:e955691; PMID:25941621; http://dx.doi.org/ 10.4161/21624011.2014.955691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stoll G, Enot D, Mlecnik B, Galon J, Zitvogel L, Kroemer G. Immune-related gene signatures predict the outcome of neoadjuvant chemotherapy. Oncoimmunology 2014; 3:e27884; PMID:24790795; http://dx.doi.org/ 10.4161/onci.27884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C et al.. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20:1301-9; PMID:25344738; http://dx.doi.org/ 10.1038/nm.3708 [DOI] [PubMed] [Google Scholar]
- 4.Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M et al.. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012; 337:1678-84; PMID:23019653; http://dx.doi.org/ 10.1126/science.1224922 [DOI] [PubMed] [Google Scholar]
- 5.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 2013; 39:74-88; PMID:23890065; http://dx.doi.org/ 10.1016/j.immuni.2013.06.014 [DOI] [PubMed] [Google Scholar]
- 6.Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, Baracco EE, Galluzzi L, Zitvogel L, Kepp O et al.. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014; 3:e28473; PMID:25050214; http://dx.doi.org/ 10.4161/onci.28473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Marino G, Lachkar S, Senovilla L et al.. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell 2012; 48:667-80; PMID:23084476; http://dx.doi.org/ 10.1016/j.molcel.2012.09.013 [DOI] [PubMed] [Google Scholar]
- 8.Concha-Benavente F, Srivastava RM, Ferrone S, Ferris RL. EGFR-mediated tumor immunoescape: The imbalance between phosphorylated STAT1 and phosphorylated STAT3. Oncoimmunology 2013; 2:e27215; PMID:24501692; http://dx.doi.org/ 10.4161/onci.27215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kida H, Ihara S, Kumanogoh A. Involvement of STAT3 in immune evasion during lung tumorigenesis. Oncoimmunology 2013; 2:e22653; PMID:23482587; http://dx.doi.org/ 10.4161/onci.22653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Pietrocola F, Zhou H, Zitvogel L, Ma Y, Kroemer G. Stat3 inhibition enhances the therapeutic efficacy of immunogenic chemotherapy by stimulating type 1 interferon production by cancer cells. Cancer Res 2015; 75; 3812-22; PMID:26208907; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-1122 [DOI] [PubMed] [Google Scholar]