

ABSTRACT
Extracellular matrix metalloproteinase inducer (EMMPRIN/CD147) mediates tumor cell–macrophage interactions, and has been shown to induce both matrix metalloproteinases (MMPs) and vascular endothelial growth factor (VEGF). However, the epitope responsible for MMP induction is controversial, and the epitope responsible for VEGF induction is yet unknown. We generated a novel anti-EMMPRIN antibody directed against a specific epitope that successfully inhibited the production of both MMP-9 and VEGF in tumor cell–macrophage in vitro co-culture systems, exhibiting a U-shaped dose response. Furthermore, this antibody efficiently inhibited in vivo tumor progression in both the RENCA renal cell carcinoma and CT26 colon carcinoma subcutaneous tumor models, and reduced tumor size and number of metastatic foci in the 4T1 orthotopic model. This was achieved by inhibiting angiogenesis as assessed by immunohistochemical staining for the endothelial marker CD31, by inhibiting tumor cell proliferation as assessed by the staining for Ki-67, and by enhancing tumor cell apoptosis as assessed in the TUNEL assay. Moreover, administration of the antibody recruited more macrophages into the tumor, and skewed the tumor microenvironment for macrophages from TGFβ-dominated anti-inflammatory microenvironment, to a less immunosuppressive one. The antibody improved the ability of stimulated macrophages to perform antibody-dependent cell cytotoxicity (ADCC) and kill tumor cells. Thus, our new antibody maps the epitope capable of inducing both MMPs and VEGF, and places EMMPRIN as a good target for cancer therapy.
KEYWORDS: ADCC, angiogenesis, anti-EMMPRIN/CD147, apoptosis, MMP-9, tumor cell–macrophage interactions, tumor microenvironment, VEGF
Introduction
Angiogenesis, which provides oxygen and nutrients, is a necessary process that enables both tumor expansion and metastasis. This process is promoted by the secretion of pro-angiogenic factors, such as the potent VEGF that drives endothelial cells to form new vessels,1 and the MMPs that degrade the extracellular matrix (ECM) to make room for the growing tumor and help in disseminating tumor cells. MMP-9 in particular has been associated with tumor progression and metastasis.2 These factors are secreted as a result of an interaction between tumor and stroma cells, particularly macrophages that infiltrate the tumor, and may consist of up to 50% of the tumor mass.3 Such interactions are mediated though the EMMPRIN (also called CD147),4 through homophilic interactions between two EMMPRIN molecules on opposing cells.5
EMMPRIN is a glycoprotein belonging to the Ig superfamily, with two heavily glycosylated extracellular domains. EMMPRIN is weakly or moderately expressed on many cell types (including monocytes, T cells, and some epithelial cells), but its expression is greatly increased on many types of malignant cells in positive correlation with higher grade and stage of tumors, and often together with increased invasiveness and poor prognosis.6,7 EMMPRIN is mostly known as an inducer of MMPs, including MMP-9, but it is also capable of inducing stromal VEGF, and thus is implicated in angiogenesis and invasiveness.8-10 EMMPRIN's ability to induce MMPs was generally mapped to a region of 20 amino acids in the first of the two highly glycosylated extracellular domains (EC-I),11-13 but the precise epitope regulating VEGF induction remained unknown. In addition to its pro-angiogenic activities, EMMPRIN is a multifunctional protein that can interact with different proteins to mediate multiple activities that are important in the tumoral context. For example, it can bind with extracellular cyclophilin A (CypA) to achieve a chemotactic activity that helps recruit leukocytes to the tumor; it is involved in the multidrug resistance phenotype via interactions with P-gp/MDR-1; and it can chaperon the lactate transporters MCT-1 and MCT-4, to facilitate lactate transport out of the cells to maintain pH and tumor cell viability (reviewed in5-7). Last, recent studies show that elevated EMMPRIN expression is associated with greater resistance to radiotherapy.14,15 Thus, EMMPRIN functions as a central hub with multiple activities, positioning it as a potential target for cancer therapy, with potential profound beneficial consequences on tumor cell metabolism, viability, proliferation, angiogenesis and metastasis.7
Although several anti-EMMPRIN antibodies are already available commercially, they were usually elicited against the entire extracellular domain of the protein, and the specific epitope that they target remained unknown. Here, we report on a novel anti-EMMPRIN antibody (designated hence forth 161-Ab) that specifically recognizes and inhibits an epitope that is responsible for the induction of both MMP-9 and VEGF, and can also inhibit tumor progression in vivo.
Results
Peptide design
In planning the immunogen to be used to elicit an anti-EMMPRIN polyclonal antibody, we used the published sequence of the mouse EMMPRIN protein (accession number NP_001070652.1), with three main considerations in mind: high immunogenicity of the peptide, maximal homology with the human EMMPRIN sequence (accession number NP_940991), and no homology with any mouse protein. High immunogenicity is usually determined in hydrophilic areas that are exposed on the surface of the protein, and we used the EMBOSS and the Open Biosystem algorithms to identify these regions. Based on the BLAST search, the entire mouse and human EMMPRIN sequences are only 58% identical, making it very difficult to identify peptides that are completely homologous to both mouse and human protein sequences. However, we identified two candidate peptides (designated 161 and 162, Fig. 1A), that demonstrated 73% and 60% identical amino acids, with three and four amino acids difference, respectively. To avoid any possible cross-reactivity, the peptides were tested by BLAST analysis and were shown not to be homologous with other mouse proteins, especially the proteins neuroplastin and embigin that belong to the same family.
Figure 1.

EMMPRIN structure and peptide design, and specificity of 161-Ab: (A) Partial amino acid sequence of the murine EMMPRIN EC-I and EC-II domains compared to human sequence, with the 161-peptide (position 52-63) and the 162-peptide (position 136–145) indicated. (B) Mouse recombinant EMMPRIN (200 ng/lane) was loaded onto a 10% SDS-PAGE, separated and transferred onto a nitrocellulose membrane. The membrane was cut into strips and each strip was probed with the rabbit anti-mouse immune (I) or pre-immune serum (P) serum diluted 1:1,000. One strip was probed with the commercial rat anti-mouse EMMPRIN and served as positive control (P.C). Representative images of EMMPRIN identification in RENCA tumor sections by immunohistochemistry using (C) the pre-immune serum, (D) the immune serum (161-Ab), or (E) a commercial anti-EMMPRIN antibody.
The 161-Ab specifically recognizes EMMPRIN
Both peptides were synthesized, conjugated to KLH and injected in three boost injections to two rabbits each, to raise two polyclonal antibodies (designated accordingly as 161-Ab and 162-Ab). Both the 161-Ab and the 162-Ab immune sera were first shown to be able to bind each to its respective immunogenic peptides with high titers (1:1:312,500 or 1:1,560,000), but not to BSA that served as a negative control (Table 1). To show that the antibodies recognize the peptide sequence within the context of the entire EMMPRIN protein, we evaluated the ability of each immune serum to bind to the protein in its denatured form by protein gel blot analysis, or to its native form by direct ELISA. Denatured mouse recombinant EMMPRIN was recognized only by the 161-Ab, and not by the 162-Ab nor by either of the pre-immune sera, indicating high specificity of the 161-Ab. A commercial antibody used in the same conditions served as a positive control (Fig. 1B). To demonstrate the ability of the 161-Ab to recognize native EMMPRIN, we used direct ELISA. Table 2 shows that again, only the 161-Ab, and not the 162-Ab nor the pre-immune sera, could specifically recognize the native EMMPRIN protein (p < 0.0002). Last, we asked if the 161-Ab can specifically identify EMMPRIN in the context of a tumor tissue. The pre-immune sera did not identify EMMPRIN protein expressed on the tumor cells (Fig. 1C), whereas the 161-Ab strongly recognized it (Fig. 1D), even more intensely than did the commercial antibody (Fig. 1E).
Table 1.
161-Ab and 162-Ab specifically recognize their immunizing peptides.a
| 161-Ab |
162-Ab |
|||
|---|---|---|---|---|
| Titer/coating peptide | 161 peptide | BSA | 162 peptide | BSA |
| 1:500 | 3.184 ± 0.146 | 0.014 ± 0.001 | 3.715 ± 0.117 | 0.025 ± 0.006 |
| 1:2,500 | 1.051 ± 0.107 | 0.004 ± 0.001 | 2.663 ± 0.207 | 0.004 ± 0.001 |
| 1:12,500 | 0.234 ± 0.025 | 0.002 ± 0.001 | 1.081 ± 0.119 | 0.001 ± 0.0001 |
| 1:62,500 | 0.146 ± 0.006 | 0.002 ± 0.001 | 0.281 ± 0.035 | 0.0001 ± 0.0001 |
| 1:312,500 | 0.009 ± 0.001 | 0.001 ± 0.001 | 0.056 ± 0.008 | 0.0001 ± 0.0001 |
| 1:1,562,500 | 0.001 ± 0.001 | 0.001 ± 0.001 | 0.006 ± 0.002 | 0.0001 ± 0.0001 |
Optical density values of the binding of each antibody to its respective peptides are indicated (n = 6).
Table 2.
161-Ab specifically recognizes mouse recombinant EMMPRIN in direct ELISAa.
| Antibody | Immune serum | Pre-Immune serum | BSA | P valueb |
|---|---|---|---|---|
| 161-Ab | 0.235 ± 0.033 | 0.064 ± 0.008 | 0.031 ± 0.003 | 0.0002 (* * * ) |
| 162-Ab | 0.076 ± 0.007 | 0.091 ± 0.002 | 0.021 ± 0.004 | 0.9538 (ns) |
Optical density values of the binding of 161-Ab or 162-Ab to the mouse recombinant EMMPRIN protein are indicated (n = 4).
p values are compared between immune and pre-immune sera.
161-Ab inhibits secretion of MMP-9 and VEGF in co-cultures
To screen for an inhibitory activity of the 161-Ab in vitro, we first determined that maximal MMP-9 and VEGF secretion was achieved after tumor cells and macrophage-like cell lines were incubated in co-culture (Fig. 2A). Relative to each of the single cultures, MMP-9 concentrations were synergistically induced by 15-folds (p < 0.0001) and 5-folds (p < 0.01) for the CT26 and TRAMP-C2, respectively, that were co-cultured with RAW 264.7 cells. Likewise, VEGF concentrations were induced by 2–3-folds in both co-culture systems (p < 0.001). Similar results were also observed in two additional tumor cell lines (the mouse renal cell carcinoma RENCA and the prostate cell carcinoma TRAMP-C1) that were co-cultured with RAW 264.7 cells, or when all four tumor cell lines were incubated with primary thioglycollate-elicited peritoneal macrophages (data not shown). These results suggest that the interaction between tumor cells and macrophages, even without the addition of any other stimulus, is necessary for the induction of high amounts of pro-angiogenic factors, and can serve as an in vitro screening platform for the identification of an antibody with the ability to inhibit MMP-9 and VEGF secretion.
Figure 2.

Screening in vitro for the inhibiting effects of 161-Ab on VEGF and MMP-9 secretion from co-cultures. (A) The tumorigenic cell lines CT26 and TRAMP-C2 were incubated alone (0.5×106 cells) or in co-culture with RAW 264.7 macrophage-like cells (0.5×106 cells) in serum-free medium for 48 h. Supernatants were collected and the concentrations of secreted MMP-9 and VEGF were determined by ELISA (n = 8). (B) CT26 or TRAMP-C2 were co-cultured with RAW 264.7 cells as before, and the effects of 161-Ab in 5-fold serial dilutions on MMP-9 and VEGF secretion were evaluated (n = 6). (C) The human tumorigenic cells A498 and MCF-7 (0.5×106 cells) were co-cultured with the monocytic-like U937 cell line (0.5×106 cells) and in the presence of TNFα (1 ng/mL), and the effects of specific dilutions of the 161-Ab on MMP-9 and VEGF secretion were tested (n = 3). *, p < 0.05, relative to the co-cultures.
We next examined the ability of both immune sera to inhibit secretion of MMP-9 and VGEF using the established screening platform. Both pre-immune and immune sera were 5-fold serially diluted (Fig. 2B), and added to the co-culture systems and after 48 h supernatants were collected and concentrations of VEGF and MMP-9 were evaluated. Whereas the 162-Ab did not inhibit MMP-9 or VEGF in any of the dilutions tested in both co-culture systems (data not shown), the immune serum containing the 161-Ab, but not the pre-immune serum, was effective in inhibiting both MMP-9 and VEGF in the two screening platforms of CT26 or TRAMP-C2 co-cultured with RAW 264.7 cells, resulting in a U-shaped curve (Fig. 2B). In the CT26 co-culture system, serial dilutions of 1:312,500 and 1:1,562,500 of the immune serum resulted in maximal inhibition of 69% and 60% in MMP-9 secretion (p < 0.05), whereas in the TRAMP-C2 co-culture system lower dilutions of 1:12,500 and 1:62,500 inhibited MMP-9 secretion by 75% and 83% (p < 0.05). VEGF was inhibited by 53% at dilution 1:1,562,500 in the CT26 co-culture system and by 62–67% at dilutions 1:312,500 and 1:1,562,500 in the TRAMP-C2 co-culture system (p < 0.05). Thus, the optimal range for inhibiting both MMP-9 and VGEF was found to be between 1:312,500 and 1:1,562,500, suggesting that our epitope is responsible for the induction of both these proteins in the EMMPRIN protein.
To establish if 161-Ab can cross-react with human EMMPRIN, as the human epitope sequence differs in only three amino acids from the mouse sequence, we next examined its ability to inhibit VEGF and MMP-9 secretion in a human co-culture system. We used two human tumor cell lines (the renal cell carcinoma A498 and the breast carcinoma MCF-7) and incubated each of them in the presence of a human monocyte-like cell line (U937) for 48 h with the addition of TNFα (1 ng/mL), as these conditions yield maximal levels of MMP-9 and VEGF,16 and with the addition of the optimal dilutions of 161-Ab previously determined for the mouse cultures. In both co-cultures, MMP-9 secretion was selectively inhibited by the addition of the immune, but not the pre-immune serum by 43% at dilution 1:312,500 (p < 0.05 for the MCF-7 co-culture), although in the A498 co-culture this did not reach significance (Fig. 2C). Likewise, the 161-Ab immune serum, but not the pre-immune serum, inhibited VEGF secretion by 31% and 25% for the A498 and MCF-7 co-cultures (p < 0.05 for both), respectively, at dilution 1:312,500.
161-Ab inhibits tumor progression
After the specificity of the 161-Ab to EMMPRIN was determined, and its ability to inhibit the pro-angiogenic MMP-9 and VEGF proteins was established, we examined its ability to inhibit tumor progression in vivo. Thus, we first injected the RENCA or CT26 tumor cells to the flanks of BALB/c mice, and only after tumors were palpable (around day 12), we i.p. injected the 161-Ab every 7 d in different concentrations (25μg, 50 μg or 100 μg in 2 mL saline). We injected either saline or used the 162-Ab which did not show any specificity to EMMPRIN, in the control groups. At the end of the experiment (day 39 for RENCA tumors and day 30 for CT26 tumors), differences between the control groups and all doses of the 161-Ab groups were visible. In RENCA tumors, the dose of 50 μg, and to a greater extent the dose of 25 μg, inhibited tumor progression by 90% (p < 0.01) or 99% (p < 0.05), respectively Fig. 3A). Likewise, for the CT26 tumors, the 161-Ab significantly inhibited tumor growth in all doses used, where the 100 μg dose inhibited tumor growth by 58% (p < 0.01), the 50 μg dose inhibited by 85% (p < 0.001) and the 25 μg dose inhibited tumor growth by 97% (p < 0.001, Fig. 3B).
Figure 3.
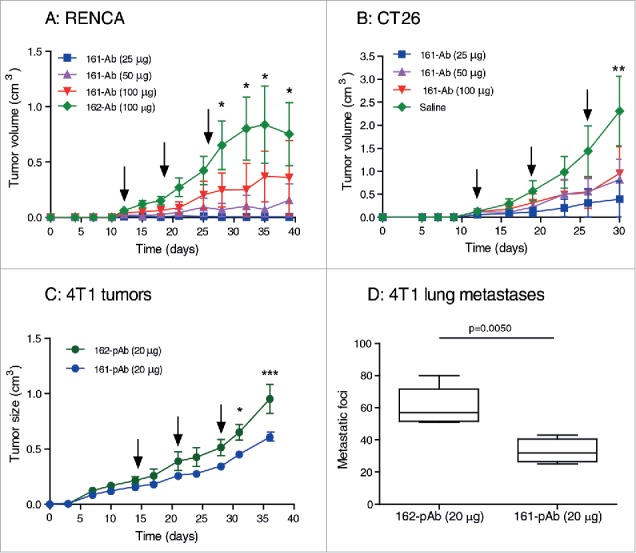
Effects of 161-Ab on tumor progression. (A) RENCA or (B) CT26 tumor cells (2×106) were injected to the flank of BALB/c mice (8 wk old). Once the tumors became palpable at day 12, mice were randomly assigned to the different groups, and received an i.p. injection of 161-Ab at different concentrations, followed by additional two more boost injections every 7 d (boost injections are indicated by black arrows). The negative control groups were injected with the 162-Ab (RENCA tumors) or with saline (CT26 tumors). Tumor volume was monitored and estimated (width ×length × 0.5). *, p < 0.05, * *, p < 0.01, the 162-Ab group relative to the 25 μg and 50 μg groups in the RENCA tumors, and relative to all groups in the CT26 tumors (n = 5–6 in each group). (C) 4T1 tumor cells (5×105) were orthotopially injected into the mammary fat pad. When tumors became well established at day 14, mice were assigned to one of the two groups and received an i.p. injection of the 161-pAb (n = 6) or the control 162-Ab (n = 5) at the concentration indicated, and additional two boost injections every 7 d (black arrows). Tumor volumes were monitored as before. *, p < 0.05 and * * *, p < 0.001. At day 36, mice were sacrificed and (D) the number of metastatic foci in their lungs was counted.
We next examined the effect of the 161-pAb in the 4T1 mammary gland orthotopic model, as tumor behavior and response to anticancerous agents have been shown to differ between subcutaneous and orthotopic tumor models.17,18 Tumor cells were injected into the mammary fat pad of BALB/c female mice, and when the tumors were well established (with an average size of 0.19 cm3) we injected three boost injections every 7 d of either the 161-pAb or the control 162-pAb to mice in the two groups. At the end of the experiment (day 36), the 161-pAb inhibited tumor progression by 37% (p < 0.001) relative to the negative control group (Fig. 3C). Moreover, the control group developed about 2-folds more metastatic foci than the treated group (means of 60 and 33 foci, respectively, p = 0.005, Fig. 3D).
Mechanisms of 161-Ab activity
We next evaluated the effects of the administration of 161-Ab on different aspects of tumor progression. Of note, since the lower dose of 161-Ab markedly inhibited RENCA tumor growth, no material was available for evaluation, and therefore, we used tumors harvested after treatment with the 50 μg dose. To demonstrate that the 161-Ab inhibited the pro-angiogenic activity of EMMPRIN, we looked at the blood vessel densities, measured by the staining of tumor tissues with anti-CD31 directed against the endothelial cell marker. The blood vessels in the control sections (162-Ab or saline) were long, continuous and dense (Fig. 4A). In contrast, blood vessels in tumors treated with the 161-Ab were short and discontinuous, with bigger gaps between them. This was clearly reflected in the significant reduction of the vessel surface area by about 2.5-folds (p < 0.0001) in both RENCA and CT26 tumors. Furthermore, the concentrations of the pro-angiogenic factors VEGF and MMP-9 were also altered by the antibody in a dose-dependent manner (Fig. 4B). The use of 50 μg 161-Ab in RENCA tumors or 25 μg 161-Ab in CT26 tumors inhibited MMP-9 production by 4.5 (p < 0.05) and 4-folds (p < 0.01), and VEGF production was inhibited by 6- and 5-folds (p < 0.05), respectively.
Figure 4.
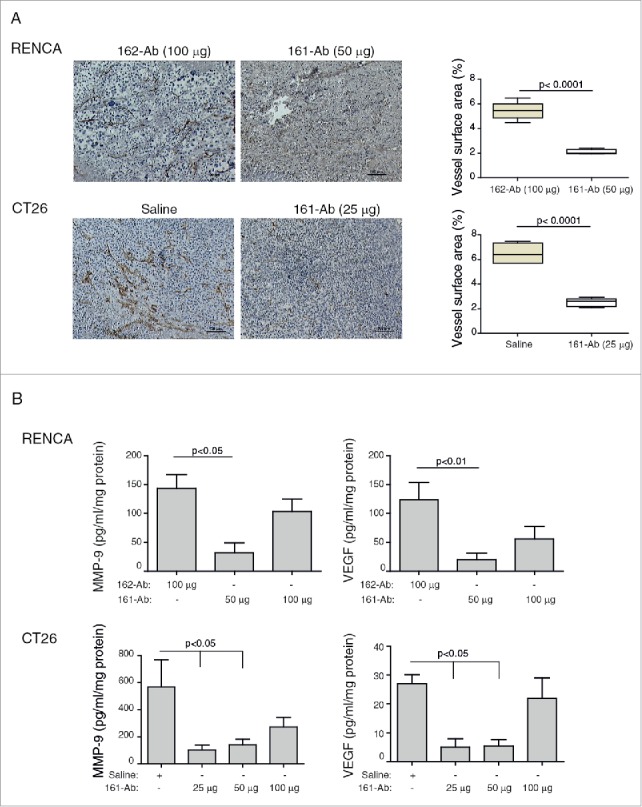
161-Ab reduces tumor angiogenesis. RENCA and CT26 s.c. tumors were harvested and were formalin fixed and paraffin embedded. (A) Representative images of tissue sections that were immunohistochemically stained for CD31, the marker of endothelial cells; Vessel density was calculated using the Weibel grid applied to at least three different fields in each slide (n = 5). Bar size is 100 μm. (B) Tumor lysates were prepared, total protein concentrations were determined by the Bradford reagent and lysate concentrations of VEGF and MMP-9 were determined by ELISA (n = 4–5).
To estimate the effects of the 161-Ab on the rate of proliferation of the tumor cells, we evaluated the proliferation index by Ki-67 staining in the tumor sections (Fig. 5A). Control sections demonstrated high proliferation rate that was reduced by 3.6-folds for the RENCA tumors (p = 0.0011), and by 3.25-folds for the CT26 tumors (p = 0.0042) by the 161-Ab. In contrast, the number of apoptotic cells (Fig. 5B) was increased by 161-Ab treatment by 46-folds for the RENCA tumors (p = 0.0019) and by 8.8-folds for the CT26 tumors (p = 0.0005). The ability of the 161-Ab to increase apoptotic death was further corroborated by the determination of the concentration of activated caspase 3 in the tumor lysates (Fig. 5C). In comparison to the control groups, activated caspases 3 was elevated by 20-folds for the RENCA tumors with the 50 μg dose (p < 0.001), and by 3.7-folds for the CT26 tumors with the 25 μg dose (p < 0.01).
Figure 5.

161-Ab reduces proliferation but increases apoptotic death. RENCA and CT26 tumor sections were prepared and analyzed for proliferation (Ki-67) and apoptosis (TUNEL). (A) Representative images of immunohistochemical staining for Ki-67 (Bar size for RENCA tumors is 100 μm and for CT26 tumor is 25 μm), and Determination of the Ki-67 positive fraction of cells (n = 4); (B) Representative images of immunohistochemical staining for apoptosis (TUNEL, bar size for RENCA tumors is 50 μm and for CT26 tumor is 25 μm), and quantification of the number of apoptotic cells per mm2 (n = 4-5); (C) Determination of the concentration of activated caspase 3 in tumor lysates by ELISA (n = 4–5).
Administration of the 161-Ab also altered the tumor microenvironment. First, the cellular infiltrate changed, and more macrophages could be seen not only in the perinecrotic areas of the tumor, but also surrounding blood vessels (Fig. 6A). In RENCA tumors administered with 50 μg of 161-Ab, the number of macrophages/mm2 increased by 1.8-folds (p = 0.0159), whereas in CT26 tumors administered with 25 μg of 161-Ab this number increased by 2.3-folds (p = 0.0331).
Figure 6.

161-Ab increases macrophage infiltration, mediates tumor cell cytotoxicity, and changes tumor microenvironment. (A) Representative images of immunohistochemical staining for F4/80 in RENCA and CT26 tumor sections. Bar size for RENCA and CT26 tumors is 50 μm. Number of macrophages per mm2 was quantified (n = 4–5). (B) CT26 cells (5×104 cells) were labeled with Cell Tracker, incubated in vitro with 1.28 ng/mL of the 161-Ab for 6 h, in the presence of complement (diluted 1:50), RAW 264.7 cells at a ratio of 2:1, or both. Percent cytotoxicity were determined as described in the methods (n = 4). (C) Cytokine concentration in tumor lysates were determined by ELISA, and normalized to the amount of total protein in each sample (n = 4–5).
Increased macrophage infiltration into the tumor upon administration of the antibody suggested that tumor cell death may be mediated by the macrophages via CDC or ADCC. To determine which of these two mechanisms better explains the increase in tumor cell death, we performed an in vitro cytotoxic assay (Fig. 6B). Macrophages (RAW 264.7 cells) incubated with the antibody but without complement, or the presence of complement without macrophages did not impact the rate of CT26 death. In contrast, when both complement and RAW 264.7 cells were present, tumor cell death significantly increased by 3.5-folds (p < 0.01) relative to the presence of complement only. As controls, we used an irrelevant antibody or incubated the CT26 cells only with the 161-Ab, and in both cases no cell death occurred (data not shown).
Changes in the cytokine microenvironment may suggest that in vivo tumor-associated macrophages (TAMs) could shift toward M1-activation in order to mediate tumor cell death. These changes were indeed demonstrated (Fig. 6C). In untreated RENCA and CT26 tumors, the pro-inflammatory cytokines TNFα and IL-1β were found in small, almost negligible amounts in the tumor lysates. However, after administration of 50 μg of 161-Ab in RENCA tumors, these cytokines were increased to 21.6 pg/mL/μg (p < 0.05), and 2.3 pg/mL/μg protein (p < 0.001), respectively. Similarly, administration of 25 μg of the 161-Ab in the CT26 tumors increased the pro-inflammatory cytokines by 4- and 11-folds (p < 0.05), respectively. The anti-inflammatory cytokine IL-10 also exhibited negligible amounts in untreated tumors in both RENCA and CT26 tumors, and addition of 50 μg or 25 μg of the 161-Ab resulted in 160-folds and 15-folds increase in IL-10 (p < 0.01) for the RENCA and CT26 tumors, respectively. The concentrations of the anti-inflammatory cytokine TGFβ in the tumor lysates reflected a mirror image to that described above for the pro-inflammatory cytokines. In control tumors, injected with the irrelevant 162-Ab or with saline, TGFβ concentrations were higher than those of the pro-inflammatory cytokines by at least two orders of magnitude. Administration of 50 μg or 25 μg of 161-Ab reduced those levels by about 11-folds (p < 0.01), in RENCA and CT26 tumors, respectively.
Discussion
We show here a novel, epitope-specific antibody directed against EMMPRIN, that inhibited secretion of the pro-angiogenic factors MMP-9 and VEGF in four in vitro co-culture systems of mouse and human tumor cells and macrophages, and that significantly reduced tumor size and its progression in two in vivo mouse model systems. This was achieved by inhibiting angiogenesis, attenuating tumor cell proliferation while enhancing their apoptosis, and by altering the microenvironment, so that more macrophages infiltrated the tumor and were then encountered by a relatively less immunosuppressive microenvironment, allowing them to become tumoricidial.
Sequences similar or adjacent to our epitope have been mentioned before in other studies as important for the ability of EMMPRIN to induce MMPs. In one study, addition of a peptide with the sequence SLNDSATEVTGHRWLK to the co-culture of human fibroblasts and cervical carcinoma SKG-II cells, interfered in a dose-dependent manner with the secretion of MMP-1 and caused up to 60% inhibition.12 The same, but slightly more elongated peptide (with the sequence SLNDSATEVTGHRWLKGGVV) was used in another study, and inhibited EMMPRIN-mediated production of MMP-2 in a dose-dependent manner by up to 80% in a co-culture of fibroblasts and different tumor cell lines.13 Both studies included part, or almost all of our epitope, and showed that the sequences chosen could inhibit MMP induction, possibly through interference with the homophilic EMMPRIN interactions. However, when the peptide was truncated into two fragments (peptides with the sequences SLNDSATEVT and GHRWLKGGVV, the latter including almost all of our epitope), no inhibition was detected, even when high concentrations of the peptides were used.13 Thus, the entire sequence of 16 amino acids could be considered crucial for the activity of MMP induction. In support of this conclusion is the study that elicited a novel anti-EMMPRIN antibody directed against the epitope TCSLNDSATEVTGHRW (including only four amino acids of our epitope), which inhibited T cell proliferation and reduced MMP-9 production, reduced T cell cytotoxicity to neurons and successfully reduced the clinical score in an EAE mouse model.19 However, in contrast to these findings, another study found that the sequence AAGTVFTTVEDLGSKILLTCSLNDSATEV (positions 22–50 in the human EMMRIN sequence), which does not include our epitope at all, was responsible for the EMMPRIN MMP induction activity and association with tumor invasion.20 We suggest that because we used an antibody to target a specific epitope of 11 amino acids, we could use a shorter sequence than the two previous studies that used peptides to directly generate a steric hindrance or to compete with the homophilic binding to EMMPRIN. More importantly, all of these epitope-mapping studies focused only on the MMP-induction activity of EMMPRIN, and showed that it might span over a stretch of about 40 amino acids, but no data so far revealed the epitope responsible for the VEGF-induction activity of the protein. Here we show for the first time, that our epitope is responsible not only for the MMP-induction activity of EMMPRIN, but also for its ability to induce VEGF. The ability to induce both of these proteins at the same time, using the same epitope, suggests a common signaling pathway that is evoked and affects both proteins, but this remains to be further studied.
Specificity of the antibody was demonstrated by the binding of 161-Ab to the peptide and to the recombinant EMMPRIN protein in protein gel blot analysis and in direct ELISA in vitro, as well as by the ability to recognize EMMPRIN in tumor sections, whereas the pre-immune serum showed no binding activity. Of note, differences in the binding intensity between the non-purified 161-pAb serum and the purified monoclonal commercial antibody to the denatured EMMPRIN protein in the protein gel blots could be explained by the different concentrations of the relative antibodies. In contrast, the opposite response observed in the staining of the tissue sections, where the 161-pAb more strongly stained tumor tissues may suggest that in the tumoral context, 161-pAb better recognizes an exposed epitope. We have previously demonstrated that targeting EMMPRIN in a human in vitro co-culture system using a commercial antibody or siRNA, inhibited the secretion of both MMP-9 and VEGF, and placed EMMPRIN as a key regulator of the interactions between tumor cells and macrophages that promote tumor angiogenesis and invasiveness.16 Based on these results, we show here similar data in two mouse in vitro co-culture systems. First, MMP-9 and VEGF secretion are maximal when tumor cells and macrophages are co-cultured, even without any additional stimulus. This allowed us to use these co-culture systems as screening platforms to identify the ability of the 161-Ab to inhibit MMP-9 and VEGF secretion. Indeed, 161-Ab, but not its pre-immune serum or the 162-Ab, could specifically inhibit both mouse and human MMP-9 and VEGF secretion, suggesting that the 161-Ab has cross-reactivity with the human EMMPRIN.
In the mouse in vitro co-culture systems, the antibody demonstrated hormetic dose-response manifested as a U-shaped curve for the inhibition of MMP-9 and VEGF secretion. Similarly, in the in vivo setting the administration of the antibody was most effective in the lower dose, rather than the higher dose, also suggesting a U-shape curve. Although the mechanisms underlying hormesis are currently unknown, this dose-response is often characteristic of anti-angiogenic agents, such as different VEGF pathway inhibitors, RGD-mimetic integrin inhibitors, endostatin and ATN-161 (reviewed in21). One possible explanation, at least for the in vivo setting, may be the development of neutralizing anti-idiotypic antibodies that may have resulted in downregulation of the 161-Ab effects at the higher concentrations used. However, this phenomenon should be further investigated.
We show that the 161-pAb works effectively to reduce tumor size both in subcutaneous and orthotopic models, even when used on well-established tumors, and also to attenuate development of metastases in the orthotopic model, thereby increasing its attractiveness as a potential therapy and opening a wide window of opportunity to use it. Monoclonal antibodies gain increasing importance as novel therapeutic tools against cancerous diseases. However, their therapeutic mode of action remains unclear. Depending on their antigen specificity, antibodies can directly induce apoptosis and inhibit proliferation by neutralizing growth factors or blocking their receptors (e.g. by anti-VEGF, anti-EGFR). Indirectly, antibodies can induce complement-dependent cytotoxicity (CDC) through classical activation of the complement, or ADCC, which is based on the expression of different Fcγ receptors on NK cells and macrophages.22 In fact, different studies that involved macrophage depletion or use of different FcγR deficient mice demonstrated that infiltrating monocytes and macrophages are the important cell population mediating antibody-dependent tumor cell killing (reviewed in23). Here, we show that our 161-Ab may act in several ways to reduce tumor progression. First, 161-Ab directly targets EMMPRIN, and so reduces angiogenesis. EMMPRIN is pro-angiogenic, as it regulates tumor cell–macrophage interactions, and induces the expression and secretion of MMPs, including MMP-9, and VEGF from macrophages. Here, using the effective low dose of the 161-Ab, we show that angiogenesis, manifested by mean vessel density (MVD) is markedly reduced, as is the tumor concentrations of MMP-9 and VEGF. Secondly, we show that proliferation is reduced while apoptosis is increased. Apoptotic death can be the direct result of disruption of EMMPRIN interactions with MCT-1 or MCT-4, which leads to accumulation of lactate in the tumor cells and increased acidosis. Additionally, it could be indirectly increased by immune-mediated cell death. In fact, we show an increase in macrophage infiltration into the tumors. We demonstrate in vitro that presence of macrophages, complement and the 161-Ab increase tumor cell death, suggesting that although some degree of CDC may be mediated by the 161-Ab, a bigger effect might be due to the presence of macrophages with their different FcγRs that enhance ADCC. Additionally, although NK cells are usually associated with ADCC, we could not find increased infiltration of these cells upon 161-Ab administration (data not shown). Last, it has been reported that binding of antibodies to TAMs can shift their activation toward cytotoxic M1-activated macrophages (reviewed in24). Here, we find evidence to support this premise, as the administration of 161-Ab shifts the TAMs toward M1-activation and change the tumor microenvironment. This may be due to a direct effect of the antibody that targets EMMPRIN and disrupts the tumor cell–macrophage interaction that leads to M2-activation of the macrophages, or to indirect effects that rely on the macrophages FcγR expression that upon engagement trigger M1-activation of the macrophages, and this phenomenon should be further investigated. Pro-inflammatory cytokines like TNFα and IL-1β are increased in a reverse dose-dependent manner, while the anti-inflammatory cytokine TGFβ is markedly reduced, in agreement with the hormetic effects of the antibody. In addition, the concentrations of nitrites are increased in the treated tumor lysates. We have shown before that RENCA tumor cells cannot express iNOS or produce NO due to elevated levels of microRNA-146a,25 and similar findings in CT26 tumor cells are now also available in our lab (unpublished data). Thus, most NO, if not all, are the product of infiltrating macrophages, and as NO production is the hallmark of M1-activated macrophages, we interpret these data to point to an in vivo shift in macrophage activation. Furthermore, the relative concentrations of the cytokines in the tumor lysates indicate that the dominant immunosuppressive cytokine is TGFβ, rather than IL-10, and that excess over the pro-inflammatory cytokines in the absence of the 161-Ab, dictates an immunosuppressive microenvironment. However, once the 161-Ab is administered, the microenvironment becomes much less immunosuppressive and more pro-inflammatory, allowing macrophages to successfully mediate tumor cell cytotoxicity.
In conclusion, we believe that EMMPRIN represents a good target for cancer therapy, as its expression is elevated on tumor cells in many types of cancerous diseases. Furthermore, it is an attractive target, because of its critical functions in tumor cell metabolism and proliferation, as well as its pro-angiogenic and pro-metastatic properties. Indeed, several attempts have already been made to inhibit EMMPRIN expression in tumors. these were achieved by either transfecting cell lines with EMMPRIN siRNA in vitro,26 or by using monoclonal antibodies that were raised against the entire extracellular fragment of EMMPRIN both in vitro27 and in vivo.28,29 All of these strategies resulted in decreased MMPs secretion, as well as in inhibition of tumor invasiveness. In fact, the F(ab')2 fragment of an anti-EMMPRIN monoclonal antibody called Licartin or Metuximab, is now being used to delay recurrence of hepatoma after transplantation in human patients.30 Our antibody, which must now be further developed into a monoclonal antibody, joins these previous strategies for cancer immunotherapy, and has demonstrated its effectiveness in inhibiting angiogenesis, proliferation, apoptosis and alteration of the inflammatory tumoral microenvironment, all of which cumulatively inhibit tumor growth, in line with the central role of EMMPRIN in tumor progression.
Methods
Cells
The human renal carcinoma A498 (ATCC HTB-44), breast carcinoma MCF-7 (ATCC HTB-22) and U937 monocyte-like cells (ATCC CRL-1593) were cultured in RPMI-1640 medium with 10% fetal calf serum (FCS), 1% L-glutamine and antibiotics. The tumorigenic mouse RENCA renal carcinoma (kind gift of Dr. Bernhard Hemmerlein, Georg-August University Hospital, Göttingen, Germany), and the CT26 colon carcinoma (ATCC CRL-2638) were cultured in RMPI-1640 medium, 10% FCS, 1% L-Glutamine and antibiotics, with addition of 100 mM HEPES buffer (pH 7.4) for the RENCA cells, and 1% sodium pyruvate for the CT26 cells. The RAW 264.7 macrophage-like (ATCC TIB-71) cell line and the mouse TRAMP-C2 prostate cancer cell line (ATCC CRL-2731) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS), 1% L-glutamine and antibiotics, with addition of 1% sodium pyruvate for the RAW 264.7 cells and 5μg/mL Insulin and 10−8 mol/L Dihydrotestosteron (Perkin-Elmer, Waltham, MA) for the TRAMP-C2 cells. Tumor cells and macrophages were co-cultured using the tumor cell line medium. To avoid possible masking of signals by exogenous stimuli, tumor cells (106 cells) were plated in 24-well plates in a serum-free medium for 48 h, alone or in co-culture with macrophages at a 2:1 ratio. Cell viability was determined using the XTT kit (Biological industries, Beit-Haemek, Israel). All cell lines were regularly tested for morphological changes and presence of mycoplasma. RAW 264.7 cells were identified as macrophages by their ability to phagocytose zymosan particles, and tumor cells were tested as cells of epithelial origin by their cytokeratin 18 expression.
ELISA
The mouse MMP-9 and VEGF concentrations were determined using ELISA kits according to the manufacturer's instructions (R&D systems, Minneapolis, MN), and the supernatant samples were diluted 1:100 according to previous calibration experiments. The concentrations of TNFα, IL-1β, IL-10 and TGFβ in tumor lysates were determined using ELISA kits (R&D systems), and normalized per 1μg of total protein that was determined by Bradford reagent.
Peptide conjugation, immunization and affinity purification of polyclonal antibodies
We identified an 11 amino acid peptide in the mouse EMMPRIN protein that exhibited high homology between the human and the mouse sequences, and showed no homology to other mouse proteins, including neuroplastin and embigin which belong to the same family of proteins. This sequence is located in the extracellular domain 1 (EC-I) of the protein (GHRWMRGGKVL, position 52–63, Fig. 1). As a control peptide we chose a different 10-amino acid sequence, located in the extracellular domain 2 (EC-II) of the protein (position 136–145 TDWFWFKTSD). The generation of the polyclonal antibody was carried out by Adar Biotech, LTD (Rehovot, Israel). Briefly, the company synthesized the requested peptides and added a cysteine residue to each peptide to facilitate its binding, conjugated them to the Keyhole Limpet Hemocyanin (KLH) carrier protein, immunized two rabbits with each peptide and performed bleeding before (pre-immune sera) and after immunization following additional boost vaccinations. To establish an immune response, the company performed an ELISA test, where the titer of the antibodies was determined. The sera were collected after the final boost injection and were sent to us, or were further purified by affinity chromatography. The in vitro assays determining specificity of the antibodies were carried out using the crude sera, whereas the in vivo experiments determining the effects of the antibodies were carried out with the purified antibodies.
Protein gel blot
Mouse recombinant EMMPRIN (200 ng/lane, R&D systems) was loaded onto a 10% SDS-PAGE, electropheretically separated and transferred onto a nitrocellulose membrane. The membrane was cut into strips and each strip was blocked with 20% skimmed milk and 1% BSA in TBST (0.1% Tween-20, 10mM Tris pH 8.0, 150mM NaCl) at room temperature overnight, and then probed for 1 h with immune or pre-immune serum (both diluted 1:1000), washed and incubated with 1:5,000 diluted HRP-conjugated donkey anti-rabbit IgG (711–035–152, Jackson ImmunoReserch Laboratories, West Grove, PA). One strip was probed with the 1:1,000 diluted commercial rat anti-mouse EMMPRIN (MAB772, R&D systems) and then with the 1:5,000 diluted HRP-conjugated goat anti-rat IgG (112–035–062, Jackson) and served as positive control (P.C). The enhanced chemiluminescence (ECL) system (Biological industries) was used for detection.
Direct ELISA
To determine if our polyclonal antibodies specifically recognize EMMPRIN in its native form, we developed a direct ELISA assay. The wells in a 96-well plate were coated with the mouse recombinant EMMPRIN (200 ng/mL, R&D systems) overnight at 4°C. The plate was washed three times with wash buffer (0.05% Tween-20 in PBS), and blocked with blocking buffer (1% BSA in PBS) for 2 h at room temperature. Strips in the plate were incubated with 100 µL of the primary antibodies (immune or pre-immune sera) diluted 1:1,000 for 2 h at room temperature, and then washed four times and incubated with 100 µL of the biotinylated donkey anti-rabbit (BAF109, R&D systems) secondary antibody diluted 1:10,000. After three more washes the streptavidin-HRP was diluted 1:200 and added for 1 h. After three additional washes, the TMB solution was added for 5 min, and the reaction was stopped with 100 µL of stop solution. The absorbance of each well was measured at 450 nm and 540 nm. The assay was repeated four times in duplicates.
In vivo mouse model
BALB/c mice (female, 8 wk old) were purchased from Harlan Laboratories (Jerusalem, Israel), and were kept with a 12 h light/dark cycle and access to food and water ad libitum. Mice were cared for in accordance with the procedures approved by the Supervision of Animal Experiments Committee at the Technion, and outlined in the NIH Guideline for the Care and Use of laboratory Animals. Tumors were generated by subcutaneously injecting 2×106 RENCA or CT26 cells suspended in matrigel in a total volume of 200 μL into the flank of BALB/c mice. After tumors became palpable (around day 12), mice were randomly assigned to five groups: the control groups were i.p. injected three times with 100 μL saline or an irrelevant antibody (162-Ab, another polyclonal anti-EMMPRIN antibody that had no effect during the screening process), and the other groups were i.p. injected with different concentrations of the 161-Ab (25, 50 and 100 μg per 25 gr body weight) in a volume of 2 ml, in 3 boosts, every 7 d Similarly, 5×105 4T1 cells were injected directly to the mammary fat pad, and a single dose (20 μg per 25 g body weight) of the 161-pAb or the 162-pAb negative control were injected i.p. three times as described above. Tumors were measured every 3–4 d and their volume was calculated for each mouse (length × width × 0.5 cm3). At the end of the experiment or when tumors were greater than 1.0 cm3, mice were euthanized. Parts of the tumor were freshly frozen for evaluation of cytokine concentrations, while other parts were fixed in 4% neutrally buffered formalin and embedded in paraffin for immunohistochemical staining.
Immunohistochemistry
Four μm thick paraffin embedded tissue sections of CT26 subcutaneous tumors were mounted on a glass slide and deparaffinized with xylene substitute K-Clear Plus (Kaltex, Padova, Italy) and rehydrated with decreasing ethanol immersions. Antigen retrieval for EMMPRIN was carried out by microwave heating for 15 min in Tris-EDTA buffer pH 9.0, for Ki-67 and F4/80 by microwave heating in citrate buffer pH 6.0, for CD31 by immersing the slides in 42 mg/mL Proteinase XXIV solution (Sigma) for 10 min at 37°C, or in 20 mg/mL of Proteinase K in Tris buffer, pH 7.4–8.0 for the TUNEL kit. Endogenous peroxidase was quenched in 3% H2O2 solution for 10 min, then the slides were blocked with 5% BSA and incubated overnight at 4°C with the following primary antibodies: the 161-Ab, the rabbit monoclonal anti-CD147 (ab108317, Abcam, Cambridge, United Kingdom) diluted 1:400, rat monoclonal anti-CD31 (BM4086, Acris Antibodies, Herford, Germany) diluted 1:50, rabbit monoclonal anti-Ki67 (ab16667, Abcam) diluted 1/140, rat monoclonal anti-F4/80 (ab6640, Abcam) diluted 1:200. After washing, the antibodies were detected with HRP-Polymer anti-rabbit (ZUC032–006, Zytomed, Berlin, Germany) or with the N-Histofine Simple Stain Mouse MAX PO (Rat) (414311F, Nichirei Bioscience, Tokyo, Japan) for 1 h and the DAB substrate Kit (Zytomed). All sections were counterstained with hematoxylin (Sigma) and coverslips were applied using Pertex mounting medium (Histolab Products AB, Gothenburg, Sweden). TUNEL staining was performed using the in situ death detection kit POD (Roche Life Science, Indianapolis, IN, USA) according to the manufacturer's instructions. All sections were viewed under the Olympus BX-60 bright field trinocular microscope equipped with a Sony DXC-950P digital camera. Images were acquired using the GrabBee X video grabber (VideoHome Technology Corp., Taipei, Taiwan). Vessel densities were assessed by CD31 staining using a Weibel grid and expressed as percent vessel surface area.31 The fraction of Ki-67-positive tumor cells was calculated by the digital image analysis web application ImageJS.32
In vitro cytotoxicity assay
Target cells (5×104 CT26 cells) were labeled with 5 μM of Cell Tracker orange™ (Molecular Probes, Invitrogen) and incubated for 6 h with 1.28 μg/mL of the purified 161-Ab, with addition of RAW 264.7 cells (2.5×104 cells) stimulated for 24 h with IFNγ (100 U/mL, R&D systems) and LPS (1 μg/mL, Sigma), or with mouse complement diluted 1:50 (MP biomedicals, Solon, OH), or with both. Release of the fluorescent stain to the supernatants indicated cell death. Percent cytotoxicity of target cells was calculated by the formula:
where RFU are the relative fluorescent units, spontaneous release (0% cytotoxicity) was measured from CT26 cells incubated alone, and maximal RFU (100% cytotoxicity) was measured from CT26 cells incubated with 5% Triton X-100.
Statistical analyses
All values are presented as means±SE. Significance between two groups was determined using the two-tailed unpaired t test. Differences between three or more experimental groups were analyzed using analysis of variance (ANOVA) and the post-hoc Bonferroni's multiple comparison tests. p values exceeding 0.05 were not considered significant.
Disclosure of potential conflicts of interest
E.S. and V.B. have declared that no conflict of interest exists. M.W., N.L., H.B. and M.A.R. are the inventors of a pending patent application related to the research described in the manuscript.
Acknowledgments
The authors wish to thank Prof. Miri Blank for her advice and critical reading of the manuscript.
Funding
This study was supported by a grant from the Rappaport Family Institute for Research in the Medical Sciences.
References
- 1.Owen JL, Iragavarapu-Charyulu V, Gunja-Smith Z, Herbert LM, Grosso JF, Lopez DM. Up-regulation of matrix metalloproteinase-9 in T lymphocytes of mammary tumor bearers: role of vascular endothelial growth factor. J Immunol 2003; 171:4340-51; PMID:14530359; http://dx.doi.org/ 10.4049/jimmunol.171.8.4340 [DOI] [PubMed] [Google Scholar]
- 2.Farina AR, Mackay AR. Gelatinase B/MMP-9 in Tumour Pathogenesis and Progression. Cancers (Basel) 2014; 6:240-96; PMID:24473089; http://dx.doi.org/ 10.3390/cancers6010240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004; 104:2224-34; PMID:15231578; http://dx.doi.org/ 10.1182/blood-2004-03-1109 [DOI] [PubMed] [Google Scholar]
- 4.Nabeshima K, Iwasaki H, Koga K, Hojo H, Suzumiya J, Kikuchi M. Emmprin (basigin/CD147): matrix metalloproteinase modulator and multifunctional cell recognition molecule that plays a critical role in cancer progression. Pathol Int 2006; 56:359-67; PMID:16792544; http://dx.doi.org/ 10.1111/j.1440-1827.2006.01972.x [DOI] [PubMed] [Google Scholar]
- 5.Yurchenko V, Constant S, Eisenmesser E, Bukrinsky M. Cyclophilin-CD147 interactions: a new target for anti-inflammatory therapeutics. Clin Exp Immunol 2010; 160:305-17; PMID:20345978; http://dx.doi.org/ 10.1111/j.1365-2249.2010.04115.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanekura T, Chen X. CD147/basigin promotes progression of malignant melanoma and other cancers. J Dermatol Sci 2010; 57:149-54; PMID:20060267; http://dx.doi.org/ 10.1016/j.jdermsci.2009.12.008 [DOI] [PubMed] [Google Scholar]
- 7.Weidle UH, Scheuer W, Eggle D, Klostermann S, Stockinger H. Cancer-related issues of CD147. Cancer Genomics Proteomics 2010; 7:157-69; PMID:20551248; http://dx.doi.org/ 10.1111/j.1365-2249.2010.04115.x [DOI] [PubMed] [Google Scholar]
- 8.Voigt H, Vetter-Kauczok CS, Schrama D, Hofmann UB, Becker JC, Houben R. CD147 impacts angiogenesis and metastasis formation. Cancer Invest 2009; 27:329-33; PMID:19160100; http://dx.doi.org/ 10.1080/07357900802392675 [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Zhu P, Jiang JL, Zhang Q, Wu ZB, Yao XY, Tang H, Lu N, Yang Y, Chen ZN. Involvement of CD147 in overexpression of MMP-2 and MMP-9 and enhancement of invasive potential of PMA-differentiated THP-1. BMC Cell Biol 2005; 6:25; PMID:15904490; http://dx.doi.org/ 10.1186/1471-2121-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bougaten F, Quemener C, Kellouche S, Naimi B, Podgorniak MP, Millot G, Gabison EE, Calvo F, Dosquet C, Lebbe C et al.. EMMPRIN promotes angiogenesis through HIF-2{alpha} mediated regulation of soluble VEGF isoforms and their receptor VEGFR-2. Blood 2009; 114:5547-56; PMID:19837976; http://dx.doi.org/ 10.1182/blood-2009-04-217380 [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Hemler ME. Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res 2001; 61:2276-81; PMID:11280798 [PubMed] [Google Scholar]
- 12.Sato T, Ota T, Watanabe M, Imada K, Nomizu M, Ito A. Identification of an active site of EMMPRIN for the augmentation of matrix metalloproteinase-1 and -3 expression in a co-culture of human uterine cervical carcinoma cells and fibroblasts. Gynecol Oncol 2009; 114:337-42; PMID:19427027; http://dx.doi.org/ 10.1016/j.ygyno.2009.04.004 [DOI] [PubMed] [Google Scholar]
- 13.Koga K, Aoki M, Sameshima T, Hamasaki M, Egawa N, Seiki M, Toole BP, Suzumiya J, Nabeshima K. Synthetic emmprin peptides inhibit tumor cell-fibroblast interaction-stimulated upregulation of MMP-2 and tumor cell invasion. Int J Oncol 2011; 39:657-64; PMID:21637915; http://dx.doi.org/ 10.3892/ijo.2011.1060 [DOI] [PubMed] [Google Scholar]
- 14.Ju XZ, Yang JM, Zhou XY, Li ZT, Wu XH. EMMPRIN expression as a prognostic factor in radiotherapy of cervical cancer. Clin Cancer Res 2008; 14:494-501; PMID:18223224; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1072 [DOI] [PubMed] [Google Scholar]
- 15.Huang XQ, Chen X, Xie XX, Zhou Q, Li K, Li S, Shen LF, Su J. Co-expression of CD147 and GLUT-1 indicates radiation resistance and poor prognosis in cervical squamous cell carcinoma. Int J Clin Exp Pathol 2014; 7:1651-66; PMID:24817962 [PMC free article] [PubMed] [Google Scholar]
- 16.Amit-Cohen BC, Rahat MM, Rahat MA. Tumor cell-macrophage interactions increase angiogenesis through secretion of EMMPRIN. Front Physiol 2013; 4:178; PMID:23874303; http://dx.doi.org/ 10.3389/fphys.2013.00178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fung AS, Lee C, Yu M, Tannock IF. The effect of chemotherapeutic agents on tumor vasculature in subcutaneous and orthotopic human tumor xenografts. BMC Cancer 2015; 15:112; PMID:25884767; http://dx.doi.org/ 10.1186/s12885-015-1091-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, Jacks T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011; 19:72-85; PMID:21251614; http://dx.doi.org/ 10.1016/j.ccr.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agrawal SM, Silva C, Wang J, Tong JP, Yong VW. A novel anti-EMMPRIN function-blocking antibody reduces T cell proliferation and neurotoxicity: relevance to multiple sclerosis. J Neuroinflammation 2012; 9:64; PMID:22480370; http://dx.doi.org/ 10.1186/1742-2094-9-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ku XM, Liao CG, Li Y, Yang XM, Yang B, Yao XY, Wang L, Kong LM, Zhao P, Chen ZN. Epitope mapping of series of monoclonal antibodies against the hepatocellular carcinoma-associated antigen HAb18G/CD147. Scand J Immunol 2007; 65:435-43; PMID:17444954; http://dx.doi.org/ 10.1111/j.1365-3083.2007.01930.x [DOI] [PubMed] [Google Scholar]
- 21.Reynolds AR. Potential relevance of bell-shaped and u-shaped dose-responses for the therapeutic targeting of angiogenesis in cancer. Dose Response 2009; 8:253-84; PMID:20877487; http://dx.doi.org/24430180 10.2203/dose-response.09-049.Reynolds [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gul N, Babes L, Siegmund K, Korthouwer R, Bogels M, Braster R, Vidarsson G, ten Hagen TL, Kubes P, van Egmond M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest 2014; 124:812-23; PMID:24430180; http://dx.doi.org/ 10.1172/JCI66776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braster R, O'Toole T, van Egmond M. Myeloid cells as effector cells for monoclonal antibody therapy of cancer. Methods 2014; 65:28-37; PMID:23811299; http://dx.doi.org/ 10.1016/j.ymeth.2013.06.020 [DOI] [PubMed] [Google Scholar]
- 24.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013; 23:277-86; PMID:23518347; http://dx.doi.org/ 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 25.Perske C, Lahat N, Levin SS, Bitterman H, Hemmerlein B, Rahat MA. Loss of inducible nitric oxide synthase expression in the mouse renal cell carcinoma cell line RENCA is mediated by microRNA miR-146a. Am J Pathol 2010; 177:2046-54; PMID:20709800; http://dx.doi.org/ 10.2353/ajpath.2010.091111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Wu G, Yu L, Yuan J, Fang F, Zhai Z, Wang F, Wang H. Inhibition of CD147 expression reduces tumor cell invasion in human prostate cancer cell line via RNA interference. Cancer Biol Ther 2006; 5:608-14; PMID:16627983; http://dx.doi.org/ 10.4161/cbt.5.6.2661 [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Ku XM, Li Y, Bian HJ, Zhang SH, Ye H, Yao XY, Li BH, Yang XM, Liao CG et al.. Regulation of matrix metalloproteinase production and tumor cell invasion by four monoclonal antibodies against different epitopes of HAb18G/CD147 extracellular domain. Hybridoma (Larchmt) 2006; 25:60-7; PMID:16704305; http://dx.doi.org/ 10.1089/hyb.2006.25.60 [DOI] [PubMed] [Google Scholar]
- 28.Tang Y, Nakada MT, Rafferty P, Laraio J, McCabe FL, Millar H, Cunningham M, Snyder LA, Bugelski P, Yan L. Regulation of vascular endothelial growth factor expression by EMMPRIN via the PI3K-Akt signaling pathway. Mol Cancer Res 2006; 4:371-7; PMID:16778084; http://dx.doi.org/ 10.1158/1541-7786.MCR-06-0042 [DOI] [PubMed] [Google Scholar]
- 29.Dean NR, Newman JR, Helman EE, Zhang W, Safavy S, Weeks DM, Cunningham M, Snyder LA, Tang Y, Yan L et al.. Anti-EMMPRIN monoclonal antibody as a novel agent for therapy of head and neck cancer. Clin Cancer Res 2009; 15:4058-65; PMID:19509148; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Shen ZY, Chen XG, Zhang Q, Bian HJ, Zhu P, Xu HY, Song F, Yang XM, Mi L et al.. A randomized controlled trial of Licartin for preventing hepatoma recurrence after liver transplantation. Hepatology 2007; 45:269-76; PMID:17256759; http://dx.doi.org/ 10.1002/hep.21465 [DOI] [PubMed] [Google Scholar]
- 31.Weibel ER, Kistler GS, Scherle WF. Practical stereological methods for morphometric cytology. J Cell Biol 1966; 30:23-38; PMID:5338131; http://dx.doi.org/ 10.1083/jcb.30.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Almeida JS, Iriabho EE, Gorrepati VL, Wilkinson SR, Gruneberg A, Robbins DE, Hackney JR. ImageJS: Personalized, participated, pervasive, and reproducible image bioinformatics in the web browser. J Pathol Inform 2012; 3:25; PMID:22934238; http://dx.doi.org/ 10.4103/2153-3539.98813 [DOI] [PMC free article] [PubMed] [Google Scholar]