

Abstract
Pseudouridine (Ψ) is the most abundant internal modification identified in RNA, and yet little is understood of its effects on downstream reactions. Yeast U2 snRNA contains three conserved Ψs (Ψ35, Ψ42, and Ψ44) in the branch site recognition region (BSRR), which base pairs with the pre‐mRNA branch site during splicing. Here, we show that blocks to pseudouridylation at these positions reduce the efficiency of pre‐mRNA splicing, leading to growth‐deficient phenotypes. Restoration of pseudouridylation at these positions using designer snoRNAs results in near complete rescue of splicing and cell growth. These Ψs interact genetically with Prp5, an RNA‐dependent ATPase involved in monitoring the U2 BSRR‐branch site base‐pairing interaction. Biochemical analysis indicates that Prp5 has reduced affinity for U2 snRNA that lacks Ψ42 and Ψ44 and that Prp5 ATPase activity is reduced when stimulated by U2 lacking Ψ42 or Ψ44 relative to wild type, resulting in inefficient spliceosome assembly. Furthermore, in vivo DMS probing analysis reveals that pseudouridylated U2, compared to U2 lacking Ψ42 and Ψ44, adopts a slightly different structure in the branch site recognition region. Taken together, our results indicate that the Ψs in U2 snRNA contribute to pre‐mRNA splicing by directly altering the binding/ATPase activity of Prp5.
Keywords: Prp5 ATPase, pseudouridylation, spliceosome assembly, splicing, U2 snRNA
Subject Categories: RNA Biology
Introduction
The removal of introns from pre‐mRNAs is catalyzed by the spliceosome, a mega‐dalton ribonucleoprotein machine consisting of five snRNAs (U1, U2, U4, U5, and U6) and more than 100 proteins (Moore et al, 1993; Staley & Guthrie, 1998; Yu et al, 1999; Fabrizio et al, 2009). In addition to its complex composition, the spliceosome is highly dynamic, requiring a series of regulated RNA–RNA and RNA–protein rearrangements during spliceosome assembly, activation, catalysis, and disassembly (Nilsen, 1994; Staley & Guthrie, 1998; Smith et al, 2009). For instance, at an early stage in spliceosome assembly, U2 folds into a structure, called the branchpoint‐interacting stem loop (BSL) (Perriman & Ares, 2007), thus presenting its branch site recognition sequence in a conformation (in the single‐stranded loop) that is capable of base‐pairing with the branch site of pre‐mRNA. This interaction leads to the formation of a pre‐splicing complex (complex A) (Moore et al, 1993). Branch site recognition involves a base‐pairing interaction between the branch site recognition region (BSRR) of U2 and the pre‐mRNA branch site (Fig 1A). Subsequently, complex A undergoes a conformational change, which is critical for the progression of spliceosome assembly and splicing.
Figure 1. Manipulation of U2 pseudouridylation.

- Schematic representation of U2‐pre‐mRNA interaction. Partial sequences/secondary structures of vertebrate and Saccharomyces cerevisiae U2 are schematically shown. Also shown are the base‐pairing interactions between the U2 branch site recognition region (BSRR) and the pre‐mRNA branch site (BS). Pseudouridines (Ψ) are indicated. Enzymes responsible for yeast U2 pseudouridylation at the three conserved sites are also indicated.
- Temperature‐sensitive growth assay. Yeast strains (wild‐type or deletion mutants, indicated on the left) were transformed with either an empty vector or a plasmid carrying an artificial guide RNA gene targeting U2 pseudouridylation at positions 42 and 44 (indicated on the top), and grown at various temperatures (indicated at the bottom).
- Blockage of pseudouridylation by deletion of specific pseudouridylase genes. RNAs isolated from wild‐type and pseudouridylase‐deletion strains (indicated on the top) were used for U2 pseudouridylation assay (CMC modification followed by primer extension). Primer extension pauses/stops correspond to Ψ sites (indicated on the left and right). Note: The above‐background signal of Ψ42 in the snr81 lane (and to some extent, in the snr81 pus7 lane) is likely caused by the presence of strong Ψ44 band. This is a primer extension artifact that often occurs when there is a strong Ψ signal in the neighboring position. Alternatively, it is possible that having Ψ at position 44 allows for an unidentified enzyme other than snR81 to modify U42 (albeit inefficiently; potentially Pus1, which is responsible for Ψ44 formation, can act in a “processive” manner). Lanes 1–8 and lanes 9–16 are from two separate gels.
- Restoration of U2 pseudouridylation by an artificial box H/ACA RNA. RNAs isolated from various yeast strains (indicated on the top), including the snr81Δ pusΔ strain that was transformed with an artificial guide RNA targeting positions 42 and 44 (lanes 9 and 10), were assayed for pseudouridylation (see legend to C).
It is known that the early conformational change in complex A is catalyzed by Prp5, a DEAD/H box ATPase. Originally discovered as an essential factor for spliceosome assembly and splicing (Lin et al, 1987), Prp5 is an RNA‐dependent ATPase, and it favors U2 sequences over other RNAs in stimulating ATP hydrolysis (O'Day et al, 1996), although this is not well understood. By hydrolyzing ATP, Prp5 is thought to closely monitor the base‐pairing interaction between the U2 BSRR and the pre‐mRNA branch site in complex A. In this regard, it has been reported that ATPase‐deficient Prp5 variants are able to suppress (i.e. improve splicing of) many branch site mutants (Perriman & Ares, 2007; Xu & Query, 2007), suggesting that Prp5 plays a role in ensuring quality control over the U2‐branch site base‐pairing interaction (Xu & Query, 2007). Recently, Liang and Cheng reported that Prp5 joins the spliceosome through interacting with the BSL of U2 and that the subsequent release of Prp5 from the spliceosome promotes the recruitment of U4/U6/U5 tri‐snRNP, allowing spliceosome assembly to move forward (Liang & Cheng, 2015).
Spliceosomal snRNAs are extensively modified, primarily by post‐transcriptional pseudouridylation (Yu et al, 2011). For instance, all six uridines in the BSRR of vertebrate U2 are converted to Ψs (Reddy & Busch, 1988; Yu et al, 1999). Previous work has shown that these Ψs are necessary for U2 snRNP assembly and pre‐mRNA splicing in Xenopus oocytes (Yu et al, 1998; Zhao & Yu, 2004) and in Saccharomyces cerevisiae (Yang et al, 2005). Interestingly, experiments using the mammalian in vitro reconstitution system suggested that only the modified nucleotides within the first 20 nucleotides of U2 (thus excluding the pseudouridines in the BSRR region) are critical (Donmez et al, 2004). Although seemingly contradictory, these different results may reflect differences in the experimental systems used in these studies (the human in vitro reconstitution system versus the Xenopus oocyte and yeast in vivo systems) (also discussed in Discussion).
Notably, three of the Ψs in the BSRR of vertebrate U2 are absolutely conserved across species (Ψ34, Ψ41, and Ψ43 in vertebrates and Ψ35, Ψ42, and Ψ44 in S. cerevisiae) (Fig 1A). Importantly, all three enzymes responsible for the formation of Ψ35, Ψ42, and Ψ44 in yeast U2 have been identified. Specifically, while Pus7 and Pus1, two stand‐alone proteins, catalyze the formation of Ψ35 and Ψ44, respectively, snR81 RNP, a box H/ACA RNP, is responsible for the formation of Ψ42 (Massenet et al, 1999; Ma et al, 2003, 2005). The identification of these enzymes has greatly facilitated our efforts at understanding the functions and mechanisms behind the role pseudouridines play in yeast pre‐mRNA splicing. In a genetic screen analysis, we found that deletion of PUS7, when coupled with U2 mutations at position 40 (U40g or U40Δ), resulted in a synthetic growth‐deficiency phenotype (Yang et al, 2005). Thus, Ψ35 appears to be important in ensuring U2's functionality during splicing under some specific conditions. Whereas some progress has been made in understanding the function of Ψs in pre‐mRNA splicing, the question of how these Ψs affect pre‐mRNA splicing mechanistically (or, in general, the effect of any Ψ on any cellular process) remains to be addressed.
Here, we block Ψ formation in yeast U2 by disrupting the three enzyme‐coding genes in all combinations and find that all three Ψs are functionally relevant. Simultaneous deletion of PUS1 and SNR81 (responsible for the formation of Ψ42 and Ψ44, respectively) has the most significant effect on splicing out of all deletion combinations. Our results suggest that these Ψs genetically interact with Prp5. Further biochemical analysis indicates that Ψ42 and Ψ44 contribute directly to Prp5 binding and Prp5's U2‐dependent ATPase activity. Thus, we identify the first example of Ψ contribution to enzyme binding and activation.
Results
Removal of Ψ(s) from yeast U2 leads to growth‐deficient phenotypes
To dissect the functions of Ψs in yeast U2 snRNA, we blocked Ψ formation by deleting the corresponding enzyme‐coding genes. Specifically, we deleted PUS7 (responsible for Ψ35 formation), SNR81 (for Ψ42), and PUS1 (for Ψ44) (Massenet et al, 1999; Ma et al, 2003, 2005), either individually or in combination, and tested the effects of these deletions on cell growth. Whereas these deletion strains grew well at 30°C, virtually all of them exhibited a temperature‐sensitive growth phenotype (Fig 1B, the first three columns). Among them, the snr81Δ pus1Δ strain, which lacks both Ψ42 and Ψ44, exhibited the most severe growth‐deficient phenotypes; no growth was observed at 37/38°C (row 2). To ensure that the deletion resulted in loss of Ψs at the target site(s), we carried out a pseudouridylation assay (CMC modification followed by primer extension) using RNAs isolated from these deletion strains. The results confirmed that Ψ formation was specifically blocked by deletion of genes coding for the corresponding enzymes (Fig 1C).
Given that Pus7 and Pus1, in addition to their role in catalyzing U2 pseudouridylation, are also responsible for the pseudouridylation of tRNA at several positions (Massenet et al, 1999; Behm‐Ansmant et al, 2003), it was necessary to test whether the observed growth‐deficiency phenotype was truly due to the loss of Ψ(s) in U2 rather than the loss of Ψ(s) in tRNA (or other RNAs). Thus, we designed several artificial box H/ACA RNAs to guide U2 pseudouridylation at positions 35, 42, and 44. Transformation of the deletion strains with their cognate guide RNAs led to growth restoration at the high temperature (38°C) (Fig 1B, column 4). For instance, when the snr81Δ pus1Δ strain was transformed with an artificial box H/ACA RNA targeting positions 42 and 44, cell growth was greatly recovered at 38°C (row 2, column 4). To ensure that the artificial box H/ACA RNA restored pseudouridylation at positions 42 and 44, we also performed the primer extension‐based pseudouridylation assay (Bakin & Ofengand, 1998). When the snr81Δ pus1Δ strain was transformed with the artificial guide RNA, U2 pseudouridylation was nearly fully restored at positions 42 and 44 (Fig 1D, compare lane 10 with lane 8).
To test whether the growth‐deficiency phenotype of the deletion strains was directly linked to splicing, we took advantage of the well‐established ACT1‐CUP1 reporter system, where the splicing of ACT1‐CUP1 fusion pre‐mRNA correlates directly to cell growth in copper‐containing media (Lesser & Guthrie, 1993). As shown in Fig 2A, while the wild‐type strain was able to grow in medium containing 1.4 mM Cu2+, all pseudouridylase‐deletion strains exhibited a growth‐deficiency phenotype in copper‐containing media even at 30°C. Again, the snr81Δ pus1Δ strain was the worst; its maximum tolerance for Cu2+ was 1 mM (row 2). Importantly, the Cu2+ tolerance for the snr81Δ pus1Δ strain was restored when cells were transformed with an artificial box H/ACA RNA targeting position 42 and/or position 44 (Fig 2B). Specifically, when the snr81Δ pus1Δ strain was transformed with an artificial box H/ACA RNA targeting either position 42 or position 44, cell growth recovered to some extent in medium containing 1.2 mM Cu2+ (compare rows 4 and 5 with rows 1 and 2). When the artificial guide RNA targeting both position 42 and position 44 was used, a near complete growth rescue was observed (compare lane 3 with lanes 1 and 2).
Figure 2. Functional importance of U2 pseudouridylation.
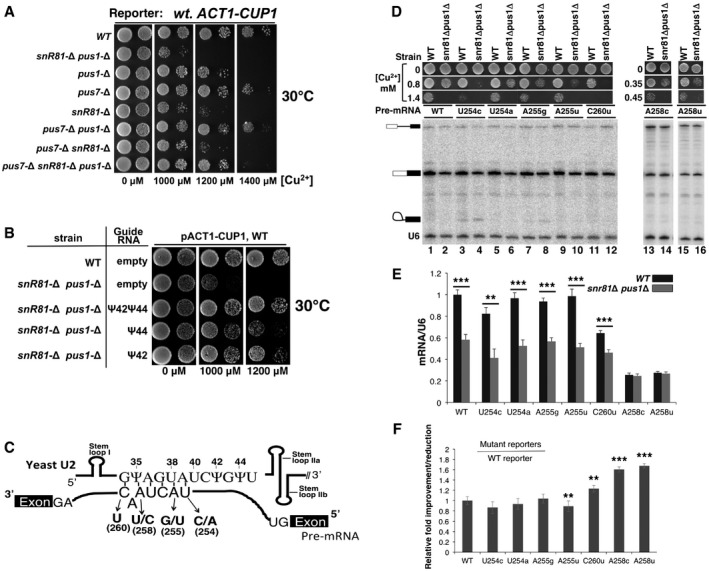
- Growth assay using the ACT1‐CUP1 reporter system. Yeast cells carrying the ACT1‐CUP1 reporter were deleted of pseudouridylase genes (indicated on the left) and were then assayed for growth at 30°C on media containing various concentrations of [Cu2+] (indicated at the bottom).
- Growth rescue by restoration of pseudouridylation. The wild‐type strain (from A) or the snr81Δ pusΔ strain (also from A), which exhibited the most severe growth‐deficiency phenotype, was transformed with an empty vector or a plasmid carrying a pseudouridylation guide RNA gene targeting either position 42, position 44, or both (indicated). The resulting cells were assayed for growth at 30°C on media containing various concentrations of [Cu2+] (indicated at the bottom).
- Schematic representation of U2–pre‐mRNA interaction. The U2 BSRR sequence and the pre‐mRNA branch site sequence are shown. The arrows indicate several point mutations at the pre‐mRNA branch site. Stem loops I, IIa, and IIb are also indicated.
- Splicing assay using the wild‐type and mutant ACT1‐CUP1 reporters. RNAs isolated from the wild‐type and the snr81Δ pusΔ strains (indicated on the top), which carry the wild‐type ACT1‐CUP1 reporter pre‐mRNA or any of the mutant ACT1‐CUP1 reporter pre‐mRNAs (indicated, see also C), were used for splicing assay (primer extension analysis). The un‐spliced pre‐mRNA, lariat intermediate, and spliced mRNA are indicated. A primer complementary to U6 was also used (as an internal control) in the assay, and the U6 band is indicated as well. In addition, the growth phenotype of each strain (in various concentrations of [Cu2+]) is also shown. Lanes 1–12, lanes 13 and 14, and lanes 15 and 16 are from separate gels.
- Quantification of ACT1‐CUP1 mRNA levels. Spliced ACT1‐CUP1 mRNA levels were calculated relative to U6 in each lane. The quantification was based on three independent experiments.
- Relative improvement/reduction for the mutant reporters. Relative improvement or reduction in splicing was calculated by normalizing the splicing efficiency (mRNA/U6) of each mutant reporter to that of WT reporter (set at 1).
We next tested splicing efficiency directly using primer extension. Both wild‐type and some well‐characterized mutant ACT1‐CUP1 pre‐mRNAs were used (Fig 2C), and the levels of spliced mRNA in the snr81Δ pus1Δ strain versus the wild‐type strain were analyzed. For most pre‐mRNA reporters tested, including the wild‐type and the very mild mutant reporters, a significant reduction in splicing was observed in the snr81Δ pus1Δ strain compared to the wild‐type strain (Fig 2D, lanes 1–12, and E); in contrast, the two relatively severe branch‐region‐mutant reporters, A258c and A258u, exhibited only a small reduction in splicing (Fig 2D, lanes 13–16). When normalized to the effects on the wild‐type reporter, this represents a relative improvement in the splicing of these two severe branch‐region‐mutant reporters (Fig 2F), similar to effects previously observed with slow Prp5 ATPase alleles (Xu & Query, 2007) (see also Discussion). In concordance with results above, when the artificial box H/ACA RNA targeting U2 snRNA at positions 42 and 44 was expressed in the snr81Δ pus1Δ strain, spliced mRNA levels were almost completely restored (data not shown).
Due to its clear effects on pre‐mRNA splicing and growth, the snr81Δ pus1Δ strain was chosen for subsequent analyses (see below).
Ψ42 and Ψ44 genetically interact with DEAD box protein Prp5
Given that Ψ42 and Ψ44 reside in the branch site recognition region (BSRR) and that the splicing factor Prp5 functions as an ATPase to monitor the base‐pairing interaction between the pre‐mRNA branch site and the U2 BSRR at early times during spliceosome assembly, we hypothesized that Ψ42 and Ψ44 are important for Prp5 function. To test this hypothesis, we examined whether blocking the formation of Ψ42 and Ψ44, when coupled with Prp5 mutation, would result in a synthetic growth‐deficiency phenotype. To do this, we created two yeast strains by plasmid shuffling. In the first strain, the chromosomal PRP5 gene was replaced by a plasmid‐borne wild‐type PRP5. The plasmid carrying wild‐type PRP5 also has the selectable marker URA, which can be used to select against cells carrying the plasmid by growing them on medium containing 5‐FOA. In the second strain, which was derived from the first strain, both SNR81 and PUS1 were further deleted. We then transformed these two strains with plasmids containing wild‐type or mutant PRP5 genes and evaluated synthetic lethality by growing cells on 5‐FOA medium.
We tested a number of Prp5 mutants, all of which bore amino acid substitutions either in motif III (SAT), a highly conserved motif that is thought to be required for coupling of ATP hydrolysis to conformational change, or in the IIb region, which is outside of the ATPase motif (Xu & Query, 2007). These mutants alone did not show growth‐deficient phenotypes under optimal growth conditions (Fig 3, row 4). Similarly, the mutant strain carrying snr81Δ pus1Δ alone grew well under the same growth conditions at 30°C (row 3). However, when the Prp5 mutations were combined with the deletion of SNR81 and PUS1 (snr81Δ pus1Δ), severe growth‐deficient phenotypes were observed (row 2). Restoration of U2 Ψ42 and Ψ44 (through expressing the previously designed box H/ACA RNA targeting positions 42 and 44) rescued growth, confirming that the synthetic growth‐deficiency phenotype was due to the absence of Ψ42 and Ψ44 in U2 (row 1). Among the three ATPase motif mutants whose ATPase activity has been measured (Xu & Query, 2007), SAT‐to‐GAR had the lowest ATPase activity (4.3% of the wild type) (Xu & Query, 2007), and it showed the greatest synthetic growth‐deficiency phenotype (row 2). SAT‐to‐GAG and SAT‐to‐TAG have relatively higher ATP hydrolysis activity (22 and 42% of the wild type, respectively) (Xu & Query, 2007), and both of these mutants displayed a relatively less severe synthetic growth‐deficiency phenotype when combined with the deletion of SNR81 and PUS1 (row 2). Taken together, it appears that the ATPase deficiencies of Prp5 mutants, which on their own were not strong enough to influence cell growth, were exacerbated upon ablation of Ψ42 and Ψ44, leading to growth‐deficient phenotypes. These results thus suggest that Ψ42 and Ψ44 indeed functionally interact with Prp5 and, as shown below, modulate its ATPase activity.
Figure 3. U2 pseudouridylation and Prp5 synthetic lethality analysis.
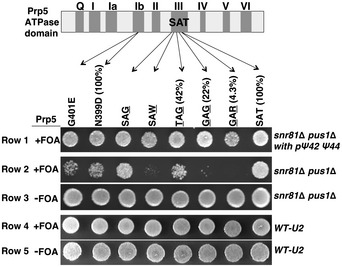
Synthetic lethality assay. The Prp5 domain structure and the mutations in some of the domains (indicated) are schematized at the top. Each of these PRP5 genes (wild‐type or mutants, plasmid‐borne) was used to replace the chromosomal PRP5. The numbers in parentheses are the relative ATPase activities of the wild‐type (SAT) Prp5 and some of the Prp5 mutants tested before (Xu & Query, 2007). The U2 status, either containing Ψ42 and Ψ44 (WT‐U2 and snR81Δ pus1Δ with pΨ42 or Ψ44) or lacking Ψ42 and Ψ44 (snR81Δ pus1Δ), is indicated (on the right). Upon transformation with the plasmids carrying PRP5 genes (wild type or any of the mutants), cells were grown on the regular medium (−FOA) or on medium containing 5‐FOA (+FOA).
Ψ42 and Ψ44 contribute to the efficiency of binding between U2 and Prp5
Given the strong evidence for functional interaction between Prp5 and Ψ42/Ψ44, we next assessed the physical interaction between Prp5 and U2 snRNA in the presence and absence of Ψ42 and Ψ44. It is possible that blocking the formation of Ψ42 and Ψ44 reduces Prp5's affinity (through SF3b) for U2 (Shao et al, 2012), thus affecting spliceosome assembly and splicing. In order to test this possibility, we prepared splicing extracts from the wild‐type strain as well as from the snR81Δ pus1Δ strain. In both strains, PRP5 was FLAG‐tagged, which allowed anti‐FLAG co‐immunoprecipitation (co‐IP) of Prp5 and U2 from the splicing extracts (Fig 4A). Our results showed that in the presence of ATP, both U1 and U2 were efficiently co‐precipitated with Prp5 (compare lane 2 with lane 1), consistent with previous observations made in S. pombe (Xu et al, 2004). However, in the snR81Δ pus1Δ strain, whereas the amount of precipitated Prp5 and co‐precipitated U1 remained essentially unchanged, a significantly reduced amount (~40%) of U2 was co‐precipitated (compare lane 3 with lane 2, quantified in Fig 4B). To ensure that U2 expression was not affected by deletion of SNR81 and PUS1, we performed primer extension to measure U2 levels in both the wild‐type and snR81Δ pus1Δ strains. Our results indicated that U2 levels were indeed comparable in both strains (Fig 4C).
Figure 4. Effect of pseudouridylation on Prp5‐U2 interaction.

- Prp5‐U2 co‐immunoprecipitation (IP). Wild‐type (lanes 1 and 2) and snR81Δ pus1Δ (lane 3) strains, each containing a FLAG‐tagged PRP5 gene (indicated on the top), were used for anti‐FLAG IP in the absence (lane 1) or presence (lanes 2 and 3) of ATP. The precipitated Prp5 (WT Prp5‐FLAG) and co‐precipitated U1 and U2, detected by primer extension, are indicated.
- Quantification of co‐precipitated U2 snRNA. For each lane (the WT lane and the snr81Δ pus1Δ lane, corresponding to lane 2 and lane 3 of A, respectively), Prp5‐co‐precipitated U2 was normalized against co‐precipitated U1 [(intensity of the U2 band)/(intensity of the U1 band)]. The snr81Δ pus1Δ lane was then further normalized against the WT lane [(the value of normalized U2 from the snr81Δ pus1Δ lane)/(the value of normalized U2 from the WT lane)]. Quantification was based on three independent experiments.
- U2 expression level in wild‐type and snR81Δ pus1Δ strains. Total RNA, isolated from the wild‐type (lane 1) or snR81Δ pus1Δ (lane 2) strains, was used for primer extension to measure the levels of U2 and U6. The U2 and U6 bands are indicated.
- Depletion of Prp5. Wild‐type (lanes 1, 3, and 4) and snR81Δ pus1Δ (lanes 2 and 5) cells containing a FLAG‐tagged PRP5 were lysed, and anti‐FLAG IP was performed at a high concentration of salt. Cell extracts, before (lanes 1 and 2) and after [lanes 3 (mock), 4 and 5] anti‐FLAG IP, were used for Western analysis using anti‐FLAG. The Prp5 band is indicated.
- IP of reconstituted Prp5 (GAR)‐U2. An equal amount of FLAG‐tagged Prp5 (GAR) was added to the wild‐type (lane 1) and snR81Δ pus1Δ (lane 2) cell extracts depleted of endogenous Prp5 (see C). After the salt concentration was brought back to its original level, anti‐FLAG IP was carried out. The Prp5 (GAR) band, detected by Western, is indicated. The co‐precipitated U1 and U2 bands, detected by primer extension, are also indicated.
- Quantification of U2 that is co‐precipitated with mutant Prp5 (GAR). For each lane (including the WT lane and the snr81Δ pus1Δ lane, corresponding to lane 1 and lane 2 of E, respectively), U2 co‐precipitated with Prp5 (GAR) was normalized against co‐precipitated U1 [(intensity of the U2 band)/(intensity of the U1 band)]. The snr81Δ pus1Δ lane was then further normalized against the WT lane [(the value of normalized U2 from the snr81Δ pus1Δ lane)/(the value of normalized U2 from the WT lane)]. Quantification was based on three independent experiments.
- Prp5‐U2‐pre‐mRNA co‐IP. Reconstitution was carried out as described in (D). Prior to IP, an equal amount of radiolabeled pre‐mRNA was also added to each reaction. Anti‐FLAG IP was then performed in the presence of ATP. The precipitated Prp5 (WT and GAR) band, detected by Western, is indicated. The co‐precipitated pre‐mRNA, directly visualized after electrophoresis, is also indicated. The pre‐mRNA input is also shown. In lanes 1 and 2, wild‐type and snR81Δ pus1Δ cell extracts, depleted of Prp5, were reconstituted with the wild‐type FLAG‐Prp5. In lanes 3 and 4, wild‐type and snR81Δ pus1Δ cell extracts, depleted of Prp5, were reconstituted with mutant FLAG‐Prp5 (GAR).
- Quantification of pre‐mRNA co‐precipitated with Prp5 and U2. For each lane in (G), pre‐mRNA co‐precipitated with Prp5 (wild‐type or GAR) was normalized against the Prp5 (WT/GAR) band in the same lane (detected by anti‐FLAG) [(intensity of pre‐mRNA)/(intensity of WT/GAR Prp5)]. Lanes 2, 3, and 4 (see G) were then further normalized against lane 1 (see G) [(the value of normalized pre‐mRNA from lane 2, 3, or 4 in G)/(the value of normalized pre‐mRNA from lane 1 in G)]. The numbers at the bottom of the bar graph correspond to the lane numbers in (G). Quantification was based on three independent experiments.
We also carried out Prp5 reconstitution and co‐IP experiments, which allowed us to test the binding of U2 by mutant Prp5 (GAR) (The SAT‐to‐GAR mutant itself alone is not sufficient to support cell growth when SNR81 and PUS1 are deleted). Here, using anti‐FLAG IP, we first depleted endogenous Prp5 from the splicing extracts (prepared from the snR81Δ pus1Δ strain or the wild‐type strain) at a high concentration of salt (1 M KCl). The depletion was highly efficient; virtually, no Prp5 was detected after depletion (Fig 4D). Escherichia coli‐expressed recombinant FLAG‐tagged mutant Prp5 (GAR) (or recombinant wild‐type FLAG‐Prp5, data not shown) was added to the depleted extracts, and salt concentrations were returned to normal (50 mM KCl). Anti‐FLAG IP was subsequently carried out in the presence of ATP. As expected, both U1 and U2 were co‐precipitated with the mutant Prp5; however, whereas an equal amount of U1 was precipitated from the wild‐type and snR81Δ pus1Δ strains, a greatly reduced amount (~40%) of U2 was pulled down from the snR81Δ pus1Δ strain (Fig 4E, compare lane 2 with lane 1; see also Fig 4F for quantification).
To address directly whether the interaction between WT Prp5 and U2 is stronger than that between mutant Prp5 (GAR) and U2, we carried out four parallel binding (anti‐FLAG IP) reactions, including co‐IP of WT Prp5 and WT U2, of WT Prp5 and mutant U2 (lacking Ψ42 and Ψ44), of mutant Prp5 (GAR) and WT U2, and of mutant Prp5 (GAR) and mutant U2 (lacking Ψ42 and Ψ44). The same amount of U1 and U2 primers was used for primer extension, thus allowing us to directly compare the levels of U1 and U2 that were co‐precipitated with Prp5 (Fig EV1). Our results indicate that while the level of co‐precipitated U1 remained virtually unchanged (lanes 1–4), WT Prp5 pulled down more U2 than the mutant Prp5 (GAR) did (compare lane 1 with lane 3, and lane 2 with lane 4); both WT Prp5 and mutant Prp5 (GAR) prefer WT U2 over mutant U2 (lacking Ψ42 and Ψ44) (compare lane 1 with lane 2, and lane 3 with lane 4). Our results suggest that the SAT motif of Prp5 contributes to U2 binding, either directly or indirectly (the RNA binding domain of Prp5 is yet to be identified).
Figure EV1. Parallel IP‐binding assay using wild‐type/mutant U2 and wild‐type/mutant Prp5.

- Prp5‐U2 co‐immunoprecipitation (IP). Wild‐type (lane 1) and snR81Δ pus1Δ (lane 2) strains, each containing a FLAG‐tagged WT PRP5 gene (indicated on the top), were lysed, and the extracts from the two strains were each divided into two equal aliquots. One aliquot was directly used for anti‐FLAG IP at a low concentration of salt (50 mM KCl) (lanes 1 and 2). The other aliquot was used for anti‐FLAG IP at a high concentration of salt (1 M KCl), thus depleting the extracts of WT Prp5 (lanes 3 and 4). Subsequently, FLAG‐tagged mutant Prp5 (GAR) were added back to the extracts, and salt concentration was brought back to the original concentration by dialysis. Anti‐FLAG IP was subsequently performed (lane 3, derived from WT; and lane 4, from snR81Δ pus1Δ). The co‐precipitated U1 and U2 in each sample were detected by primer extension using an equal amount of the same primers (anti‐U1 and anti‐U2 primers).
- Quantification of co‐precipitated U2 snRNA. For each lane [lane 1, WT U2 and WT Prp5; lane 2, U2 lacking Ψ42 and Ψ44 and WT Prp5; lane 3, WT U2 and mutant Prp5 (GAR); lane 4, U2 lacking Ψ42 and Ψ44 and mutant Prp5 (GAR)], Prp5‐co‐precipitated U2 was normalized against co‐precipitated U1 [(intensity of the U2 band)/(intensity of the U1 band)]. All lanes were then further normalized against lane 1 (WT U2 and WT Prp5) [(the value of normalized U2 from each lane)/(the value of normalized U2 from lane 1)]. Quantification was based on three independent experiments. Error bars, mean ± SD. **P < 0.01, ***P < 0.001 (Student's t‐test).
By adding radiolabeled ACT1 pre‐mRNA along with either wild‐type recombinant FLAG‐tagged Prp5 or mutant recombinant FLAG‐tagged Prp5 (GAR) to the cell extracts pre‐depleted of endogenous Prp5, we further analyzed the amount of co‐precipitated pre‐mRNA (presumably pulled down via the interaction between the U2 BSRR and the pre‐mRNA branch site) (Fig 4G and H). As expected, the amount of co‐precipitated pre‐mRNA correlated well with the amount of U2 co‐precipitated with Prp5 (compare lanes 1 and 2 of Fig 4G with lanes 2 and 3 of Fig 4A, lanes 3 and 4 of Fig 4G with lanes 1 and 2 of Fig 4E). Taken together, our data suggest that, while the overall level of U2 is comparable in both strains, wild‐type U2 (along with its associated pre‐mRNA), when compared with U2 lacking Ψ42 and Ψ44, is favored for binding with Prp5.
Ψ42 and Ψ44 directly impact Prp5's U2‐dependent ATPase activity
To further dissect the effect of Ψ42 and Ψ44 on Prp5 function, we next carried out a Prp5 ATPase activity assay in the presence of wild‐type U2 or U2 lacking Ψ42 and Ψ44. All reactions contained an equal amount of recombinant Prp5 (wild‐type or GAR mutant), an equal amount of [α‐32P]ATP, and an equal amount of either U2 (pseudouridylated or un‐pseudouridylated) or an irrelevant RNA. Conversion (hydrolysis) of [α‐32P]ATP to [α‐32P]ADP was analyzed by TLC. As shown in Fig 5A, when no RNA was added, recombinant Prp5 (FLAG‐tagged) essentially failed to hydrolyze ATP (lane 3). The same result was generated when LiCl‐precipitated RNA (lane 1) or tRNA (lane 2) was present. In contrast, in the presence of a short in vitro transcribed U2 fragment (nucleotides 1–76, including the BSRR) a clear ATP‐to‐ADP conversion was detected (lane 4). Likewise, a much longer U2 containing the first 120 nucleotides was also able to activate Prp5's ATPase activity (lane 6). These results are consistent with the notion that Prp5 favors U2 over other irrelevant RNAs in catalyzing ATP hydrolysis (O'Day et al, 1996). More interestingly, when the uridines within the U2 fragments were completely substituted with Ψs (UTP was substituted with ΨTP during in vitro transcription), a ~three‐fold increase in Prp5 ATPase activity was observed (compare lane 5 with lane 4, and lane 7 with lane 6; quantified in Fig 5B), indicating that Prp5 favors pseudouridylated U2. Direct binding between U2 (1–76 nts) and Prp5 was then assessed by anti‐FLAG IP (Fig 5C and D). Whereas a small fraction of un‐pseudouridylated U2 was co‐precipitated (Fig 5C and D, compare lane 2 with lane 1), Prp5 pulled down a majority of the pseudouridylated U2 (compare lane 4 with lane 3). Thus, Prp5 is capable of interacting directly with U2, preferably pseudouridylated U2, under the conditions used here.
Figure 5. Effect of U2 pseudouridylation on Prp5's ATPase activity.

- Prp5's ATPase activity assay using synthetic U2. The 5' sequence of Saccharomyces cerevisiae U2 is shown (top). The three Ψs (35, 42, and 44) and the branch site recognition sequence (underlined) are indicated. In the ATPase assay (bottom), no RNA (lane 3), or an equal amount of LiCl‐precipitated RNA (lane 1) or tRNA (lane 2), a short U2 fragment (nts 1–76) (lane 4), a fully pseudouridylated short U2 fragment (nts 1–76) (lane 5), a long U2 fragment (nts 1–120) (lane 6), or a fully pseudouridylated long U2 fragment (nts 1–120) (lane 7), was used. The un‐hydrolyzed ATP and hydrolyzed product ADP are indicated. Lanes 1 and 2, lanes 3–5, and lanes 6 and 7 are from separate gels.
- Quantification of Prp5's ATPase activity activated by in vitro transcribed U2. Prp5's ATPase activity shown in (A) was calculated using the formula (ADP)/(ADT+ATP). The numbers at the bottom of the bar graph correspond to the lane numbers in (A). Quantification was based on three independent experiments.
- U2‐Prp5 binding assay. FLAG‐Prp5 and a radiolabeled U2 fragment (nts 1–76), either un‐pseudouridylated (lanes 1 and 2) or pseudouridylated (lanes 3 and 4), were incubated under the conditions used for ATPase activity assay. Anti‐FLAG IP was then carried out. Co‐precipitated (lanes 2 and 4) and un‐precipitated (lanes 1 and 3) U2 RNAs were analyzed by electrophoresis. The U2 band is indicated.
- Quantification of Prp5‐U2 binding. Relative intensities of U2 bands shown in (C) were calculated [setting the un‐precipitated uridine‐containing U2 (lane 1 and 5) to 1]. The numbers at the bottom of the bar graph correspond to the lane numbers in (C). Quantification was based on three independent experiments.
- Prp5's ATPase activity assay using cellular U2. U2 RNA isolated from the wild‐type strain (lanes 2 and 4) or from the snR81Δ pus1Δ strain (lanes 6 and 8) was used for the ATPase activity assay. In odd‐numbered lanes, no RNA was added. Both the wild‐type Prp5 (lanes 1–4) and the mutant Prp5 (GAR) (lanes 5–8) were tested.
- Quantification of Prp5's ATPase activity activated by U2 from cells. Prp5's ATPase activity shown in (E) was calculated using the formula (ADP)/(ADT+ATP). The numbers at the bottom of the bar graph correspond to the lane numbers in (E). Quantification was based on four independent experiments.
- U2 level in the ATPase activity reactions. RNAs from lanes 2 and 4 in (C) were recovered, and U2 level was measured using primer extension. Lanes 1‐5, a titration of U2 snRNA. Lane 6, U2 from the wild‐type strain (lane 2 in C). Lane 7, U2 from the snR81Δ pus1Δ strain (lane 4 in C).
To confirm the IP results, we also carried out gel‐shift assay, where low concentrations (nM) of Prp5 and U2 were used (Fig EV2). While Prp5‐pseudouridylated U2 shifted nicely and efficiently (right panel), bands of Prp5‐un‐pseudouridylated U2 complexes smeared (left panel) (the reason for this is unclear). Nonetheless, the gel‐shift experiments also indicate that Prp5 has a higher binding affinity for pseudouridylated U2 than for un‐pseudouridylated U2 (quantified in Fig EV2B). Thus, the results from both assays (co‐IP and gel‐shift) are highly consistent. While it is possible that Ψ42 and Ψ44 might have a direct impact on Prp5 catalysis, it is equally possible that the observed effect of pseudouridylated U2 on Prp5's ATPase activity is a consequence of tighter binding. Our current work cannot distinguish between these two possibilities.
Figure EV2. Prp5‐U2 binding affinity assessed by gel‐shift assay.
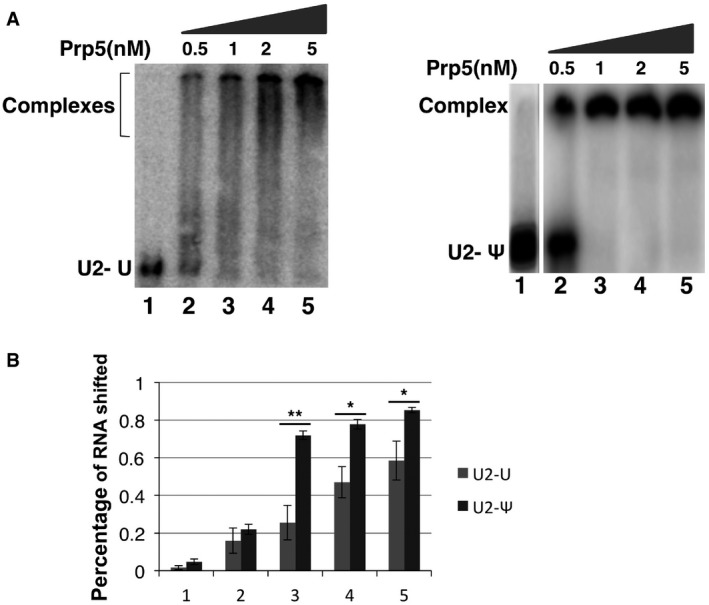
- Prp5‐U2 gel‐shift assay. In vitro transcription was used to synthesize a radiolabeled and un‐pseudouridylated 5' fragment of U2 and a radiolabeled and pseudouridylated 5' fragment of U2 (UTP was replaced by ΨTP during transcription). A small fixed amount of U2 fragment was used for Prp5‐U2 gel‐shift assay. The un‐pseudouridylated U2 fragment (left panel) and the pseudouridylated U2 fragment were each mixed with 0 (lane 1), 0.5, 1, 2, and 5 nM of WT Prp5 (right panel). After a ∼10‐min incubation in a low‐salt buffer, the reactions were loaded onto a 5% native polyacrylamide gel, electrophoresed at a low voltage, and autoradiographed. Lane 1 and lanes 2‐5 are from two separate gels. Interestingly, while Prp5‐pseudouridylated U2 shifted nicely and efficiently (right panel), the Prp5‐un‐pseudouridylated U2 complex bands smeared (left panel). It is possible that un‐pseudouridylated U2 adopted alternative folding structures (with similar G), allowing Prp5 to bind (perhaps simultaneously but loosely) at multiple sites (non‐specific binding sites). On the other hand, incorporation of pseudouridines may rigidify or stabilize the most favorable U2 structure (Charette & Gray, 2000; Ge & Yu, 2013), promoting efficient and site‐specific binding with Prp5. It is worth noting that the conditions used for IP [binding at low‐salt concentration (50 mM KCl), and washing with a buffer of higher concentration of salt (150 mM), see Materials and Methods] were more stringent than the conditions used for gel‐shift assay (binding at 50 mM KCl, and no washing). Thus, the loosely bound complexes between Prp5 and un‐pseudouridylated U2 might be detected by gel‐shift assay but not by IP (see Fig 5C and D). Nonetheless, the gel‐shift results indicate that Prp5 has a higher binding affinity for pseudouridylated U2 than for un‐pseudouridylated U2 (quantified in B). The results from both assays (co‐IP and gel‐shift) are highly consistent.
- Quantification of gel‐shifted Prp5‐U2 complexes. The percentage of Prp5‐U2 complexes detected in (A) was calculated [(intensity of the complex band)/(intensity of the complex band + intensity of U2 band)]. Quantification was based on three independent experiments. Error bars, mean ± SD. *P < 0.05, **P < 0.01 (Student's t‐test).
To analyze specifically the effects of Ψ42 and Ψ44, and in the context of full‐length U2 snRNA, we isolated U2 from the wild‐type strain (containing Ψ42 and Ψ44) and from the snR81Δ pus1Δ strain (lacking Ψ42 and Ψ44) and subsequently carried out the U2‐dependent Prp5 ATPase activity assay (Fig 5E). Remarkably, Prp5 favors U2 snRNA containing Ψ42 and Ψ44 over U2 RNA lacking Ψ42 and Ψ44 when catalyzing ATP hydrolysis; a ~two‐fold difference in ATP‐to‐ADP conversion was detected between the two (Fig 5E, compare lane 2 with lane 4; also quantified in Fig 5F). When the wild‐type Prp5 was substituted with the mutant Prp5 (GAR), we detected a similar level of difference of ATPase activity in favor of pseudouridylated U2 (Fig 5E, compare lane 6 with lane 8; also quantified in Fig 5F), although the overall activity of the mutant Prp5 was much lower than that of the wild‐type Prp5 (Fig 5E, compare lanes 6 and 8 with lanes 2 and 4). To ensure that the same amount of U2 was used, we performed primer extension analysis, and our data confirmed that the same amount of U2 with or without Ψ42 and Ψ44 was in the reaction (Fig 5G). Thus, our data demonstrate that Ψ42 and Ψ44 contribute directly to U2 function in modulating the ATPase activity of Prp5.
Ψ42 and Ψ44 contribute to the assembly of early splicing complexes
Since ATP‐dependent association of Prp5 with U2 snRNP and pre‐mRNA, as well as the ATPase activity of Prp5, is important at the early stage of spliceosome assembly, we further tested whether blockage of Ψ42 and Ψ44, which lowers the efficiency of U2‐Prp5 binding and Prp5‐catalyzed ATP hydrolysis, affects splicing complex formation. Splicing extracts were prepared from the wild‐type and snR81Δ pus1Δ strains. Endogenous FLAG‐tagged wild‐type Prp5 was subsequently removed through anti‐FLAG IP at high salt concentration. Salt concentration was then restored to its original level, and the extracts were reconstituted with recombinant wild‐type Prp5 or mutant Prp5 (GAR). Upon addition of 32P‐radiolabeled pre‐mRNA to the reconstituted splicing extracts, splicing complex formation was analyzed on a native polyacrylamide gel (Ansari & Schwer, 1995). When Prp5 was depleted, essentially no splicing complexes were detected (Fig 6A, lane 1). When the depleted extracts were reconstituted with wild‐type Prp5, splicing complexes A and B were detected. Importantly, the splicing complexes formed more efficiently in the extracts containing wild‐type U2 (containing Ψ42 and Ψ44) than in the extracts containing U2 lacking Ψ42 and Ψ44 (Fig 6A, compare lane 1 with lane 2). Likewise, extracts reconstituted with the mutant Prp5 (GAR) also favored wild‐type U2 over the mutant U2 (lacking Ψ42 and Ψ44) when forming complexes A and B (Fig 6A, compare lane 3 with lane 4). These results are consistent with our observations that Ψ42 and Ψ44 contribute to U2‐Prp5 binding and Prp5's ATPase activity (Figs 4 and 5), which are known to occur in splicing complex A.
Figure 6. Effect of U2 pseudouridylation on spliceosome assembly and U2 structure.
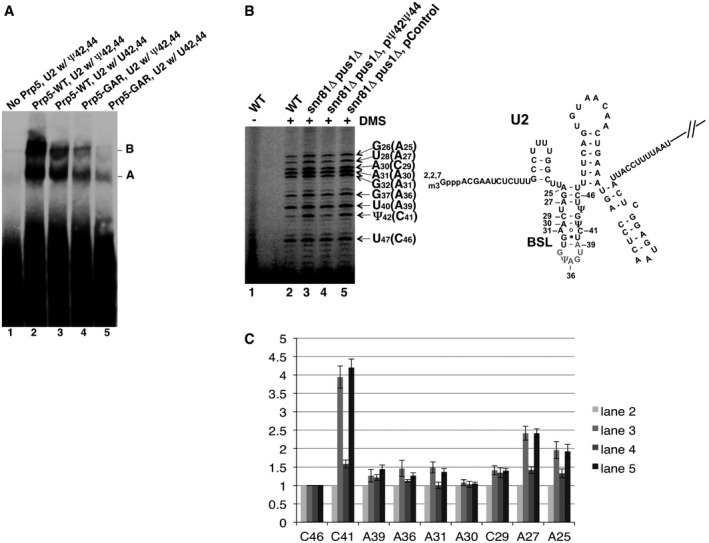
- Native gel analysis of pre‐splicing complexes. Pre‐splicing complex assembly was carried out in the test tube with Prp5‐reconstituted cell extracts and labeled pre‐mRNA. Lane 1, Prp5‐depleted wild‐type cell extract. Lane 2, Prp5‐depleted wild‐type cell extract, plus wild‐type Prp5. Lane 3, Prp5‐depleted snR81Δ pus1Δ cell extract, plus wild‐type Prp5. Lane 4, Prp5‐depleted wild‐type cell extract, plus mutant Prp5 (GAR). Lane 5, Prp5‐depleted snR81Δ pus1Δ cell extract, plus mutant Prp5 (GAR). The pre‐splicing complexes A and B are indicated.
- DMS in vivo probing. After being exposed to DMS, yeast cells were lysed, total RNA collected, and primer extension analysis carried out (left panel). Lane 1, cells that were not exposed to DMS. Lane 2, wild‐type cells exposed to DMS. Lane 3, snR81Δ pus1Δ cells exposed to DMS. Lane 4, snR81Δ pus1Δ cells, transformed with a plasmid carrying an artificial guide RNA gene targeting positions 42 and 44, and exposed to DMS. Lane 5, snR81Δ pus1Δ cells, transformed with a plasmid carrying a control guide RNA gene with random guide sequences, and exposed to DMS. The nucleotides in parentheses are DMS‐modified C and A residues that are one nucleotide after the actual primer extension stops (indicated by the arrows). A partial U2 sequence (along with the BSL structure) is shown (right panel). DMS‐modified adenosine and cytosine in the BSL are also indicated.
- Residue accessibility quantification. Relative accessibility (relative band intensity) was quantified based on three independent experiments. The intensity of every band in each lane was normalized against the C46 band (the least changed band; compare lanes 2–5 in B) in its respective lane [(intensity of a band)/(intensity of C46 in the same lane)]. Each C46‐normalized band in lanes 3–5 (refer to B) was then further normalized against its respective counterpart in lane 2 [(the value of a C46‐normalized band in lanes 3–5)/(the value of its counterpart in lane 2)]. The error bars represent the standard deviations of the measurements (based on three independent experiments).
Blockage of the formation of Ψ42 and Ψ44 alters the local structure of U2
Our experiments thus far revealed genetic and biochemical interactions between U2 Ψs (i.e. Ψ42 and Ψ44) and Prp5 during spliceosome assembly; however, the detailed molecular mechanisms behind these interactions remained unclear. While it is possible that Ψs (carrying an extra hydrogen bond donor) directly contribute to U2‐Prp5 binding and Prp5's U2‐dependent ATPase activity, it is equally possible that Ψs may alter the local structure of U2, thereby indirectly affecting Prp5 function. To test the latter possibility, we carried out in vivo chemical (DMS) probing experiments to examine the structure of U2 in the presence and absence of Ψ42 and Ψ44. Wild‐type yeast cells (containing Ψ42 and Ψ44) or snR81Δ pus1Δ yeast cells (lacking Ψ42 and Ψ44) were briefly exposed to DMS, which is known to specifically modify accessible C and A residues of an RNA. Cells were collected and total RNA purified. Primer extension (with an anti‐sense U2 primer) was subsequently carried out. It is well established that primer extension will stop one nucleotide before DMS‐modified C and A residues (Wells et al, 2000). As shown in Fig 6B, when U2 from the wild‐type cells was probed, DMS modified most C and A residues in the BSRR at relatively high intensity except for C41 and to a lesser extent, A25 and A27 (lane 2). C41 is immediately upstream of Ψ42, suggesting that this residue is normally buried inside some structure and inaccessible. Interestingly, however, when Ψ42 and Ψ44 were absent, C41, and to a lesser extent, A25, and A27, became accessible and were modified (lane 3). When the artificial box H/ACA RNA targeting positions 42 and 44 was introduced into the snR81Δ pus1Δ strain, C41 (to a lesser extent, A25 and A27 as well) was once again rendered inaccessible (lane 4), and this change in accessibility was not observed when an irrelevant box H/ACA RNA was expressed (lane 5). The changes in accessibility (band intensity) in response to pseudouridylation at positions 42 and 44 are quantified for all residues in this region (Fig 6C). Our data suggest that the local chemical environment of C41 (and to a lesser extent, the local chemical environment of A25 and A27), rather than the overall BSRR structure (i.e. BSL (Perriman & Ares, 2010); see also Discussion), depends on the presence or absence of Ψ42 and Ψ44. Our results also suggest that the U‐to‐Ψ change (U and Ψ have distinct chemical properties), together with the concurrent changes in the geometry of surrounding nucleotide(s), form a key component modulating U2‐Prp5 interaction (perhaps through SF3b) and Prp5's ATPase activity.
Discussion
Here, we have studied the function and mechanisms by which U2 pseudouridylation affects pre‐mRNA splicing in S. cerevisiae. Our experimental results demonstrate that Ψs (especially Ψ42 and Ψ44 in combination) within the BSRR of U2 snRNA play an important role in pre‐mRNA splicing and cell growth. Using genetic and biochemical approaches, we have also shown that Ψ42 and Ψ44 take part in genetic and direct biochemical interactions with Prp5, a U2‐dependent ATPase known to play a critical role in monitoring U2‐intron branch site interactions at early stages of spliceosome assembly (Xu & Query, 2007). This work thus assigns a specific function to the Ψs in yeast U2 snRNA.
We note that the previously reported in vitro results (Donmez et al, 2004) suggested that the Ψs in the first 20 nts of U2 were required for splicing and that the ones in the BSRR were not. It is likely that their in vitro system was sensitive to what the first 20 nts contribute; the other Ψs are also important, but the in vitro system (which is probably a 100 times slower than in vivo) is perhaps not sensitive to them. In fact, now there are data from three different systems: the human in vitro system (Donmez et al, 2004), the Xenopus in vivo system (Zhao & Yu, 2004), and the yeast in vivo system (Yang et al, 2005; and this work). The BSRR Ψs have detectable effects in both in vivo systems, suggesting that it really is the nature of the in vitro system that fails to be sensitive to the BSRR Ψs.
How do Ψ42 and Ψ44 contribute to U2 function?
It has long been known that Ψ has chemical properties that are distinct from any other known nucleotides (Charette & Gray, 2000; Ge & Yu, 2013). When compared with uridine, Ψ has an extra hydrogen bond donor. In addition, unlike the other nucleotides in which there is a nitrogen–carbon linkage between the base and sugar, Ψ contains a carbon–carbon (~10% longer) glycosidic bond. Such unique properties appear to be sufficient to allow Ψ to alter RNA function. For instance, it has been reported that Ψ, when incorporated into RNA, often enhances base stacking and pairing affinity to A (relative to a U‐A pair) (Davis, 1995; Newby & Greenbaum, 2001). Additionally, it has been shown that Ψ can rigidify the RNA backbone and alter the local structure of an RNA (Arnez & Steitz, 1994; Charette & Gray, 2000; Newby & Greenbaum, 2002a,b). In this regard, we have consistently detected (by DMS in vivo probing) a change in the local structure of U2 at or surrounding position 41, when both Ψ42 and Ψ44 are removed (Fig 6B and C). It is conceivable that the local structure/geometry change, coupled with Ψ's other unique chemical properties (e.g. the extra hydrogen bond donor that may interact with bound protein), enables Ψ42 and Ψ44 to impact Prp5 binding and subsequent U2‐dependent ATPase activity, thus influencing pre‐mRNA splicing. Relatedly, it has been reported that, when a specific uridine in the U7 Sm binding site is pseudouridylated, U7 snRNP assembly is greatly compromised (Kolev & Steitz, 2006). Likewise, it has also been shown that when a specific uridine in the pre‐mRNA polypyrimidine tract near the 3′ splice site is converted to Ψ, no pre‐mRNA‐U2AF binding is detected, resulting in failure to splice the pre‐mRNA (Chen et al, 2010).
Ψs within the U2 BSRR and the local structure contribute directly to Prp5 binding
Given that U2 is distinct from any other RNAs in both primary sequence and secondary structure, it is possible that Prp5/SF3b specifically recognizes the unique sequence/structure of U2 and then hydrolyzes ATP to drive a conformational change in the pre‐spliceosome complex (Complex A). The Ares group has proposed a branch site recognition model wherein the U2 BSRR and its surrounding sequence form a stem loop (BSL), presenting the branch site recognition sequence to base pair with the branch site of pre‐mRNA (Perriman & Ares, 2010). Importantly, when the branch site is recognized and complex A forms, the BSL is then disrupted, presumably by Prp5's ATPase activity, leading to a conformational change that drives spliceosome assembly moving forward (Perriman & Ares, 2010). Since the Prp5‐catalyzed BSL disruption is a critical step in this process, this model nicely explains why a relatively less stable BSL favors spliceosome assembly, and why a hyperstabilized BSL is unfavorable for spliceosome formation (Perriman & Ares, 2010). A less stable BSL can be equated with a more active Prp5, and a more stable BSL can be equated with a less active Prp5 (Xu & Query, 2007; Perriman & Ares, 2010).
Given that Ψs can slightly stabilize double‐stranded RNA (Newby & Greenbaum, 2001), it is possible that blockage of the formation of Ψ42 and Ψ44, both of which reside within the BSL stem, might result in a less stable BSL and more efficient splicing. However, our experimental data show otherwise: splicing was inhibited upon ablation of Ψ42 and Ψ44 (Fig 2). It is thus likely that the BSL structure was not destabilized when the formation of Ψ42 and Ψ44 was blocked. Consistent with this hypothesis, our in vivo DMS probing experiments showed that blockage of U‐to‐Ψ conversion at positions 42 and 44 led to a local structure change in U2 at or surrounding C41; however, the overall BSL structure seemed unchanged (band intensities, with the exception of the intensity of C41 band and to a lesser extent, the intensities of A25 and A27 bands, were unchanged, see Fig 6B and C). Thus, we propose that Ψ42 and Ψ44 (each of which has an extra hydrogen bond donor that may directly help recruit Prp5), together with the unique geometry of the surrounding nucleotides (not the whole BSL structure), participate in binding to U2‐Prp5/SF3b. Removal of Ψ42 and Ψ44, which is always accompanied by a structural alteration at position 41 (resulting in a local structure alteration, but not necessarily a destabilization, of the BSL), hinders Prp5 binding and thus results in inefficient spliceosome assembly and splicing (Fig 7).
Figure 7. Model for the function of Ψ42 and Ψ44 during spliceosome assembly.

In wild type, Ψ42 and Ψ44, the surrounding nucleotides, and perhaps the whole BSL structure (Perriman & Ares, 2010), stimulate the interaction of U2 with Prp5 (perhaps through SF3b) and activation of Prp5's ATPase activity, resulting in a conformational change and Complex A formation. In the snr81Δ pus1Δ mutant, the absence of Ψ42 and Ψ44 leads to a U2 local structural change. The lack of Ψs coupled with the local structural change negatively impacts Prp5 binding and Prp5's ATPase activity. Consequently, Complex A formation or spliceosome assembly becomes less efficient.
Does U2 directly interact with Prp5?
It has been previously reported that the U2‐Prp5 association is mediated by splicing factor SF3b (Shao et al, 2012). However, results from our current work indicate that Prp5 is truly capable of interacting directly with U2 snRNA, especially pseudouridylated U2 snRNA, under binding conditions where low concentrations (nM) of Prp5 and U2 were used and where no other cellular proteins were present (Fig 5C). This result is consistent with the fact that Prp5 exhibits U2‐dependent ATPase activity, that Prp5 favors binding to pseudouridylated U2 over un‐pseudouridylated U2 or other RNAs, and that pseudouridylated U2, when compared with un‐pseudouridylated U2 or other RNAs, is the most effective in activating Prp5's ATPase activity (Figs 4 and 5). This result is also consistent with the most recent report indicating that Prp5 directly interacts with the BSL element of U2 (Liang & Cheng, 2015). Based on the data obtained from the current work and previously published studies, we suggest that the Prp5‐SF3b interaction helps recruit U2 snRNP to the U1‐5′‐splice site complex, and then, a direct Prp5‐U2 BSL RNA interaction, enhanced by Ψ42 and Ψ44, stimulates Prp5 ATPase activity (shown here) and facilitates opening of the BSL.
Ψ42 and Ψ44, like prp5 mutants, alter splicing of suboptimal branch‐region introns
Ablation of Ψ42 and Ψ44 resulted in inhibited splicing of all splicing reporters tested, consistent with the hypothesized importance of these modified nucleotides in the splicing process; however, the degree of inhibition for different reporters varied significantly. WT and some very mild mutant ACT1‐CUP1 reporters were strongly inhibited; in contrast, the relatively more severe branch‐flanking mutant reporters (including A258c and A258u) that were impaired for base‐pairing with U2 snRNA, suffered much less inhibition (see Fig 2C, D and E, and data not shown). This is directly parallel to results observed with Prp5 mutants, where there was a relative improvement of the A258c and A258u reporters compared to WT (Xu & Query, 2007). Thus, the new results are consistent with our hypothesis that Ψ42 and Ψ44 are strong activators of Prp5 ATPase activity; their ablation strongly reduces Prp5 binding and ATPase activity (as shown in Figs 4 and 5), and their effects on splicing are consistent with a role in activating Prp5 ATPase.
Complex effects of Ψs in the U2 BSRR
Whereas it is now clear that the Ψs in yeast U2 are important for U2 function, the effect of these Ψs on pre‐mRNA splicing and cell growth appears to be far more complex than expected. For instance, the degree to which each Ψ affects cell growth varies (Figs 1B and 2A). More interestingly, while blocking Ψ formation at positions 42 and 44 has a significant impact on U2 function and cell growth, further blockade of pseudouridylation at position 35 rescues, to some extent, U2 function and cell growth (Figs 1B and 2A). The simplest explanation is that perhaps all three Ψs, with Ψ35 being a negative regulator, and Ψ42 and Ψ44 positive activators, contribute to U2‐Prp5 binding (presumably through SF3b) and Prp5's U2‐dependent ATPase activity. However, given that Ψ35 is directly involved in base‐pairing with the branch site of pre‐mRNA (see Fig 1A) and that blockage of Ψ35 formation also results in mild defects in splicing and growth (Fig 2A), how removal of this Ψ counters the effects of ablation of Ψ42 and Ψ44 on U2 function and cell growth is complicated. Further analysis will be necessary to clarify this issue.
Materials and Methods
Strains and plasmids
Parental S. cerevisiae strains used in this work were YCL46 (MATa, cup1::ura3 leu2 trp1 lys2 ade2 his3) (Lesser & Guthrie, 1993), yYZX02 (MATa, ade2 cup1::ura3 his3 leu2 lys2 prp5::loxP trp1, pRS316‐PRP5[PRP5 URA3 CEN ARS]) (Xu & Query, 2007), and BJ2168 (MATa leu2 trp1 ura3‐52 prb1‐1122 pep4‐3 prc1‐407 gal2) (Jones, 1991). Single or multiple gene deletions from these strains were generated according to standard gene deletion protocol (Baudin et al, 1993). In brief, the genes for HIS, kanamycin, and hygromycin resistance were PCR amplified with at least 45 bp overhangs homologous to the chromosomal flanking regions of PUS7, SNR81, and PUS1 genes, respectively. The PCR products were transformed into respective strains for homologous recombination. Successful gene deletion was verified by PCR and CMC‐primer extension analysis (see below). ACT1‐CUP1 wild‐type reporter was a kind gift from C. Guthrie (Lesser & Guthrie, 1993). All mutated ACT1‐CUP1 reporter constructs were made by an improved site‐directed mutagenesis method (Zheng et al, 2004). Constructs containing guide RNAs with altered pseudouridylation pockets were made by four‐piece overlapping PCR (Huang et al, 2012). Essentially four oligos, each containing mutated pocket sequences in the middle, had ~15 nts overlapping sequences with adjacent oligos, so that they served as templates for each other. The oligos were added with a 20:1:1:20 ratio so that the two oligos at both ends greatly exceeded the two in the middle, thus maximizing production of full‐length guide RNA. PCR products were enzyme digested and subcloned into yEPlac195[URA 2μ] or yEPplac181[LEU 2μ] vectors that were modified to contain a GPD or Gal promoter between EcoRI and BamHI sites (Gietz & Sugino, 1988). Prp5 wild‐type (WT) and mutant (GAR, GAG, TAG, SAW, SAP, N399D, G401E) expression vectors (pRS314‐PRP5[PRP5 TRP1 CEN ARS]) are as described (Xu & Query, 2007). To generate TAP‐tagged PRP5 in BJ2168 and its derived strain, a PCR fragment containing the TAP tag flanked by 350 bp 5′ UTR and 100 bp of 3′ URT of PRP5 was generated from a PRP5‐TAP stain (a kind gift from Eric Phizicky). The PCR fragment is transformed into the strains and selected with the URA marker in the TAP tag. For recombinant Prp5 expression, WT or mutant PRP5 gene were amplified from pRS314‐PRP5 vectors and subcloned into pGEX‐6p‐2 vector between BamHI and EcoRI. The GST tag upstream of BamHI was fused to Prp5. A FLAG tag was further added to the C‐terminus before the stop codon.
RNA isolation and pseudouridylation assay
Typically, 25 ml of yeast cells grown in YPD (yeast extract, peptone, dextrose) was harvested at OD 2 for RNA isolation. RNA was isolated using a modified method from Invitrogen. Briefly, 0.5‐mm glass beads (Cat. No. 11079105) were added in 1:1 ratio to yeast cell pellet in 2‐ml screw cap tubes. Trizol (1 ml per 50 OD cells) was added, and the tubes were vigorously vortexed using beadbeater (20 sec, 8 times on ice). The lysates were spun twice at 15,600 g for 2 min to remove debris and were added to 0.2 volume of phenol. After vigorous vortex (> 1 min) and phase separation, the upper phase was again extracted using phenol/chloroform/isoamyl alcohol (25:24:1). RNA was precipitated using 2.5 volumes of 200 proof ethanol and washed with 70% ethanol. For CMC‐primer extension pseudouridylation assay, 8–10 μg of total cellular RNA was treated with CMC (Sigma, cat. no. 29469) at a final concentration of 50 mM Bicine, 0.15 M CMC, 4 mM EDTA, and 7 M urea at 37°C for 20 min. RNA was then precipitated and washed with ethanol, then resuspended in sodium carbonate buffer (50 mM, pH 10.4) and incubated at 37°C for 2 h. RNA was then extracted by phenol/chloroform/isoamyl alcohol and precipitated and washed by ethanol. The resulting RNA pellet was used for primer extension (see below). The primer used for mapping Ψs in U2 snRNA was 5′‐GTGCCAAAAAATGTGTATTGTA‐3′, which annealed to nucleotides 102–123 of U2.
Copper sensitivity assay
Cells were transformed with ACT1‐CUP1 reporters alone or cotransformed with ACT1‐CUP1 reporters and guide RNA expression vectors. All transformations are carried out by incubating early log phase cells (OD between 0.5 and 1.0) and 200 ng plasmid (1 μg of each for cotransformation) at 42°C for 30 min in 30 μl of OST buffer (200 mM lithium acetate, 40% (w/v) PEG3350). Cells were then incubated in liquid YPD media at 30°C for 1 h and spread on selective plates. To assay copper sensitivity, yeast strains were grown in selective media to saturation, diluted to OD 0.2 and grown another 6 h. Resulting cultures were then serially diluted (5×) with corresponding selective media. Equal volumes of each strain were spotted on selective plates with varying copper concentrations. The plates were incubated at 30°C (unless otherwise indicated) for 3 days and photographed.
Primer extension assay
Splicing products were analyzed by primer extension using total RNA as template (Wu et al, 2011). Total RNAs were isolated from cells grown to mid‐log phase. Typically, 5 μg of RNA was suspended in annealing buffer (50 mM Tris–HCl, 60 mM NaCl, 10 mM DTT, pH 8.3) with 50 nM gel‐purified RT primer labeled by 32P at the 5′ end. The mixture was heated at 90°C for 3 min, then 55°C for 1 min, then chilled on ice. To the mixture 0.25 unit of AMV reverse transcriptase (promega) and 0.4 mM dNTP were added in a final concentration of 35 mM Tris–HCl, 40 mM NaCl, 7 mM DTT, and 5 mM magnesium acetate (pH 8.3). The RT reaction was incubated at 42°C for 20 min. The reaction was stopped by adding 2× RNA loading dye (fermentas) and heated at 95°C for 4 min. Primer extension products were loaded on 8% polyacrylamide gel (acrylamide/bisacrylamide 19:1) containing 8 M urea. Radioactive reverse transcription products were visualized by autoradiography. The primer 5′‐GGCACTCATGACCTTC‐3′ was complementary to the CUP1 exon and thus reverse‐transcribed pre‐mRNA, mRNA, and lariat intermediate. U6 primer 5′‐CGGTTCATCCTTATGCAGG‐3′ and U1 primer 5′‐TTGCATCAATGACTTCAATGAAC‐3′ were used as controls. Relative levels of reverse transcription stop bands were quantified by ImageQuant 5.2.
Co‐immunoprecipitation and native gel assays
A flag tag was inserted at the C‐terminus of Prp5 in the pRS314‐PRP5 vectors to generate pRS314‐PRP5‐FLAG. yXYZ‐02 and its derivative snR81Δ pus1Δ strain were transformed with pRS314‐PRP5‐FLAG and selected on 5‐FOA plates to remove untagged PRP5. Resulting colonies were grown to OD 2 to make splicing extracts (Ansari & Schwer, 1995). In brief, cell pellets from 2 l culture were suspended in 7.5 ml of AGK buffer (10 mM HEPES‐KOH pH 7.9, 1.5 mM MgCl2, 200 mM KCl, 0.5 mM DTT, and 10% glycerol), and pippetted dropwise into liquid nitrogen. Frozen pellets were vigorously ground using pestle and mortar to a fine power; the powder was slowly thawed and cleared by ultracentrifugation at 140,000 g for 1 h. Extracts were dialyzed twice against buffer D (20 mM HEPES (pH 8), 20% glycerol, 50 mM KCl, 0.2 mM EDTA (pH 8), 0.5 mM DTT), aliquoted and stored at −80°C. For Co‐IP, anti‐FLAG M2 resin pre‐washed with buffer D was rotated with splicing extracts, then washed with IPP150 buffer (10 mM Tris–Cl pH 8.0, 150 mM NaCl, 0.1% NP‐40) three times. Co‐IP'd RNA was eluted by boiling beads in 2× SDS–PAGE buffer (4% SDS, 10% 2‐mercaptoethanol, 20% glycerol, 0.125 M Tris–HCl, 0.004% bromophenol blue). RNA was extracted twice with phenol/chloroform and precipitated by ethanol. Co‐precipitated U2 was measured by primer extension analysis. To carry out mutant Prp5 IP, wild‐type Prp5 was depleted by anti‐FLAG M2 immunoprecipitation at high concentration of salt (1 M KCl) as described (Xu et al, 2004), except that anti‐FLAG M2 resin was used instead of IgG sepharose. FLAG‐tagged mutant Prp5 proteins were added back to the extracts, and salt concentration was brought back to the original concentration by dialysis using buffer D. For spliceosome assembly assay, PRP5 TAP‐tagged BJ2168 and its derivative snR81Δ pus1Δ strain were used to make splicing extracts. Prp5 was depleted by IgG sepharose beads at high concentration of salt (Xu et al, 2004). Recombinant Prp5 was added to the depleted extract, and salt concentration was brought back to the original level by dialysis using buffer D. In vitro splicing reactions (40% splicing extract, 2 mM ATP, 2.5 mM MgCl2, 60 mM KPO4, 3% PEG8000, 5,000 cpm mRNA) were assembled and incubated at 28°C for 10 min (Ansari & Schwer, 1995), mixed with native gel loading dye and separated on 4% (79:1 acrylamide:bisacrylamide) native gel at 80 V for overnight (Konarska, 1989).
Plasmid shuffling assay using 5‐FOA
Yeast strain yYu (GB‐53) (MATa, ade2 cup1‐Δ::ura3 leu2 lys2 trp1 prp5‐Δ::loxP pus‐Δ::HIS3 snR81‐Δ::kanMX pus1‐Δ::hphMX6, pRS316‐PRP5[PRP5 URA3 CEN ARS]) (made in this study) and yYZX02 were cotransformed with pRS314‐PRP5 variants and yEPplac181 variants expressing mutant snR81 or empty vector. Transformants were selected on solid media lacking tryptophan and leucine. Colonies were picked and grown in selective media, and equal amounts of cells were dropped on 5‐FOA plates. Plates were incubated at 30°C for 4 days and photographed.
Expression of recombinant protein and ATPase assay
The recombinant construct was transformed into Rosetta 2 competent cell. Cells (1 l) were grown at 37°C to reach OD 0.7, then transferred to 16°C and grown for 16 h after induction with 0.4 mM IPTG. Cell pellets were washed twice in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7.2), then resuspended in PBS and sonicated. Cell lysates were cleared by centrifugation at 26,891.8 g using a Sorval SS‐34 rotor and then incubated with 0.5 ml of glutathione agarose pre‐washed with PBS. After washing with PBS twice, the beads were further washed once with PBS with additional NaCl (final 500 mM). GST proteins were eluted using 20 mM glutathione and then dialyzed twice using 1 l buffer D. ATPase assays were carried out in a 10 μl reaction incubated at 30°C for 45 min (Xu et al, 2004). The reaction contained final concentrations of 45 mM Tris–HCl (pH 8.0), 25 mM NaCl, 2.2 mM DTT, 1 mM MgCl2, 0.1 mM ATP, 0.04 μCi/μl [α‐32P] ATP, 0.01% Triton X‐100, 0.1 mM EDTA, and 220 nM Prp5. After incubation, 2 μl from each reaction was spotted onto a microcrystalline cellulose coated TLC plate (analtech) and developed for 8 h in the buffer containing 22 ml of isobutyric acid, 6 ml of ddH2O, and 1 ml of 30% ammonium hydroxide (Fang et al, 2010). The TLC plate was dried in a chemical hood overnight and visualized by phosphoimaging.
In vivo DMS mapping of U2
DMS mapping of yeast U2 snRNA was carried out essentially as described (Wells et al, 2000). Yeast cells were grown in synthetic media to log phase at 30°C. Ten OD cells were transferred to 50‐ml falcon tube and resuspended in 2 ml of YPD. Five hundred μl of DMS (1:4 diluted with 200 proof ethanol) was added to the tube. The tube was immediately mixed and vigorously vortexed for 2 min in a 30°C shaker. The reaction was stopped by adding 5 ml of 0.6 M 2‐mercaptoethanol and 5 ml of isoamyl alcohol. Cells were collected and washed with 0.6 M 2‐mercaptoethanol twice. RNA was extracted for primer extension using standard protocol as described (see above). The oligo used for U2 was 5′‐TTGAGGTCATTTCAGTTGTTAC‐3′.
Author contributions
GW and YTY designed experiments and interpreted data. GW, HA, JG, and DS carried out experiments. CCQ and YTY wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We would like to thank members of the Yu laboratory for valuable discussions. This work was supported by grants GM104077 (to Y.‐T.Y.) and GM57829 (to C.C.Q.) from the National Institute of Health.
The EMBO Journal (2016) 35: 654–667
References
- Ansari A, Schwer B (1995) SLU7 and a novel activity, SSF1, act during the PRP16‐dependent step of yeast pre‐mRNA splicing. EMBO J 14: 4001–4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnez JG, Steitz TA (1994) Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl‐tRNA synthetase and ATP suggests a possible role for pseudo‐uridines in stabilization of RNA structure. Biochemistry 33: 7560–7567 [DOI] [PubMed] [Google Scholar]
- Bakin AV, Ofengand J (1998) Mapping of pseudouridine residues in RNA to nucleotide resolution. Methods Mol Biol 77: 297–309 [DOI] [PubMed] [Google Scholar]
- Baudin A, Ozier‐Kalogeropoulos O, Denouel A, Lacroute F, Cullin C (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae . Nucleic Acids Res 21: 3329–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm‐Ansmant I, Urban A, Ma X, Yu YT, Motorin Y, Branlant C (2003) The Saccharomyces cerevisiae U2 snRNA:pseudouridine‐synthase Pus7p is a novel multisite‐multisubstrate RNA:Psi‐synthase also acting on tRNAs. RNA 9: 1371–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette M, Gray MW (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49: 341–351 [DOI] [PubMed] [Google Scholar]
- Chen C, Zhao X, Kierzek R, Yu YT (2010) A flexible RNA backbone within the polypyrimidine tract is required for U2AF65 binding and pre‐mRNA splicing in vivo . Mol Cell Biol 30: 4108–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DR (1995) Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res 23: 5020–5026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Hartmuth K, Luhrmann R (2004) Modified nucleotides at the 5′ end of human U2 snRNA are required for spliceosomal E‐complex formation. RNA 10: 1925–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Luhrmann R (2009) The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell 36: 593–608 [DOI] [PubMed] [Google Scholar]
- Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, Wang X (2010) The ER UDPase ENTPD5 promotes protein N‐glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 143: 711–724 [DOI] [PubMed] [Google Scholar]
- Ge J, Yu YT (2013) RNA pseudouridylation: new insights into an old modification. Trends Biochem Sci 38: 210–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A (1988) New yeast‐Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six‐base pair restriction sites. Gene 74: 527–534 [DOI] [PubMed] [Google Scholar]
- Huang C, Wu G, Yu YT (2012) Inducing nonsense suppression by targeted pseudouridylation. Nat Protoc 7: 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EW (1991) Tackling the protease problem in Saccharomyces cerevisiae . Methods Enzymol 194: 428–453 [DOI] [PubMed] [Google Scholar]
- Kolev NG, Steitz JA (2006) In vivo assembly of functional U7 snRNP requires RNA backbone flexibility within the Sm‐binding site. Nat Struct Mol Biol 13: 347–353 [DOI] [PubMed] [Google Scholar]
- Konarska MM (1989) Analysis of splicing complexes and small nuclear ribonucleoprotein particles by native gel electrophoresis. Methods Enzymol 180: 442–453 [DOI] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C (1993) Mutational analysis of pre‐mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WW, Cheng SC (2015) A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev 29: 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Lustig AJ, Abelson J (1987) Splicing of yeast nuclear pre‐mRNA in vitro requires a functional 40S spliceosome and several extrinsic factors. Genes Dev 1: 7–18 [DOI] [PubMed] [Google Scholar]
- Ma X, Zhao X, Yu YT (2003) Pseudouridylation (Psi) of U2 snRNA in S. cerevisiae is catalyzed by an RNA‐independent mechanism. EMBO J 22: 1889–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Yang C, Alexandrov A, Grayhack EJ, Behm‐Ansmant I, Yu YT (2005) Pseudouridylation of yeast U2 snRNA is catalyzed by either an RNA‐guided or RNA‐independent mechanism. EMBO J 24: 2403–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenet S, Motorin Y, Lafontaine DL, Hurt EC, Grosjean H, Branlant C (1999) Pseudouridine mapping in the Saccharomyces cerevisiae spliceosomal U small nuclear RNAs (snRNAs) reveals that pseudouridine synthase pus1p exhibits a dual substrate specificity for U2 snRNA and tRNA. Mol Cell Biol 19: 2142–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JM, Query CC, Sharp PA (1993) Splicing of precursors to mRNA by the spliceosome In The RNA World, Gesteland RF, Atkins JF. (eds), pp 303–357. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Newby MI, Greenbaum NL (2001) A conserved pseudouridine modification in eukaryotic U2 snRNA induces a change in branch‐site architecture. RNA 7: 833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby MI, Greenbaum NL (2002a) Investigation of Overhauser effects between pseudouridine and water protons in RNA helices. Proc Natl Acad Sci USA 99: 12697–12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby MI, Greenbaum NL (2002b) Sculpting of the spliceosomal branch site recognition motif by a conserved pseudouridine. Nat Struct Biol 9: 958–965 [DOI] [PubMed] [Google Scholar]
- Nilsen TW (1994) RNA‐RNA interactions in the spliceosome: unraveling the ties that bind. Cell 78: 1–4 [DOI] [PubMed] [Google Scholar]
- O'Day CL, Dalbadie‐McFarland G, Abelson J (1996) The Saccharomyces cerevisiae Prp5 protein has RNA‐dependent ATPase activity with specificity for U2 small nuclear RNA. J Biol Chem 271: 33261–33267 [DOI] [PubMed] [Google Scholar]
- Perriman RJ, Ares M Jr (2007) Rearrangement of competing U2 RNA helices within the spliceosome promotes multiple steps in splicing. Genes Dev 21: 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Ares M Jr (2010) Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell 38: 416–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy R, Busch H (1988) Small nuclear RNAs: RNA sequences, structure, and modifications In Structure and Function of Major and Minor Small Nuclear Ribonucleoprotein Particles, Birnstiel ML. (ed.), pp 1–37. Heidelberg: Springer‐Verlag Press; [Google Scholar]
- Shao W, Kim HS, Cao Y, Xu YZ, Query CC (2012) A U1‐U2 snRNP interaction network during intron definition. Mol Cell Biol 32: 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Konarska MM, Query CC (2009) Insights into branch nucleophile positioning and activation from an orthogonal pre‐mRNA splicing system in yeast. Mol Cell 34: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C (1998) Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326 [DOI] [PubMed] [Google Scholar]
- Wells SE, Hughes JM, Igel AH, Ares M Jr (2000) Use of dimethyl sulfate to probe RNA structure in vivo . Methods Enzymol 318: 479–493 [DOI] [PubMed] [Google Scholar]
- Wu G, Xiao M, Yang C, Yu YT (2011) U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J 30: 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Newnham CM, Kameoka S, Huang T, Konarska MM, Query CC (2004) Prp5 bridges U1 and U2 snRNPs and enables stable U2 snRNP association with intron RNA. EMBO J 23: 376–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Query CC (2007) Competition between the ATPase Prp5 and branch region‐U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28: 838–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, McPheeters DS, Yu YT (2005) Psi35 in the branch site recognition region of U2 small nuclear RNA is important for pre‐mRNA splicing in Saccharomyces cerevisiae . J Biol Chem 280: 6655–6662 [DOI] [PubMed] [Google Scholar]
- Yu YT, Shu MD, Steitz JA (1998) Modifications of U2 snRNA are required for snRNP assembly and pre‐mRNA splicing. EMBO J 17: 5783–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YT, Scharl EC, Smith CM, Steitz JA (1999) The growing world of small nuclear ribonucleoproteins In The RNA World, Gesteland RF, Cech TR, Atkins JF. (eds), 2nd edn, pp 487–524. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Yu AT, Ge J, Yu YT (2011) Pseudouridines in spliceosomal snRNAs. Protein Cell 2: 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yu YT (2004) Pseudouridines in and near the branch site recognition region of U2 snRNA are required for snRNP biogenesis and pre‐mRNA splicing in Xenopus oocytes. RNA 10: 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Baumann U, Reymond JL (2004) An efficient one‐step site‐directed and site‐saturation mutagenesis protocol. Nucleic Acids Res 32: e115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File