

Abstract
Human-associated microbiota form and stabilize communities based on interspecies interactions. We review how these microbe-microbe and microbe-host interactions are communicated to shape communities over a human’s lifespan, including periods of health and disease. Modeling and dissecting signaling in host-associated communities is crucial to understand their function, and will open the door to therapies that prevent or correct microbial community dysfunction to promote health and treat disease.
Introduction
Throughout history, humans decried the wrath of gods or the configuration of the stars as the cause of ravaging diseases, such as bubonic plague (Yersinia pestis), Cholera (Vibrio cholerae), Typhus (Rickettsia typhi), or smallpox (Variola major). The microbiology era began when Antonie Van Leeuwenhoek crafted fine lenses and microscopes to visualize environmental and human-associated ‘animalcules’ or microorganisms, a finding which amazed both the scientific community and general public. An equally stunning revolution has been underway over the last two decades as scientists use DNA sequencing to observe and classify the trillions of microbes that cover our planet and our bodies.
Rather than trying to maintain sterility at the interface with the environment, hosts evolve mechanisms to influence the composition and function of their associated microbial communities. Scientific studies have transformed our thinking of all microbes as enemies to highlight the fundamental roles human-associated microbes play in promoting health throughout a lifetime.
The bacteria, fungi, viruses, and archaea that reside in and on the human body constitute our microbiota, and their genes are our microbiome. These complex communities contain taxa from across the tree of life, with deep lineages of multiple genera, species and strains. Although techniques to culture these microbes have improved, as we understand more about their metabolic requirements, only a small minority has been cultured. Culture-independent high-throughput sequencing has greatly expanded the repertoire of microbes known to reside in our bodies and in the environment.
A growing body of research seeks to uncover the ‘rules’ by which microbes interact with each other in multispecies communities and cohabitate with their multicellular hosts such as humans. A corollary goal is to understand how these rules are communicated or enforced. Here, we begin by considering the assembly and stability of human-associated microbial communities, including both inter- and intra-individual variation. We highlight studies focused on the acquisition of strains in the early childhood years, which examine this time as a critical set-point to shape microbial communities and prime the immune system. Next we explore microbe-host and microbe-microbe interactions, with host pruning of the gut community as an elegant example of collaborations and competitions played out through mucus and antimicrobial peptides acting in close proximity and diffusible metabolites acting at a distance. These studies span the spectrum from basic to clinical, from one microbe colonizing a gnotobiotic mouse to donor feces infused into a human patient with intractable Clostridium difficile diarrhea. While myriad attributes of microbes and microbial communities have been elucidated, this review is limited primarily to human-associated communities and admittedly mainly on bacteria. Topics such as the virome or host immune-microbe interactions were generally neglected by us, but definitively reviewed by experts in these fields (Belkaid and Hand, 2014; Virgin, 2014).
We look forward in two ways: by surveying emerging topics in microbial community signaling, including metabolic ‘outsourcing’ from microbes to humans; and by considering the growing need to understand microbe-microbe and microbe-host interactions at the level of molecular mechanism. Ultimately, a central aim is to achieve therapeutic benefit by redirecting microbial communication signals to increase community resilience or promote colonization by organisms that can treat or prevent disease.
1. Assembly and stability of human-associated microbial communities
Overview
Since molecular tools were developed to interrogate species and strains of bacteria present in a community, a driving theme has been to determine how human-associated microbial communities are formed, and to determine at what level – if any – these communities are stable. A tremendous amount of data obtained at ever greater resolution has been collected and analyzed to address fundamental questions of microbial community structure: How much do communities vary between individuals? Do people share a core microbiome at the level of species, strains or functional units? As baseline data is collected, scientists and the general public are asking whether the composition and function of these communities depend on race, ethnicity, place of birth, mode of delivery, age, health status, diet, and myriad other genetic and environmental factors.
Heritability of human-associated microbial communities
To address questions about microbial inheritance, scientists have first grappled with the enormity of the datasets. It would be impractical to perform an all-against-all comparison of every microbial taxa with every piece of clinical metadata. Data complexity has to be reduced either by defining a core microbiome and then aggregating microbes into community sub-structures, or by controlling for genetic and environmental heterogeneity.
In terms of reducing data complexity, the search for microbial community types is akin to arranging single nucleotide polymorphisms (SNPs) into blocks of human genetic linkage disequilibrium. These blocks simplified the task of associating regions of the human genome with inherited disease susceptibilities. However, SNPs are arranged linearly along a strand of DNA, which provides the organizational structure to cluster SNPs (International HapMap Consortium, 2005). No similar organizing principle has been identified for the units of function within a microbial community (Knights et al., 2014; Koren et al., 2013). Even the concept of a core microbiome has remained elusive as discussions continue about whether “core” should be defined based on species, strains or functional units. Although enterotypes or vaginal community types may obscure important elements of microbial community structure, they have proved useful for analyzing longitudinal studies of disease (Arumugam et al., 2011; Nobel et al., 2015; Ravel et al., 2011).
The other approach to reduce gene/environment complexity is to study mono-and dizygotic twins (MZT and DZT, respectively). Twin studies provide baseline data to quantify heritability of microbial taxa attributed to shared genetic (and environmental) features. Based on limited bacterial 16S rRNA (used as a signature of bacterial genus or species) sequencing of the gut microbial communities of 31 MZT and 23 DZT, an early study concluded that gut microbial communities have limited heritability with a comparable degree of co-variation between adult monozygotic and dizygotic twin pairs (Turnbaugh et al., 2009). A larger study of 171 MZT and 245 DZT, including 98 longitudinal samples sequenced with higher resolution concurred that relatively little of the human microbiome is heritable. However, with their large dataset, they could identify several linked microbial taxa whose abundances were influenced by host genetics (Goodrich et al., 2014). Ley and colleagues focused on the bacterial family Christensenellaceae, which had the highest heritability and formed a co-occurrence network with other closely associated heritable gut bacterial families. Phenotypically, Christensenellaceae was enriched in individuals with low body mass index. In a mouse model, addition of a strain of Christensella reduced recipient weight gain, completing the loop by showing that host genetics influences the composition of the gut microbial community, which in turn can affect host metabolism. This study demonstrates how taxa such as Christensenellaceae may be recruited and retained for benefits they provide the host. However, while these findings set the stage for deciphering the complexity of host-microbial community associations, they leave open the possibility that stochastic features might contribute to early strain acquisition followed by host selection and community pruning.
Opstal and Bordenstein expand this intellectual framework, challenging the host-centric interpretation of microbiome heritability as unidirectional and encouraging the use of models that encompass microbial interspecies interactions that drive the assembly and stability of the community (van Opstal and Bordenstein, 2015). Community heritability analyses treat the host as a foundational species, but still part of the ecosystem, and then use data clustering methods to identify the fraction of total variation in the microbial community that relates to host genetic variation. Emerging experiments, many discussed below, point to microbial community interactions as self-organizing or self-selecting, so these concepts are clearly valid. The issue remains how to power human studies to encompass community heritability. One might turn to a mouse model to try to scale the complexity of controlling for microbial community structure and genetic relatedness among individuals. Rich studies with different inbred mouse strains showed diversity in the gut microbiota composition and provided evidence for variation determined by host genetic background (Org et al., 2015). Importantly for these studies genetic relatedness was determined as shared SNPs amongst the 100 sequenced inbred strains of mice that constitute the hybrid mouse diversity panel. Rather than relying upon traditional mouse genetic crosses with physical transmission of microbiota between generations, this experimental strategy better mirrors human population structures. Within these studies, cross-fostering of pups between two strains of mothers affected the pups microbial composition and response to high-fat, high-sucrose diet. And as such the authors were able to triangulate between host-encoded genetic variation and candidate microbiota to take more of a community heritability approach to assess how gut microbial communities are assembled each generation, influenced by maternal seeding, environmental factors, and host genetics.
Stability of strains in human-associated communities
Beyond strain acquisition due to heritability and environment exposure, an equally fundamental question is the extent and level of microbial community stability. Several studies have explored microbial community longitudinal stability of healthy volunteers to explore ecological principles and to power clinical studies. 16S rRNA sequencing studies across body sites have shown a community stability with a range of temporal variability dependent on body site and individual. Fierer’s comprehensive study of 4 body sites (forehead, gut, palm, tongue) of 85 adults sampled weekly for 3 months concluded that the inter-individual variability exceeds intra-individual variability observed over time (Flores et al., 2014). So while an individual monitored over the course of a disease may still serve as his/her own best control, Fierer and colleagues caution that the variability across time can still be high even if focusing on abundant taxa or community diversity. And thus clinical studies that survey chronic disorders will need to consider the ‘personal microbiome’ composition in the context of temporal variability. As for the ecological principles that shape and govern stability, Gilbert and colleagues provided a first look at the maintenance or acquisition of prevalent microbes from family members and the home environment (Lax et al., 2014).
To explore the organizing principles of microbial community stability, several groups have considered whether strains are stably maintained within diverse communities. Bork and Weinstock analyzed gut bacterial strain stability by analyzing two large shotgun stool metagenomic datasets at the SNP level. In contrast to the variation observed between individuals, strains appeared to colonize stably, with low SNP variation for 43 subjects sampled one year later (Schloissnig et al., 2013). Tackling the same question of gut microbial strain stability, Gordon used low error amplicon sequencing of the 16S rRNA gene to study the gut communities of 37 healthy adults over a five-year period (Faith et al., 2013). They found that each person harbored ∼100 bacterial species and ∼200 bacterial strains, with the majority stably maintained over the study period, and by extrapolation perhaps over a lifetime. Gordon concomitantly sequenced the genomes of 500 anaerobic bacteria to associate the 16S rRNA sequences to a genome of origin. With a nod to heritability, Gordon found that strains (defined as isolates sharing >96% of their genome content) of Methanobrevibacter smithii and Bacteroides thetaiotaomicron were shared between close family members including siblings/twins and mother-daughter pairs.
Similar to the gut, multiphyletic communities of skin commensal Staphylococcus epidermidis and Propionibacterium acnes strains are stably maintained by healthy volunteers over at least one year as assessed by strain tracking and SNP analysis. This study proposes that functional saturation across the full gene encoding potential of the species (known as the pan-genome) might drive the acquisition and stability of these communities composed of multiple strains from one species. ((Oh et al., 2014); Oh, Byrd, Kong, Segre, in press). Consistent with Gilbert’s study (Lax et al., 2014), this strain level analysis dispels the popular misconception that skin-associated strains are washed off during a shower, only to be repopulated by the environment.
An important implication of these analyses is that they empower clinical studies of chronic relapsing disorders in which a patient will be used as his/her own control.
Early childhood acquisition of strains
These studies show that a strain, once acquired, can exist within a community for years, likely having an outsize impact on host biology. As such, important studies have focused on early childhood exploring strain acquisition and the consequent important effects on immune development and health into adulthood (Figure 2).
Fig. 2. Modes of signaling discussed in the review.
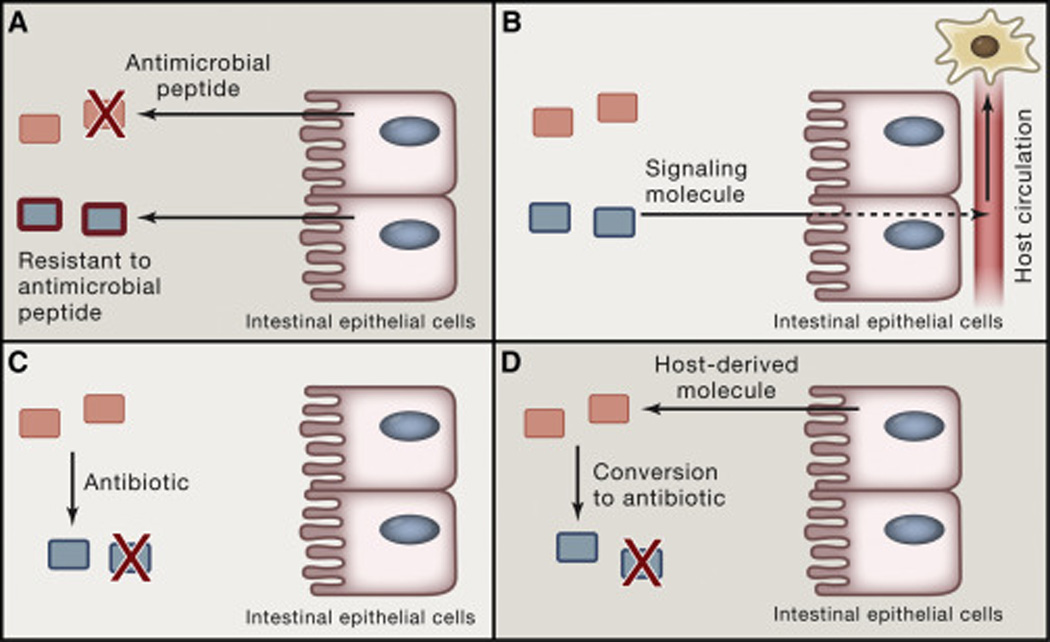
(A) Host pruning of a bacterial community. In the example shown, the host produces an antimicrobial peptide to which one species is sensitive and the other resistant. (B) Bacterially derived molecules acting at a distance. A bacterial species produces a signaling molecule that traverses the intestinal epithelium, enters circulation, and acts on a distal cellular target. (C) Direct microbe-microbe interaction, for example by the production of an antibiotic to which a neighboring species is sensitive. (D) Conversion of a host-derived molecule (e.g., a primary bile acid) to a derivative that inhibits the growth of a neighboring species.
To gain insight into the process of strain acquisition, Gordon and colleagues studied healthy children (including MZT and DZT) and adults from the Amazonas of Venezuela, rural Malawi and US metropolitan areas. The authors found distinct features of microbial composition among the three ethnic groups, and yet all three groups reached functional maturation or community stability by three years of age (Yatsunenko et al., 2012). This finding highlights early childhood as a critical set point: perturbations during this period could have lifelong effects on community composition. Of importance to global health, severe acute malnutrition (SAM) during early childhood leads to alterations in the gut community, which may have profound lifelong effects (Subramanian et al., 2014). SAM is associated with relative microbial immaturity, which is only partially ameliorated with nutritional interventions. This microbial deficiency is, consistent with the incomplete restoration of healthy growth observed in children who have experienced early SAM and later receive nutritional supplements. Understanding more about microbial communication would help to predict whether delivery of probiotic strains with food interventions might produce better long-term outcomes for malnutrition.
Picking up on Gordon’s observation that Amazonas and Malawi have more diverse gut-associated microbial communities, Sonnenburg and colleagues explore how over time, strains may be lost by individuals and by extension even from human societies when diets are poor in bacterial nutrients, such as carbohydrates found in dietary fiber (Sonnenburg et al., 2016). Specifically, Sonnenburg colonizes mice with a human fecal community and then feed mice either the Western diet (high in fat and simple carbohydrates, low in fibre) or a more traditional diet and determines that over several generations the gut communities of Western-diet mice lose bacterial diversity that cannot be restored simply by reverting the diet. Earlier, Blaser and Falkow had proposed that medical practices and lifestyle changes might have had a dramatic, poorly appreciated and likely devastating effect on ancestral beneficial microbes (Blaser and Falkow, 2009). Blaser and Falkow’s postulate that this loss of microbial diversity or “disappearing microbiota” might exacerbate and underlie the recent rise of modern conditions such as asthma and obesity.
Faith, Colombel and Gordon contextualize these findings by proposing two basic tenets: the majority of resident strains are acquired in the first three years of life, primarily from family members; and strains can colonize for decades, and may only affect host phenotype later in life (Faith et al., 2015). This point is crucial in teasing apart where to place microbes in the context of diseases with gene-environment interactions. For example, perhaps early childhood strain inheritance from a family member (e.g. affected parent or sibling) impacts the relative risk or severity for a complex adult-onset disorders such as inflammatory bowel disease (Figure 1). An intriguing question is whether inherited microbes might be part of the “missing heritability” that cannot be mapped to either common or rare variants in the human genome. The authors argue that the failure of numerous studies seeking to find association of a microbe to a complex disease may be the long latency between strain/community colonization and disease-associated manifestations.
Fig. 1. Microbial inheritance, strain stability and late onset disease manifestation.
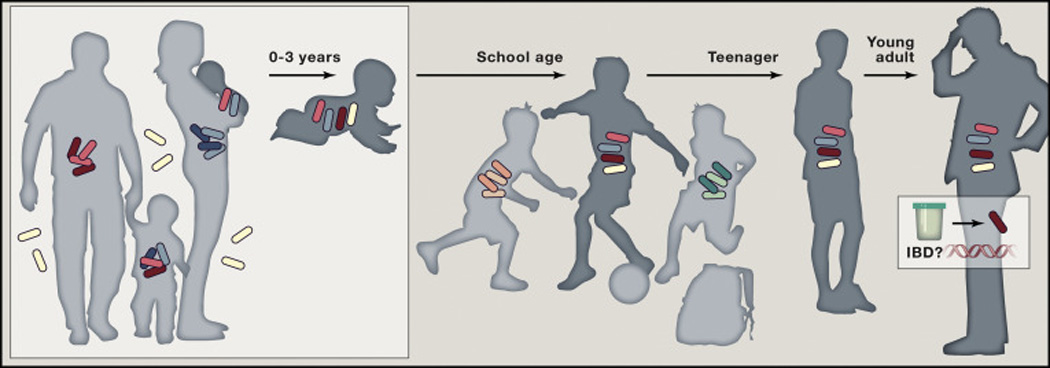
During the first three years of life children acquire microbes from closely relatives and environment. Heritance may act on individual species and/or microbial communities. These strains are stably maintained and may impact the relative risk or severity for complex adult-onset disorders such as inflammatory bowel disease.
The importance of early childhood exposure in priming and establishing immune responses has been reviewed extensively (Belkaid and Hand, 2014; Hegazy and Powrie, 2015). Specific to microbial communication, tolerance established in early life has been an unexpected feature of microbial communities. Honda and colleagues demonstrated that oral inoculation of Clostridium in early life promoted accumulation of specific regulatory T cells and resulted in resistance to induced gut inflammation in adult mice (Atarashi et al., 2013). Similarly, Rosenblum and colleagues demonstrated that S. epidermidis exposure across an impaired skin barrier elicits a skin inflammatory response in adult mice. However, an initial exposure to S. epidermidis in the first week of life leads to tolerance in adult mice through induction of specific regulatory T cells (Scharschmidt et al., 2015). These mouse experiments suggest new potential therapeutic options for microbial associated gut and skin diseases such as inflammatory bowel disease and atopic dermatitis, respectively. A fascinating clinical trial underscored the possible relevance of “tolerance” to human disease. Whereas avoidance has been the common recommendation to avoid peanut allergy, the LEAP study team established a large cohort of 530 at-risk infants and demonstrated that active consumption of peanuts reduced the prevalence of peanut allergy at 5 years from 13.7% to 1.9% (Toit et al., 2015). Even for the ∼100 participants with an initial positive skin-prick test to allergen, the prevalence of peanut allergy was 35.3% in the avoidance group and only 10.6% in the consumption group.
Together these findings underscore the critical nature of early childhood for strain acquisition, microbial community stabilization, and appropriate priming of the immune system. We now explore the mechanisms by which these communities might be acquired, selected and stabilized.
2. Microbe-host interactions
Host pruning of the gut community
Nearly all internal and external surfaces of multicellular organisms are colonized by bacteria. Rather than trying to maintain sterility at the interface with the environment, hosts have evolved signaling mechanisms by which they can influence the composition and function of their associated communities by controlling the molecular composition of the surface to be colonized. All of these features can be interpreted as an effort on the part of the host to control the composition and/or function of its colonists: 1) the chemical composition of mucus and secretions; 2) which molecules are sequestered and not secreted; and 3) how mucus and secretions change in response to a stimulus (Figure 2).
One common class of molecular signals for controlling community composition are antimicrobial peptides (AMPs), secreted by epithelial cells and typically associated with inflammation. The intricate relationship of these gut-induced AMPs and commensal microbes was uncovered by Goodman and colleagues, who set out to solve the mystery of why Bacteroides, the most common genus in the human gut community, is markedly resistant to cationic antimicrobial peptides (Cullen et al., 2015). A genetic screen for increased AMP sensitivity amongst a gut Bacteroides revealed an endogenous phosphatase that dephosphorylates lipid A (the lipopolysaccharide core, normally di-phosphorylated in most Gram-negative species (Christian R H Raetz, 2002)) to make it less anionic; the phosphatase mutant is unable to stably colonize the murine intestine. By showing that the predominant commensal Gram-negative gut bacterium has evolved to grow in the presence of high concentration AMPs, this work reveals why the host can then produce AMPs to restrict the growth of other likely pathogenic Gram-negative bacteria. Cationic antimicrobial peptides are not the only host factors that shape the gut community; Hooper and coworkers have shown elegantly that secreted antibacterial lectins help to maintain a largely bacterium-free mucus layer in the gut by forming hexameric pores in the membrane of Gram-positive bacteria (Cash et al., 2006; Mukherjee et al., 2014; Vaishnava et al., 2011). Together, these studies demonstrate the means by which epithelial cells prune nearby bacterial communities by secreting factors that selectively inhibit taxa that have not evolved means of evasion. In turn, these evasion mechanisms can also be understood as ‘colonization factors’ and constitute remarkable examples of microbe-host co-evolution.
Another means by which the host can modulate community composition is to secrete diffusible metabolites that promote the growth of some taxa and inhibit others. A recent example comes from the bacterial communities associated with plant roots, which resemble inside-out intestines (Lebeis et al., 2015). Arabidopsis roots secrete the hormone salicylic acid, which acts by two mechanisms: 1) direct stimulation or suppression of microbial growth in a taxon-specific manner, and 2) indirect sculpting of the community by modulating the host immune response. Other molecules exuded by plant roots are thus candidate mediators of community composition and function.
Hansson and colleagues highlighted the profound influence the commensal microbiota exert on the dynamics of mucus layer development (Johansson et al., 2015). There are major differences between the mucosal surfaces of wild-type and germ-free mice: for example, the ileal mucus of germ-free mice is just slightly thicker than that of wild-type mice, but 85% of the wild-type mucus could be dislodged with gentle aspiration as compared to only 23% of the mucus in germ-free mice. Mechanistically, the mucus of the small intestine keeps the epithelium clean by detaching and expelling trapped bacteria in the feces. The mucus of the inner colon only becomes impenetrable after 4–6 weeks of colonization, during which time the bacterial community composition shifted, as new niches were formed. These data show the extent to which the host can remodel the coating on its ‘consumer facing’ surface, and they reveal gaps in our knowledge of how the composition of mucus is dynamically regulated in response to microbial colonists.
Finally, a key non-host but still microbiota-extrinsic factor that shapes the gut community is diet. Several studies (for example, see (David et al., 2014)) have shown that a dietary shift has a rapid but reversible effect on the composition of the gut microbiome; indeed, diet appears to have a much stronger influence on gut community composition than host genetics (Carmody et al., 2015). Although a complete mechanistic picture of how diet shapes the microbiome has not yet emerged, early studies have associated Bacteroides gene clusters with the catabolism of a specific oligosaccharide and the ability to bloom in the presence of that substrate in vivo (Larsbrink et al., 2014; Sonnenburg et al., 2010). If this principle can be generalized, then diet could come to be viewed as a collection of microbially accessible substrates that reach the small and large intestine, each one of which may enable a corresponding microbial taxon to bloom. More speculatively, the process of ‘unwrapping’ a food particle to release its contents could represent a mechanical mutualism between a species that eats, e.g., the polysaccharide coat and another that ferments the proteinaceous innards of a food particle (Fischbach and Sonnenburg, 2011a).
Bacterially derived molecules acting at a distance
Through the lens of chemical engineering, the gut is a stopped flow reactor with a selectively permeable membrane. Inside this vessel, gut bacterial convert food into high concentrations into dozens of diffusible metabolites at high concentration (mid-micromolar to low-millimolar). Many of these molecules accumulate in host circulation, which gives them the opportunity to signal at a distance in the host.
Perhaps the best-known example is the short-chain fatty acids (SCFAs) that are produced at high concentration by a wide variety of gut bacteria. In addition to serving as an energy source for enterocytes (Wilson, 2005), these molecules signal through a pair of G-protein coupled receptors (GPR41/GPR43), modulating a diverse range of host processes including regulatory T cell function (Atarashi et al., 2013; Furusawa et al., 2013; Smith et al., 2013). It is likely that other GPCRs and nuclear receptors that are orphan or which have likely been assigned a non-physiological ligand actually respond to a microbiota-derived metabolite. For example, the vitamin D receptor binds more tightly to the secondary (bacterially produced) bile acid lithocholic acid (LCA) than to vitamin D (Makishima et al., 2002), suggesting a role in sensing gut bacterial metabolism of primary bile acids.
Another molecule that is produced exclusively by the gut microbiota is trimethylamine-N-oxide (TMAO), which derives from dietary cholines (both free and phospholipid-conjugated) and carnitine. Gut bacteria harboring one of several related catabolic gene clusters (Martínez-del Campo et al., 2015) extrude trimethylamine from the dietary precursor, which is then hydroxylated in the liver to TMAO. TMAO appears to promote atherogenesis, and small molecules that inhibit the bacterial trimethylamine lyase block atherosclerosis in apoE-null mice (Wang et al., 2015). The molecular mechanism by which TMAO signals has begun to emerge; platelets exposed to the molecule become hyperreactive by a mechanism that involves the induced release of Ca2+ from intracellular stores, increasing the risk of thrombosis (Zhu et al, Cell 2016, in press) (Figure 3).
Fig. 3. Microbe-host signaling at a distance.
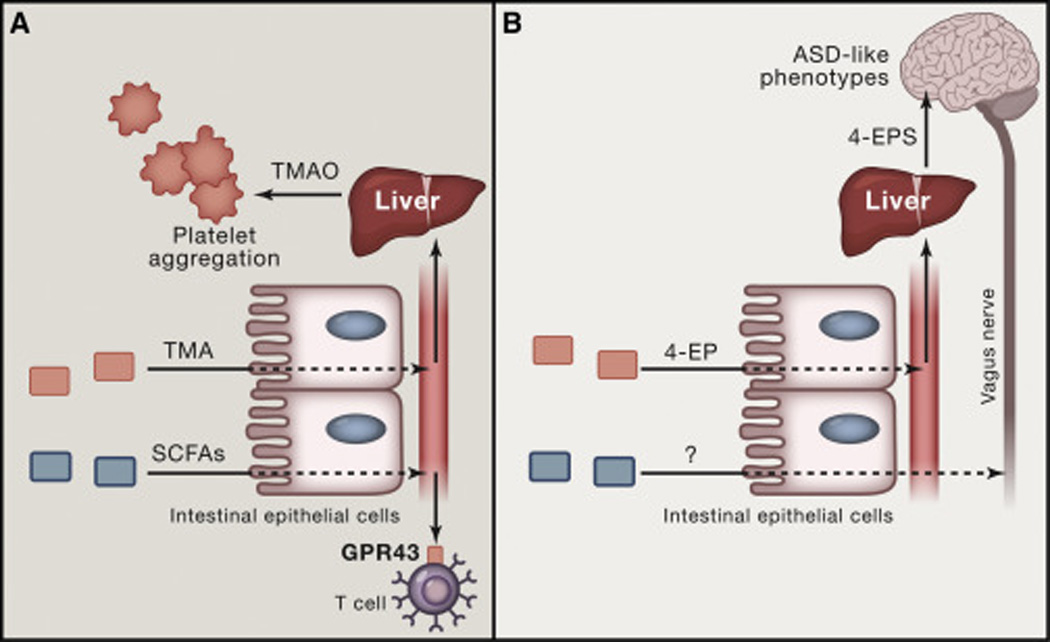
(A) Two examples are shown in which a molecule produced by a gut bacterium acts at a distance in the host. Trimethylamine (TMA) is produced by gut bacterial metabolism of choline and carnitine, enters the bloodstream, and is N-hydroxylated in the liver to trimethylamine-N-oxide (TMAO). TMAO then acts directly on platelets, causing them to aggregate. Short-chain fatty acids (SCFAs) produced by gut bacteria are sensed by a variety of cells that produce GPR41 or GPR43, including T cells. (B) Two mechanisms by which gut bacteria can signal to the brain. In the first example, a bacterial species in the gut produces 4-ethylphenol (4-EP), which is sulfated in the liver to 4-EPS. 4-EPS is then thought to cross the blood-brain barrier, where it can alter host cognition, resulting in some of the phenotypes characteristic of autism spectrum disorder (ASD). Another route by which gut bacteria can signal directly to the brain is less well understood, but involves signal transmission via the vagus nerve.
An observation about the metabolic niche created by trimethylamine-containing dietary molecules shows the therapeutic promise of microbial ecology: when vegans are fed carnitine, no TMA (and thus no TMAO) is produced, due to a lack of TMA-producing bacteria in the community (Koeth et al., 2013). This shows that healthy gut communities exist that enable humans to eat meat and eggs without suffering the ill effects of carnitine-derived TMAO; even if the community needed daily replenishment to prevent the otherwise inevitable outgrowth of TMA producers, this would undoubtedly be more effective (and palatable) than convincing at-risk patients to modify their diet.
A wide variety of ligands for receptors in immune cells act at a distance. These include LPS, which signals through TLR4 (Park et al., 2009) and an intracellular sensor, caspase-4/5/11 (Yang et al., 2015); and ligands for TLR2, including polysaccharide A from Bacteroides fragilis (Round et al., 2011) and lipoproteins (Jin et al., 2007). Medzhitov and coworkers were among the first to show that immune response to the microbiota can be beneficial (Rakoff-Nahoum et al., 2004); they showed that TLR4 signaling, which had been a hallmark of the pro-inflammatory response to Gram-negative pathogens, was paradoxically protective against DSS-induced colitis. Among the many outstanding questions in this area, three stand out: What are the microbiota-derived molecules that induce naïve T cells to differentiate into regulatory T cells? In what form do lipid-bearing ligands get to immune cells and how do they traverse the mucus layer: outer membrane vesicles, membrane sloughing from dead cells, or bile acid solubilization, with analogy to solubilizing dietary lipids? And what role is played by contact-dependent signaling by microbes that adhere to the intestinal epithelium (Atarashi et al., 2013; Sano et al., 2015)?
Gut-brain and gut-metabolism axes
A growing number of recent studies (reviewed in (Ridaura and Belkaid, 2015)) have shown an effect of the gut microbiota on host behavior and cognition. In the story that has gone furthest into molecular mechanistic detail, Mazmanian and coworkers used the murine maternal immune activation (MIA) model of autism spectrum disorder (ASD) to show that the gut microbiota of MIA mice can confer on germ-free mice some behavioral deficits characteristic of ASD (Hsiao et al., 2013). A metabolomic analysis pointed to ethyl phenyl sulfate as a microbiota-derived molecule whose level was increased in the serum of MIA mice, and this molecule alone was sufficient to induce a similar phenotype (Figure 3).
Importantly, this manuscript shows that a microbially-produced molecule can get absorbed into the bloodstream, cross the blood-brain barrier and alter host cognition – in this way, it establishes a plausible case for a molecular mechanism by which the microbiota can directly alter host behavior. Although the study does not show that the microbiota play a causative role in ASD, it opens the door to more detailed studies that seek to determine how microbiota-derived metabolites impinge directly on signaling in the brain, a theme that is extended by recent work from Hsiao and coworkers showing that the gut microbiota can induce serotonin production by the host (Yano et al., 2015).
Satiety (the sense of feeling full) is another important thread linking gut-brain signaling to the control of host metabolism. Initial work showed that induction of intestinal gluconeogenesis is the mechanism by which protein feeding leads to a satiety response involves (Mithieux et al., 2005). With subsequent investigation Mithieux and colleagues showed that this effect involves the sensing of protein-derived peptides by µ-opioid receptors in portal vein nerves (Duraffourd et al., 2012). And finally, connecting to the microbiome, the same group showed that intestinal gluconeogenesis can be induced by microbiota-derived short-chain fatty acids, with propionate and butyrate activating the same circuit by distinct signaling mechanisms (De Vadder et al., 2014). Together, these studies show that a host physiological response critical for metabolic homeostasis responds to chemical signals from the gut, including directly from the microbiota. The gut microbiota influence host metabolism in many other ways; interested readers are directed to a recent review from Tremaroli and Backhed (Tremaroli and Bäckhed, 2012).
3. Microbe-microbe interactions
Why are bacterial communities stable?
Community stability is a basic ecological phenomenon with broad implications for community composition and microbe-host interactions. Several studies have investigated both theoretically and experimentally how microbial communities are stabilized. Schluter and Foster leverage network models from theoretical ecology to identify general principles underlying microbiome stability (Coyte et al., 2015). The authors extend these theories to include unstructured ecological networks, and measure stability or resilience as the probability that a community will return to its original state after a perturbation. Most of the existing models suggest that high species diversity (such as observed in human microbiome datasets) would be unstable, an apparent contrast between theory and experiment. The authors propose a resolution to this conflict by developing models demonstrating that a diverse and competitive community leads to a stable microbiome. While increasing species or strains is a priori a destabilizing force, in fact the competition introduces negative-feedback loops that have a stabilizing effect. In contrast to cooperation, which can lead to species loss, competition dampens positive-feedback loops and promotes stability. Remarkably, these models can also account for how adaptive immunity promotes stability by suppressing outgrowth of pathogenic species. In closing, they describe hosts as “ecosystem engineers that manipulate general, system-wide properties of the microbial communities to their benefit”. Discovering and testing many of these stabilizing modes of communication will require tractable experimental systems.
Competitive exclusion
One mechanism by which community stability is maintained is competitive exclusion, a process in which a resident species prevents an incipient colonist from gaining a foothold in the community. Mazmanian and colleagues sought to define the molecular processes used by symbiotic bacteria to stably colonize the gastrointestinal tract, using the prevalent gut genus Bacteroides as their example. Germ-free mice mono-associated with a single Bacteroides species resist colonization by another strain of this same species, while still retaining the capacity to be co-colonized with other Bacteroides species. To identify the molecular mechanism underlying colonization saturation, the authors devised a clever scheme to identify dominant-acting factors by mono-associating mice with Bacteroides vulgatus, and then challenging with them a library of B. vulgatus containing ∼10 kb fragments of genomic DNA from Bacteroides fragilis (Lee et al., 2013) (Figure 4). Of the thousands of clones that were used to challenge B. vulgatus mono-associated mice, only two stably colonized the animals, and the cloned inserts both mapped to the same locus, subsequently named commensal colonization factors (ccf). ccf genes encode proteins that bind and import glycans, potentially creating both a physical and metabolic niche for the bacterial species expressing them. This study highlights how clever observations combined with defined, reductionist systems are essential to generate and test functional hypothesis about communication between strains, species and hosts. Moreover, it raises the question of how the substrates and form of the mucosal surface promotes stable and resilient colonization.
Fig. 4. Competition exclusion, colonization resistance, and decolonization via microbial interactions.
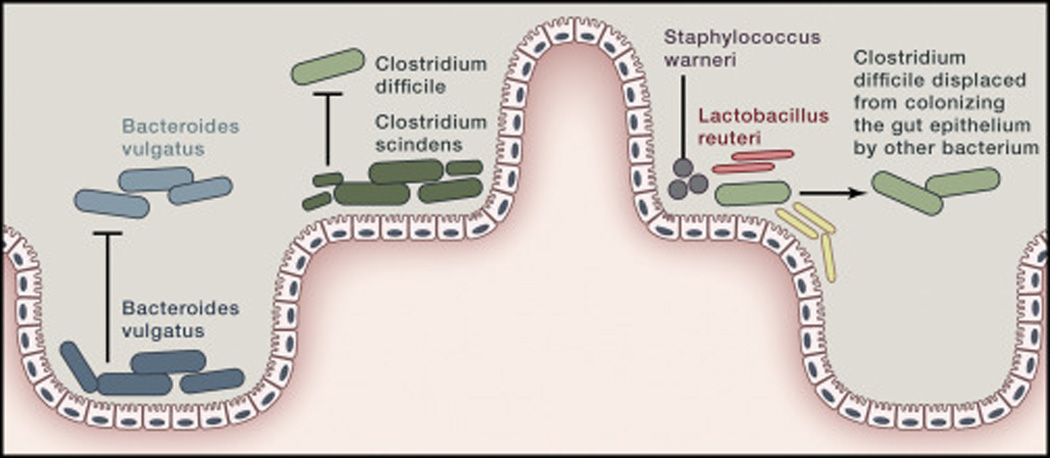
An example of competition exclusion is one strain of Bacteroides vulgatus (dark blue) excluding another (lighter blue) from colonizing the gut epithelium. Similarly, colonization resistance is demonstrated when Clostridium scindens in combination with a stable microbial community excludes the pathogen C. difficile from colonizing the gut. If C. difficile colonizes the gut epithelium, a mixture of Staphylococcus warneri, Enterococcus hirae, Lactobacillus reuteri, and novel species of Anaerostipes, Bacteroidetes and Enterorhabdus can displace the C. difficile resulting in decolonization.
While Mazmanian presents one example of how commensals may develop an intimate physical relationship with the more protected crypts, Kishony’s work explores antibiotic production as a specific mode of microbial communication, and proposes general theories of how microbial communities support species diversity (Kelsic et al., 2015). Simplistic models assume pairwise species relationships in which antibiotic producing organisms inhibit sensitive species. These pairwise inhibitory interactions can support multiple species coexisting with a model of cyclic dominance, best understood by the rock-paper-scissors game (Durrett and Levin, 1998; Kerr et al., 2002). Kishony and colleagues introduce into the model a third “modulator” species that can attenuate the effect of the antibiotic-producing species on the antibiotic-sensitive species (for example, by enzymatically degrading the antibiotic). Incorporating modulator species enables stable communities to form even with large differences in growth rates and tight intermixing of species. Experimentally, mixing organisms capable of producing and degrading multiple antibiotics led to robust coexistence in a well-mixed chemostat. Interestingly, these communities are robust to cheaters, species who take a small growth advantage by ceasing production or degradation of antibiotics. These models provide a starting point to model and engineer multi-species microbial consortia that include additional means of shaping a community, such as resource competition, secondary fermentation, and predator-prey relationships.
Colonization resistance in the gut community
Human and animal models studies have begun to uncover the molecular mechanisms underlying a related phenomenon, colonization resistance, or the community’s susceptibility to a colonizing pathogen, often in the context of antibiotic-induced instability. Pamer and colleagues treated mice with various antibiotics, triggering susceptibility to infection by Clostridium difficile, a major cause of antibiotic-induced diarrhea (Buffie et al., 2014). In parallel, they performed a similar analysis with a cohort of patients undergoing stem-cell transplant, who due to antibiotic treatment and compromised immune function are particularly susceptible to C. difficile infection. The authors correlated the abundance of different bacterial species to increased or reduced susceptibility to C. difficile infection, revealing Clostridium scindens as a commensal associated with colonization resistance. Consistent with the computational prediction, pre-colonization of mice with C. scindens ameliorated the symptoms associated with C. difficile infection. Noting that genes carried by C. scindens are known to 7-dehydroxylate primary bile acids cholic acid and chenodeoxycholic acid to the secondary bile acids deoxycholic acid and lithocholic acid, the authors hypothesized that secondary bile acids might impair C. difficile growth in vivo, as has been shown in vitro (Sorg and Sonenshein, 2008) (Figure 4). Ultimately, Pamer and colleagues provide a well-validated example of how a single organism can confer colonization resistance against a common pathogen. This is important in both scientific and translational arenas as researchers strive to create artificial communities capable of recapitulating the positive effects of fecal transplant in patients with recurrent C. difficile infections.
Colonization resistance in the skin community
Similarly, skin microbes contribute to colonization resistance, shaping microbial communities and modulating innate defense both directly and indirectly (via the host). In the nares, Mizunoe and colleagues showed that strains of the common commensal Staphylococcus epidermidis provide colonization resistance against Staphylococcus aureus in human challenge experiments. The authors started with the epidemiologic finding that approximately one-third of the human population is colonized asymptomatically with S. aureus in the nares, which is a risk factor for subsequent infection (Eiff et al., 2001). In vitro, they showed that a subset of S. epidermidis strains can inhibit S. aureus biofilm formation, an activity they biochemically purified and identified as a serine protease, ESP. In isogenic strains of S. epidermidis, ESP was both necessary and sufficient for the destruction of S. aureus biofilms. Notably, S. aureus was unable to develop resistance to ESP even after one year of co-culturing, suggesting this protein targets an essential function. When healthy S. aureus-colonized human volunteers were colonized with S. epidermidis, only strains expressing ESP eliminated S. aureus colonization. Moreover, purified ESP also cleared S. aureus colonization, cementing the key role of this protein in colonization resistance. Finally, as an example of host and microorganism cooperating to combat invasion, ESP works synergistically with the human antimicrobial peptide beta-defensin 2 to kill S.aureus. Together with Gallo’s pioneering work demonstrating that commensal S. epidermidis strains enhance antimicrobial peptide expression to augment the skin’s defense against infection (Lai et al., 2009), these microbial findings present tremendous possibility for therapeutic potential to target both colonization and clearance of the human pathogen S. aureus, without having to rely on increase antibiotic usage.
Perturbing microbial communities with fecal microbiota transplant
With the potential that microbial communities can be modified for therapeutic applications, manipulation of these consortia represents an attractive new treatment modality. The lead indication has been enteric infections by Clostridium difficile, the major cause of antibiotic-associated diarrhea. First-line treatment for C. difficile infection has been the antibiotics vancomycin or metronidazole, although roughly one-quarter of patients will relapse when treatment is stopped. In 2013, Keller and colleagues reported that infusion of donor feces was more successful than standard antibiotic treatment in a randomized human clinical study, and the results were so definitive that the trial was stopped mid-way to offer all patients donor-feces infusion (van Nood et al., 2013). 15 of the first 16 (94%) patients treated with donor-feces infusion were cured, as compared to only 23–31% receiving standard of care vancomycin. The enormous clinical success of fecal microbial transplant to cure intractable Clostridium difficile diarrhea shines a very bright light on the need to understand gut microbial colonization resistance, pathogen invasion and decolonization. And as questions about microbial transplants capture the imagination of scientists and the public, basic research is exploring the ecologic principles governing the complexity of microbial communities in this context of human disease.
Before donor-feces infusion was used in a human clinical study, Lawley and others had developed these ideas in animal models. Mice infected with an epidemic strain of C. difficile, receiving the antibiotic clindamycin, developed highly contagious, chronic intestinal disease that was refractory to vancomycin treatment (Lawley et al., 2012). Administration of healthy donor feces via oral gavage to C. difficile-infected mice ameliorated clinical symptoms and cleared the pathogenic invader. To define a tractable resistant community, the authors then cultured the feces and combined individual isolates into phylogenetically distinct mixtures until a six-member community was found that reproducibly reduced C. difficile infection and bacterial load. These experiments laid the ground-work for the human clinical trial, and the hunt to identify defined strains or species of gut commensals that either promote decolonization or provide colonization resistance against C. difficile.
New frontiers in microbe-microbe signaling in communities
Quorum sensing has been well studied in the context of pathogen infection (O’Loughlin et al., 2013; Parsek and Greenberg, 2005); however, its role in the commensal microbiome is not well understood. A second class of signaling interactions is likely more common in bacterial communities, especially those that grow under anaerobic conditions. The major metabolic output of an individual organism (e.g., acetate or propionate) can be present at millimolar levels, which very likely alters the physiology of microbes in the vicinity and induces a transcriptional response. These interactions, which are bidirectional, are inevitable consequences of primary metabolism; in order to generate sufficient ATP to grow and divide, cells need to excrete large quantities of molecules to which other cells cannot help but to respond. Sometimes this establishes a syntrophic metabolic network; for example, secondary fermenters in the gut can convert Bacteroides-derived succinate to acetate or consume H2 (Fischbach and Sonnenburg, 2011b), and pairs of organisms living in the same niche can exchange oxidized and reduced sulfur compounds (Woyke et al., 2006). Secondary fermenters are often keystone species in a community – their presence enables the primary fermenters to thrive. However, the broader consequences of metabolism-related signaling interactions in bacterial communities are not well understood.
Emerging topics in microbial community signaling
Outsourcing biochemical functions to microbes
Throughout evolutionary time, humans have taken advantage of gut microbial colonization to outsource basic biochemical functions involved in food digestion to our myriad bacteria. As a key example, dietary polysaccharides supply the body with energy, but the human genome lacks the carbohydrate active enzymes to extract these nutrients from plants. Instead, humans rely on diverse gut microbes to break down oligosaccharides, including predominant Bacteroides species, many of which harbor hundreds of genes involved in glycosidic bond cleavage (Martens et al., 2014; Sonnenburg et al., 2005; Xu et al., 2003). A clear symbiotic relationship has been forged between the host and its resident Bacteroides species to access the rich nutrients of plant material that human cells cannot metabolically process. While the mutual benefit is clear, experiments are still underway to functionally identify the modes of communication whereby the microbe senses the luminal environment and the host manipulates its gene expression to establish and maintain this partnership.
A recent human evolutionary adaptation in this process was revealed when Czjzek, Michel and colleagues were looking for proteins that break down algal biomass, and identified a new class of glycoside hydrolases (Hehemann et al., 2010). With the gene in hand for enzymes capable of agarose degradation, the authors searched bacterial databases and found predicted porphyranase sequences in multiple marine Bacteroides and also, intriguingly, in Japanese stool metagenomes and the commensal gut bacterium Bacteroides plebeius. As this porphyranase gene is found uniquely in Japanese and not North American gut communities, the authors hypothesized that the Porphyra (nori) used to wrap sushi was both the genetic source and the evolutionary force underlying the recent acquisition of these genes. Since the efficiency with which the microbiota degrades polysaccharides relates to the calories the host can extract from its diet, this microbial adaptation may have recently accompanied dietary changes.
Another role that has been outsourced to microbial communities is their ability to produce antimicrobials, evolved through eons of within- and cross-kingdom competition. Through computational searches, Mougous and colleagues found that homologs of the type VI secretion amidase effector (Tae) proteins, potent bactericidal proteins, are found in eukaryotic genomes ranging from protozoa to multicellular metazoans (Chou et al., 2015). These bacterial genes now contain introns and are flanked by other eukaryotic genes, dispelling any idea that these sequences are the result of contaminating bacterial matter. Phylogenetic analyses would suggest that these multiple Tae genes have been acquired from diverse bacteria in at least 6 horizontal gene transfer events. The eukaryote-domesticated Tae genes appear to be expressed and function to provide benefit to their hosts, such as the Tae-like protein expressed by deer ticks to limit proliferation of Borrelia burgdorferi, the Lyme disease pathogen. Imagine the therapeutic potential if humans could similarly domesticate a Tae protein to recognize and cleave a B. burgdorferi peptidoglycan, before bacterial proliferation and onset of Lyme disease symptoms.
An extreme form of metabolic outsourcing surrounds fungus-farming ants. Currie and coworkers discovered that a set of crypts under the mandible of these ants serves as a bacteria-cultivating organ (Currie et al., 1999; Oh et al., 2009). The actinomycete mutualists that grow in a mat over the mycangium produce an antifungal agent that inhibits the growth of a fungal pest that can overgrow the crop fungus. More recently, this story has been extended in three directions: by showing that different clades of fungus farming ants farm distinct fungal cultivars and grow different actinomycete mutualists, whose interactions form a rich web of interactions (Cafaro et al., 2011); by identifying additional symbionts in the community (Aylward et al., 2014); and by showing that actinomycete mutualists are essential to controlling the growth of pests on other insects that cultivate fungi, including pine beetles (Scott et al., 2008).
Instead of waiting tens of thousands of years for the bacteria to find beneficial genes, could we identify or engineer beneficial microbes to provide biochemical functions the human genome lacks either for general health of for patients with rare metabolic disorders?
The microbiome and personalized medicine
There is a growing appreciation that the microbiome will be integral to personalized health and precision medicine (Collins and Varmus, 2015). An early appreciation of the personalized nature of the microbiome was the recognition that at least 40 human drugs are directly metabolized by the gut microbiome (Carmody and Turnbaugh, 2014; Patterson and Turnbaugh, 2014). Recent studies continue to impress the scientific community with unexpected roles for the microbiome. For example, one of the great breakthroughs in cancer treatment over the last decade has been immunotherapy, stimulating one’s own immune system to search out and destroy tumor cells. One puzzling feature of these new treatments has been the varying immune and clinical responses observed among patients. Papers from the groups of Gajewski and Zitvogel (Sivan et al., 2015; Vétizou et al., 2015) have now shown that commensal gut microbiota influence the immune responses against tumors and the therapeutic benefit of checkpoint blockade drugs to stimulate immunotherapeutic interventions for treating advanced melanoma. Gajewski and colleagues observe differential tumor growth in a mouse model of melanoma, depending on the vendor from which the mice were acquired. By co-housing the mice, performing fecal transfers, and isolating strains, they showed that live Bifidobacterium alters dendritic cell activity, which improves tumor-specific T cell function and thus leads to better clinical outcomes. Similarly, Zitvogel and coworkers deduced a microbial role after showing that treating mice with broad-spectrum antibiotics dampened the checkpoint blockade efficacy, and showed that gut colonization with Bacteroides species boosts the efficacy of immunotherapy. They also note that checkpoint blockade may alter the intestinal microbial composition, again suggesting a role for augmenting or preserving beneficial strains while undergoing treatment.
The gut microbial community affects how the host metabolizes food and converts it to, inter alia, blood glucose. Previous studies have shown how the relative abundance of bacteria in a defined community is altered by dietary perturbations (David et al., 2014; Faith et al., 2011). Nevertheless, most people calculate protein, carbohydrates, or simply calories of a meal and use this as a universal measure when trying to reduce their weight or blood glucose levels. Segal, Elinav and colleagues devised an algorithm that integrated a large number of personalized features with gut microbial composition to predict personalized glycemic responses, an important risk factor for cardiovascular disease and type 2 diabetes (Zeevi et al., 2015). While it does not identify key levers or keystone species, this study sets the stage for tailoring nutritional advice to individuals based on factors that include modifying their gut microbial communities.
Studying microbial signaling at the level of molecular mechanism
The ‘mechanism’ gap in current microbiome studies
Moving forward, there is a compelling need to understand signaling in microbial communities at the level of molecular mechanism. Doing so will transform a discipline that has been predominantly descriptive and correlative into one that is understood in terms of molecular interactions that drive biology. Of course, the level of detail required to prove mechanism depends on the context. In oncology, one can show that a mutation in a proto-oncogene is necessary and sufficient to transform a non-cancerous cell into a cancer cell; that genetic ablation or pharmacological inhibition reverses this phenotype; and that resistance to such a small molecule inhibitor is mediated through a mutation in the target itself or by mutational activation of a downstream component of the signaling pathway in question. In infectious disease, Koch’s postulates set the standard for proving that a microorganism or virus is the etiologic agent of disease, and a recent perspective has put forward criteria adapting these strict criteria to microbiome studies (Byrd and Segre, 2016).
A small but growing set of microbiome studies have begun approaching molecular mechanism in a way that bodes well for the future. Several have been covered earlier in the review: Goodman and colleagues showed convincingly that lipid A dephosphorylation enables Bacteroides to colonize the gut in the presence of cationic antimicrobial peptides (Cullen et al., 2015); Pamer and colleagues singled out secondary bile acids produced by C. scindens as a mechanism for colonization resistance against C. difficile (Buffie et al., 2014); Mazmanian and coworkers identified ethyl phenyl sulfate as a microbiota-derived molecule that can give rise to some of the behavioral phenotypes characteristic of the MIA model of ASD (Hsiao et al., 2013); and Hazen and colleagues have accumulated convincing evidence that TMAO plays a causative role in atherosclerosis (Wang et al., 2015). Other notable examples include landmark work from Sonnenburg and Martens that has associated Bacteroides gene clusters with the catabolism of a specific oligosaccharide and the ability to bloom in the presence of that substrate in vivo (Larsbrink et al., 2014; Sonnenburg et al., 2010), and pioneering efforts by Littman and Belkaid that have shown the extraordinary specificity with which the immune system recognizes commensals, even in the absence of an epithelial barrier breach (Naik et al., 2015; Yang et al., 2014). These studies are terrific examples of how understanding microbe-microbe and microbe-host interactions at the molecular level advance the field immeasurably and raise the prospect of new treatments for a variety of microbiome-related diseases.
Among the near-term opportunities for exploring mechanism in the microbiome, inflammatory bowel disease (particularly Crohn’s disease) stands out. Multiple studies, including two large efforts to enumerate treatment-naïve pediatric Crohn’s patients, have shown that the gut communities of a subset of Crohn’s patients are characterized by a bloom of E. coli (Gevers et al., 2014; Lewis et al., 2015). Although the disease is known to involve overactive immune signaling, and animal studies have linked adherent invasive E. coli (AIEC) to disease pathogenesis (Craven et al., 2012; Darfeuille-Michaud et al., 2004; Dogan et al., 2014; Martin et al., 2004), remarkably little is known definitively about whether E. coli (or any of the other species that are known to bloom in the Crohn’s gut) cause, trigger, or contribute to disease. Even if E. coli (or another organism) eventually satisfies Koch’s postulates for Crohn’s disease – perhaps as a necessary microbial trigger that requires a host genetic predisposition for sufficiency – the question of molecular mechanism will still remain. Which bacterial molecules help drive disease, and how? It will be necessary to identify the molecule; construct non-producing bacterial mutants that fail to cause disease in an animal model; complement this mutation with the purified molecule; identify the receptor and show that its deficiency interrupts disease initiation or progression; show biochemically that the molecule engages its receptor both in vitro and in vivo; and obtain data showing that all of this is true not just in animal models, but also in patients. This will no doubt be a steep hill to climb, but the degree to which it will advance our understanding of the disease and the ability to develop new treatments would be tremendous.
Conclusion
One of the most salient features of the relationship between host and microbial communities is the high level of integration of the microbiome with almost every organ and tissue of the human body. Even tissues considered “sterile” are still integrated with host circuits that are fueled by products of microbial degradation of energy, and under the influence of metabolic signals that result from microbial processes. Human-associated microbial communities may turn out to directly impact many of the features that we consider most “human”, underscoring the view that we must start to understand ourselves as super-organisms.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Aylward FO, Suen G, Biedermann P, Adams AS. Convergent bacterial microbiotas in the fungal agricultural systems of insects. MBio. 2014 doi: 10.1128/mBio.02077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the Microbiota in Immunity and Inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nature Reviews. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 1–16. 2014 doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, Segre JA. Adapting Koch’s postulates. Science (New York, N.Y. 2016;351:224–226. doi: 10.1126/science.aad6753. [DOI] [PubMed] [Google Scholar]
- Cafaro MJ, Poulsen M, Little AEF, Price SL, Gerardo NM, Wong B, Stuart AE, Larget B, Abbot P, Currie CR. Specificity in the symbiotic association between fungus-growing ants and protective Pseudonocardia bacteria. Proc. Biol. Sci. 2011;278:1814–1822. doi: 10.1098/rspb.2010.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Turnbaugh PJ. Host-microbial interactions in the metabolism of therapeutic and diet-derived xenobiotics. J Clin Invest. 2014;124:4173–4181. doi: 10.1172/JCI72335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM, Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host & Microbe. 2015;17:72–84. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic Bacteria Direct Expression of an Intestinal Bactericidal Lectin. Science (New York, N.Y. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Daugherty MD, Peterson SB, Biboy J, Yang Y, Jutras BL, Fritz-Laylin LK, Ferrin MA, Harding BN, Jacobs-Wagner C, et al. Transferred interbacterial antagonism genes augment eukaryotic innate immune function. Nature. 2015;518:98–101. doi: 10.1038/nature13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian RH, Raetz CW. Lipopolysaccharide Endotoxins. Annual Review of Biochemistry. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Varmus H. A New Initiative on Precision Medicine. The New England Journal of Medicine. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: Networks, competition, and stability. Science (New York, N.Y. 2015;350:663–666. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, Bowman D, Scherl EJ, Simpson KW. Inflammation Drives Dysbiosis and Bacterial Invasion in Murine Models of Ileal Crohn’s Disease. PLoS ONE. 2012;7:e41594. doi: 10.1371/journal.pone.0041594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science (New York, N.Y. 2015;347:170–175. doi: 10.1126/science.1260580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie CR, Scott JA, Summerbell RC, Malloch D. Fungus-growing ants use antibiotic-producing bacteria to control garden parasites. Nature. 1999;398:701–704. [Google Scholar]
- Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Bäckhed F, Mithieux G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell. 2014;156:84–96. doi: 10.1016/j.cell.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. Inflammation-associated Adherent-invasive Escherichia coli Are Enriched in Pathways for Use of Propanediol and Iron and M-cell Translocation. Inflammatory Bowel Diseases. 2014;20:1919–1932. doi: 10.1097/MIB.0000000000000183. [DOI] [PubMed] [Google Scholar]
- Duraffourd C, De Vadder F, Goncalves D, Delaere F, Penhoat A, Brusset B, Rajas F, Chassard D, Duchampt A, Stefanutti A, et al. Mu-Opioid Receptors and Dietary Protein Stimulate a Gut-Brain Neural Circuitry Limiting Food Intake. Cell. 2012;150:377–388. doi: 10.1016/j.cell.2012.05.039. [DOI] [PubMed] [Google Scholar]
- Durrett R, Levin S. Spatial Aspects of Interspecific Competition. Theoretical Population Biology. 1998;53:30–43. doi: 10.1006/tpbi.1997.1338. [DOI] [PubMed] [Google Scholar]
- Eiff von C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. The New England Journal of Medicine. 2001;344:11–16. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- Faith JJ, Colombel J-F, Gordon JI. Identifying strains that contribute to complex diseases through the study of microbial inheritance. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:633–640. doi: 10.1073/pnas.1418781112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The Long-Term Stability of the Human Gut Microbiota. Science (New York, N.Y. 2013;341:1237439–1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, Gordon JI. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science (New York, N.Y. 2011;333:101–104. doi: 10.1126/science.1206025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Sonnenburg JL. Eating For Two: How Metabolism Establishes Interspecies Interactions in the Gut. Cell Host & Microbe. 2011a;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Sonnenburg JL. Cell Host & Microbe. 2011b;10:336. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores GE, Caporaso JG, Henley JB, Rideout JR. Temporal variability is a personalized feature of the human microbiome. Genome …. 2014 doi: 10.1186/s13059-014-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host & Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human Genetics Shape the Gut Microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegazy AN, Powrie F. Microbiota RORgulates intestinal suppressor T cells. Science (New York, N.Y. 2015;349:929–930. doi: 10.1126/science.aad0865. [DOI] [PubMed] [Google Scholar]
- Hehemann J-H, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik S-G, Lee H, Lee J-O. Crystal Structure of the TLR1-TLR2 Heterodimer Induced by Binding of a Tri-Acylated Lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Johansson MEV, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodriguez-Piñeiro AM, Arike L, Wising C, Svensson F, Bäckhed F, et al. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host & Microbe. 2015;18:582–592. doi: 10.1016/j.chom.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsic ED, Zhao J, Vetsigian K, Kishony R. Counteraction of antibiotic production and degradation stabilizes microbial communities. Nature. 2015;521:516–519. doi: 10.1038/nature14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr B, Riley MA, Feldman MW, Bohannan BJM. Local dispersal promotes biodiversity in a real-life game of rock|[ndash]|paper|[ndash]|scissors. Nature. 2002;418:171–174. doi: 10.1038/nature00823. [DOI] [PubMed] [Google Scholar]
- Knights D, Ward TL, McKinlay CE, Miller H, González A, McDonald D, Knight R. Rethinking “Enterotypes.”. Cell Host & Microbe. 2014;16:433–437. doi: 10.1016/j.chom.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. . 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Knights D, González A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A Guide to Enterotypes across the Human Body: Meta-Analysis of Microbial Community Structures in Human Microbiome Datasets. PLOS Comput Biol. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu Z-R, Hooper LV, Schmidt RR, Aulock von S, et al. Commensal bacteria regulate Toll-like receptor 3|[ndash]|dependent inflammation after skin injury. Nat Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 2014;506:498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, et al. Targeted Restoration of the Intestinal Microbiota with a Simple, Defined Bacteriotherapy Resolves Relapsing Clostridium difficile Disease in Mice. PLoS Pathog. 2012;8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, Gibbons SM, Larsen P, Shogan BD, Weiss S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science (New York, N.Y. 2014;345:1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, Malfatti S, del Rio TG, Jones CD, Tringe SG, et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science (New York, N.Y. 2015;349:860–864. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013;501:426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host & Microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- Martens EC, Kelly AG, Tauzin AS, Brumer H. The Devil Lies in the Details: How Variations in Polysaccharide Fine-Structure Impact the Physiology and Evolution of Gut Microbes. Journal of Molecular Biology. 2014;426:3851–3865. doi: 10.1016/j.jmb.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- Martínez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio. 2015;6:e00042–15. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithieux G, Misery P, Magnan C, Pillot B, Gautier-Stein A, Bernard C, Rajas F, Zitoun C. Portal sensing of intestinal gluconeogenesis is a mechanistic link in the diminution of food intake induced by diet protein. Cell Metabolism. 2005;2:321–329. doi: 10.1016/j.cmet.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch CL, Rollins D, Propheter DC, Rizo J, Grabe M, Jiang Q-X, et al. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature. 2014;505:103–107. doi: 10.1038/nature12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520:104–108. doi: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, Chung J, Sohn J, Barber CM, Goldfarb DS, et al. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nature Communications. 2015;6:1–15. doi: 10.1038/ncomms8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DC, Poulsen M, Currie CR, Clardy J. Dentigerumycin: a bacterial mediator of an ant-fungus symbiosis. Nature Chemical Biology. 2009 doi: 10.1038/nchembio.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Deming C, Conlan S, Program NCS, Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Org E, Parks BW, Joo JWJ, Emert B, Schwartzman W, Kang EY, Mehrabian M, Pan C, Knight R, Gunsalus R, et al. Genetic and environmental control of host-gut microbiota interactions. Genome Research. 2015;25:1558–1569. doi: 10.1101/gr.194118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17981–17986. doi: 10.1073/pnas.1316981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4|[ndash]|MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends in Microbiology. 2005;13:27–33. doi: 10.1016/j.tim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Patterson AD, Turnbaugh PJ. Microbial Determinants of Biochemical Individuality and Their Impact on Toxicology and Pharmacology. Cell Metabolism. 2014;20:761–768. doi: 10.1016/j.cmet.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of Commensal Microflora by Toll-Like Receptors Is Required for Intestinal Homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura V, Belkaid Y. Gut Microbiota: The Link to Your Second Brain. Cell. 2015;161:193–194. doi: 10.1016/j.cell.2015.03.033. [DOI] [PubMed] [Google Scholar]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-Like Receptor 2 Pathway Establishes Colonization by a Commensal of the Human Microbiota. Science (New York, N.Y. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee J-Y, Ziel JW, Miraldi ER, Domingos AI, et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell. 2015;163:381–393. doi: 10.1016/j.cell.2015.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Truong H-A, Gearty SV, Pauli ML, Nosbaum A, Gratz IK, Otto M, Moon JJ, Liese J, et al. A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity. 2015;43:1011–1021. doi: 10.1016/j.immuni.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JJ, Oh DC, Yuceer MC, Klepzig KD, Clardy J, Currie CR. Bacterial protection of beetle-fungus mutualism. Science (New York, N.Y. 2008;322:63. doi: 10.1126/science.1160423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre M-L, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (New York, N.Y. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (New York, N.Y. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen C-H, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan Foraging in Vivo by an Intestine-Adapted Bacterial Symbiont. Science (New York, N.Y. 2005;307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. Journal of Bacteriology. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;510:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toit Du G, Roberts G, Sayre PH, Bahnson HT, Radulovic S, Santos AF, Brough HA, Phippard D, Basting M, Feeney M, et al. Randomized Trial of Peanut Consumption in Infants at Risk for Peanut Allergy. The New England Journal of Medicine. 2015;372:803–813. doi: 10.1056/NEJMoa1414850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The Antibacterial Lectin RegIIIγ Promotes the Spatial Segregation of Microbiota and Host in the Intestine. Science (New York, N.Y. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. The New England Journal of Medicine. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- van Opstal EJ, Bordenstein SR. Rethinking heritability of the microbiome. 2015 doi: 10.1126/science.aab3958. [DOI] [PubMed] [Google Scholar]
- Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (New York, N.Y. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW. The Virome in Mammalian Physiology and Disease. Cell. 2014;157:142–150. doi: 10.1016/j.cell.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163:1585–1595. doi: 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. Microbial inhabitants of humans : their ecology and role in health and disease. New York: Cambridge University Press; 2005. [Google Scholar]
- Woyke T, Teeling H, Ivanova NN, Huntemann M, Richter M, Gloeckner FO, Boffelli D, Anderson IJ, Barry KW, Shapiro HJ, et al. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature. 2006;443:950–955. doi: 10.1038/nature05192. [DOI] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science (New York, N.Y. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83. doi: 10.1016/j.coi.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, Nagler CR, Ismagilov RF, Mazmanian SK, Hsiao EY. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell. 2015;161:264–276. doi: 10.1016/j.cell.2015.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M, et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell. 2015;163:1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]