

ABSTRACT
Two major challenges facing cancer immunotherapy are the relatively low therapeutic efficacy and the potential side effects. New drug delivery system and efficient drug combination are required to overcome these challenges. We utilize an alginate hydrogel system to locally deliver 2 FDA-approved drugs, celecoxib and programmed death 1 (PD-1) monoclonal antibody (mAb), to treat tumor-bearing mice. In two cancer models, B16-F10 melanoma and 4T1 metastatic breast cancer, the alginate hydrogel delivery system significantly improves the antitumor activities of celecoxib (CXB), PD-1 mAb, or both combined. These effects are associated with the sustained high concentrations of the drugs in peripheral circulation and within tumor regions. Strikingly, the simultaneous dual local delivery of celecoxib and PD-1 from this hydrogel system synergistically enhanced the presence of CD4+inteferon (IFN)-γ+ and CD8+IFN-γ+ T cells within the tumor as well as in the immune system. These effects are accompanied with reduced CD4+FoxP3+ regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) in the tumor, reflecting a weakened immuosuppressive response. Furthermore, this combinatorial therapy increases the expression of two anti-angiogenic chemokines C-X-C motif ligand (CXCL) 9 and CXCL10, and suppresses the intratumoral production of interleukin (IL)-1, IL-6, and cycloxygenase-2 (COX2), suggesting a dampened pro-tumor angiogenic and inflammatory microenvironment. This alginate-hydrogel-mediated, combinatorial therapy of celecoxib and PD-1 mAb provides a potential valuable regimen for treating human cancer.
KEYWORDS: Alginate hydrogel, angiogenesis, antitumor immunity, cancer immunotherapy, celecoxib, effector T cells, inflammation, MDSC, PD-1, 20 blockade, Treg
Abbreviations
- PD-1
programmed death 1
- mAb
monoclonal antibody
- CXB
celecoxib
- IFN
interferon
- Tregs
regulatory T cells
- MDSCs
myeloid derived suppressor cells
- CXCL
C-X-C motif ligand
- IL
interleukin
- COX2
cycloxygenase-2
- TILs
tumor-infiltrating lymphocytes
Introduction
Ineffective immune responses are commonly found in patients with established tumors as tumor cells develop various immunosuppressive mechanisms coping with immune surveillance, leading to tumor immune evasion.1 Immunotherapy has proven to be a promising way to improve cancer treatment outcomes.2 However, the overall results of cancer immunotherapeutic trials suggest an urgent need of further improving current immunotherapy.3,4 The therapeutic efficacy of the immunotherapy is thought to be affected by the immunosuppressive network and chronic inflammation milieu.5,6 Simultaneously targeting these two aspects within a tumor would likely improve the treatment efficacy.
The PD-L1 and PD-1 signaling pathway is an important component of immunosuppressive networks.1,7 Programmed death 1 (PD-1) is highly expressed on tumor-infiltrating lymphocytes (TILs). By expressing PD-1 ligands, PD-L1 and PD-L2, tumor cells and antigen presenting cells suppress T cell immune responses via PD-1/PD-L1 interaction, leading to T cell apoptosis, anergy, or exhaustion.8,9 Antibody blockade of PD-1 converts anergic T cells into functional effector T cells,10 and has been shown to enhance antitumor immune responses in patients with advanced melanoma, lung carcinoma, and renal cell carcinoma.11-13 However, PD-1 blockade can increase the expression of pro-tumor inflammatory cytokines, which potentially offsets the therapeutic effects of PD-1 blockade.14,15 Thus, inhibiting PD-1-blockade-induced inflammation might maximize the therapeutic effects of PD-1 blockade.
Celecoxib (CXB), 4-[5-(4-methylphenyl)-3 -trifluoromethyl-1H-pyrazol-1-yl] benzenesulfonamide, is a specific inhibitor of cycloxygenase-2 (COX2) and widely used to treat autoimmune diseases.16,17 Interestingly, celecoxib has been shown to have certain antitumor activities in various human cancers.18,19 Given its anti-inflammatory property, celecoxib may subvert the pro-tumor inflammatory effects induced by PD-1 blockade.
In this study, we have used an alginate hydrogel to simultaneously and locally deliver anti-PD-1 mAb and celecoxib to treat tumor-bearing mice. The dual delivery of anti-PD-1 mAb and celecoxib from the alginate polymer matrices elicits a potent and sustained antitumor effect, which is accompanied with enhanced effector T cell immunity, reduced immunosuppression, and lessened inflammation and tumor angiogenesis. Thus, simultaneously targeting the immune suppressive network and inflammation via a biomaterial-based delivery system may be a novel anti-cancer regime and warrant further investigation and clinical trials in patients with cancer.
Results
Alginate hydrogel individual delivery of celecoxib and anti-PD-1 mAb enhances their antitumor effects
Alginate hydrogels are used as a drug and/or cell delivery vehicle due to its good biocompatibility and other features.20 Using a B16-F10 melanoma mouse model, we first determined the optimal encapsulation dosages of celecoxib and anti-PD-1 mAb for the alginate hydrogel delivery system with respect to the antitumor effects. Because of celecoxib's low water solubility, the celecoxib powder was mixed into the alginate solution by sonication before the gelation. Three dosages of celecoxib (10, 25, and 50mg/kg) delivered by the alginate hydrogels injected subcutaneously into the vicinity of the melanoma were compared. While 10mg/kg celecoxib mediated marginal antitumor effects, the 25mg/kg and 50mg/kg dosages significantly inhibited the tumor growth and extended the survival time in a similar way (Fig. 1A). The dosage of 25mg/kg celecoxib was thus chosen for further experiments.
Figure 1.
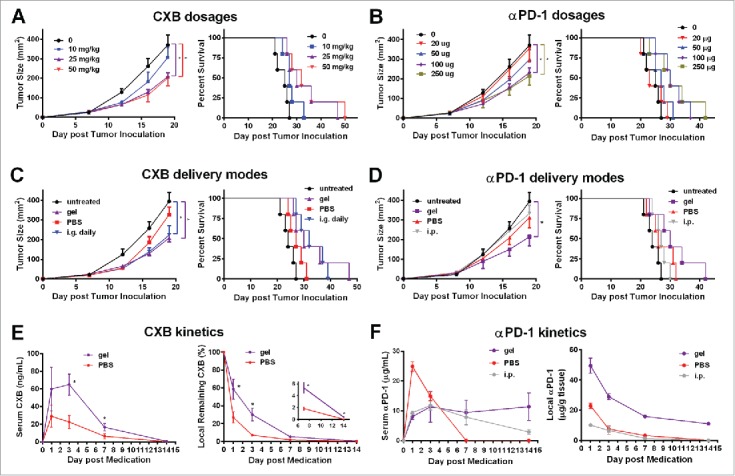
Alginate hydrogel delivery enhances the antitumor efficacy of celecoxib (CXB) and anti-PD-1 mAb (αPD-1). C57BL/6 mice received the different treatments at Day 7 after the inoculation of 1.0 × 105 B16-F10 cells. (A and B) Quantification of the tumor sizes (left) and the survival percentage (right) of the animals treated with the hydrogels encapsulated with the indicated dosages of CXB (A) or αPD-1 (B). (C) Quantification of the tumor sizes (left) and the survival percentage (right) of the animals receiving CXB (25mg/kg) delivered in three ways: one-time subcutaneous injection of the CXB-encapsulated hydrogel (gel), one-time subcutaneous injection of PBS-dissolved CXB (PBS), and daily intragastrical administration (i.g. daily). (D) Quantification of the tumor sizes (left) and the survival percentage (right) of the animals receiving αPD-1 (100µg per animal) in three ways: one-time subcutaneous injection of the αPD-1-encapsulated hydrogel (gel), one-time subcutaneous injection of PBS-dissolved αPD-1 (PBS), and one-time αPD-1 intraperitoneal injection (i.p.). n = 5 animals per group in A–D. (E) The CXB serum concentrations (left) and CXB amount within the tumors (right) in the animals treated with CXB (25mg/kg) delivered via gel or PBS. (F) αPD-1 concentrations in serum (left) and within the tumors (right) in the animals treated with 100µg αPD-1 given in three ways described in (D). n = 3 animals per time point. *P < 0.05, Student's t-tests. Error bars represent the standard error of the mean.
Next we tested four dosages of anti-PD-1 mAb (20, 50, 100, and 250 µg/mouse) in the B16-F10 melanoma-bearing mice. The treatments with the hydrogels encapsulated with anti-PD-1 mAb at the dosage of 20μg/mouse or 50μg/mouse did not significantly inhibit tumor growth and extend survival time (Fig. 1B). In contrast, the 100μg/mouse and 250μg/mouse dosages drastically reduced the tumor growth and improved the survival (Fig. 1B). 100μg/mouse of anti-PD-1 mAb was then chosen for further studies.
We next compared the therapeutic effects of celecoxib given in three different ways for treating the B16-F10 bearing mice: (a) the daily intragastrical administration (i.g. daily), (b) one-time direct subcutaneous injection of PBS-dissolved celecoxib in the region adjacent to the tumor (PBS), and (c) one-time subcutaneous injection of the alginate hydrogel with encapsulated celecoxib in the region adjacent to the tumor (gel). The untreated group and the subcutaneous injection of PBS-dissolved celecoxib group showed no obvious differences in tumor growth (Fig. 1C). As expected, the daily intragastrical administration (i.g.) of 25mg/kg celecoxib, an effective dosage for animal experiments,21 inhibited the tumor growth (Fig. 1C). This inhibition was similarly achieved by the one-time injection of the hydrogel carrying celecoxib at the same dosage (25mg/kg) (Fig. 1C). More importantly, this hydrogel-mediated celecoxib delivery significantly extended the survival time compared to the daily intragastrical administration and the subcutaneous injection of PBS-dissolved celecoxib (Fig. 1C). These observations indicate that the hydrogel enables celecoxib to reach more potent antitumor effects.
Similarly, we compared the therapeutic effects in the melanoma-bearing animals receiving the treatments with the same dosage of anti-PD-1 mAb (100μg/mouse) delivered in three different ways: (a) the intraperitoneal administration (i.p.) of anti-PD-1 mAb,14 (b) one-time direct subcutaneous injection of PBS-dissolved anti-PD-1 mAb in the region adjacent to the tumor (PBS), and (c) one-time subcutaneous injection of the alginate hydrogel with encapsulated anti-PD-1 mAb in the region adjacent to the tumor (gel). Compared to the untreated controls, the intraperitoneal treatment and the direct subcutaneous injection of PBS-dissolved anti-PD-1 mAb did not inhibit the tumor growth (P > 0.05) (Fig. 1D). In contrast, the subcutaneous injection of the alginate hydrogel with encapsulated anti-PD-1 mAb in the region adjacent to the tumor reduced tumor size by 50% at Day 18 and significantly increased the animal survival (Fig. 1D). Together, the results indicate that the hydrogel delivery system improves the therapeutic efficacy of anti-PD-1 mAb.
Next we examined the release profiles of celecoxib and anti-PD-1 mAb delivered from the hydrogel system by measuring the serum and intratumoral concentrations of these two drugs. The serum concentration of celecoxib delivered by the subcutaneous injection of the hydrogel into the vicinity of the tumors was maintained within a range from 10 to 70ng/mL for a week, which was approximately 2-fold higher at all the time points examined than that in the animals receiving the subcutaneous injection of the same amount of PBS-mixed celecoxib (Fig. 1E). In these two groups, the complete clearance of the serum celecoxib took slightly more than 14 days, significantly longer than the oral administration resulting in a 24 h transient serum high concentration (Fig. S1). We next measured celecoxib at the injection site by recovering the tumor and adjacent tissue containing the hydrogel at different time points. Compared to the local injection of PBS-suspended celecoxib, the alginate hydrogel preserved significantly higher percentages of celecoxib within the local region for 2 weeks (Fig. 1E). By Day 14, 0.4% of the encapsulated celecoxib still remained locally, which equaled to the concentration of 3μg per gram of tumor tissue weight, an effective concentration according to previous studies.22 Similarly, the hydrogel delivery of anti-PD-1 mAb led to relatively high serum concentrations of anti-PD-1 for 2 weeks, ranging from 8 to 11μg/mL (Fig. 1F), which was in contrast to the one-time intraperitoneal administration resulting in high serum concentration for one week, and the one-time subcutaneous injection causing a burst release that was cleared within a week (Fig. 1F). Importantly, the hydrogel delivery also maintained a significantly higher antibody concentra-tion in the tumor local region (Fig. 1F). Together, these results indicate that the hydrogel-mediated local delivery is an effective approach of maintaining the high levels of celecoxib or anti-PD-1 mAb in serum and the tumor microenvironment.
Hydrogel dual delivery of celecoxib and anti-PD-1 mAb augments their individual antitumor effects
Next we assessed the antitumor effects of the hydrogel-mediated dual delivery of celecoxib and anti-PD-1 mAb. The treatments were given via the subcutaneous injection at the site immediately adjacent to the tumor on the 7th day after 2.5 × 104 B16-F10 cells were inoculated (Fig. 2A). Compared to the blank hydrogel treated animals, the hydrogel individually delivering celecoxib or anti-PD-1 mAb resulted in an approximate 50% or 67% reduction in tumor size by Day 22, respectively. Strikingly, the dual delivery of celecoxib and anti-PD-1 mAb led to a 90% reduction, indicating a significant enhancement (Fig. 2A). Notably, 56% (n = 5/9) of the treated mice had no visible tumors during the subsequent 3-month follow-up, suggesting a complete tumor regression (Fig. 2B). To further validate the effectiveness of this hydrogel-mediated celecoxib and anti-PD-1 mAb dual delivery therapy, we injected 1.0 × 105 B16-F10 cells into mice, 4-fold more than the above experiment. This dual delivery exhibited the consistent tumor growth inhibition (Fig. S2A). Compared to the individual delivery of celecoxib or anti-PD-1 mAb, the dual delivery markedly extended the mouse survival (Fig. S2B). More importantly, the dual delivery extended the mouse survival much more significantly than the administration of simply combined celecoxib and anti-PD-1 mAb without a hydrogel (Fig. 2B, Fig. S2B), revealing an important role of the hydrogel vehicle in improving the combined antitumor efficacy of celecoxib and anti-PD-1 mAb, likely due to the slow release of these drugs from the hydrogel (Fig. 1E and F). These results indicate that the combination of celecoxib and anti-PD-1 mAb delivered by a hydrogel efficiently inhibits tumor growth, extends mouse survival, and potentially leads to complete tumor regression.
Figure 2.
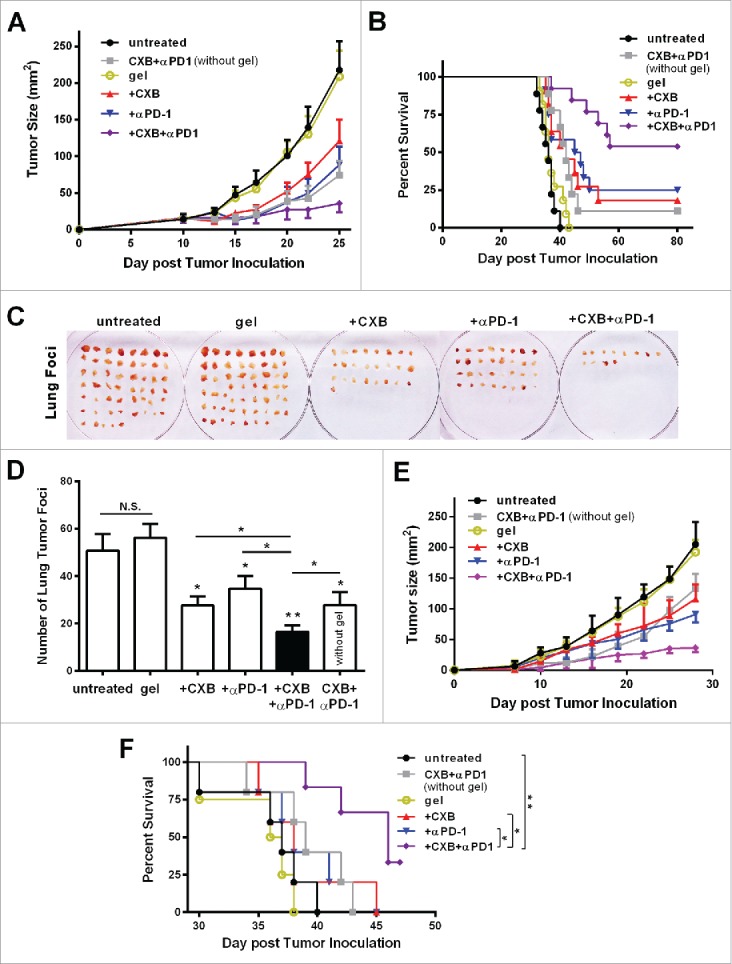
Simultaneous delivery of celecoxib and anti-PD-1 mAb augments their individual inhibitory effects on tumor growth and metastasis. (A and B) C57BL/6 mice received the different treatments at Day 7 after the inoculation of 2.5 × 104 B16-F10 cells. Quantification of the tumor sizes (A) and the survival percentage (B) over time in the animals receiving the non-hydrogel combined therapy of CXB and anti-PD-1 mAb (CXB + αPD-1), the blank hydrogel treatment (gel), and the treatments with the hydrogels delivering CXB (+ CXB), anti-PD-1 mAb (+ αPD-1), or both (+ CXB + αPD-1). n = 9–12 animals per group. Four independent experiments were performed. (C–F) BALB/c mice received the different treatments at Day 7 after the inoculation of 1.0 × 106 4T1 cells. The representative images (C) and quantification (D) of the pulmonary metastatic nodules isolated at Day 32 after tumor cell inoculation from the 4T-1-breast-cancer bearing mice receiving the indicated treatments as in (A) and (B) at Day 7 after tumor inoculation. The primary tumor sizes (E) and the survival percentage (F) in the corresponding treatment groups described in (C). n = 4–6 animals per group. Three independent experiments were performed. *P < 0.05, **P < 0.01, N.S., not significant, Student's t-tests. The asterisk without a line underneath indicates the comparison to the blank hydrogel group. Error bars represent the standard error of the mean.
We further examined the antitumor effects of this hydrogel/celecoxib/anti-PD-1 mAb system in the 4T1 breast tumor metastasis model inoculated with the large number of 4T1 cells (1.0 × 106 per mouse). Consistent with the B16-F10 melanoma model, the hydrogel dual delivery of celecoxib and anti-PD-1 mAb caused a drastic reduction in the number of metastatic lung foci and the primary tumor size, and an increase in survival, compared to the hydrogel individual delivery of celecoxib or anti-PD-1 mAb, and the combination of these two drugs without a gel (Fig. 2C-F).
Taken together, the results from both the melanoma model and the metastatic breast cancer model demonstrate that when used in combination and delivered via a hydrogel celecoxib and anti-PD-1 mAb can generate potent antitumor effects.
Dual delivery of celecoxib and anti-PD-1 mAb elicits a synergistic antitumor immunity
Next we studied the potential mechanisms by which the hydrogel-mediated dual delivery of celecoxib and anti-PD-1 mAb produced potent antitumor effects. PD-1 interacting with its ligand PD-L1 leads to T cell exhaustion.2,12 IFN-γ is a cytokine crucial for T-cell activation and effector function.23 As antitumor T cell priming occurs in tumor draining lymph nodes,24 we first examined IFN-γ-expressing T cells in tumor draining lymph nodes. Compared to the animals receiving the blank hydrogel treatment, the treatments using the hydrogels carry-ing either celecoxib or anti-PD-1 mAb did not significantly increase the percentage of IFN-γ-expressing CD4+ T cells in the tumor draining lymph nodes (Fig. 3A). In sharp contrast, the simultaneous delivery of celecoxib and anti-PD-1 mAb from the hydrogel dramatically increased the percentage of IFN-γ-expressing CD4+ T cells (Fig. 3A). We also examined IFN-γ positive CD8+ T cells. While the treatment with the hydrogel carrying celecoxib had the similar percentage of IFN-γ-expressing CD8+ T cells to the blank hydrogel treatment, the treatment with the hydrogel carrying anti-PD-1 mAb resulted in a mild increase, 1.8 folds (Fig. 3B). The hydrogel dual delivering celecoxib and anti-PD-1 mAb led to a 6.4-fold increase, reaching 49% (Fig. 3B). These results reveal a synergistic effect between celecoxib and anti-PD-1 mAb on upregulating IFN-γ+ subsets in both CD4+ and CD8+ T cells in the draining lymph nodes.
Figure 3.
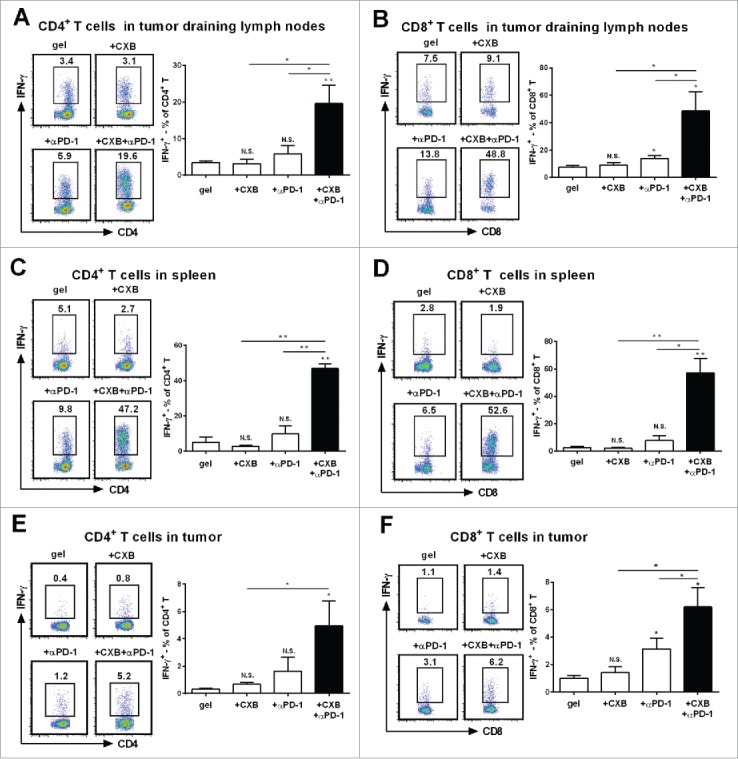
Synergistic effects of dually delivered celecoxib (CXB) and anti-PD-1 mAb (αPD-1) on increasing the presence of INFγ-expressing CD4 and CD8 T cells. C57BL/6 mice received the treatments with the blank hydrogel (gel) and the hydrogels delivering CXB (+ CXB), anti-PD-1 mAb (+ αPD-1), or both (+ CXB + αPD-1) at Day 7 after the inoculation of 1.0 × 105 B16-F10 cells. (A and B) The representative flow cytometric analysis images (left) and the corresponding quantification (right) of IFN-γ positive CD4+ T cells (A) and CD8+ T cells (B) from the tumor draining lymph nodes of the mice 7 days after the treatments. (C and D) The similar T cell analyses for spleens. (E and F) The similar T cell analyses for tumor tissues. Each column represents 3 independent experiments (n = 6–8 mice per group per experiment). *P < 0.05, **P < 0.01, N.S., not significant, Student's t-tests. The asterisk or “N.S.” without a line underneath indicates the comparison to the blank hydrogel group. Error bars represent the standard error of the mean.
We next analyzed the spleen T lymphocytes. Compared to the blank hydrogel treatment, the individual delivery of celecoxib or anti-PD-1 mAb from the hydrogels did not obviously increase IFN-γ+ subsets in CD4+ and CD8+ T cells (Fig. 3C and D). Strikingly, the dual delivery of celecoxib and anti-PD-1 mAb from the hydrogels increased IFN-γ-expressing CD4+ and CD8+ T cells by 8.1 folds and 21.3 folds, respectively (Fig. 3C, D), indicating a strong synergistic interaction between celecoxib and anti-PD-1 mAb in promoting IFN-γ+ subsets in CD4+ and CD8+ T cells in spleens. Together, these results suggest that the combinatorial usage of celecoxib and anti-PD-1 mAb can upregulate IFN-γ+ subsets and improve T-cell viability in a synergistic manner within the immune system.
To eliminate tumor cells, effector T cells have to infiltrate tumor tissue.1 We then analyzed whether the synergistic improvement on IFN-γ-expressing T cells also occurred within the tumor microenvironment. The blank hydrogel group had the low level presence of IFN-γ-expressing CD4+ (Fig. 3E) and CD8+ T cells (Fig. 3F) within the tumor. The individual delivery of celecoxib or anti-PD-1 mAb resulted in a slight increase of IFN-γ-expressing CD4+ and CD8+ T cells (Fig. 3E, F). In contrast, the dual delivery of celecoxib and anti-PD-1 mAb caused a 5-6-fold increase in IFN-γ-expressing CD4+ T cells (Fig. 3E) and IFN-γ-expressing CD8+ T cells (Fig. 3F), suggesting an enhanced antitumor immunity in the tumor microenvironment. Together, these results indicate that celecoxib and anti-PD-1 mAb synergistically promote local T cell effector presence.
Dual delivery of celecoxib and anti-PD-1 mAb abrogates the immunosuppressive mechanisms
Tumor-infiltrating regulatory T cells (Tregs) suppress antitumor immunity and weaken therapeutic efficacy of immunotherapy.25 We further studied the role of the combinatorial therapy in Tregs. Compared to the blank hydrogel treatment, the individual delivery of celecoxib reduced Tregs by 25%, whereas anti-PD-1 mAb delivery did not change the percentage of Tregs (Fig. 4A). Surprisingly, when celecoxib and anti-PD-1 mAb were delivered simultaneously, a 50% decrease in Tregs was observed (Fig. 4A). The ratio between intratumoral effector T cells (IFN-γ+CD8+) and Tregs, reflecting the therapeutic efficacy of tumor immunotherapy,26 was 3-4-fold higher in the animals receiving the dual delivery than those receiving the individual delivery (Fig. 4B). These results indicate that the combined therapy effectively targets Tregs and tilts the balance to effector T cells.
Figure 4.
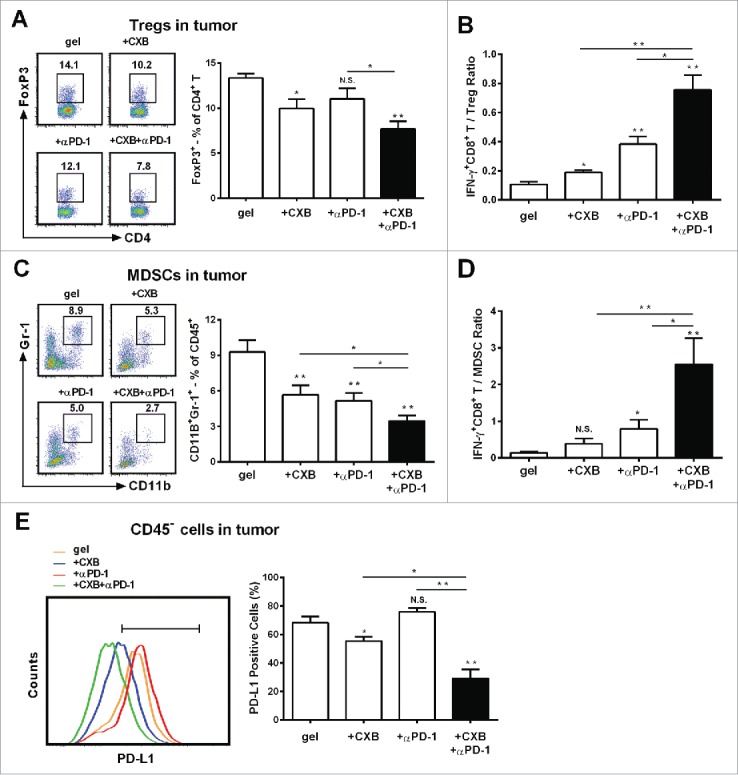
The enhanced effects of celecoxib (CXB) and anti-PD-1 mAb (αPD-1) on decreasing the presence of intratumoral Tregs, MDSCs, and PD-L1 positive tumor cells. C57BL/6 mice received the treatments with the blank hydrogel (gel) and the hydrogels delivering CXB (+ CXB), anti-PD-1 mAb (+ αPD-1), or both (+ CXB + αPD-1) at Day 7 after the inoculation of 1.0 × 105 B16-F10 cells. Single-cell suspensions made from digested tumor tissues were subject to flow cytometric analyses 7 days after the treatments. (A) The representatives flow cytometric analysis images (left) and the corresponding quantification (right) of FoxP3+ analyses of CD4+ T cells. (B) The ratios of IFNγ+CD8+ T cells to Tregs. Each column represents three independent experiments (n = 6–8 animals per group per experiment). (C) The representative flow cytometric analysis images (left) and the corresponding quantification (right) of MDSCs (CD11b+Gr-1+) in CD45+ cells. (D) The ratios of IFN-γ+CD8+ T cells to MDSCs. (E) The representative flow cytometric analysis images (left) and the corresponding quantification (right) of PD-L1 analyses within the CD45− cells. Each column represents three independent experiments (n = 8–12 animals per group per experiment). *P < 0.05, **P < 0.01, N.S., not significant, Student's t-tests. The asterisk or “N.S.” without a line underneath indicates the comparison to the blank hydrogel group. Error bars represent the standard error of the mean.
In addition to Tregs, MDSCs potently dampen antitumor immunity.27 We assessed intratumoral MDSCs in our therapeutic regimen. In agreement with the previous studies showing that celecoxib and anti-PD-1 mAb inhibit MDSCs,14,28 the individual delivery of celecoxib or anti-PD-1 mAb decreased the amount of MDSCs by 39% and 47%, respectively, within the tumors in comparison to the blank hydrogel treatment (Fig. 4C). The hydrogel dual delivery of celecoxib and anti-PD-1 mAb led to a further reduction by 65% (Fig. 4C). Remarkably, the ratio between IFN-γ+ CD8+ T cells and MDSCs within the tumors in the animals receiving the dual delivery of celecoxib and anti-PD-1 mAb was 3-6-fold higher than those in the individually delivered treatment (Fig. 4D). These observations indicate that the combined usage of celecoxib and anti-PD-1 mAb greatly reduces MDSCs in the tumor microenvironment.
Immunotherapy-associated inflammation upregulates the expression of PD-L1 in tumors, a transmembrane protein acting as the ligand for PD-1 to mediate cancer evasion.29,30 We next assessed PD-L1 expression within tumors. Compared to the blank hydrogel control, the hydrogel-delivered celecoxib reduced PD-L1 positive cells by 13%, whereas anti-PD-1 mAb delivered from the hydrogels did not significantly affect PD-L1 expression (Fig. 4E). However, when celecoxib was added into the hydrogel carrying anti-PD-1 mAb, PD-L1 positive cells were reduced by 57% in comparison to the control. These results suggest that the blockade of PD-1 significantly enhances celecoxib's suppressive role in the presence of PD-L1 positive cells within tumors.
Dual delivery of celecoxib and anti-PD-1 mAb inhibits angiogenesis and inflammation
Antitumor effects of celecoxib are in part ascribed to its ability of inhibiting vascularization.21 We evaluated the angiostatic effects of the hydrogel-based treatments by staining CD31, a marker for the endothelium of microvessels. While celecoxib delivered from the hydrogel inhibited microvessel density and total surface by 38% in comparison to the blank hydrogel treatment, anti-PD-1 mAb delivered from the hydrogel did not affect microvessel density and total surface (Fig. 5A, B and C). However, the combined delivery of celecoxib and anti-PD-1 mAb further reduced microvessel density and total surface by 56% compared to the blank hydrogel control, suggesting that PD-1 blocking enhances celecoxib's angiostatic effect (Fig. 5A, B and C). The CXCR3 ligands, monokine induced by IFN-γ (Mig)/CXCL9 and IFN-inducible protein 10 (IP-10)/CXCL10, are the key angiostatic factors inhibiting tumor vasculature.31-33 To examine whether these cytokines were involved in the angiostatic mechanisms, the melanoma tumors receiving the treatments for 1 week were recovered for gene expression analysis. Compared to the control, the expression of these two chemokines, CXCL10 and CXCL9, was increased in the tumors receiving the dual delivery of celecoxib and anti-PD-1 mAb (Fig. 5D-G). This similar change was also observed for IFN-γ production (Fig. 5H).
Figure 5.
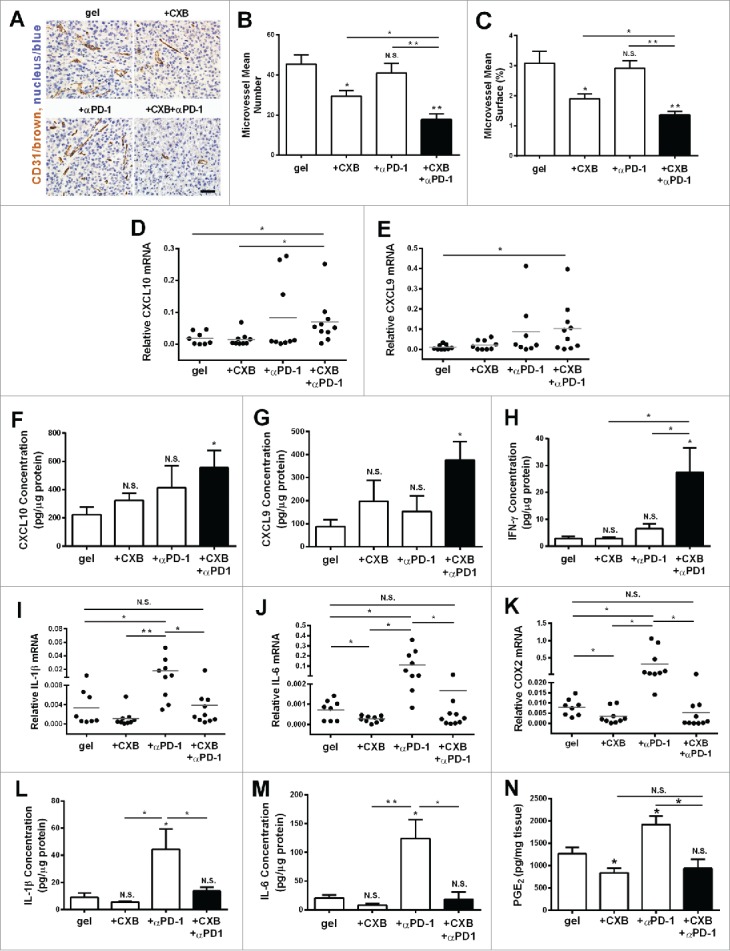
The inhibition on angiogenesis and inflammation in the tumors resulting from the dual delivery of celecoxib (CXB) and anti-PD-1 mAb (αPD-1). C57BL/6 mice received the treatments with the blank hydrogel (gel) and the hydrogels delivering CXB (+ CXB), anti-PD-1 mAb (+ αPD-1), or both (+ CXB + αPD-1) at Day 7 after the inoculation of 1.0 × 105 B16-F10 cells. The tumors were surgically taken from the mice 7 d after the treatments. (A) The representative images of blood vessel CD31 immunohistochemical staining for the tumors. (B) Quantification of the number of microvessels. (C) Quantification of the microvessel surface percentage. (D–N) The relative mRNA levels of CXCL10 (D), CXCL9 (E), IL-1β (I), IL-6 (J), COX2 (K), the protein levels of CXCL10 (F), CXCL9 (G), IFN-γ (H), IL-1β (L) and IL-6 (M) and PGE2 concentrations (N) within the tumors in the corresponding treatment groups. *P < 0.05, **P < 0.01, N.S., not significant, Student's t-tests. The asterisk or “N.S.” without a line underneath indicates the comparison to the blank hydrogel group. Error bars represent the standard error of the mean.
PD-1 blockade treatment increases the expression of pro-inflammatory cytokines.14,15 We next examined the expression of typical inflammatory molecules COX2, IL-1β, IL-6, iNOS and TNF-α, and of the anti-inflammatory molecule IL-10, and VEGF. While anti-PD-1 mAb delivery alone did not change the mRNA levels of iNOS, TNF- α, IL-10 and VEGF (data not shown), and the mRNA levels of COX2, IL-1β, IL-6, were increased (Fig. 5I-K). This increase was completely abolished when celecoxib was delivered simultaneously with anti-PD-1 mAb from the hydrogel (Fig. 5I-K). Similar results were observed in the protein levels of IL-1β and IL-6 (Fig. 5L and M). We assessed the level of PGE2 in the tumor specimens. The gel-delivered celecoxib alone significantly inhibited the production of PGE2 in tumors, in line with specific inhibitory role of celecoxib on COX2. Of note, the anti-PD-1 mAb treatment caused an increase of PGE2, which was completely abolished by celecoxib (Fig. 5N). These observations provide the additional evidence justifying the combined utilization of these two drugs for treating tumors.
Given that celecoxib is one of nonsteroidal anti-inflammatory drugs (NSAIDs) that inhibit COX1 and COX2, we tested another NSAID, SC-560, a specific inhibitor of COX1.34 In B16 melanoma and 4T1 breast cancer models, SC-560 did not inhibit tumor growth in vivo (Fig. S3A and B). Notably, the inclusion of SC-560 did not enhance the inhibitory effect of anti-PD-1 mAb on tumor growth (Fig. S3A and B). Similarly, the inclusion of SC-560 to anti-PD-1 mAb treatment did not yield synergistic effects on IFNγ+CD8+ T cells and Treg cells in the B16-F10 melanoma, compared to the combination of celecoxib and anti-PD-1 mAb (Fig. S3C and D). These results demonstrate that the inhibition of COX2, but not COX1, enhances the antitumor effect of anti-PD-1 mAb treatment.
Discussion
PD-1 blockade has been used in combination with various therapeutic agents to improve immunotherapeutic effects, such as another immunosuppressive blocking antibody cytotoxic T-lymphocyte antigen-4 (CTLA-4) mAb,14 the immune stimulating factor IL-2,35 the blocking antibody of human epidermal growth factor receptor-2 (HER-2 / ErbB-2),36 and radiotherapy.37,38 In this study, we have explored two strategies to improve therapeutic efficacy of the high-profile PD-1 blocking mAb treatment and the potential side effects associated with PD-1 blockade: (a) an alginate hydrogel delivery system to deliver therapeutic agents to tumor local regions; (b) in combination with celecoxib.
The alginate hydrogel is chosen due to its good biocompatibility, biodegradation, nontoxicity and fully demonstrated safety20 and the fact that various alginate gel formulations have been used in clinical products.39,40 The hydrogel delivery of celecoxib greatly improves its absorption and efficacy as evidenced by the high and sustained levels of celecoxib in serum and tumor microenvironment, providing an effective alternative to bypass celecoxib's poor bioavailability when administered orally due to its low water solubility.41 Celecoxib is an FDA approved, non-steroidal anti-inflammatory drug for treating arthritis. Although celecoxib is thought to be useful for cancer therapy given its anti-inflammatory nature,21,28 its antitumor effects on established tumors were poor.42 Our work reveals that the alginate hydrogel delivery system improves celecoxib's antitumor activity, offering a potential way of broadly utilizing it in anti-cancer therapy. Similarly, the application of the hydrogel delivery system also enhances the therapeutic effect of anti-PD-1 mAb. The hydrogel delivery decreases the effective dosage required for cancer treatment and maintains an over 2-week high level of anti-PD-1 mAb in serum, and more importantly, in tumor local regions, a major place where anti-PD-1 mAb protects cytotoxic T lymphocytes from immunosuppressive factors produced by cancer and cancer-associated cells. This dosage reduction and the long sustained high concentration would bring various benefits including avoiding frequent administration, minimizing potential toxicity of PD-1 mAb systemic administration demonstrated by other studies,43,44 and reducing the cost of antibody treatment.
We have further validated the alginate hydrogel delivery system in the combined therapy of celecoxib and PD-1 blocking mAb. Hydrogel dual delivered celecoxib and PD-1 mAb significantly improve local and systemic antitumor immunity as evidenced by the synergistically increased CD4+ and CD8+ effector T cells in spleens, tumor draining lymph nodes, and tumors. This synergy is accompanied by reduced Tregs and MDSCs in the tumor microenvironment, which may be explained by several possible mechanisms. Celecoxib inhibits COX2, which blocks the CXCL12-CXCR4 pathway and reduces the recruitment of MDSCs to the cancer region.28 Meanwhile, prostaglandin (PG) E2, a COX2 product, increases Tregs,45,46 which would be partly abrogated by celecoxib in our regimen. Further, when combined with radiotherapy, anti-PD-1 mAb reduces Tregs and MDSCs due to apoptosis induced by TNF-α from CD8+ T cells.38 Although unconfirmed, this might be another contributing mechanism underlying Tregs and MDSCs reduction observed in our study. More importantly, the reduction in Tregs and MDSCs shifts the immune balance toward antitumor effects.14,47 Taken together, the synergistic antitumor effects of PD-1 mAb and celecoxib combined therapy may be largely attributed to the removal of immunosuppressive mechanisms and the rescue of exhausted T cells.
The relief on immunosuppression mediated by PD-1 blockade is often accompanied by the upregulation of the inflammatory gene expression, which is thought to offset the therapeutic effects of PD-1 blockade.14,15 The combinatorial use of celecoxib with PD-1 mAb completely suppresses the upregulation of the inflammatory genes, including IL-1β and IL-6, both of which play critical roles in promoting tumor progression.48,49 This observation provides the additional justification for the combined utilization of these two drugs for treating tumors. Further, celecoxib coordinated with anti-PD-1 mAb promotes the expression of two anti-angiogenic chemokines, CXCL9 and CXCL10. While the angiogenesis inhibition resulted from the hydrogel delivery of celecoxib alone is independent of CXCL9 and CXCL10, the angiogenesis inhibition observed for the dual delivery of celecoxib and anti-PD-1 mAb is likely dependent on both CXCL9 and CXCL10 as their mRNA and protein levels were significantly upregulated. Given IFN-γ's known role in promoting the expression of CXCL9 and CXCL10,50 this upregulation may be explained by the synergistically increased IFN-γ+ T cells. Thus, targeting cancer inflammation and angi-ogenesis contributes to the enhanced and sustained antitumor effects of the celecoxib and PD-1 mAb combined therapy.48
In summary, our study has demonstrated that the alginate hydrogel dual delivered celecoxib and PD-1 mAb therapy elicits the potent antitumor effects by remodeling immune, inflammatory and angiogenic microenvironments within the tumors. Given that celecoxib and PD-1 mAb are the FDA-approved agents, our work provides a scientific rationale for possible clinical trials in treating patients with cancer.
Materials and methods
Cell lines and animals
Mouse B16-F10 melanoma cell line and mouse 4T-1 breast cancer cell line were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences. B16-F10 cells were cultured in Dulbecco's Modified Eagle medium (Gibco). 4T-1 cells were cultured in RPMI 1640 medium (Gibco), supplemented with 10% (v/v) fetal bovine serum (Gibco) and 1% antibiotics (penicillin and streptomycin, Hyclone) with 5% CO2 at 37°C. Both cell lines were verified to be mycoplasma free and showed appropriate pathologic morphology during the experiments. These cells did not undergo further testing. C57BL/6 mice (6–8-week old male) and BALB/c (6–8-week old female) were purchased from Beijing HFK Bioscience Co. Ltd.
Encapsulation of celecoxib and anti-PD-1 mAb
The low molecular weight (LMW) alginate was generated by gamma irradiation on high molecular weight (HMW) LF 20/40 alginate (FMC Biopolymer) at 5.0 Mrad for 4 h with a cobalt-60 source. Both LMW and HMW alginates were diluted to 2% (w/v) in PBS and then filtered. To prepare gels, the premixed alginate solution (LMW:HMW = 3:1, w/w) was cross-linked with aqueous slurries of a calcium sulfate solution using two syringes connected through a connector as described previously.51 Before gelation, the anti-PD-1 monoclonal antibody (RMP1-14, Bioxcell) was added into the alginate solution, while the celecoxib powder (LC Labs) was suspended in the alginate solution by ultrasonicating.
In vivo tumor therapy
C57BL/6 mice were injected in the flank subcutaneously at Day 0 with 1.0 × 105 or 2.5 × 104 B16-F10 cells. The mice were treated at Day 7 with a total volume of 200µL of the alginate hydrogel containing celecoxib, anti-PD-1 mAb, or both. The hydrogel was injected through a 25G needle (BD biosciences) into the region adjacent to the B16-F10 melanoma. The tumor size was measured and indicated as length × width (mm2).
For the lung metastatic model of 4T-1 breast cancer cells, BALB/c mice were injected in the fourth mammary pad at Day 0 with 1.0 × 106 4T-1 cells. The alginate hydrogels encapsulated with celecoxib, anti-PD-1 mAb or both were injected to the vicinity of the tumors at Day 7 after tumor cell inoculation. To examine the pulmonary metastasis, the mice were sacrificed at Day 32. The lungs were isolated and fixed in 4% formalin overnight to distinguish white tumor colonies from yellowish lung parenchyma. The metastatic foci on the surface of the lungs were isolated, counted and imaged with a regular digital camera (IXUS 155, Canon).
High-performance liquid chromatography-mass spectrometry (hplc-ms) for quantification of celecoxib concentration
For the analysis of celecoxib in mouse serum, 20µL of 2.5µg/mL pioglitazone hydrochloride (Sigma) and 600µL HPLC grade acetonitrile (Sigma) were added to 200µL serum. The resulting mixture was thoroughly mixed using a vortex for 2 min and then centrifuged at 12,000rpm for 10 min. 50µL supernatant was injected into the HPLC unit. To analyze celecoxib at tumor sites, the tumors were weighed and ground in PBS 8 times the volume of the tumor. 150µL of the mixture was extracted three times with 450µL ethylacetate. The supernatants were combined and dried completely at 65°C. The residue was dissolved in 900µL of acetonitrile. 50µL of the solution filtered through a 0.22µm microporous membrane (Shimadzu) was analyzed using the HPLC unit with LC-30AD pump (Shimadzu) and a SIL-30AC autosampler (Shimadzu) coupled to an API QTRAP 5500 triple quadrupole mass spectrometer equipped with an electrospray ionization source (AB/MDSSciex, Ontario) with the C18 MG III column (100mm × 2.1mm, 3μm, 6L science). The mobile phase consisted of acetonitrile (35% volume), 20mM ammonium acetate containing 0.2% formic acid (65% volume) at an isocratic flow rate of 0.3mL/min. The injection volume of 10μL was analyzed at the wavelength 380nm with approximately 5min run time. A calibration curve was obtained from the celecoxib reference solutions ranging from 0.5ng/mL to 50µg/mL. Good linear correlation was achieved over the entire concentration range.
ELISA for quantification of αPD-1 concentration
C57BL/6 mice were injected with 1.0 × 105 B16-F10 cells and received the different treatments at Day 7. The melanoma mice were sacrificed at Day 8, 11, 14, and 21. Peripheral blood was obtained from an eye vein, agglutinated, and centrifuged at 2000rpm for 5 min. The serum samples were collected. The tumors were isolated from the mice, weighed, and homogenized in PBS premixed with protease inhibitors (Merck Millipore). Then the supernatant was collected by centrifugation. The αPD-1 concentrations were quantified by ELISA. Briefly, the ELISA plates (Costar) were coated with mouse recombinant PD-1 protein (ACRO Biosystems, M5228) overnight, washed three times with PBST, blocked with 1mg/mL bovine serum albumin in PBST overnight and washed three times. The titrated sera, the tumor samples and αPD-1 standard samples were incubated in the pre-coated wells for 1 h at 37°C, followed by washing 6 times with PBST. The plates were incubated with 0.25μg/mL of HRP-conjugated goat-anti-rat IgG antibody (141612, KPL) for 30 min, washed 6 times with PBST and developed with the SureBlue TMB Microwell Peroxidase Substrate (KPL) following the manufacturer's instructions. The absorption at 450nm was measured on a tunable microplate reader (TECAN, infinite F50). The actual concentrations were calculated by comparing the results to the standard curve.
Cell isolation and FACS analysis
For flow cytometric analysis, spleens, double inguinal lymph nodes, and tumors were surgically taken from the mice at Day 14 after the tumor cell inoculation. Single-cell suspensions were made as previously described.52,53 For intracellular IFN-γ staining, the isolated cells were stimulated with 1µL/mL phorbol-12-myristate-13-acetate (PMA, Sigma), 1µL/mL ionomycin (Sigma), 1µL/mL brefeledin (Sigma) and 0.67µL/mL GolgiStop (BD Biosciences) for 4 h before staining. The cells were first stained extracellularly with the specific antibodies, including mouse CD3-PerCP/Cy5.5 (17A2, Biolegend), CD8-FITC (H35-17.2, eBioscience), CD4-PE/Cy7 (GK1.5, Biolegend), PD-L1-APC (10F.9G2, Biolegend ), CD45-APC/Cy7 (30-F11, Biolegend), CD11b-FITC (M1/70, Biolegend) or Gr-1-PE (RB6-8C5, Biolegend). The cells then were fixed and permeabilized with the Perm/Fix solution (eBioscience), and were stained intracellularly with IFN-γ-APC (XMG1.2, eBioscience) and Foxp3-PE (NRRF-30, eBioscience). These cell samples were analyzed by Canto II (BD).
Immunohistochemistry (IHC)
The melanoma tumors were removed from the mice at the time of sacrifice (Day 14 after B16-F10 cell inoculation) and then were fixed in 4% formaldehyde. IHC staining was performed as described previously,7 using the mouse monoclonal CD31 antibody (M-20, Santa Cruz Biotechnology). Microvessels within tumors were assessed by quantifying the microvessel number and the percentage of microvessel-occupied surface area with a field of 105 μm2 area (8–10 random fields per sample, 8 mice per group). Quantification was independently performed by two pathologists in a double-blind manner.
Quantitative real-time PCR (qRT-PCR) analysis
Tumors were harvested at the time of sacrifice (14 days after B16-F10 cell inoculation) for RNA extraction using the Trizol reagent (Invitrogen). Total RNA was reversely transcribed into complementary DNA (cDNA) using the First Strand cDNA Synthesis kit (Fermentas) and oligo-dT primers (TSINGKE). qRT-PCR was performed on the ABI StepOne Plus Detector System (Applied Biosystem) using SYBR Green I Assay (Takara). The relative mRNA level was normalized to that of the housekeeping gene hypoxanthine phosphoribosytransferase (HPRT). The primers used were listed in Table S1.
Luminex assay for quantification of cytokines
C57BL/6 mice were injected with 1.0 × 105 B16-F10 cells and received the different treatments at Day 7. The melanoma mice were sacrificed at Day 14. Peripheral blood was obtained from an eye vein, and centrifuged at 2000 rpm for 5 min. The resulting serum samples were collected. Meanwhile, the tumors were isolated from the mice, weighed, and homogenized in lysis buffer (Merck Millipore). The supernatant was collected by centrifugation. The concentration of proteins was determined using the Pierce BCA protein assay kit (Thermo Scientific). The concentrations of the cytokines (IL-1β, IFN-γ, CXCL10, CXCL9, and IL-6) in these samples were detected by the MILLIPLEX® MAP Multiplex Immunoassay kits (Merck Millipore) following the manufacturer's instructions. The median fluorescent intensity was read on the Luminex 200™ IS and analyzed using the logistic curve-fitting method to determine cytokine concentrations.
Quantification of PGE2 concentration
C57BL/6 mice were injected with 1.0×105 B16-F10 cells and received the different treatments at Day 7. The melanoma mice were sacrificed at Day 14. The tumors were isolated from the mice, weighed, and homogenized in PBS. The supernatant was collected by centrifugation. The concentration of PGE2 of tumor tissue was detected by the ParameterTM PGE2 assay kit (R&D) following the manufacturer's instructions.
Statistical analysis
All statistical comparisons were performed using two-tailed Student's t-tests. P < 0.05 was considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank N. Xu, S. Qi and C. Zhang for the assistance with animal experiments, and L. Shi for the technical support on FACS assays. We thank the members of the Research Center for Tissue Engineering and Regenerative Medicine for helpful discussions throughout this study.
Funding
This work is supported by the National Natural Science Foundation of China Programs 81272559 and 81441077, the International Science and Technology Corporation Program of Chinese Ministry of Science and Technology S2014ZR0340, the Science and Technology Program of Chinese Ministry of Education 113044A, the Frontier Exploration Program of Huazhong University of Science and Technology 2015TS153, and the Natural Science Foundation Program of Hubei Province 2015CFA049.
Supplemental Material
Supplemental Material may be downloaded here: publisher's website
References
- 1.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/ 10.1038/nrc1586 [DOI] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al.. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010; 33:828-33; PMID:20842054; http://dx.doi.org/ 10.1097/CJI.0b013e3181eec14c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jandus C, Speiser D, Romero P. Recent advances and hurdles in melanoma immunotherapy. Pigment Cell Melanoma Res 2009; 22:711-23; PMID:19735459; http://dx.doi.org/ 10.1111/j.1755-148X.2009.00634.x [DOI] [PubMed] [Google Scholar]
- 5.Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors. Cancer Immunol Immunother 2014; 63:11-20; PMID:23990173; http://dx.doi.org/ 10.1007/s00262-013-1468-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanterman J, Sade-Feldman M, Baniyash M. New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol 2012; 22:307-18; PMID:22387003; http://dx.doi.org/ 10.1016/j.semcancer.2012.02.008 [DOI] [PubMed] [Google Scholar]
- 7.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC et al.. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med 2003; 9:562-7; PMID:12704383; http://dx.doi.org/ 10.1038/nm863 [DOI] [PubMed] [Google Scholar]
- 8.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002; 99:12293-7; PMID:12218188; http://dx.doi.org/ 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008; 8:467-77; PMID:18500231; http://dx.doi.org/ 10.1038/nri2326 [DOI] [PubMed] [Google Scholar]
- 10.Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, Tamada K, Pardoll DM, Chen L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood 2007; 110:180-5; PMID:17289811; http://dx.doi.org/ 10.1182/blood-2006-11-060087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS et al.. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369:134-44; PMID:23724846; http://dx.doi.org/ 10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL et al.. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28:3167-75; PMID:20516446; http://dx.doi.org/ 10.1200/JCO.2009.26.7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010; 107:4275-80; PMID:20160101; http://dx.doi.org/ 10.1073/pnas.0915174107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dirks J, Egli A, Sester U, Sester M, Hirsch HH. Blockade of programmed death receptor-1 signaling restores expression of mostly proinflammatory cytokines in anergic cytomegalovirus-specific T cells. Transpl Infect Dis 2013; 15:79-89; PMID:23176118; http://dx.doi.org/ 10.1111/tid.12025 [DOI] [PubMed] [Google Scholar]
- 16.Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T et al.. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946-52; PMID:10874062; http://dx.doi.org/ 10.1056/NEJM200006293422603 [DOI] [PubMed] [Google Scholar]
- 17.Tindall E. Celecoxib for the treatment of pain and inflammation: the preclinical and clinical results. J Am Osteopath Assoc 1999; 99:S13-7; PMID:10643176. [DOI] [PubMed] [Google Scholar]
- 18.Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res 1998; 58:409-12; PMID:9458081 [PubMed] [Google Scholar]
- 19.Gendy AS, Lipskar A, Glick RD, Steinberg BM, Edelman M, Soffer SZ. Selective inhibition of cyclooxygenase-2 suppresses metastatic disease without affecting primary tumor growth in a murine model of Ewing sarcoma. J Pediatr Surg 2011; 46:108-14; PMID:21238650; http://dx.doi.org/ 10.1016/j.jpedsurg.2010.09.074 [DOI] [PubMed] [Google Scholar]
- 20.Lee KY, Mooney DJ. Alginate: properties and biomedical applications. Prog Polym Sci 2012; 37:106-26; PMID:22125349; http://dx.doi.org/ 10.1016/j.progpolymsci.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leahy KM, Ornberg RL, Wang Y, Zweifel BS, Koki AT, Masferrer JL. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res 2002; 62:625-31; PMID:11830509 [PubMed] [Google Scholar]
- 22.Cao H, Yu R, Tao Y, Nikolic D, van Breemen RB. Measurement of cyclooxygenase inhibition using liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal 2011; 54:230-5; PMID:20817448; http://dx.doi.org/ 10.1016/j.jpba.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol 2013; 25:214-21; PMID:23298609; http://dx.doi.org/ 10.1016/j.coi.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fransen MF, Arens R, Melief CJ. Local targets for immune therapy to cancer: tumor draining lymph nodes and tumor microenvironment. Int J Cancer 2013; 132:1971-6; PMID:22858832; http://dx.doi.org/ 10.1002/ijc.27755 [DOI] [PubMed] [Google Scholar]
- 25.Facciabene A, Santoro S, Coukos G. Know thy enemy: why are tumor-infiltrating regulatory T cells so deleterious? Oncoimmunology 2012; 1:575-7; PMID:22754792; http://dx.doi.org/ 10.4161/onci.19401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest 2006; 116:1935-45; PMID:16778987; http://dx.doi.org/ 10.1172/JCI27745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/ 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res 2011; 71:7463-70; PMID:22025564; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL et al.. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012; 4:127ra37; PMID:22461641; http://dx.doi.org/ 10.1126/scitranslmed.3003689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Upregulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013; 5:200ra116; PMID:23986400; http://dx.doi.org/ 10.1126/scitranslmed.3006504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arenberg DA, Kunkel SL, Polverini PJ, Morris SB, Burdick MD, Glass MC, Taub DT, Iannettoni MD, Whyte RI, Strieter RM. Interferon-gamma-inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J Exp Med 1996; 184:981-92; PMID:9064358; http://dx.doi.org/ 10.1084/jem.184.3.981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahin H, Borkham-Kamphorst E, Kuppe C, Zaldivar MM, Grouls C, Al-samman M, Nellen A, Schmitz P, Heinrichs D, Berres ML et al.. Chemokine Cxcl9 attenuates liver fibrosis-associated angiogenesis in mice. Hepatology 2012; 55:1610-9; PMID:22237831; http://dx.doi.org/ 10.1002/hep.25545 [DOI] [PubMed] [Google Scholar]
- 33.Ruehlmann JM, Xiang R, Niethammer AG, Ba Y, Pertl U, Dolman CS, Gillies SD, Reisfeld RA. MIG (CXCL9) chemokine gene therapy combines with antibody-cytokine fusion protein to suppress growth and dissemination of murine colon carcinoma. Cancer Res 2001; 61:8498-503; PMID:11731434 [PubMed] [Google Scholar]
- 34.Perrone MG, Scilimati A, Simone L, Vitale P. Selective COX-1 inhibition: a therapeutic target to be reconsidered. Curr Med Chem 2010; 17:3769-805; PMID:20858219; http://dx.doi.org/ 10.2174/092986710793205408 [DOI] [PubMed] [Google Scholar]
- 35.West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, Youngblood B, Freeman GJ, Smith KA, Ahmed R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest 2013; 123:2604-15; PMID:23676462; http://dx.doi.org/ 10.1172/JCI67008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, Teng MW, Smyth MJ. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A 2011; 108:7142-7; PMID:21482773; http://dx.doi.org/ 10.1073/pnas.1016569108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, Stratford IJ, Poon E, Morrow M, Stewart R et al.. Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Res 2014; 74:5458-68; PMID:25274032; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1258 [DOI] [PubMed] [Google Scholar]
- 38.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014; 124:687-95; PMID:24382348; http://dx.doi.org/ 10.1172/JCI67313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selmi TA, Verdonk P, Chambat P, Dubrana F, Potel JF, Barnouin L, Neyret P. Autologous chondrocyte implantation in a novel alginate-agarose hydrogel: outcome at two years. J Bone Joint Surg Br 2008; 90:597-604; PMID:18450625; http://dx.doi.org/ 10.1302/0301-620X.90B5.20360 [DOI] [PubMed] [Google Scholar]
- 40.Lee LC, Wall ST, Klepach D, Ge L, Zhang Z, Lee RJ, Hinson A, Gorman JH 3rd, Gorman RC, Guccione JM. Algisyl-LVR with coronary artery bypass grafting reduces left ventricular wall stress and improves function in the failing human heart. Int J Cardiol 2013; 168:2022-8; PMID:23394895; http://dx.doi.org/ 10.1016/j.ijcard.2013.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulson SK, Vaughn MB, Jessen SM, Lawal Y, Gresk CJ, Yan B, Maziasz TJ, Cook CS, Karim A. Pharmacokinetics of celecoxib after oral administration in dogs and humans: effect of food and site of absorption. J Pharmacol Exp Ther 2001; 297:638-45; PMID:11303053 [PubMed] [Google Scholar]
- 42.Dirix LY, Ignacio J, Nag S, Bapsy P, Gomez H, Raghunadharao D, Paridaens R, Jones S, Falcon S, Carpentieri M et al.. Treatment of advanced hormone-sensitive breast cancer in postmenopausal women with exemestane alone or in combination with celecoxib. J Clin Oncol 2008; 26:1253-9; PMID:18323548; http://dx.doi.org/ 10.1200/JCO.2007.13.3744 [DOI] [PubMed] [Google Scholar]
- 43.Marabelle A, Kohrt H, Levy R. Intratumoral anti-CTLA-4 therapy: enhancing efficacy while avoiding toxicity. Clin Cancer Res 2013; 19:5261-3; PMID:23965900; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJ. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res 2011; 17:2270-80; PMID:21389097; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2888 [DOI] [PubMed] [Google Scholar]
- 45.Trinath J, Hegde P, Sharma M, Maddur MS, Rabin M, Vallat JM, Magy L, Balaji KN, Kaveri SV, Bayry J. Intravenous immunoglobulin expands regulatory T cells via induction of cyclooxygenase-2-dependent prostaglandin E2 in human dendritic cells. Blood 2013; 122:1419-27; PMID:23847198; http://dx.doi.org/ 10.1182/blood-2012-11-468264 [DOI] [PubMed] [Google Scholar]
- 46.Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, Dubinett SM. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 2005; 65:5211-20; PMID:15958566; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0141 [DOI] [PubMed] [Google Scholar]
- 47.Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 2013; 73:6900-12; PMID:23975756; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 2007; 67:10019-26; PMID:17942936; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Q, Russell MR, Shahriari K, Jernigan DL, Lioni MI, Garcia FU, Fatatis A. Interleukin-1beta promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res 2013; 73:3297-305; PMID:23536554; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3970 [DOI] [PubMed] [Google Scholar]
- 50.Piali L, Weber C, LaRosa G, Mackay CR, Springer TA, Clark-Lewis I, Moser B. The chemokine receptor CXCR3 mediates rapid and shear-resistant adhesion-induction of effector T lymphocytes by the chemokines IP10 and Mig. Eur J Immunol 1998; 28:961-72; PMID:9541591; http://dx.doi.org/ 10.1002/(SICI)1521-4141(199803)28:03%3c961::AID-IMMU961%3e3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- 51.Silva EA, Mooney DJ. Spatiotemporal control of vascular endothelial growth factor delivery from injectable hydrogels enhances angiogenesis. J Thromb Haemost 2007; 5:590-8; PMID:17229044; http://dx.doi.org/ 10.1111/j.1538-7836.2007.02386.x [DOI] [PubMed] [Google Scholar]
- 52.Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S, Banerjee M, Mao Y, Kotarski J, Wicha MS et al.. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer 2012; 130:29-39; PMID:21480217; http://dx.doi.org/ 10.1002/ijc.25967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou W, Machelon V, Coulomb-L'Hermin A, Borvak J, Nome F, Isaeva T, Wei S, Krzysiek R, Durand-Gasselin I, Gordon A et al.. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med 2001; 7:1339-46; PMID:11726975; http://dx.doi.org/ 10.1038/nm1201-1339 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.