

Abstract
A key step of Wnt signaling activation is the recruitment of β‐catenin to the Wnt target‐gene promoter in the nucleus, but its mechanisms are largely unknown. Here, we identified FoxM1 as a novel target of Wnt signaling, which is essential for β‐catenin/TCF4 transactivation. GSK3 phosphorylates FoxM1 on serine 474 which induces FoxM1 ubiquitination mediated by FBXW7. Wnt signaling activation inhibits FoxM1 phosphorylation by GSK3–Axin complex and leads to interaction between FoxM1 and deubiquitinating enzyme USP5, thereby deubiquitination and stabilization of FoxM1. FoxM1 accumulation in the nucleus promotes recruitment of β‐catenin to Wnt target‐gene promoter and activates the Wnt signaling pathway by protecting the β‐catenin/TCF4 complex from ICAT inhibition. Subsequently, the USP5–FoxM1 axis abolishes the inhibitory effect of ICAT and is required for Wnt‐mediated tumor cell proliferation. Therefore, Wnt‐induced deubiquitination of FoxM1 represents a novel and critical mechanism for controlling canonical Wnt signaling and cell proliferation.
Keywords: FoxM1, ICAT, ubiquitination, USP5, Wnt/β‐catenin signaling
Subject Categories: Development & Differentiation, Signal Transduction
Introduction
The canonical Wnt/β‐catenin signaling pathway is transduced through Wnt receptors of the Frizzled family and stabilizes β‐catenin, which enters the nucleus and forms a complex with TCF4/LEF‐1 transcription factors (Behrens et al, 1996; Bhanot et al, 1996; Molenaar et al, 1996; Liu et al, 1999). Activation of TCF4/LEF‐1 by binding to β‐catenin induces the transcription of various target genes, including c‐Myc and cyclin D1 (He et al, 1998; Shtutman et al, 1999; Tetsu & McCormick, 1999). The pathway is also persistently activated in many types of tumors and critically regulates tumor growth, invasion, and angiogenesis (Weeraratna et al, 2002; Clevers, 2006; Anastas & Moon, 2013). Unlike colorectal cancer, in which constitutive activation of β‐catenin is due to inactivation of the tumor suppressor APC or mutations in β‐catenin, many sporadic tumors have no such mutations (Morin et al, 1997; Paraf et al, 1997; Bafico et al, 2004). Therefore, it is important to elucidate whether aberration in other components of the Wnt signaling pathway causes the activation of β‐catenin/TCF4‐mediated transcription in such tumors, including glioma.
We recently found that FoxM1 is a novel component of Wnt signaling (Zhang et al, 2011). FoxM1 is a member of the forkhead box (Fox) transcription factor family. Growing evidence indicates that FoxM1 plays important roles in mammal development and human tumorigenesis (Kalin et al, 2011; Raychaudhuri & Park, 2011). FoxM1 is a key cell cycle regulator of both the transition from G1 to S phase and progression to mitosis (Ye et al, 1997; Korver et al, 1998). FoxM1 is substantially elevated in most human tumors and contributes to oncogenesis in many tissue types, including liver, prostate, brain, breast, lung, colon, and pancreatic tumors (Kalin et al, 2011; Raychaudhuri & Park, 2011; Gong & Huang, 2012). Our recent studies also showed that expression of FoxM1 in high‐grade glioma is significantly higher than that in low‐grade glioma and that FoxM1 expression contributes to the growth of glioma, as well as pancreatic, colon, and breast cancer cells by promoting uncontrolled cell proliferation, invasion, and angiogenesis (Liu et al, 2006; Zhang et al, 2008; Li et al, 2013; Xue et al, 2014). Moreover, we showed that FoxM1 critically regulates stemness and tumorigenicity of glioma stem cells (Zhang et al, 2011).
Although FoxM1 is commonly overexpressed in most human tumors, the molecular mechanisms for its overexpression remain unknown. Furthermore, it is well known that protein degradation can be regulated through both E3 ubiquitin ligases and deubiquitinases (DUBs). However, although a E3 ubiquitin ligase, APC/C‐Cdh1, which regulates FoxM1 has been reported (Laoukili et al, 2008; Park et al, 2008), the role of DUB in FoxM1 protein stability has been virtually unexplored.
The Wnt signaling pathway is one of the major signaling pathways in stem cells and cancer cells. Aberrant activation of the Wnt/β‐catenin pathway is widespread in human cancers. A crucial but unclear step of Wnt/β‐catenin pathway activation is the assembly of a β‐catenin/TCF transcription activation complex to the promoter of Wnt target gene. β‐Catenin contains 12 armadillo repeats to form a superhelix that features a positively charged groove that can serve as a binding surface for its many partners, such as TCF4, FoxM1, and inhibitor of β‐catenin and TCF4 (ICAT) (Huber et al, 1997; Tutter et al, 2001; Daniels & Weis, 2002; Graham et al, 2002). TCF4 binds to armadillo repeats 3–9 of β‐catenin to form β‐catenin/TCF transcription activation complex (Daniels & Weis, 2002).
In contrast, ICAT is a newly identified physiological Wnt inhibitor that prevents the binding of TCF4 to β‐catenin and thus the assembly of a β‐catenin/TCF transcription activation complex. ICAT contains an N‐terminal helical domain that binds to repeats 11 and 12 of β‐catenin, and an extended C‐terminal region that binds to repeats 5–10 in a manner similar to that of TCF4 (Daniels & Weis, 2002; Graham et al, 2002). Therefore, full‐length ICAT dissociates complexes of β‐catenin and TCF4, thus inhibiting the Wnt/β‐catenin pathway (Daniels & Weis, 2002; Graham et al, 2002).
In this study, we examined whether aberration of FoxM1 and/or ICAT is an important molecular mechanism for the activation of Wnt/β‐catenin signaling in glioma cells. Our data revealed that FoxM1 is a novel downstream target of Wnt signaling and that USP5 is the first deubiquitylase that regulates FoxM1 deubiquitination and protein stabilization induced by Wnt activation. Wnt activation increased FoxM1 protein stabilization and thus nuclear accumulation. Nuclear FoxM1 then directly interacted with nuclear β‐catenin, which released β‐catenin from ICAT and enhanced recruitment of β‐catenin to the promoter of Wnt target gene, hence increasing the expression of Wnt target gene. Furthermore, USP5 increased Wnt‐mediated cell proliferation through FoxM1, which abrogated the inhibitory effect of ICAT. In human glioma tissues, high levels of FoxM1 and cyclin D1 are associated with high levels of USP5. Therefore, our study provides a novel mechanism for Wnt magnification of its own signaling activation.
Results
Wnt activation increases the stability of FoxM1 protein
Recently, we reported that Wnt activation increased the expression level of FoxM1 (Zhang et al, 2011). To determine whether Wnt regulates FoxM1 protein stability, we first treated glioma cell lines LN229 and U87 with Wnt‐3a at various doses (0–100 ng/ml) for 6 h. Wnt‐3a treatments increased the levels of β‐catenin (Fig 1A), indicating that Wnt signaling had been activated. The FoxM1 expression level was also increased by Wnt treatments in a dose‐dependent manner in both cell lines (Fig 1A). Moreover, Wnt‐3a treatment increased the half‐life of FoxM1, measured by a pulse–chase assay (Fig 1B). However, Wnt‐3a treatment for 6 h did not increase the mRNA level of FoxM1 in the U87 cells (Appendix Fig S1A), which excludes that FoxM1 transcription is regulated by Wnt‐3a in the 6‐h time frame. Furthermore, we determined the effect of inhibition of endogenous Wnt signaling on FoxM1 expression with use of a broadly efficacious Wnt antagonist, DKK1 (Glinka et al, 1998). DKK1 increased FoxM1 degradation compared with the control in both LN229 and U87 cells (Fig 1C). The above results indicated that activation of Wnt signaling increases the protein stability of FoxM1.
Figure 1. Wnt signaling regulates the stability of FoxM1 and GSK3 kinase regulates the degradation of FoxM1 protein.
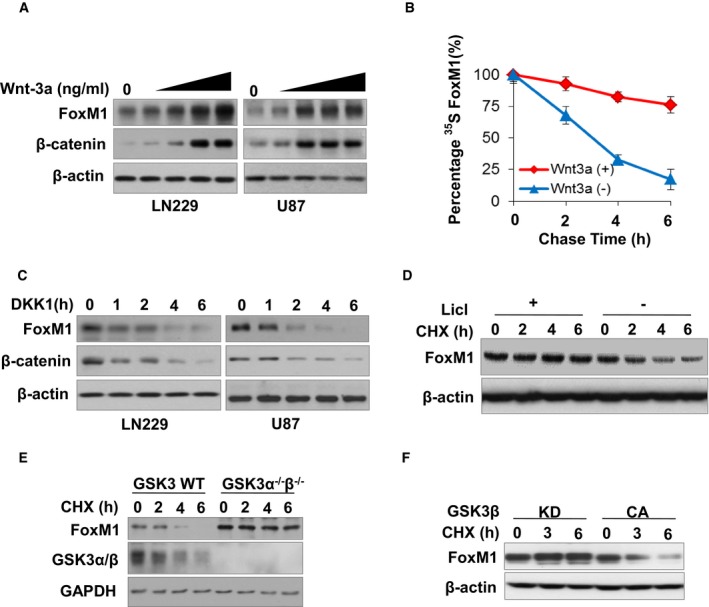
- LN229 and U87 cells treated with Wnt‐3a (0, 10, 20, 50, 100 ng/ml) for 6 h. The indicated proteins were analyzed by Western blotting.
- LN229 cells treated with or without Wnt‐3a (50 ng/ml) were pulsed with [35S] methionine for 30 min and chased for the times indicated. Protein extracts were used for IP with anti‐FoxM1 antibodies and subjected to SDS–PAGE and autoradiography. The intensities of the FoxM1 bands in autoradiography were then quantified by using NIH Image software, and the densities of the FoxM1 bands at time 0 were set as 100%. Values are mean ± SD from two independent experiments.
- Levels of FoxM1 were analyzed by Western blotting in LN229 and U87 cells treated with Wnt inhibitor DKK1 (100 ng/ml) for the indicated times. Note that the basal levels of FoxM1 and β‐catenin in (A) and (C) are similar but look different due to variations in the exposure time of the Western blots.
- FoxM1 expression levels were determined in LN229 cells treated with or without GSK3 inhibitor LiCl (10 mM) and CHX (100 μg/ml) for the indicated times.
- GSK3 wild‐type (WT) and knockout (KO) cells were treated with CHX (100 μg/ml) for the indicated times, and FoxM1 and GSK3α/β expression levels were determined by Western blotting.
- GSK3βCA or GSK3βKD plasmids were transfected into 293T cells for 48 h. The cells were then treated with CHX (100 μg/ml) for the indicated times. Endogenous FoxM1 expression level was determined by Western blotting.
GSK3 kinase regulates the degradation of FoxM1 protein
Since inhibition of GSK3‐mediated β‐catenin degradation is a key process in canonical Wnt signaling (Clevers, 2006; Kim et al, 2013), we explored whether GSK3 also mediates FoxM1 degradation. We first treated LN229 cells with a GSK3 inhibitor, LiCl, along with a protein synthesis inhibitor, cycloheximide. LiCl stabilized FoxM1 protein in the presence of CHX (Fig 1D).
To confirm that GSK3 regulates FoxM1 stability, the protein degradation of endogenous FoxM1 was measured in GSK3 wild‐type (GSK3α+/+;GSK3β+/+) and knockout (GSK3α−/−;GSK3β−/−) mouse embryonic stem cells. The levels of FoxM1 in GSK3α−/−;GSK3β−/− cells were increased compared with those in GSK3α+/+;GSK3β+/+cells; also, the FoxM1 underwent degradation with time in GSK3α+/+;GSK3β+/+cells but was quite stable in GSK3α−/−;GSK3β−/− cells (Fig 1E). Furthermore, to determine whether GSK3 kinase activity impacts FoxM1 protein stability, FoxM1 protein degradation was analyzed in 293T cells transfected with GSK3β‐CA (constitutively active) or GSK3β‐KD (kinase‐inactive) plasmid. FoxM1 protein degradation was inhibited by GSK3β‐KD but not by GSK3β‐CA transfection in the cells (Fig 1F). The above results indicated that GSK3 kinase activation increases the degradation of FoxM1 protein.
GSK3 phosphorylates FoxM1 protein at the S474 site, which promotes ubiquitination of FoxM1
To understand the mechanisms responsible for the regulation of FoxM1 protein expression by the Wnt pathway through inhibition of GSK3, we determined whether GSK3 interacts with and phosphorylates FoxM1 protein. We constructed deletion mutants of Flag‐tagged FoxM1 (Fig 2A). Subsequently, with use of the Flag‐tagged FoxM1 full‐length or deletion mutant proteins (Fig 2B) in 293T cells by co‐IP assay, we found that full‐length and C‐terminal of FoxM1 interact with endogenous GSK3 (Fig 2B).
Figure 2. Phosphorylation of FoxM1 by GSK3β at S474 promotes the ubiquitination of FoxM1.
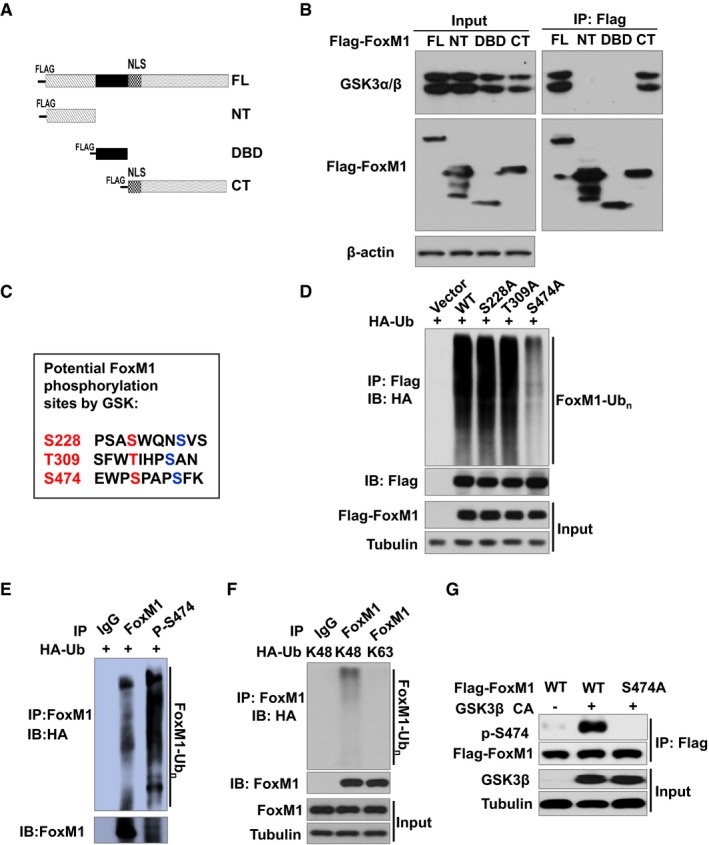
- Schematic of Flag‐FoxM1 deletion mutants in a mammalian expression system. FL: full length; NT: NH 2‐terminal domain; DBD: DNA‐binding domain; CT: COOH‐terminal domain.
- Lysates from 293T cells expressing Flag‐FoxM1 or its mutants were subjected to IP using mouse anti‐Flag antibody, followed by IB with anti‐GSK3α/β antibody (upper panel) and rabbit anti‐Flag antibody (middle panel).
- Sequence analysis of human FoxM1 identified three putative GSK3β target sites at S228, T309, and S474.
- HA‐ubiquitin and Flag‐tagged FoxM1 (WT) or mutants FoxM1S228A, FoxM1T309A, or Flag‐FoxM1S474A plasmids were co‐transfected into 293T cells. After 36 h, cells were treated with 25 nM MG132 for 6 h. Cell lysates were subjected to IP with anti‐Flag antibody, followed by IB with anti‐Flag and anti‐HA antibody.
- HA‐ubiquitin was co‐transfected into 293T cells. After 36 h, cells were treated with 25 nM MG132 for 6 h. Cell lysates were subjected to IP with IgG or anti‐FoxM1 phospho‐S474 antibody, followed by IB with anti‐FoxM1 and anti‐HA antibody.
- HA‐tagged K48‐only ubiquitin or K63‐only ubiquitin construct was transfected into 293T cells. After 36 h, cells were treated with 25 nM MG132 for 6 h. Cell lysates were subjected to IP with IgG or anti‐FoxM1 antibody, followed by IB with anti‐FoxM1 and anti‐HA antibody.
- GSK3β CA or vector and Flag‐tagged FoxM1 (WT) or mutant S474A were co‐transfected into 293T cells. Lysates of the cells were subjected to IP with anti‐Flag antibody and followed by IB with anti‐FoxM1 phospho‐S474 antibody.
We then sought to determine whether FoxM1 protein contains consensus sequence for GSK3 substrates by using the GSK3 substrates consensus sequence S/T–X–X–X‐S/T, in which the first S (serine) or T (threonine) is the target residue (Cohen & Frame, 2001; Doble & Woodgett, 2003). Analysis of the FoxM1 coding sequence identifies three motifs that resemble the GSK target sequence: S228‐XXX‐S232, T309‐XXX‐S313, and S474‐XXX‐S478 (Fig 2C). To determine the importance of these putative sites in protein stability, we generated T309A, S474A, and S228A mutants by mutating the serine or threonine at the sites of Flag‐FoxM1 plasmid to alanine.
Meanwhile, we determined whether the ubiquitin proteasome pathway is involved in Wnt signaling‐mediated FoxM1 stability by detecting the FoxM1 level in LN229 cells treated with DKK1 and MG132 (a proteasome inhibitor). MG132 inhibited FoxM1 degradation induced by DKK1 (Appendix Fig S2). Thus, we analyzed ubiquitination of FoxM1 wild‐type (WT) and S228A, S309A, or S474A mutants in 293T cells and found that S474A had the lowest ubiquitination level among the FoxM1 mutants and WT (Fig 2D), suggesting that phosphorylation at the S474 site is important for the ubiquitination of FoxM1. To confirm this point, we generated an antibody that specifically recognizes FoxM1 phosphorylated at S474 residues, named p‐S474 antibody. Then, we used this antibody to pull down the ubiquitinated FoxM1 and found that most of the S474 phosphorylated FoxM1 protein was ubiquitinated compared with wild‐type FoxM1 (Fig 2E).
Next, we determine the type of ubiquitin chains for FoxM1 ubiquitination. Previous studies showed that ubiquitination through lysine‐48 (K48)‐linked poly‐ubiquitin chains generally targets proteins for degradation, whereas ubiquitination through K63‐linked poly‐ubiquitin chains play a critical role in signaling activation (Nathan et al, 2013). Thus, we transfected 293T cells with HA‐tagged K48‐only or K63‐only ubiquitin constructs and then analyzed the ubiquitination of FoxM1. We found that only poly‐ubiquitin‐K48 in FoxM1 was detected by anti‐HA antibody, suggesting that FoxM1‐ubiquitin chains are K48‐linked poly‐ubiquitin chains (Fig 2F). Furthermore, consistent with the above results, mutation in the S474 site prevented GSK3β‐CA‐mediated phosphorylation of FoxM1, which was recognized by the p‐S474 antibody in 293T cells (Fig 2G). These results suggest that phosphorylation of FoxM1 by GSK3 at the S474 site promotes ubiquitination of FoxM1. Moreover, these results suggest that serine 474 followed by a proline is a classical targeted site for GSK3 which is established as a proline‐directed serine/threonine kinase (Hooper et al, 2008).
Wnt activation inhibits the phosphorylation of FoxM1 mediated by Axin–GSK3 complex
It is known that Wnt activation disrupts the interaction of β‐catenin with Axin which scaffolds between β‐catenin and GSK3, thereby inhibiting phosphorylation of β‐catenin by GSK3 and causing β‐catenin stabilization (Clevers & Nusse, 2012; Kim et al, 2013). We thus determined whether Axin is a scaffold protein between FoxM1 and GSK3. First, we found that FoxM1 interacted with Axin and formed a complex with Axin and GSK3 (Fig 3A). Knockdown of Axin inhibited the interaction of FoxM1 with GSK3 (Fig 3B). Knockdown of Axin also inhibited the phosphorylation of FoxM1 at S474 (Fig 3C). These results suggest that Axin plays a scaffold role in the interaction of FoxM1 with GSK3. Moreover, we found that Wnt‐3a treatment induced the dissociation of Axin with FoxM1 (Fig 3D), suggesting that Wnt‐3a regulates FoxM1 phosphorylation by GSK3 through Axin–GSK3 complex. Furthermore, we found that knockdown of Axin led to significantly reduction of FoxM1 poly‐ubiquitination (Fig 3E). Knockdown of Axin also further enforced the Wnt‐3a‐induced reduction of FoxM1 poly‐ubiquitination (Fig 3E). These results indicated that Wnt activation inhibits the phosphorylation of FoxM1 mediated by Axin–GSK3 complex which is required for the FoxM1 ubiquitination.
Figure 3. Wnt activation inhibits the phosphorylation of FoxM1 mediated by Axin–GSK3 complex and the ubiquitination of FoxM1 mediated by FBXW7.

- Cell lysates of LN229 cells were subjected to IP using anti‐Axin antibody or control IgG, followed by IB with the indicated antibodies.
- Axin siRNA or control siRNA was transfected into LN229 cells and, after 36 h, co‐IP was performed with lysates of the cells using anti‐FoxM1 antibody followed by IB with the indicated antibodies.
- Axin siRNA or control siRNA was transfected into LN229 cells for 36 h. Lysates of the cells were subjected to IB with anti‐FoxM1 phospho‐S474 antibody.
- Cell lysates of LN229 cells with or without Wnt‐3a treatment for 4 h were subjected to IP using anti‐Axin antibody or control IgG, followed by IB with anti‐FoxM1 and anti‐Axin antibodies.
- Axin siRNA or control siRNA with HA‐ubiquitin were transfected into 293T cells and, after 36 h, cells were treated with MG132 with or without Wnt‐3a (50 ng/ml) for 6 h. Then, IP was performed with lysates of the cells using anti‐FoxM1 antibody followed by IB with the indicated antibodies.
- Wild‐type Flag‐FoxM1 or S474A mutant was transfected into 293T cells after 36 h, and cells were treated with MG132 for 6 h. Then, IP was performed with lysates of the cells using anti‐Flag antibody followed by IB with the indicated antibodies.
- HA‐ubiquitin and/or FBXW7, GSK3β CA, Flag‐FoxM1 were co‐transfected into 293T cells and, after 36 h, cells were treated with MG132 for 6 h. Then, IP was performed with lysates of the cells using anti‐Flag antibody followed by IB with the indicated antibodies.
- FBXW7 siRNA or control siRNA and HA‐ubiquitin were transfected into 293T cells and, after 36 h, cells were treated with MG132 with or without Wnt‐3a (50 ng/ml) for 6 h. Then, IP was performed with lysates of the cells using anti‐FoxM1 antibody followed by IB with the indicated antibodies.
FBXW7 mediates FoxM1 ubiquitination, which is inhibited by Wnt/GSK3 signaling
It has been reported that Wnt activation leads to stabilization of proteins by β‐catenin‐independent Wnt/STOP (Wnt‐dependent stabilization of proteins) signaling (Acebron et al, 2014; Koch et al, 2015). The roles of GSK3 as well as Axin/APC complex in Wnt/STOP signaling have been explored previously (Taelman et al, 2010; Stolz et al, 2015). Once phosphorylated by GSK3, proteins can be ubiquitinated by E3 ubiquitin ligases, including FBW7 (F box/WD repeat‐containing protein 7) and NEDD4L (neural precursor cell expressed, developmentally down‐regulated 4‐like), which lead to degradation of substrate proteins (Kim et al, 2009; Taelman et al, 2010; Acebron et al, 2014; Stolz et al, 2015). When FoxM1 protein was examined to identify putative degrons for the above E3 ubiquitin ligases, the motif LWEWPS(474)PAPS of FoxM1 was found to be closed to a consensus degron for FBXW7: ΩxΩΩΩ(S/T)Pxx(S/T/E) [Ω = hydrophobic] (Welcker & Clurman, 2008). Thus, we determined whether FBXW7 is an E3 ligase for FoxM1. We found that FBXW7 interacted with wild type of FoxM1 but not with S474A mutant of FoxM1 in the presence of MG132 (Fig 3F). Moreover, overexpression of FBXW7 induced FoxM1 ubiquitination as compared with control (Fig 3G, lane 4 versus lane 2). Next, since S474 is phosphorylated by GSK3, we determined whether FBXW7‐mediated FoxM1 ubiquitination is regulated by GSK3 activation, by using GSK3β CA construct. We found that FBXW7‐induced FoxM1 ubiquitination was increased by GSK3β CA transfection (Fig 3G), suggesting that GSK3 activity enhances FBXW7‐mediated FoxM1 ubiquitination. Furthermore, we found that knockdown of FBXW7 by siRNA inhibited FoxM1 ubiquitination as compared with control siRNA (Fig 3H). More importantly, knockdown of FBXW7 reversed the inhibitory effect of Wnt treatment on FoxM1 ubiquitination (Fig 3H). Collectively, these results suggest that FBXW7‐mediated FoxM1 ubiquitination is regulated by Wnt signaling and that Wnt induced FoxM1 stabilization via a Wnt/STOP mechanism.
USP5 regulates stabilization of FoxM1 by deubiquitination in response to Wnt stimulation
Ubiquitin–ubiquitin and ubiquitin–protein bonds can be cleaved by the action of DUB enzymes including ubiquitin‐specific proteases (USPs). Since the S478A mutation suppresses ubiquitination of FoxM1, we hypothesized that Wnt activation may function in cooperation with an unknown DUB. To identify a DUB that targets the ubiquitin of FoxM1, we screened a panel of USPs in which a total of 32 USP cDNA plasmids were transfected into 293T cells. Strikingly increased endogenous FoxM1 expression under Wnt‐3a treatment was observed in the cells transfected with USP5 (Fig 4A). Moreover, when USP5 was overexpressed in 293T cells, FoxM1 degradation was inhibited (Fig 4B), suggesting a role of USP5 in FoxM1 protein stability.
Figure 4. USP5 deubiquitylates and stabilizes FoxM1 in response to Wnt stimulation.
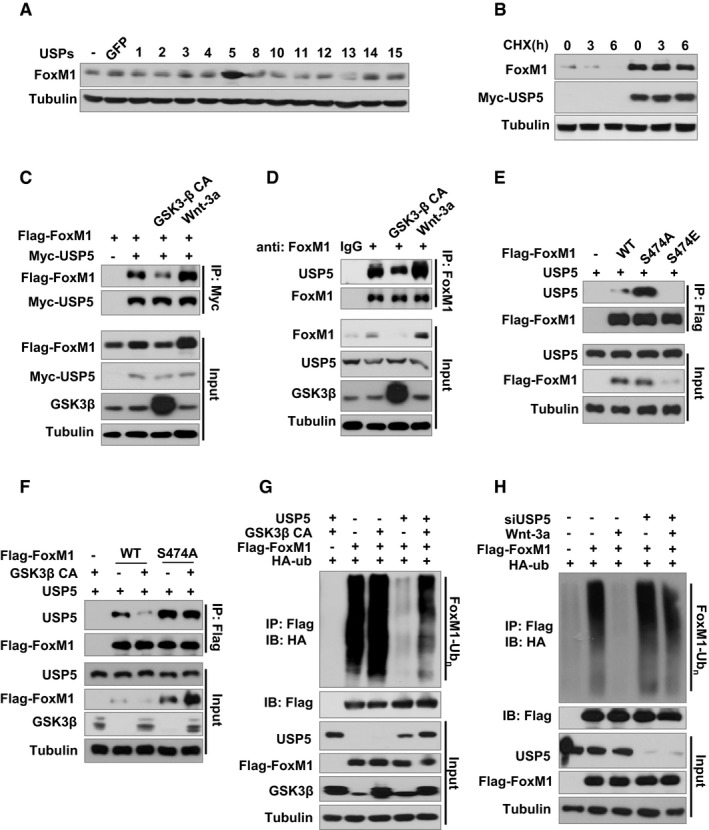
- 293T cells were transfected with USPs cDNA or control plasmid. After 36 h of transfection, cells were treated with 50 ng/ml Wnt‐3a for 4 h. FoxM1 protein levels in the cells were then determined by Western blotting.
- 293T cells were transfected with 1 μg control plasmid or USP5 cDNA in 6‐well plates. After 36 h of transfection, cells were treated with 100 μg/ml CHX for 0, 3, or 6 h. FoxM1 protein levels in the cells were then determined by Western blotting.
- GSK3β CA, USP5, or control plasmid was co‐transfected with Flag‐FoxM1 into 293T cells. After 36 h of transfection, cells were treated with 50 ng/ml Wnt‐3a or control PBS for 4 h. Cell lysates were subjected to IP with anti‐Myc antibody and followed by IB with anti‐Flag or anti‐Myc antibody.
- GSK3β CA, USP5, or control plasmid was co‐transfected with Flag‐FoxM1 into 293T cells. After 36 h of transfection, cells were treated with 50 ng/ml Wnt‐3a or control PBS for 6 h. Cell lysates were subjected to IP with anti‐FoxM1 antibody and followed by IB with anti‐FoxM1 or USP5 antibody.
- USP5 and Flag‐FoxM1 (WT) or mutants Flag‐FoxM1S474A or Flag‐FoxM1S474E plasmids were co‐transfected into 293T cells for 36 h. Cell lysates were subjected to IP with anti‐Flag antibody and followed by IB with anti‐Flag or anti‐USP5 antibody.
- GSK3β CA, USP5, or control plasmid was co‐transfected with Flag‐FoxM1 (WT) or mutant Flag‐FoxM1S474A into 293T cells for 36 h. Cell lysates were subjected to IP with anti‐Flag antibody and followed by IB with anti‐Flag or anti‐USP5 antibody.
- HA‐ubiquitin and Flag‐FoxM1 were co‐transfected with or without GSK3β CA into 293T cells. After 36 h, cells were treated with 25 nM MG132 for 6 h. Cell lysates were subjected to IP with anti‐Flag followed by IB with anti‐HA or anti‐FoxM1 antibody.
- HA‐ubiquitin and Flag‐FoxM1 were co‐transfected with or without siUSP5 into U87 cells. After 36 h, cells were treated with 50 ng/ml Wnt‐3a and 25 nM MG132 for 6 h. Cell lysates were subjected to IP with anti‐Flag antibody followed by IB with anti‐HA or anti‐FoxM1 antibody.
We sought to determine whether USP5 directly interacts with FoxM1 and functions as a bona fide FoxM1 deubiquitylase. Co‐IP assays confirmed that ectopically expressed Flag‐FoxM1 could be detected in Myc‐tagged USP5 immunoprecipitates in 293T cells (Fig 4C), indicating that USP5 interacts with FoxM1 in vivo. Endogenous USP5 also interacts with endogenous FoxM1 in LN229 cells (Fig 4D). Moreover, Wnt‐3a treatment enhanced the interaction between USP5 and FoxM1, while constitutive activated GSK3β weakened such interaction in both 293T (Fig 4C) and LN229 (Fig 4D) cell lines. Consistent with this finding, mutation of Ser474 to Ala (S474A) on FoxM1 increased the interaction of USP5 and FoxM1 (Fig 4E). In contrast, mutation of Ser474 to Glu (S474E), which mimics phosphorylation of Ser474, abolished the interaction of USP5 and FoxM1 (Fig 4E). S474A mutation was also resistant to GSK3β‐CA‐induced reduction of USP5–FoxM1 interaction compared with WT FoxM1 (Fig 4F). Therefore, these results indicated that inhibition of GSK3β‐mediated phosphorylation of FoxM1 at S474 is required for the interaction of USP5–FoxM1.
We reasoned that USP5 regulates Wnt‐induced FoxM1 stabilization by affecting deubiquitination. Indeed, overexpression of USP5 reduced the endogenous ubiquitination of FoxM1 as well as GSK3β‐CA‐induced ubiquitination of FoxM1 (Fig 4G). Furthermore, Wnt‐3a reduced ubiquitination of FoxM1, and this reduction was reversed by silencing USP5 expression with a specific siRNA (Fig 4H), suggesting that Wnt‐3a‐induced deubiquitination of FoxM1 is dependent on USP5.
Nuclear FoxM1 induced by Wnt signaling enhances recruitment of β‐catenin to TCF‐binding elements by disrupting ICAT–β‐catenin interaction
FoxM1 has both cytosolic and nuclear locations, and cytosolic FoxM1 functions in β‐catenin nuclear translocation (Zhang et al, 2011). Wnt activation increased the nuclear level of FoxM1 (Appendix Fig S3A). Our previous study suggested that nuclear FoxM1 appears to govern another critical aspect of β‐catenin nuclear function: recruitment of β‐catenin to the β‐catenin/TCF4 transcription activation complex in Wnt target‐gene promoter (Zhang et al, 2011). However, the mechanism for this function of nuclear FoxM1 is unknown. As well known, β‐catenin contains 12 armadillo repeats to serve as a binding surface for its many partners, such as TCF4, FoxM1, and ICAT (Appendix Fig S3B). ICAT binds to 5–12 armadillo repeats of β‐catenin, which overlaps the binding site for TCF4 (Appendix Fig S3B) and prevents β‐catenin/TCF4 interaction, thereby excluding β‐catenin's recruitment to Wnt target‐gene promoter (Huber et al, 1997; Tutter et al, 2001; Daniels & Weis, 2002; Graham et al, 2002). Moreover, β‐catenin repeats 10–12 are the only repeats required for ICAT binding (Tago et al, 2000; Graham et al, 2002); FoxM1 also interacts with β‐catenin via binding its repeats 11–12 (Zhang et al, 2011).
We have found that the levels of ICAT in glioma cell lines (Hs683 and SW1783), glioma stem cell lines (GSC11, 20 and 23), and breast cancer cell lines (BT474 and MDA‐MB‐231) were similar to those in nontumor cell lines (HaCaT and 293T) (Appendix Fig S3C). However, our previous studies showed that the above tumor cells expressed much higher levels of FoxM1 than nontumor cells expressed (Zhang et al, 2011; Xue et al, 2014). Moreover, levels of ICAT in human glioblastoma multiforme (GBM) were similar to those in adjacent normal brain tissue (Fig 5A and B). In contrast, levels of FoxM1 in human GBM were significantly up‐regulated compared with those in normal brain tissue (Fig 5A and C). Therefore, these results suggested that up‐regulated Wnt/β‐catenin activity in GBM cells is likely due to increased FoxM1 expression but not due to decreased ICAT expression. Based on these observations, we hypothesized that FoxM1–β‐catenin interaction prevents ICAT–β‐catenin interaction, hence up‐regulating Wnt/β‐catenin activity, because it is clear that both FoxM1 and ICAT interact with the 11–12 repeats of β‐catenin.
Figure 5. Wnt promotes FoxM1 nuclear translocation to enhance interaction of β‐catenin/TCF4 via FoxM1 competing with ICAT .

- Expression of FoxM1 and ICAT was examined via IHC staining in 30 GBM and paired adjacent normal tissues. Representative ICAT and FoxM1 expressions are shown in a section of a GBM tumor with adjacent normal brain. Scale bar: 50 µm.
- ICAT stainings were scored as 1–12 according to the intensity and percentage in the 30 GBM and paired adjacent normal tissues. Boxes indicate interquartile range. Bars from each box extend to largest and smallest observations.
- FoxM1 stainings were scored as 1–12 according to the intensity and percentage in the 30 GBM and paired adjacent normal tissues. Boxes indicate interquartile range. Bars from each box extend to largest and smallest observations.
- Increased amounts of FoxM1 plasmids were transfected into 293T cells. After 36 h, cells were treated with 50 ng/ml Wnt‐3a for 6 h. Cell lysates were then subjected to IP with anti‐β‐catenin antibody, followed by IB with anti‐β‐catenin, anti‐ICAT, anti‐FoxM1, or IgG antibodies.
- Increased amounts of ICAT plasmids were transfected into 293T cells. After 36 h, cells were treated with 50 ng/ml Wnt‐3a for 6 h. Cell lysates were then subjected to IP with anti‐β‐catenin antibody, followed by IB with anti‐β‐catenin, anti‐ICAT, anti‐FoxM1, or IgG antibodies.
- FoxM1 siRNA or control siRNA was transfected into U87 cells, and lysates were subjected to IP with anti‐β‐catenin antibody, followed by IB with anti‐β‐catenin, anti‐ICAT, anti‐FoxM1, or IgG antibodies.
- ICAT siRNA or control siRNA was transfected into LN229 cells, and lysates were subjected to IP with anti‐β‐catenin antibody, followed by IB with anti‐β‐catenin, anti‐ICAT, anti‐FoxM1, or IgG antibodies.
- ChIP analyses of endogenous β‐catenin binding at the TCF‐binding site of the Axin2 promoter were performed in vector‐, FoxM1‐, and/or ICAT plasmid‐transfected U87 cells. Fold was calculated relative to that in cells transfected with the vector, which was set as 1.
- ChIP analyses of endogenous β‐catenin binding at the TCF‐binding site of the Axin2 promoter were performed in FoxM1 siRNA‐, ICAT siRNA‐, control siRNA‐, or combination of FoxM1 siRNA and ICAT siRNA‐transfected U87 cells as described in (H).
- ChIP analyses of endogenous β‐catenin binding at the TCF‐binding site of the Axin2 promoter were performed in vector‐, β‐catenin‐NLS‐, sh‐FoxM1‐, FoxM1‐shR‐, and/or ICAT plasmid‐transfected U87 cells as described in (H).
We first tested whether FoxM1 overexpression blocks exogenous ICAT–β‐catenin interaction in 293T cells. ICAT plasmid and increasing amounts of FoxM1 plasmids were co‐transfected into the cells; the cells were then treated with Wnt‐3a, and Co‐IP assays were conducted with use of nuclear protein from the cells. We found that β‐catenin binding to ICAT decreased with increasing FoxM1 expression (Fig 5D). In contrast, when FoxM1 plasmid and increasing amounts of ICAT plasmid were co‐transfected into the 293T cells and then the cells were treated with Wnt‐3a, β‐catenin binding to FoxM1 decreased with increasing ICAT expression (Fig 5E).
Next, we silenced FoxM1 with use of a specific siRNA in U87 cells to analyze endogenous ICAT, β‐catenin, and FoxM1 interactions. Silencing FoxM1 increased the interaction between ICAT and β‐catenin (Fig 5F). Silencing ICAT by its specific siRNA in LN229 increased the interaction between FoxM1 and β‐catenin (Fig 5G).
It is well established that nuclear β‐catenin associates with TCF4/LEF‐1 transcription factors on TCF‐binding elements (TBEs) to regulate Wnt target‐gene expression (Behrens et al, 1996; Molenaar et al, 1996). ICAT prevents binding between β‐catenin and TCF4, thereby suppressing the recruitment of β‐catenin to TBE (Daniels & Weis, 2002). To provide direct evidence that FoxM1 enhances recruitment of β‐catenin to TBE by antagonizing ICAT function, we chose Axin2, a prototypic Wnt/β‐catenin/TCF4 target gene. Axin2 promoter has a TBE located between −108 and −102 bp (Leung et al, 2002). We performed ChIP assays via the β‐catenin, IgG (control), and specific PCR primers flanking the TBE of the Axin2 promoter. ICAT overexpression inhibited the recruitment of β‐catenin to TBE of Axin2 promoter in U87 cells (Fig 5H). In contrast, FoxM1 overexpression increased the recruitment of β‐catenin to TBE of Axin2 promoter, and the effect of FoxM1 overexpression on the recruitment of β‐catenin was inhibited by ICAT overexpression (Fig 5H). Silencing FoxM1 inhibited the recruitment of β‐catenin to the TBE (Fig 5H), whereas silencing ICAT increased the recruitment of β‐catenin to the TBE (Fig 5I). Moreover, the effect of FoxM1 silencing on the recruitment of β‐catenin was overridden by silencing of ICAT (Fig 5J).
To further distinguish the role of nuclear FoxM1 from cytoplasmic FoxM1 in the β‐catenin activation, we used β‐catenin‐NLS construct which can translocate into the nucleus constitutively. Expression of β‐catenin‐NLS induced the recruitment of β‐catenin to TBE of Axin2 promoter, and the effect of β‐catenin‐NLS expression on the recruitment of β‐catenin was inhibited by FoxM1 silencing (Fig 5J). This result confirms that in nuclear, FoxM1 enhances the recruitment of β‐catenin to the β‐catenin/TCF4 transcription activation complex in Wnt target‐gene promoter. Moreover, the effect of FoxM1 silencing on the recruitment of β‐catenin was overridden by FoxM1‐shR (shRNA‐resistant) but not by ICAT (Fig 5J). Collectively, these results indicated that in the nuclear, FoxM1–β‐catenin interaction prevents ICAT–β‐catenin interaction, thereby promoting the recruitment of β‐catenin to the Wnt target gene.
FoxM1–β‐catenin interaction is required for β‐catenin transcriptional activity by antagonizing ICAT's function
We next determined whether β‐catenin binding to the TBE of the target gene that is increased by FoxM1 leads to increased transcription activity of β‐catenin. Overexpression of FoxM1 increased the activity of Axin2 promoter, but overexpression of ICAT reduced the activity of Axin2 promoter induced by FoxM1 by a factor of 10 (Fig 6A). Moreover, knocking down FoxM1 inhibited the activity of Axin2 promoter (Fig 6B), whereas knocking down ICAT enhanced the activity of Axin2 promoter (Fig 6C).
Figure 6. FoxM1 regulates β‐catenin transcription activity and promotes Wnt target‐gene expression.
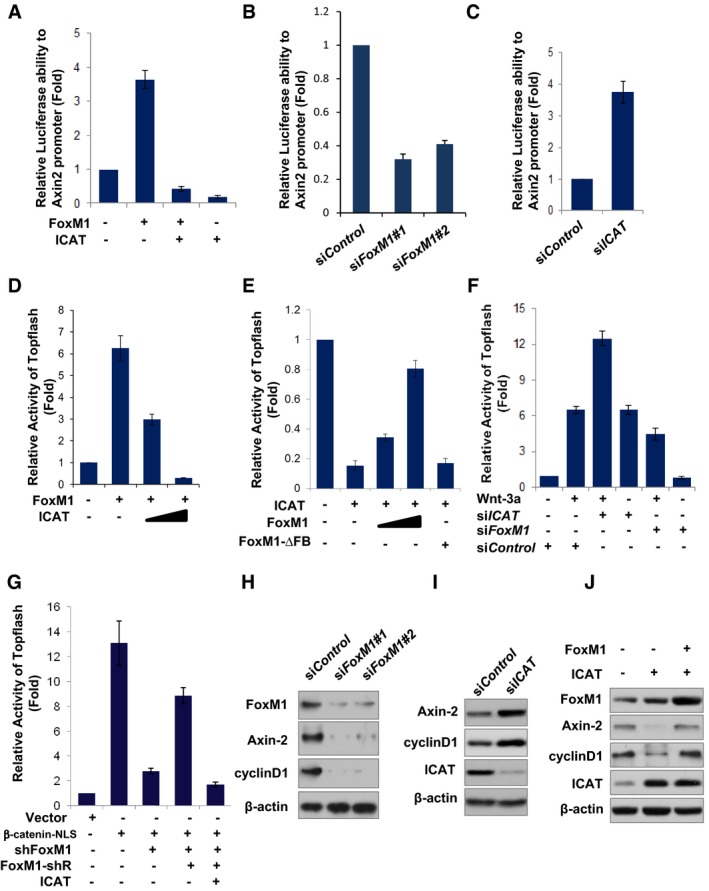
- FoxM1 and/or ICAT plasmids were co‐transfected with Axin2 promoter luciferase and TK‐Renilla plasmid into U87 cells. Luciferase activity was measured with the Dural Luciferase reporter assay, and relative luciferase activity was normalized to that of the vector group. Values are mean ± SD for triplicate samples.
- FoxM1 siRNA#1, #2, or control siRNA was co‐transfected to U87 cells with Axin2 promoter reporter. Luciferase activity was measured with the Dural Luciferase reporter assay, and relative luciferase activity was normalized to that of the control siRNA group. Values are mean ± SD for triplicate samples.
- ICAT siRNA or control siRNA was co‐transfected into LN229 cells with Axin2 promoter reporter. Luciferase activity was measured as in (A).
- FoxM1 and/or ICAT plasmids were co‐transfected with TOP‐Flash reporter and TK‐Renilla plasmids into U87 cells. Luciferase activity of TOP‐Flash in the cells was measured as in (A). Values are mean ± SD for triplicate samples.
- ICAT, FoxM1, or FoxM1‐∆FB (FoxM1 mutant lacking the forkhead box domain) plasmid or their combinations were co‐transfected with TOP‐Flash reporter and TK‐Renilla plasmids into LN229 cells. Luciferase activity of TOP‐Flash in the cells was measured as in (A). Values are mean ± SD for triplicate samples.
- FoxM1 siRNA and/or ICAT siRNA were co‐transfected with TOP‐Flash reporter and TK‐Renilla plasmids into LN229 cells, and the cells were treated with Wnt‐3a (50 ng/ml) or control PBS. Luciferase activity of TOP‐Flash in the cells was measured as in (A). Values are mean ± SD for triplicate samples.
- Vector, β‐catenin‐NLS, sh‐FoxM1, FoxM1‐shR, and/or ICAT plasmids were co‐transfected with TOP‐Flash reporter and TK‐Renilla plasmids into U87 cells. Luciferase activity of TOP‐Flash in the cells was measured as in (A). Values are mean ± SD triplicate samples.
- Levels of Axin‐2 and cyclin D1 protein in LN229 cells transfected with FoxM1 siRNA#1 or #2 were determined by Western blotting.
- Levels of Axin‐2 and cyclin D1 protein in LN229 cells transfected with ICAT siRNA were determined by Western blotting.
- Levels of Axin‐2 and cyclin D1 protein in LN229 cells transfected with FoxM1 and/or ICAT plasmids were determined by Western blotting.
We further determined whether FoxM1 is required for β‐catenin transcriptional activity by using the TOP‐Flash report assay, a Wnt/β‐catenin‐responsive reporter assay. Overexpression of FoxM1 increased the endogenous β‐catenin transcriptional activity, whereas overexpression of ICAT inhibited the FoxM1‐induced β‐catenin transcriptional activity in a dose–response manner (Fig 6D). In contrast, overexpression of ICAT inhibited the endogenous β‐catenin transcriptional activity, whereas overexpression of FoxM1 antagonized the inhibitory effect of ICAT in a dose–response manner (Fig 6E). However, overexpression of FoxM1‐∆FB mutant, which does not interact with β‐catenin since it lacks the forkhead box domain of FoxM1, did not antagonize the inhibitory effect of ICAT (Fig 6E). Conversely, knocking down ICAT enhanced both endogenous and Wnt‐induced β‐catenin transcriptional activity, whereas knocking down FoxM1 did the opposite (Fig 6F). Moreover, knocking down FoxM1 antagonized the effect of ICAT knockdown on Wnt‐induced β‐catenin transcriptional activity (Fig 6F). Furthermore, knocking down FoxM1 inhibited β‐catenin‐NLS‐induced β‐catenin transcriptional activity (Fig 6G), whereas the effect of FoxM1 silencing on the recruitment of β‐catenin was overridden by FoxM1‐shR (shRNA‐resistant) but not by ICAT (Fig 6G). These results confirmed the role of FoxM1 on β‐catenin nuclear function.
Finally, we detected the expression of Wnt/β‐catenin target genes including Axin‐2 and cyclin D1 to confirm the above findings. Knocking down FoxM1 reduced the expression of Axin‐2 and cyclin D1 (Fig 6G), whereas knocking down ICAT increased their expression (Fig 6H). Furthermore, overexpression of FoxM1 revised the inhibitory effect of ICAT overexpression of the target genes (Fig 6I). Therefore, these results indicated that FoxM1 enhances β‐catenin transcriptional activity by antagonizing the inhibitory effect of ICAT.
USP5–FoxM1 axis abrogates the inhibitory effect of ICAT and is required for Wnt/β‐catenin signaling‐mediated cell proliferation
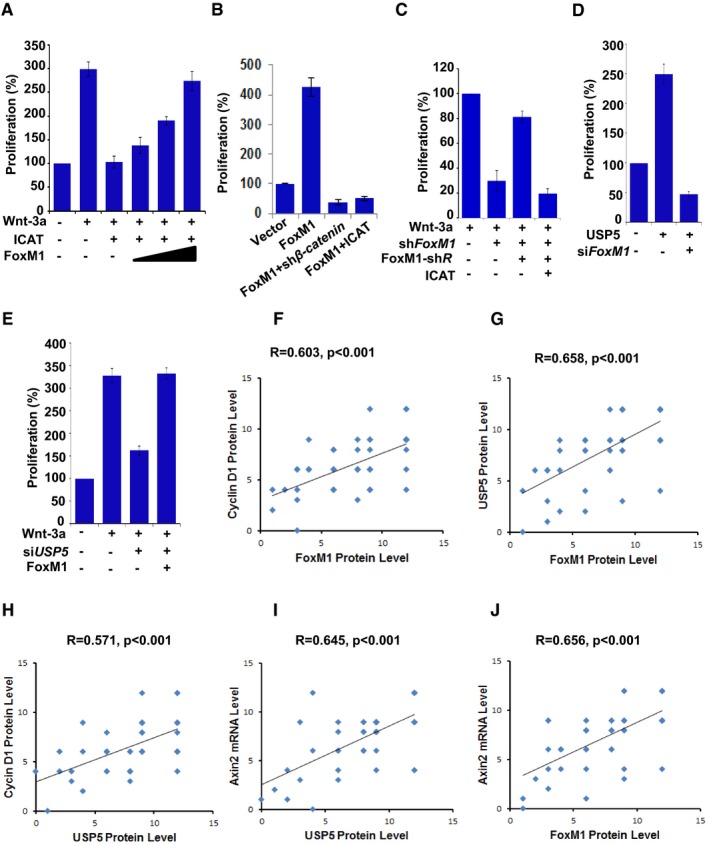
Cyclin D1 expression is required for G1/S phase transition and cell proliferation (Resnitzky & Reed, 1995), thus, we examined whether Wnt‐dependent FoxM1 deubiquitination by USP5, which promotes cyclin D1 expression, regulates cell proliferation. We found that Wnt‐induced cell proliferation in LN229 cells is inhibited by overexpression of ICAT (Fig 7A). Overexpression of FoxM1 overrode the inhibitory effect of ICAT on cell proliferation induced by Wnt‐3a in a dose–response manner (Fig 7A). In addition, overexpression of FoxM1 in LN229 cells increased cell proliferation, but the effect of FoxM1 on cell proliferation was inhibited by β‐catenin depletion as well as by ICAT overexpression (Fig 7B), suggesting that β‐catenin plays an important role in FoxM1‐induced cell proliferation. In contrast, depletion of FoxM1 reduced the cell proliferation induced by Wnt‐3a (Fig 7C); the inhibitory effect of FoxM1 depletion, however, could be rescued by expression of shRNA‐resistant FoxM1, but the rescuing effect of shRNA‐resistant FoxM1 was overridden by expression of ICAT (Fig 7C). Furthermore, overexpression of USP5 increased cell proliferation, but depletion of FoxM1 suppressed the cell proliferation induced by USP5 (Fig 7D). Conversely, depletion of USP5 inhibited Wnt‐induced cell proliferation, but overexpression of FoxM1 overrode the inhibitory effect of USP5 depletion (Fig 7D). Together, these results strongly suggest that the Wnt‐dependent USP5–FoxM1 axis, which abrogates the inhibitory effect of ICAT, is required for Wnt/β‐catenin signaling‐mediated cell proliferation.
Figure 7. USP5–FoxM1 axis regulated Wnt/β‐catenin signaling‐mediated cell proliferation.
-
AA total of 1 × 104 LN229 cells were transfected with control vector, FoxM1, and/or ICAT plasmids and treated with Wnt‐3a (50 ng/ml) and were then plated and counted 48 h after seeding. The proliferation percentage was calculated relative to that in cells transfected with the control vector, which was set as 100%. Data represent the mean ± SD of three independent experiments.
-
BLN229 cells were transfected with vector control, FoxM1, FoxM1 plus sh‐β‐catenin or FoxM1 plus ICAT plasmid. The proliferation percentage was determined as described in (A).
-
CLN229‐shFoxM1 cells were transfected with FoxM1‐shR (siRNA‐resistant FoxM1), or FoxM1‐shR plus ICAT plasmid. The cells were then treated with Wnt‐3a (50 ng/ml). The proliferation percentage was determined as described in (A).
-
DLN229 cells were transfected with USP5 and/or FoxM1 siRNA. The proliferation percentage was determined as described in (A).
-
ELN229 cells were transfected with USP5 siRNA and/or FoxM1 plasmid. The cells were then treated with Wnt‐3a (50 ng/ml). The proliferation percentage was determined as described in (A).
-
F–HExpression of FoxM1, cyclin D1, and USP5 proteins was examined via IHC staining in 50 GBM specimens. Staining of FoxM1, cyclin D1, or USP5 was scored as 1–12 according to the intensity and percentage. The significance of the correlations was determined by Pearson's correlation test.
-
I, JAxin2 mRNA levels in the above 50 GBM specimens were examined through RNAscope technology. The staining was scored as 1–12 according to the intensity and percentage. The significance of the correlations was determined by Pearson's correlation test.
FoxM1, cyclin D1, and Axin‐2 expression in human GBM correlates with levels of USP5
We analyzed the significance of FoxM1‐mediated Wnt/β‐catenin activation in human GBM with use of a panel of 50 GBM samples by determining the expression levels of FoxM1 and the expression levels of Wnt target cyclin D1. The expression levels of FoxM1 protein directly correlated with those of cyclin D1 protein (Fig 7F and Appendix Fig S4), further supporting the critical role of FoxM1 in Wnt/β‐catenin activation in human GBM. Next, we analyzed the significance of USP5‐modulated Wnt‐FoxM1 activation in human GBM by determining the expression levels of USP5 in the above GBM samples. USP5 was highly expressed in 34 samples and moderately expressed in 11 samples. Notably, the expression level of USP5 protein directly correlated with expression levels of FoxM1 and cyclin D1 proteins (Fig 7G and H, Appendix Fig S4). We also analyzed the Wnt‐specific target Axin2 mRNA levels in the same GBM samples (Appendix Fig S4, panel 4). The mRNA level of Axin2 highly correlated with expression levels of USP5 and FoxM1 proteins (Fig 7I and J, Appendix Fig S4). Together, our data underscore the clinical relevance of USP5‐mediated deubiquitination of FoxM1 in regulating activation of Wnt/β‐catenin signaling in GBM.
Discussion
Aberrantly activated Wnt signaling occurs in association with the development and progression of human cancers (Clevers & Nusse, 2012). The Wnt signaling pathway regulates various processes that are important for cancer progression, including tumor initiation and growth, as well as cell senescence, cell death, differentiation, and metastasis. Wnt signaling in the nucleus is transmitted through the key transcriptional co‐regulator β‐catenin. However, the mechanism for β‐catenin recruitment to the Wnt target‐gene promoter is largely unknown. Herein, we demonstrate that Wnt signaling is required for protein level maintenance of FoxM1 via blocking phosphorylation of FoxM1 at S474 by GSK3–Axin complex. Stabilized FoxM1 enters the nuclear and directly interacts with β‐catenin, which releases β‐catenin from ICAT, a major inhibitor of β‐catenin/TCF4 complex, thereby promoting the recruitment of β‐catenin to the Wnt target gene (Fig 8).
Figure 8. Working model.
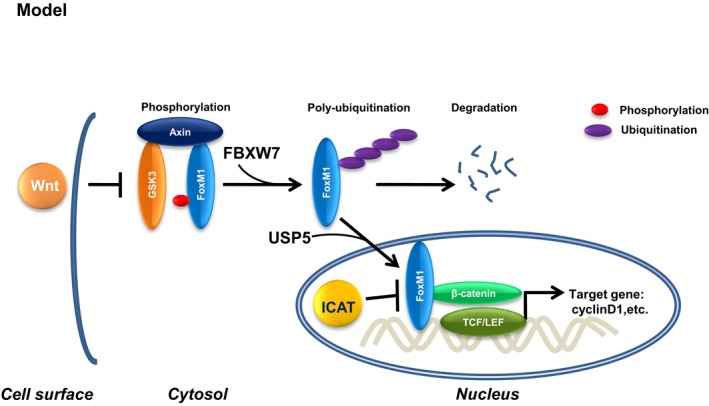
Wnt‐induced deubiquitination and stabilization of FoxM1 abolishes the inhibitory effect of ICAT leading to transactivation of β‐catenin, hence promoting cell proliferation.
Previous studies demonstrated that Wnt activation stabilizes β‐catenin protein by inhibition of the phosphorylation/degradation of β‐catenin mediated by a dual‐kinase mechanism, in which phosphorylation of β‐catenin by CKIa leads to phosphorylation of β‐catenin by GSK3. The phosphorylated β‐catenin is then recognized by the ubiquitin E3 ligase β‐TrCP, triggering ubiquitination and degradation of β‐catenin (Liu et al, 1999, 2002). In the current study, we provided compelling evidence that Wnt activation also stabilizes FoxM1 protein by inhibition of the phosphorylation/degradation of FoxM1 mediated by GSK3. However, unlike phosphorylation of β‐catenin by GSK3 required priming by CKIα, phosphorylation of FoxM1 at S474 by GSK3 does not need priming because S474 is followed by a proline. Furthermore, the experiments using GSK3α−/−β−/− and GSK3 wild‐type cells provided unequivocal evidence for the regulation of FoxM1 by GSK3.
Previous studies indicate that FoxM1 is degraded in late mitosis and early G1 phase by the anaphase‐promoting complex/cyclosome E3 ubiquitin ligase (Laoukili et al, 2008; Park et al, 2008). However, DSPs or DUBs, which play direct role in the process of deubiquitination of FoxM1 protein, have not been reported before; in the only previous study linking USP with FoxM1, USP22 was shown to regulate FoxM1 RNA transcription indirectly (Ning et al, 2014). In this study, we identified USP5 as a specific USP for FoxM1. USP5 is a DSP recently identified as part of the 26S proteasome (Besche et al, 2009). USP5 regulates unanchored poly‐ubiquitin Ub chains (Reyes‐Turcu et al, 2006). USP5 can also specifically regulate protein deubiquitination by binding to a protein. Indeed, a recent study showed that USP5 interacted with Cav3.2 and removed ubiquitin groups from the protein (Garcia‐Caballero et al, 2014). Knockdown of USP5 increases Cav3.2 protein ubiquitination and decreases Cav3.2 protein levels (Garcia‐Caballero et al, 2014). In the current study, we found that USP5 directly interacts with FoxM1. Overexpression of USP5 reduced the endogenous ubiquitination of FoxM1 as well as GSK3β CA‐induced ubiquitination of FoxM1. This deubiquitination process then increased FoxM1 expression. Therefore, the results demonstrated that FoxM1 is a substrate for USP5.
Most signaling pathways require both phosphorylation and ubiquitination for full regulatory control, and ubiquitination is usually regulated by phosphorylation. However, whether deubiquitination is regulated by phosphorylation is unknown. Here, we demonstrated for the first time that dephosphorylation of FoxM1 at S474 is required for the interaction of USP5–FoxM1. Indeed, Wnt‐3a treatment enhanced the interaction between USP5 and FoxM1 by inhibiting phosphorylation of FoxM1 at S474, whereas constitutive activated GSK3β weakened such interaction by mediating the phosphorylation.
There is evidence that β‐catenin is capable of recruiting co‐activators and co‐repressors to regulate its ability to activate Wnt target gene. These include the β‐catenin/TCF complex co‐activators Legless/Bcl9 and Pygopus (Pygo) (Kramps et al, 2002; Townsley et al, 2004), the histone acetyl transferase CBP (Takemaru & Moon, 2000), and the co‐repressors ICAT (Tago et al, 2000) and Chibby (Takemaru et al, 2003; Li et al, 2008). Nuclear antagonist ICAT binds to β‐catenin and disrupts β‐catenin/TCF interactions (Huber et al, 1997; Tutter et al, 2001; Daniels & Weis, 2002; Graham et al, 2002). In this study, we found that FoxM1 serves as a co‐activator to interact with the β‐catenin 11–12 Arm‐repeats portion and to inhibit ICAT binding with β‐catenin, thus promoting TCF/β‐catenin‐dependent transcription. Therefore, simultaneous increases in levels of both FoxM1 and β‐catenin by Wnt ligand stimulation would ensure full activation of Wnt signaling.
It was reported that APC loss led to tumor formation in liver, colon, and kidney but not in pancreas islet which has high level of ICAT (Strom et al, 2007). The mechanism for increased ICAT expression in pancreas of APC loss mice is unknown (Strom et al, 2007), since ICAT protein levels have been shown not to be altered by activation or inhibition of Wnt signaling (Gottardi & Gumbiner, 2004). Other studies have shown that somatic mutations of the ICAT gene in various human cancers are infrequent and that the expression level of ICAT in human colon cancers is not decreased (Koyama et al, 2002; Imai et al, 2004). On the other hand, FoxM1 is expressed in proliferating epithelium at the base of the crypt (which has high level of Wnt activity), but not in the intestinal villus (Ye et al, 1997). Moreover, a study of the role of FoxM1 in colon cancer development using conditional FoxM1 knockout and overexpression transgenic mice has shown that FoxM1 affects β‐catenin/TCF‐4 signaling in colon cancers (Yoshida et al, 2007). Our results in glioma are consistent with the finding that the level of ICAT is not the cause of aberrant β‐catenin/TCF activity. Instead, in combination with the investigation of ICAT and FoxM1 expression in human glioma tissues, we found that FoxM1 overexpression is a critical molecular mechanism for aberrant β‐catenin/TCF activation in glioma. Given that FoxM1 is overexpressed in most tumors, our study uncovered a novel mechanism for the persistent activation of β‐catenin/TCF‐mediated transcription in tumors such as glioma, in which APC or β‐catenin mutations are not common; this mechanism has novel therapeutic strategies against malignant tumors.
Materials and Methods
Cell lines and culture conditions
Human glioma cell lines LN229 and U87 as well as the 293T cell line were obtained from the American Type Culture Collection and cultured in DMEM with 10% FBS. Wild‐type (WT) and GSK3α/β double knockout (KO) mouse stem cells were cultured as described previously (Doble et al, 2007). LN229‐sh‐FoxM1 cells were cultured in DMEM with 10% FBS.
siRNA treatment
Human siFoxM1 #1 target sequence has been described previously (Zhang et al, 2011). siFoxM1 #2 target sequence is GGACCACUUUCCCUACUUU. siRNA pools for ICAT (sc‐43858), AXIN (sc‐41449), FBXW7 (sc‐37547), USP5 (sc‐76869), and control siRNA (sc‐37007) were obtained from Santa Cruz Biotechnology. siRNA oligos were transfected into subconfluent cells with Oligofectamine or Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's instructions.
Immunoblots and immunoprecipitation
Cells were lysed in EBC buffer (50 mM Tris [pH 7.5], 120 mM NaCl, and 0.5% NP‐40) supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem). The lysates were then resolved by SDS–PAGE and immunoblotted with the indicated antibodies. For immunoprecipitation, 800 μg of lysates was incubated with the appropriate antibody (1–2 μg) overnight followed by a 4‐h incubation with Protein G Sepharose beads (GE Healthcare). Immunocomplexes were washed five times with EBC buffer before being resolved by SDS–PAGE and immunoblotted with the indicated antibodies. Information regarding the antibodies used in the study is presented in Appendix Table S1.
Phospho‐FoxM1 (Ser474) rabbit polyclonal antibody
To prepare phospho‐FoxM1 (Ser474) rabbit polyclonal antibody (pAb), we designed and synthesized two peptides that contain phosphorylated or nonphosphorylated Ser474 and the flanking 4–6 amino acid residues (EEWPSPAPSFK or EEWPS(PO4)PAPSFK) plus a Cys (C) at the amino‐terminal. After conjugating to KLH by the amino‐terminal C, the two KLH‐peptide conjugates were respectively injected into 11‐ or 12‐week‐old female New Zealand white rabbits at multiple intradermal sites. After the conjugates were injected 3 or 4 times within a 2‐week interval, we collected rabbit blood to separate serum. The phospho‐FoxM1 (Ser474) rabbit pAb was prepared from the phosphorylated conjugate immunized serum by a 3‐step purification method: (i) The serum was purified by a protein A affinity column and (ii) passed through SulfoLink beads (Pierce, Rockford, IL) immobilized with the nonphosphorylated peptide. (iii) The flow‐through was collected and finally purified by the phosphorylated peptide‐coupled SulfoLink beads. The nonphospho‐FoxM1 rabbit pAb was purified from the nonphosphorylated conjugate immunized serum by using the first two purification steps above. The specificity of the phospho‐FoxM1 (Ser474) rabbit pAb was identified by Peptide‐ELISA and Western blotting with prepared nonphospho‐FoxM1 rabbit pAb as a control.
Immunofluorescence cell staining
Cells were grown on chamber slides pre‐coated with poly‐L‐ornithine and fibronectin. Cells were fixed with 4% paraformaldehyde, permeabilized for 5 min with PBS containing 0.1% Triton X‐100 (PBS‐T), quenched with 50 mM NH4Cl in PBS‐T, and blocked with 1% BSA in PBS‐T. Immunostaining was performed by using the appropriate primary and secondary antibodies, and images were acquired with use of an Olympus FluoView FV1000 confocal microscope.
Promoter reporters and dual‐luciferase assays
For the luciferase reporter assay, cells were transfected with TOP‐Flash or Axin2 promoter reporter (Addgene). Transfection efficiency was normalized by co‐transfection with a TK‐Renilla reporter. The activities of firefly luciferase and Renilla luciferase were quantified by using the dual‐luciferase reporter assay system (Promega).
Chromatin immunoprecipitation assays
For the ChIP assay, 2 × 106 cells were prepared with the ChIP assay kit (Cell Signaling Technology) according to the manufacturer's instructions. The resulting precipitated DNA samples were analyzed by PCR to amplify the putative TCF4 binding region 1 (−200 to −53 bp) of Axin2 promoter using the primers 5′‐TCATCTGAACCTCCTCTC‐3′ and 5′‐GTTGCTTGATTTGAATTTGAG‐3′.
Site‐specific mutagenesis
Site‐specific mutagenesis of the FoxM1 was performed with the QuikChange site‐directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Mutations were confirmed by DNA sequencing.
Chase and pulse assay
LN229 cells treated with or without Wnt‐3a (50 ng/ml) were incubated in methionine and cysteine‐free MEM for 15 min. Cells were pulsed with 0.5 mCi [35S] methionine in methionine and cysteine‐free MEM for 30 min. Cells were washed three times in PBS and incubated in chase medium (MEM supplemented with 10% FCS and 10‐fold molar excess of Cys and Met) for the times indicated (0, 2, 4, 6 h). Whole‐cell protein extracts were used for IP with anti‐FoxM1 antibodies and subjected to SDS–PAGE and auto‐radiography.
In situ hybridization analysis of Axin2 mRNA
For detection of Axin2 mRNA in human GBM specimens, RNAscope Intro Pack 2.0 HD Reagent Kit BROWN‐Hs and RNAscope Probe‐Hs‐AXIN2 (Advanced Cell Diagnostics, #300055 and #400241) were used according to the manufacturer's instructions. Briefly, tissue slides were pre‐treated with heat and protease. Then, the slides were incubated with the Axin2 probe for 2 h at 40°C. Diaminobenzidine was used as the chromogen. The slides were counterstained with hematoxylin. Two investigators blinded to the clinical data scored Axin2 mRNA staining by incorporating the intensity of staining and the percentage of cells that were positive. The intensity of staining was scored as follows: no staining (score 0), 1–3 dots/cell (score 1), 4–10 dots/cell (score 2), and > 10 dots/cell (score 3). The percentage of positive cells was divided into five percentage scores: < 10% (score 0), 10–25% (score 1), 26–50% (score 2), 51–75% (score 3), and > 75% (score 4). The samples for IHC studies were randomized to be examined. The use of human GBM specimens was approved by the institutional review board at The University of Texas MD Anderson Cancer Center.
Human tissue samples and IHC analysis
The tissue slides from paraffin‐embedded human GBM specimens were stained with antibodies against phosphor USP5, FoxM1, ICAT, and cyclin D1. Immunohistochemical analyses were performed on the tissue slides using a standard immunostaining protocol. Briefly, deparaffinized slides were treated with 0.3% hydrogen peroxide and then placed into a citrate buffer antigen‐retrieval solution. Slides were then blocked with 10% normal horse serum with 0.02% Tween‐20. Slide was incubated for overnight at 4°C with the indicated antibody. Slides were next incubated with a secondary antibody. Slides were then counterstained with hematoxylin. The intensity of immunostaining was scored as previously described (Xue et al, 2015).
Statistical analysis
The sample sizes were justified by statistical considerations and statistical power analyses. The significance of the data from patient specimens was determined by the Pearson correlation coefficient test. The significance of the in vitro data and in vivo data between experimental groups was determined by 2‐tailed Student's t‐test. P < 0.05 was considered to be significant.
Author contributions
YC, YL, and SH designed the experiments. YC and SH analyzed data and wrote the manuscript. YC, YL, JX, AG, GY, AZ, KL, SZ, and NZ performed the experiments. CJG provided reagents and contributed to the discussion of the results.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank Tamara Locke in MD Anderson's Department of Scientific Publications for editing the manuscript. We thank Dr. James R. Woodgett (Mount Sinai Hospital, Toronto) for providing the wild‐type GSK3 and GSK3α−/−β−/− mouse stem cells. We thank Dr. Xi He (Children's Hospital Boston, Harvard Medical School, Boston) for critically discussing the data in the manuscript. This work was supported in part by U.S. National Cancer Institute grants R01CA157933, R01CA182684, R01CA152309, P50CA127001, and P30CA016672 (Cancer Center Support Grant).
The EMBO Journal (2016) 35: 668–684
References
- Acebron SP, Karaulanov E, Berger BS, Huang YL, Niehrs C (2014) Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell 54: 663–674 [DOI] [PubMed] [Google Scholar]
- Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13: 11–26 [DOI] [PubMed] [Google Scholar]
- Bafico A, Liu G, Goldin L, Harris V, Aaronson SA (2004) An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell 6: 497–506 [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W (1996) Functional interaction of beta‐catenin with the transcription factor LEF‐1. Nature 382: 638–642 [DOI] [PubMed] [Google Scholar]
- Besche HC, Haas W, Gygi SP, Goldberg AL (2009) Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin‐like domain from HHR23B reveals novel proteasome‐associated proteins. Biochemistry 48: 2538–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R (1996) A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 382: 225–230 [DOI] [PubMed] [Google Scholar]
- Clevers H (2006) Wnt/beta‐catenin signaling in development and disease. Cell 127: 469–480 [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R (2012) Wnt/beta‐catenin signaling and disease. Cell 149: 1192–1205 [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S (2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776 [DOI] [PubMed] [Google Scholar]
- Daniels DL, Weis WI (2002) ICAT inhibits beta‐catenin binding to Tcf/Lef‐family transcription factors and the general coactivator p300 using independent structural modules. Mol Cell 10: 573–584 [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR (2003) GSK‐3: tricks of the trade for a multi‐tasking kinase. J Cell Sci 116: 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR (2007) Functional redundancy of GSK‐3alpha and GSK‐3beta in Wnt/beta‐catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 12: 957–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, Francois A, Bourinet E, Zamponi GW (2014) The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83: 1144–1158 [DOI] [PubMed] [Google Scholar]
- Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C (1998) Dickkopf‐1 is a member of a new family of secreted proteins and functions in head induction. Nature 391: 357–362 [DOI] [PubMed] [Google Scholar]
- Gong A, Huang S (2012) FoxM1 and Wnt/beta‐catenin signaling in glioma stem cells. Cancer Res 72: 5658–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottardi CJ, Gumbiner BM (2004) Role for ICAT in beta‐catenin‐dependent nuclear signaling and cadherin functions. Am J Physiol Cell Physiol 286: C747–C756 [DOI] [PubMed] [Google Scholar]
- Graham TA, Clements WK, Kimelman D, Xu W (2002) The crystal structure of the beta‐catenin/ICAT complex reveals the inhibitory mechanism of ICAT. Mol Cell 10: 563–571 [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c‐MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S (2008) The GSK3 hypothesis of Alzheimer's disease. J Neurochem 104: 1433–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AH, Nelson WJ, Weis WI (1997) Three‐dimensional structure of the armadillo repeat region of beta‐catenin. Cell 90: 871–882 [DOI] [PubMed] [Google Scholar]
- Imai M, Nakamura T, Akiyama T, Horii A (2004) Infrequent somatic mutations of the ICAT gene in various human cancers with frequent 1p‐LOH and/or abnormal nuclear accumulation of beta‐catenin. Oncol Rep 12: 1099–1103 [PubMed] [Google Scholar]
- Kalin TV, Ustiyan V, Kalinichenko VV (2011) Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle 10: 396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lee YI, Seo M, Kim SY, Lee JE, Youn HD, Kim YS, Juhnn YS (2009) Calcineurin dephosphorylates glycogen synthase kinase‐3 beta at serine‐9 in neuroblast‐derived cells. J Neurochem 111: 344–354 [DOI] [PubMed] [Google Scholar]
- Kim SE, Huang H, Zhao M, Zhang X, Zhang A, Semonov MV, MacDonald BT, Zhang X, Garcia Abreu J, Peng L, He X (2013) Wnt stabilization of beta‐catenin reveals principles for morphogen receptor‐scaffold assemblies. Science 340: 867–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Acebron SP, Herbst J, Hatiboglu G, Niehrs C (2015) Post‐transcriptional Wnt signaling governs epididymal sperm maturation. Cell 163: 1225–1236 [DOI] [PubMed] [Google Scholar]
- Korver W, Schilham MW, Moerer P, van den Hoff MJ, Dam K, Lamers WH, Medema RH, Clevers H (1998) Uncoupling of S phase and mitosis in cardiomyocytes and hepatocytes lacking the winged‐helix transcription factor Trident. Curr Biol 8: 1327–1330 [DOI] [PubMed] [Google Scholar]
- Koyama T, Tago K, Nakamura T, Ohwada S, Morishita Y, Yokota J, Akiyama T (2002) Mutation and expression of the beta‐catenin‐interacting protein ICAT in human colorectal tumors. Jpn J Clin Oncol 32: 358–362 [DOI] [PubMed] [Google Scholar]
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Zullig S, Basler K (2002) Wnt/wingless signaling requires BCL9/legless‐mediated recruitment of pygopus to the nuclear beta‐catenin‐TCF complex. Cell 109: 47–60 [DOI] [PubMed] [Google Scholar]
- Laoukili J, Alvarez‐Fernandez M, Stahl M, Medema RH (2008) FoxM1 is degraded at mitotic exit in a Cdh1‐dependent manner. Cell Cycle 7: 2720–2726 [DOI] [PubMed] [Google Scholar]
- Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER (2002) Activation of AXIN2 expression by beta‐catenin‐T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem 277: 21657–21665 [DOI] [PubMed] [Google Scholar]
- Li FQ, Mofunanya A, Harris K, Takemaru K (2008) Chibby cooperates with 14‐3‐3 to regulate beta‐catenin subcellular distribution and signaling activity. J Cell Biol 181: 1141–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Wei P, Peng Z, Huang C, Tang H, Jia Z, Cui J, Le X, Huang S, Xie K (2013) The critical role of dysregulated FOXM1‐PLAUR signaling in human colon cancer progression and metastasis. Clin Cancer Res 19: 62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X (1999) beta‐Trcp couples beta‐catenin phosphorylation‐degradation and regulates Xenopus axis formation. Proc Natl Acad Sci USA 96: 6273–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X (2002) Control of beta‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 108: 837–847 [DOI] [PubMed] [Google Scholar]
- Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, Aldape KD, Xie TX, Pelloski CE, Xie K, Sawaya R, Huang S (2006) FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res 66: 3593–3602 [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson‐Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H (1996) XTcf‐3 transcription factor mediates beta‐catenin‐induced axis formation in Xenopus embryos. Cell 86: 391–399 [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of beta‐catenin‐Tcf signaling in colon cancer by mutations in beta‐catenin or APC. Science 275: 1787–1790 [DOI] [PubMed] [Google Scholar]
- Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL (2013) Why do cellular proteins linked to K63‐polyubiquitin chains not associate with proteasomes? EMBO J 32: 552–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Wang A, Liang J, Xie Y, Liu J, Feng L, Yan Q, Wang Z (2014) USP22 promotes the G1/S phase transition by upregulating FoxM1 expression via beta‐catenin nuclear localization and is associated with poor prognosis in stage II pancreatic ductal adenocarcinoma. Int J Oncol 45: 1594–1608 [DOI] [PubMed] [Google Scholar]
- Paraf F, Jothy S, Van Meir EG (1997) Brain tumor‐polyposis syndrome: two genetic diseases? J Clin Oncol 15: 2744–2758 [DOI] [PubMed] [Google Scholar]
- Park HJ, Costa RH, Lau LF, Tyner AL, Raychaudhuri P (2008) Anaphase‐promoting complex/cyclosome‐CDH1‐mediated proteolysis of the forkhead box M1 transcription factor is critical for regulated entry into S phase. Mol Cell Biol 28: 5162–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri P, Park HJ (2011) FoxM1: a master regulator of tumor metastasis. Cancer Res 71: 4329–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D, Reed SI (1995) Different roles for cyclins D1 and E in regulation of the G1‐to‐S transition. Mol Cell Biol 15: 3463–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes‐Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng X, Wilkinson KD (2006) The ubiquitin binding domain ZnF UBP recognizes the C‐terminal diglycine motif of unanchored ubiquitin. Cell 124: 1197–1208 [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben‐Ze'ev A (1999) The cyclin D1 gene is a target of the beta‐catenin/LEF‐1 pathway. Proc Natl Acad Sci USA 96: 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Neufeld K, Ertych N, Bastians H (2015) Wnt‐mediated protein stabilization ensures proper mitotic microtubule assembly and chromosome segregation. EMBO Rep 16: 490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom A, Bonal C, Ashery‐Padan R, Hashimoto N, Campos ML, Trumpp A, Noda T, Kido Y, Real FX, Thorel F, Herrera PL (2007) Unique mechanisms of growth regulation and tumor suppression upon Apc inactivation in the pancreas. Development 134: 2719–2725 [DOI] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, De Robertis EM (2010) Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143: 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tago K, Nakamura T, Nishita M, Hyodo J, Nagai S, Murata Y, Adachi S, Ohwada S, Morishita Y, Shibuya H, Akiyama T (2000) Inhibition of Wnt signaling by ICAT, a novel beta‐catenin‐interacting protein. Genes Dev 14: 1741–1749 [PMC free article] [PubMed] [Google Scholar]
- Takemaru KI, Moon RT (2000) The transcriptional coactivator CBP interacts with beta‐catenin to activate gene expression. J Cell Biol 149: 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemaru K, Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT (2003) Chibby, a nuclear beta‐catenin‐associated antagonist of the Wnt/Wingless pathway. Nature 422: 905–909 [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F (1999) Beta‐catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426 [DOI] [PubMed] [Google Scholar]
- Townsley FM, Cliffe A, Bienz M (2004) Pygopus and Legless target Armadillo/beta‐catenin to the nucleus to enable its transcriptional co‐activator function. Nat Cell Biol 6: 626–633 [DOI] [PubMed] [Google Scholar]
- Tutter AV, Fryer CJ, Jones KA (2001) Chromatin‐specific regulation of LEF‐1‐beta‐catenin transcription activation and inhibition in vitro . Genes Dev 15: 3342–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM (2002) Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1: 279–288 [DOI] [PubMed] [Google Scholar]
- Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8: 83–93 [DOI] [PubMed] [Google Scholar]
- Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M, Feng XH, Sawaya R, Medema RH, Hung MC, Huang S (2014) Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF‐beta‐dependent cancer metastasis. J Clin Invest 124: 564–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Chen Y, Wu Y, Wang Z, Zhou A, Zhang S, Lin K, Aldape K, Majumder S, Lu Z, Huang S (2015) Tumour suppressor TRIM33 targets nuclear beta‐catenin degradation. Nat Commun 6: 6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, Roebuck KA, Costa RH (1997) Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol 17: 1626–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH (2007) The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology 132: 1420–1431 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang N, Dai B, Liu M, Sawaya R, Xie K, Huang S (2008) FoxM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res 68: 8733–8742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, Sawaya R, Xie K, Yung WK, Medema RH, He X, Huang S (2011) FoxM1 promotes beta‐catenin nuclear localization and controls Wnt target‐gene expression and glioma tumorigenesis. Cancer Cell 20: 427–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File