

Abstract
More than 46 million people worldwide suffer from Alzheimer’s disease. A large number of potential treatments have been proposed; among these, the inhibition of the aggregation of amyloid β-peptide (Aβ), considered one of the main culprits in Alzheimer’s disease. Limitations in monitoring the aggregation of Aβ in cells and tissues restrict the screening of anti-amyloid drugs to in vitro studies in most cases. We have developed a simple but powerful method to track Aβ aggregation in vivo in real-time, using bacteria as in vivo amyloid reservoir. We use the specific amyloid dye Thioflavin-S (Th-S) to stain bacterial inclusion bodies (IBs), in this case mainly formed of Aβ in amyloid conformation. Th-S binding to amyloids leads to an increment of fluorescence that can be monitored. The quantification of the Th-S fluorescence along the time allows tracking Aβ aggregation and the effect of potential anti-aggregating agents.
Amyloid aggregation is linked to an increasing number of human diseases, including both non-neurologic and neurodegenerative disorders1. These human disorders, grouped under the term “conformational diseases”, include Alzheimer’s (AD), Parkinson’s (PD) and Huntington’s (HD) diseases, frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS) or type II diabetes, among others1. Nowadays, more than 46 million people worldwide suffer from AD and the number is predicted to exceed 130 million by 20502,3. AD is a multifactorial and highly complex process, whose pathogenesis involves multiple mechanisms4. However, the appearance of both, amyloid plaques –consequence of the accumulation of amyloid β-peptide (Aβ)– and neurofibrillary tangles –mainly formed of hyperphosphorylated forms of tau protein from neuronal microtubules– are the most prominent pathological hallmarks in the brain of AD patients, leading to neuronal cell death and tissue loss throughout the brain5. For years it has been discussed which is the main cause of Alzheimer’s disease. Currently Aβ aggregation is widely accepted to be one of the main culprits of the illness6,7. In this light, in the last few years the search for potential inhibitors of amyloid aggregation has become one of the most pursued therapeutic strategies in the fight against AD8,9,10,11.
A large number of methods to track the amyloid aggregation have been recently proposed12,13,14,15,16. The evaluation of potential anti-amyloid drugs is usually hampered by the lack of physiologically relevant methods that can be easily implemented in high-throughput screening. Monitoring of amyloid aggregation in cells and tissues suffers from important drawbacks arising from low protein concentration, slow aggregation process and low reproducibility17. These limitations have restricted the screening of anti-amyloid compounds to in vitro studies using expensive synthetic peptides17. Currently, only a handful of methods, which usually track the amyloid associated toxicity in cell lines or the amyloid aggregation in vitro, are used to screen anti-Alzheimer anti-amyloid drugs18,19,20,21. However, the in vitro Aβ aggregation is far from in vivo conditions. Moreover, the cellular toxicity is not directly related to the final amyloid amount, but to the type of amyloid-like aggregates formed during the aggregation process. In fact, soluble Aβ oligomers, generated at the early stages of the fibrillation process, are considered the primary cytotoxic species1,22,23,24. Because in vivo amyloid kinetics may provide key information about both the amyloid aggregation process, still essentially uncharacterized, and inhibition mechanisms, the development of fast, simple, reproducible in vivo methods could represent a breakthrough in the understanding of the amyloid aggregation process and eventually in the search for potential anti-Alzheimer anti-amyloid drugs. Bacteria represent a simple but quantitative method, which will always be closer to the in vivo conditions in mammals than in vitro and toxicity assays.
The proposed method uses bacteria as an in vivo reservoir to track in real-time amyloid aggregation kinetics. The use of prokaryotic systems as microbial cell factories in the production of recombinant proteins has become an essential tool for the biotechnological industry and biomedical research25. Over-expression of amyloid-prone proteins in bacteria entails the formation of insoluble protein aggregates called inclusion bodies (IBs)26,27. Compelling evidence has unequivocally demonstrated that recombinant amyloid-prone proteins are folded in amyloid-like conformations into IBs26,27. Indeed, IBs formed after the over-expression in bacteria of the major Aβ peptides, namely Aβ40 and Aβ42, have been shown to display amyloid-like structures28. In this context, we have recently shown the usefulness of the amyloid specific dye Thioflavin-S (Th-S) to track the amyloid deposits of different amyloid-prone proteins in bacteria29,30. The facts that (1) Th-S staining of amyloid-like structures leads to an increase in it specific fluorescence –when excited under blue light– which can be easily monitored without interfering the bacteria growth and (2) Th-S crosses membranes and penetrates into the cell without affecting amyloid aggregation, make Th-S the dye of choice for tracking amyloid aggregation in vivo29,30. Despite the noticeable applicability of our previously reported methods in anti-amyloid drug discovery, they do not allow the quantification of the amyloid amount in cell or the amazing possibility to track in-real time the in vivo aggregation of amyloid-prone proteins. Herein, we describe an alternative method that compiles the capacity of bacteria as physiologically relevant models for the in vivo study of amyloid aggregation and the ability of Th-S to stain amyloid structures, thereby enabling the quantitative in vivo screening of anti-amyloid drugs, both in kinetic and thermodynamic terms. Importantly, the proposed method allows to track simply but effectively the inhibitory capacity of potential anti-amyloid drugs at each stage of the aggregation process, providing direct information of the behavior of each inhibitor along the aggregation time.
Results and Discussion
Aβ40 and Aβ42 are the main components of the senile plaques. However, the physicochemical properties of Aβ40 e.g. higher solubility and lower aggregation propensity relative to Aβ42 – have made it the peptide of choice in kinetic in vitro assays. Because the over-expression of Aβ42 in bacteria results in the formation of a large amount of oligomeric species28, we propose here to use the Aβ40 variant, which does not form oligomers in bacteria28, to screen the anti-aggregation effect of two potential inhibitors of the amyloid fibril polymerization. However, Aβ42 aggregation kinetics have been additionally analyzed to assess the effect of the inhibitors in both Aβ variants. We show herein the high efficiency and performance of our ultra rapid in vivo screening method for anti-Alzheimer anti-amyloid drugs in bacteria over-expressing Aβ40 and Aβ42 in the presence or absence of two potential inhibitors of Aβ aggregation; DP-128, a chlorotacrine-based inhibitor of Aβ42 and tau aggregation, and apigenin, a natural flavone with demonstrated activity against insulin amyloids (structures in Supplementary Fig. 1)31,32. In order to show the differences in the Th-S spectra of bacteria over-expressing or not Aβ (viz. with or without IBs containing Aβ in amyloid conformation, respectively) as well as the potential tracking of amyloid formation in vivo, we measured the kinetics of induced (over-expressing Aβ40) and non-induced (non-overexpressing Aβ40) bacterial cultures in the presence and absence of inhibitors, by tracking the Th-S fluorescence spectra along the time in a range from 460 to 600 nm, exciting at 440 nm. Under these conditions, the formation of Aβ40 and Aβ42 IBs with time leads to unequivocal changes between non-induced and induced cultures as a consequence of the Th-S staining of IBs (Supplementary Fig. 2). In the non-induced samples the maximal fluorescence of Th-S as a consequence of the interaction with bacteria is at ~523 nm (Supplementary Fig. 2a,c,e). However, in the induced samples without inhibitor we observe the progressive appearance of a new peak (non-observable in the non-induced samples) at ~495 nm, arising from the Th-S binding to gradually formed Aβ IBs (Supplementary Fig. 2b); Interestingly, this peak is reduced in the induced samples in the presence of inhibitors as a result of the reduction of Aβ aggregation (Supplementary Fig. 2d,f). When the spectra of non-induced cultures were subtracted from those of Aβ over-expressing cultures along the time-course, we obtained the typical amyloid band displaying a maximal peak at ~485 nm (Fig. 1 and Supplementary Fig. 3). Importantly, we observed a diminution of the amyloid band in the samples with inhibitor (Fig. 1b,c) in comparison with the samples without inhibitor (Fig. 1a); with this fact being indicative of a reduction of the amyloid structures in the bacterial cells that grow in the presence of inhibitors (Supplementary Fig. 3a). The relative fluorescence data at 485 nm can be transformed into amyloid concentration. To quantify the obtained peaks we built a Th-S–Aβ40 standard curve using in vitro preformed Aβ40 amyloid fibrils (Supplementary Fig. 4b,c)33. Then, the Aβ40 amount in amyloid-like conformation that is present in IBs can be easily and precisely quantified by simple interpolation of the Th-S relative fluorescence data from the time-course reaction at 485 nm in the standard curve (Fig. 2). In order to discard differences in bacterial growth and protein expression level as a consequence of the presence of the inhibitors, we evaluated Aβ40 concentration along the time (by tricine-SDS-PAGE) (Fig. 3a and Supplementary Fig. 5) and bacterial growth (by OD at 600 nm) in the presence and absence of each inhibitor (Fig. 3b). The SDS-PAGE band corresponding to Aβ peptide has been previously identified by western blotting using 6E10 Aβ antibody28. As shown in Fig. 3a,b, no significant differences in expression and growth patterns were observed, hence confirming that these inhibitors do not interfere with these parameters. In the case that the potential drug interfered with these parameters the differences would have to be taken into account to normalize further amyloid aggregation kinetics. Complementarily, we checked the Aβ40 distribution between soluble and insoluble fractions (i.e. between soluble and aggregated Aβ40) at the end-point of kinetics. As shown in Fig. 3c, Aβ40 is present almost exclusively in the insoluble fraction. Interestingly, no significant differences in the Aβ40 distribution between soluble and insoluble fractions were observed in the absence and presence of inhibitors (Fig. 3c).
Figure 1. Th-S relative fluorescence of the amyloid band along the time-course.

(a) In the absence of inhibitor. (b) In the presence of 10 μM DP-128. (c) In the presence of 10 μM apigenin. The dotted arrows show the maximal amyloid peak ~485 nm. In green and red, the initial (0 min) and final (480 min) time-course. Th-S relative fluorescence measurements were performed in triplicate and the standard errors were less than 5%.
Figure 2. Aβ40 amyloid concentration along the time-course kinetics and amyloid concentration at end-point of the time-course.

In black, red and green, in the absence (control) and presence of 10 μM DP-128 and apigenin, respectively. Aβ40 concentrations were measured in triplicate and the standard errors were less than 5%.
Figure 3. Aβ40 expression and bacterial growth in the absence and presence of inhibitors.

(a) Protein expression in bacterial cultures along the time tracked by tricine-SDS-PAGE. (b) Bacterial growth monitored by optical density at 600 nm (OD600); the ODs were measured in triplicate and the standard errors were less than 5%. In black, red and green, in the absence (control) and presence of 10 μM DP-128 and apigenin, respectively. Solid and dashed lines show induced and non-induced cultures, respectively. (c) Aβ40 distribution in bacteria: Total (T), soluble (S) and insoluble (I) fraction of Aβ40 at end-point of kinetics.
Amyloid kinetics can be mechanistically described as a nucleation dependent polymerization process34, wherein initially soluble species are associated forming nuclei in a thermodynamically disfavored stage, with these preformed nuclei acting as templates of soluble species that are favorably added to form first amyloid fibrils and eventually mature fibers34,35. In vitro and in vivo systems are being extensively used for exploring and modeling the kinetics of Aβ aggregation, which is one of the most important target for drug discovery and development13,15,35,36,37,38,39,40,41,42,43,44,45. Within a wide range of models, this kind of processes can be mathematically addressed as an autocatalytic reaction, considering the nucleation (kn) and elongation (ke) constants of the process, and associated kinetic times: lag time of the thermodynamically disfavored phase (t0), maximal growth (t1/2) and final time of the amyloid aggregation (t1)35. When the obtained data from the aggregation kinetics are analyzed using the autocatalytic model, the effect of each inhibitor can be easily quantified in kinetic and thermodynamic terms. Thus, kn displays reductions of ~1.7 and ~1.5-fold in the presence of DP-128 and apigenin, respectively, leading to elongations of ∼25% in t0 for both inhibitors. Reductions in kn rate denote an inhibitory action in the nucleation phase, i.e. the formation of amyloid nuclei is partially hindered as a consequence of the presence of the inhibitor delaying the appearance of the first amyloid-like species. In contrast, keapp is increased ∼1.6 and ∼2-fold in the presence of DP-128 and apigenin, respectively, but without significant changes in t1/2 and t1. Increments in the keapp should be interpreted as an increment in the elongation rate, i.e. as an acceleration of the elongation of the fibrils. However, this is only due to the fact that keapp has been calculated using the final Aβ40 amyloid amount of each aggregation kinetic, it being 6.0, 4.0, and 3.2 μM for the control without inhibitor, DP-128, and aginenin, respectively (Table 1). In brief, at a same elongation rate, the less monomers are available to form fibrils, the sooner the reaction ends. However, as shown in Table 1, when [Aβ40fibrils]·ke is calculated similar ke are obtained (~0.017 min−1). In consequence, t1/2 and t1 remain essentially unaltered in the presence of inhibitors. In kinetic terms, the obtained data suggest that both inhibitors act by delaying the amyloid aggregation and hindering the nuclei formation. In addition, the final amounts of amyloid-like Aβ40 were reduced in 33 and 46% for DP-128 and apigenin, respectively (Fig. 2 and Table 1). The obtained data show that DP-128 and apigenin act essentially in the nucleation whereas elongation is almost not altered. However, both inhibitors led to a remarkable reduction in the final amount of amyloid aggregates under our experimental conditions. As shown in Supplementary Fig. 6 Th-S staining could be directly assessed by optical fluorescence microscopy. Whereas non-induced cells do not show any Th-S fluorescence (Supplementary Fig. 6a), induced cells in the absence of inhibitor (Supplementary Fig. 6b) display high fluorescence as a consequence of Th-S staining so that the Aβ40 IBs can be easily detected. In the presence of active inhibitors the fluorescence is drastically reduced (Supplementary Fig. 6c,d) denoting a reduction of amyloid amount in IBs. Interestingly, similar inhibitions are obtained when the Aβ42 variant has been tested. Indeed, as shown in Supplementary Fig. 7, DP-128 and apigenin lead to inhibitions of Aβ42 aggregation of 28 and 45%, respectively.
Table 1. Kinetic and thermodynamic parameters of Aβ40 amyloid aggregation.
| Inhibitor | Without | DP-128 a | Apigenin a |
|---|---|---|---|
| kn (106·min−1) | 200.5 | 118.5 | 135.0 |
| keapp (M−1·min−1)b,c | 2723.5 | 4427.8 | 5336.5 |
| [Aβ40fibrils]·ke (min−1) | 0.0164 | 0.0179 | 0.0173 |
| t0 (min) | 112.0 | 140.6 | 144.3 |
| t1/2 (min) | 268.3 | 279.4 | 279.4 |
| t1 (min) | 424.5 | 418.3 | 414.6 |
| [Aβ40fibrils] (μM)d | 6.0 | 4.0 | 3.2 |
aInhibition parameters at 10 μM of inhibitor.
bSince Aβ40 concentration is not constant along the aggregation process, the ke are apparent.
cIn order to calculate the keapp, the final Aβ40 amyloid amount of each aggregation kinetic has been considered.
dAβ40 concentration in amyloid conformation at end-point of the time-course.
In summary, the proposed method, directly tracking the Th-S relative fluorescence in bacterial cells, allows the in vivo monitoring of the activity of potential anti-amyloid drugs in an extremely short time (<24 h), shedding light on the inhibitor behavior and enabling the determination of IC50 values if different inhibitor concentrations are used. Interestingly, the present method allows simultaneous acquisition of the kinetic and thermodynamic parameters of each process, enabling the fast classification of inhibitors by accurately quantifying their inhibitory activity in each stage of the amyloid aggregation process. The reported method could be easily adapted to real-time micro-plate readers, thereby enabling high-throughput screenings with the simultaneous tracking of up to 1536 samples. Importantly, the method can be potentially used for the screening of anti-amyloid drugs against all conformational diseases by simply changing the amyloid-prone protein over-expressed in bacteria.
Methods
Chemicals and bacterial media
DP-128, N-{8-[(6-chloro-1,2,3,4-tetrahydroacridin-9-yl)amino]octyl}-5-(4-chlorophenyl)-1,2,3,4-tetrahydrobenzo[h][1,6]naphthyridine-9-carboxamide, was prepared as previously reported31. The natural flavone apigenin (4′,5,7-trihydroxyflavone) and all other general chemicals were purchased from Sigma-Aldrich. Compounds for bacterial media were purchased from Pronadisa. M9 minimal medium; For 100 mL: 10 mL salts 10 × (0.68 g Na2HPO4, 0.30 g KH2PO4, 0.05 g NaCl, 0.10 g NH4Cl), 0.2 mL 1 M MgSO4, 0.2 mL 50 mM CaCl2, 2.5 mL 20% glucose and 87.1 mL H2O.
Preparation of soluble Aβ40 peptide
Aβ40 (trifluoroacetic acid salt) was obtained from Bachem. A stock solution was prepared by dissolving 1 mg of Aβ40 in 0.5 mL of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), which had been dried at 4 °C over molecular sieves Type 4A and then centrifuged (15,000 g for 15 min) to remove molecular sieve dust. The solution was incubated under stirring at room temperature for 1 h in sealed vials, bath-sonicated for 30 min, and stirred for an additional hour at room temperature. Then, the solution was kept at 4 °C for 30 min to avoid solvent evaporation when aliquoting. Once the sample was aliquoted, HFIP was removed by evaporation under a gentle stream of nitrogen, leaving a slightly yellow film. The samples of soluble Aβ40 peptide were frozen at −80 °C. Frozen aliquots were re-suspended in 50 μL of anhydrous dimethyl sulfoxide (DMSO) and bath-sonicated for 10 min. Sonication was crucial to remove any traces of un-dissolved seeds that may resist solubilization. Aliquots of Aβ were re-suspended in 950 μL of PBS at pH 7.4. The final buffer contained 5% (v/v) DMSO46. The final concentration of Aβ was adjusted by UV absorbance considering a molar absorptivity of 1490 mol−1·dm3·cm−1 at 280 nm using a UV-2401 PC UV-Vis spectrophotometer from Shimadzu.
In vitro Aβ40 aggregation
Aggregation of initially soluble Aβ40 was carried out for 48 h at 37 °C and 1,400 rpm using a Thermomixer from Eppendorf. The amount of insoluble amyloid fibrils at the end-time of the reaction was quantified after centrifugation at 14,000 g for 30 min. As previously observed, the insoluble fraction represented 90% of the initial Aβ40, remaining 10% as soluble33. This correction was taken into account in the determination of the amyloid stock concentration.
The standard curve was built by sequential dilution of the stock solution. The relative fluorescence of the samples was determined using a final concentration of Th-S of 250 μM and 75, 50, 25, 12.5, 6.25, 3.125, 1.56, 0.8, 0.4 and 0 μM of in vitro fibrils of Aβ40 preformed under the same conditions as the kinetics.
Cloning and expression of Aβ peptide
Escherichia coli competent cells BL21 (DE3) were transformed with the pET28a vector from Novagen carrying the DNA sequence of Aβ40 or Aβ42. Because of the addition of the initiation codon ATG in front of gene, the over-expressed peptide contains an additional methionine residue at its N terminus (MDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV for Aβ40 and MDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA for Aβ42). For overnight culture preparation, 10 mL of M9 minimal medium containing 50 μg·mL−1 of kanamycin were inoculated with a single colony of BL21 (DE3) bearing the plasmid to be expressed at 37 °C. For expression of the Aβ peptide, 100 μL of overnight culture (providing a starting OD600 of 0.1–0.2) was used to inoculate 9.9 mL of fresh M9 minimal medium containing kanamycin and Th-S for a final concentration of 50 μg·mL−1 and 250 μM, respectively, and 10 μL of the inhibitors in 10 mM stock solution DMSO. Note that in the samples without inhibitor 10 μL of DMSO had to be added. Then, bacterial cultures were grown at 37 °C and 250 rpm. When OD600 reached ~0.7, 10 μL of isopropyl 1-thio-β-D-galactopyranoside (IPTG) at 1 M were added to induce Aβ over-expression. Then, 200 μL of sample were collected every 30 min during all time-course of the kinetics and fluorescence and absorbance were determined.
As “positive control” we used induced bacterial cells bearing Aβ plasmid (over-expressing Aβ) without inhibitor, displaying the maximal potential aggregation of the Aβ peptide in bacteria. As “negative control” we used non-induced bacterial cells bearing Aβ plasmid without inhibitor, showing the minimal expression of Aβ in the cells. However, to discard potential effects of the inhibitor in the Th-S signal (e.g. when the inhibitor also displays certain fluorescence in Th-S range), a “negative control” of each inhibitor was performed using non-induced bacterial cells bearing Aβ plasmid in the presence of each inhibitor. Importantly, this is highly useful to discard potential effects of the inhibitors in the cell growth (by OD600).
Quantification of Aβ40 in amyloid conformation in bacterial cells
Because non-induced cultures display relative fluorescence in the Th-S emission range (460–600 nm) when exciting at 440 nm (Supplementary Fig. 2a,c,e), these spectra would have to be removed from Th-S fluorescence spectra of induced cultures (Supplementary Fig. 2b,d,f) to quantify the amount of Aβ40 in amyloid-like conformation. Since OD600 are known at each kinetic time, induced cultures (over-expressing Aβ40) could be easily corrected by subtracting the spectrum of non-induced cells with similar OD600, thus obtaining Th-S amyloid band (Fig. 1).
As shown in Supplementary Fig. 4b,c, the standard curve of in vitro Aβ40 fibrils shows a linear relationship between Aβ40 fibril concentration and Th-S relative fluorescence at 485 nm (the maximum of the amyloid band). The interpolation of Th-S fluorescence of in vivo kinetics at 485 nm in the standard curve allows the easy transformation from fluorescence into amyloid concentration.
Aβ40 expression levels in bacterial cells
Aβ40 concentration in cell cultures in the presence and absence of inhibitor was assessed by tricine-SDS-PAGE. 300 μL of induced bacterial cells were collected each hour during all time-course of the kinetics. Then, the samples were concentrated by gentle centrifugation obtaining OD600 of 8. 30 μL of concentrated sample were added to 4× loading buffer (250 mM Tris-HCl pH 6.8, 10% SDS, 0.008% Bromophenol Blue, 40% glycerol and 2.86 M β-mercaptoethanol). Finally, the samples were placed at 95 °C for 10 min. Denatured samples were analyzed using 16% tricine SDS-PAGE (Fig. 3a and Supplementary Fig. 5).
In order to check the Aβ40 aggregation in bacteria, soluble and insoluble protein fractions were required. Then, 1 mL of concentrated sample from the end-point of each kinetic was pelleted by centrifugation at 14,000 rpm (20 min, 4 °C). The pellet containing the cells was frozen at −80 °C for at least 2 h. Then, cell pellets were re-suspended in 1 mL of phosphate buffer saline 1× (PBS) and sonicated using a SONICS Vibra CellTM from Vibra-Cell-Sonics & Materials, Inc. at 30% during 1 min (with intervals of 1 seg sonication/0.5 seg stand-by). Soluble and insoluble fractions were separated by centrifugation at 14,000 rpm (20 min, 4 °C). The insoluble fraction was re-suspended in 1 mL PBS 1 ×. Finally, the samples were analyzed by tricine-SDS-PAGE (Fig. 3c).
Thioflavin-S (Th-S) Steady-State Fluorescence
Relative fluorescence of Th-S binding to Aβ was checked using an Aminco Bowman Series 2 luminescence spectrophotometer from Aminco-Bowman with an excitation wavelength of 440 nm and emission range from 460 to 600 nm. The emission at 485 nm was recorded for further determination of the amyloid-like concentration. Excitation and emission slit widths of 4 nm were used and spectra were acquired at 1 nm intervals and 500 nm·min−1 rates and 875 V47. Importantly, in order to compare the obtained data, the voltage must always be known and not changed.
Aggregation assay analysis
The amyloid aggregation in bacteria may be studied as an autocatalytic reaction using the equation:
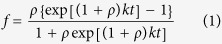 |
under the boundary condition of t = 0 and f = 0, where k = kea (when a is the protein concentration) and ρ represents the dimensionless value to describe the ratio of kn to k. By non-linear regression of f against t, values of ρ and k are obtained, and from them the rate constants, ke (elongation constant) and kn (nucleation constant)35. Note that because the concentration of Aβ is not constant in the bacterial cells during the protein over-expression, we have arbitrarily considered as a the Aβ concentration of the final time-course, obtaining apparent values for ke, which allow quantitative comparison of kinetics. The extrapolation of the linear portion of the sigmoid curve to abscissa (f = 0), and to the highest ordinate value of the fitted plot, afforded two values of time (t0 and t1), which correspond to the lag time and to the end-time reaction. The time at which half of the protein was aggregated (i.e., when f = 0.5) was considered the time of half-aggregation (t1/2)35.
Optical fluorescence microscopy
Bacterial cells overexpressing Aβ40 were incubated 1 h with 125 μM of Th-S. Th-S was removed by centrifugation and the cells were re-suspended in PBS twice and placed on a microscope slide. Th-S fluorescence was detected using a Leitz DMIRB microscope under UV light using a GFP filter (excitation filter BP480/40 and emission filter BP527/30)48.
Additional Information
How to cite this article: Espargaró, A. et al. Ultra rapid in vivo screening for anti-Alzheimer anti-amyloid drugs. Sci. Rep. 6, 23349; doi: 10.1038/srep23349 (2016).
Supplementary Material
Acknowledgments
This work was supported by Ministerio de Economía y Competitividad (MINECO) (SAF2014-57094-R (D.M.-T.), start-up grant of the Ramón y Cajal program RYC-2011-07987 (R.S.)), and Generalitat de Catalunya (GC) (2014SGR52 (D.M.-T. and O.D.P.) and 2014SGR938 (R.S. and A.E.)). A contract from the Juan de la Cierva program of MINECO to A.E. (JCI-2012-12193) is gratefully acknowledged.
Footnotes
Author Contributions A.E., A.M. and O.D.P. performed the research. D.M.-T. drafted the manuscript and R.S. supervised the research, drafted the manuscript and edited the draft. All authors edited the final manuscript.
References
- Chiti F. & Dobson C. M. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75, 333–366 (2006). [DOI] [PubMed] [Google Scholar]
- Reitz C., Brayne C. & Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol 7, 137–152, doi: 10.1038/nrneurol.2011.2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M., Wimo A., Guerchet M., Ali G.-C., Wu Y.-T. & Prina M. World Alzheimer Report 2015. The global impact of dementia. An analysis of prevalence, incidence, cost & trends; Alzheimer’s Disease International: London, http://www.alz.co.uk, (2015) (Accessed:15/02/2016)
- Huang Y. & Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222, doi: 10.1016/j.cell.2012.02.040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C. et al. Alzheimer’s disease. Lancet 377, 1019–1031, doi: 10.1016/S0140-6736(10)61349-9 (2011). [DOI] [PubMed] [Google Scholar]
- Skaper S. D. Alzheimer’s disease and amyloid: culprit or coincidence? Int Rev Neurobiol 102, 277–316, doi: 10.1016/B978-0-12-386986-9.00011-9 (2012). [DOI] [PubMed] [Google Scholar]
- Bucciantini M. et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511, doi: 10.1038/416507a (2002). [DOI] [PubMed] [Google Scholar]
- Godyn J., Jonczyk J., Panek D. & Malawska B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68, 127–138, doi: 10.1016/j.pharep.2015.07.006 (2016). [DOI] [PubMed] [Google Scholar]
- Jia Q., Deng Y. & Qing H. Potential therapeutic strategies for Alzheimer’s disease targeting or beyond β-amyloid: insights from clinical trials. Biomed Res Int 2014, 837157, doi: 10.1155/2014/837157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aisen P. S. The development of anti-amyloid therapy for Alzheimer’s disease : from secretase modulators to polymerisation inhibitors. CNS Drugs 19, 989–996, doi: 19122 (2005). [DOI] [PubMed] [Google Scholar]
- Lannfelt L. et al. Perspectives on future Alzheimer therapies: amyloid-β protofibrils-a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6, 16, doi: 10.1186/alzrt246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M. et al. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724, doi: 10.1038/nature06616 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuraku K., Marquardt M. & Ikezu T. Real-time imaging and quantification of amyloid-β peptide aggregates by novel quantum-dot nanoprobes. PLoS One 4, e8492, doi: 10.1371/journal.pone.0008492 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgold S., Filser S., Dorostkar M. M., Schmidt B. & Herms J. In vivo imaging reveals sigmoidal growth kinetic of β-amyloid plaques. Acta Neuropathol Commun 2, 30, doi: 10.1186/2051-5960-2-30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G. et al. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc Natl Acad Sci USA 111, 9384–9389, doi: 10.1073/pnas.1401564111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah G., Demuro A., Parker I. & Pearson J. E. Analyzing and Modeling the Kinetics of Amyloid Β Pores Associated with Alzheimer’s Disease Pathology. PLoS One 10, e0137357, doi: 10.1371/journal.pone.0137357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar-Pique A., Espargaro A., Ventura S. & Sabate R. In vivo amyloid aggregation kinetics tracked by time-lapse confocal microscopy in real-time. Biotechnol J, doi: 10.1002/biot.201500252 (2015). [DOI] [PubMed] [Google Scholar]
- Xu X. et al. Prevention of β-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem Cell Res 10, 213–227, doi: 10.1016/j.scr.2012.11.005 (2013). [DOI] [PubMed] [Google Scholar]
- Hou X. Q. et al. A novel assay for high-throughput screening of anti-Alzheimer’s disease drugs to determine their efficacy by real-time monitoring of changes in PC12 cell proliferation. Int J Mol Med 33, 543–549, doi: 10.3892/ijmm.2013.1608 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini M. et al. Insight into the kinetic of amyloid β (1–42) peptide self-aggregation: elucidation of inhibitors’ mechanism of action. ChemBioChem 8, 2152–2161, doi: 10.1002/cbic.200700427 (2007). [DOI] [PubMed] [Google Scholar]
- Bartolini M. et al. Kinetic characterization of amyloid-β 1–42 aggregation with a multimethodological approach. Anal Biochem 414, 215–225, doi: 10.1016/j.ab.2011.03.020 (2011). [DOI] [PubMed] [Google Scholar]
- Haass C. & Selkoe D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8, 101–112, doi: nrm2101 (2007). [DOI] [PubMed] [Google Scholar]
- Prangkio P., Yusko E. C., Sept D., Yang J. & Mayer M. Multivariate analyses of amyloid-β oligomer populations indicate a connection between pore formation and cytotoxicity. PLoS One 7, e47261, doi: 10.1371/journal.pone.0047261 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein S. L. et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat Chem 1, 326–331, doi: 10.1038/nchem.247 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura S. & Villaverde A. Protein quality in bacterial inclusion bodies. Trends Biotechnol 24, 179–185, doi: S0167-7799(06)00052-7 (2006). [DOI] [PubMed] [Google Scholar]
- Wang L., Maji S. K., Sawaya M. R., Eisenberg D. & Riek R. Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol 6, e195, doi: 10.1371/journal.pbio.0060195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot N. S., Sabate R. & Ventura S. Amyloids in bacterial inclusion bodies. Trends Biochem Sci 34, 408–416 (2009). [DOI] [PubMed] [Google Scholar]
- Dasari M. et al. Bacterial inclusion bodies of Alzheimer’s disease β-amyloid peptides can be employed to study native-like aggregation intermediate states. ChemBioChem 12, 407–423, doi: 10.1002/cbic.201000602 (2011). [DOI] [PubMed] [Google Scholar]
- Pouplana S. et al. Thioflavin-S staining of bacterial inclusion bodies for the fast, simple, and inexpensive screening of amyloid aggregation inhibitors. Curr Med Chem 21, 1152–1159, doi: CMC-EPUB-56032 (2014). [DOI] [PubMed] [Google Scholar]
- Espargaro A., Sabate R. & Ventura S. Thioflavin-S staining coupled to flow cytometry. A screening tool to detect in vivo protein aggregation. Mol Biosyst 8, 2839–2844, doi: 10.1039/c2mb25214g (2012). [DOI] [PubMed] [Google Scholar]
- Di Pietro O. et al. Tetrahydrobenzo[h][1,6]naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur J Med Chem 84, 107–117, doi: 10.1016/j.ejmech.2014.07.021 (2014). [DOI] [PubMed] [Google Scholar]
- Amini R., Yazdanparast R. & Bahramikia S. Apigenin reduces human insulin fibrillation in vitro and protects SK-N-MC cells against insulin amyloids. Int J Biol Macromol 60, 334–340, doi: 10.1016/j.ijbiomac.2013.06.013 (2013). [DOI] [PubMed] [Google Scholar]
- Sabate R. & Estelrich J. Pinacyanol as effective probe of fibrillar β-amyloid peptide: comparative study with Congo Red. Biopolymers 72, 455–463, doi: 10.1002/bip.10485 (2003). [DOI] [PubMed] [Google Scholar]
- Jarrett J. T. & Lansbury P. T. Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73, 1055–1058 (1993). [DOI] [PubMed] [Google Scholar]
- Sabate R., Gallardo M. & Estelrich J. An autocatalytic reaction as a model for the kinetics of the aggregation of β-amyloid. Biopolymers 71, 190–195, doi: 10.1002/bip.10441 (2003). [DOI] [PubMed] [Google Scholar]
- Cohen S. I., Vendruscolo M., Dobson C. M. & Knowles T. P. Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J Chem Phys 135, 065106, doi: 10.1063/1.3608917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. et al. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys 135, 065105, doi: 10.1063/1.3608916 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A., Chung D. S., Benedek G. B., Kirschner D. A. & Teplow D. B. On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA 93, 1125–1129 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A., Teplow D. B., Kirschner D. A. & Benedek G. B. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc Natl Acad Sci USA 94, 7942–7947 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallitto M. M. & Murphy R. M. A mathematical model of the kinetics of β-amyloid fibril growth from the denatured state. Biophys J 81, 1805–1822, doi: S0006-3495(01)75831-6 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. M. & Pallitto M. M. Probing the kinetics of β-amyloid self-association. J Struct Biol 130, 109–122, doi: 10.1006/jsbi.2000.4253 (2000). [DOI] [PubMed] [Google Scholar]
- Murphy R. M. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim Biophys Acta 1768, 1923–1934, doi: S0005-2736(06)00491-3 (2007). [DOI] [PubMed] [Google Scholar]
- Knowles T. P. et al. An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537, doi: 10.1126/science.1178250 (2009). [DOI] [PubMed] [Google Scholar]
- Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol 309, 256–274 (1999). [DOI] [PubMed] [Google Scholar]
- Cohen S. I., Vendruscolo M., Dobson C. M. & Knowles T. P. Nucleated polymerization with secondary pathways. III. Equilibrium behavior and oligomer populations. J Chem Phys 135, 065107, doi: 10.1063/1.3608918 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabate R. & Estelrich J. Evidence of the existence of micelles in the fibrillogenesis of β-amyloid peptide. J Phys Chem B 109, 11027–11032 (2005). [DOI] [PubMed] [Google Scholar]
- Sola I. et al. Novel Levetiracetam Derivatives That Are Effective against the Alzheimer-like Phenotype in Mice: Synthesis, in Vitro, ex Vivo, and in Vivo Efficacy Studies. J Med Chem 58, 6018–6032, doi: 10.1021/acs.jmedchem.5b00624 (2015). [DOI] [PubMed] [Google Scholar]
- Perez-Areales F. J. et al. Shogaol-huprine hybrids: dual antioxidant and anticholinesterase agents with β-amyloid and tau anti-aggregating properties. Bioorg Med Chem 22, 5298–5307, doi: 10.1016/j.bmc.2014.07.053 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.