

Abstract
BACKGROUND & AIMS
Augmenter of liver regeneration (ALR, encoded by GFER) is a widely distributed pleiotropic protein originally identified as a hepatic growth factor. However, little is known about its roles in hepatic physiology and pathology. We created mice with liver-specific deletion of ALR to study its function.
METHODS
We developed mice with liver-specific deletion of ALR (ALR-L-KO) using the albumin-Cre/LoxP system. Liver tissues were collected from ALR-L-KO mice and ALRfloxed/floxed mice (controls) and analyzed by histology, reverse-transcription PCR, immunohistochemistry, electron microscopy, and techniques to measure fibrosis and lipids. Liver tissues from patients with and without advanced liver disease were determined by immunoblot analysis.
RESULTS
Two weeks after birth, livers of ALR-L-KO mice contained low levels of ALR and ATP; they had reduced mitochondrial respiratory function and increased oxidative stress, compared with livers from control mice, and had excessive steatosis, and hepatocyte apoptosis. Levels of carbamyl-palmitoyl transferase 1a and ATP synthase subunit ATP5G1 were reduced in livers of ALR-L-KO mice, indicating defects in mitochondrial fatty acid transport and ATP synthesis. Electron microscopy showed mitochondrial swelling with abnormalities in shapes and numbers of cristae. From weeks 2–4 after birth, levels of steatosis and apoptosis decreased in ALR-L-KO mice, whereas numbers of ALR-expressing cells increased, along with ATP levels. However, at weeks 4–8 after birth, livers became inflamed, with hepatocellular necrosis, ductular proliferation, and fibrosis; hepatocellular carcinoma developed by 1 year after birth in nearly 60% of the mice. Hepatic levels of ALR were also low in ob/ob mice and alcohol-fed mice with liver steatosis, compared with controls. Levels of ALR were lower in liver tissues from patients with advanced alcoholic liver disease and nonalcoholic steatohepatitis than in control liver tissues.
CONCLUSIONS
We developed mice with liver-specific deletion of ALR, and showed that it is required for mitochondrial function and lipid homeostasis in the liver. ALR-L-KO mice provide a useful model for investigating the pathogenesis of steatohepatitis and its complications.
Keywords: Augmenter of liver regeneration, mouse model, NASH, ALD
INTRODUCTION
Augmenter of liver regeneration (ALR) (encoded by Gfer, Growth Factor ERV1 homolog of Saccharomyces cerevisiae) protein was originally identified and purified from weanling and regenerating rat livers, and its gene cloned (1–4). Expression of ALR in unmodified adult rat liver (5) suggested that it might also be functionally significant in this setting. ALR is known to function as a sulfhydryl oxidase (6), cytochrome c reductase (7,8), and inducer of cytosolic protein Fe/S maturation (9) and has additional effects such as suppression of hepatic NK cell cytotoxicity (10) and Kupffer cell activation (11). Inhibition of ALR synthesis in cultured hepatocytes leads to mitochondrial dysfunction/damage and cell death (12).
In order to dissect the roles that ALR plays in normal hepatocyte physiology, we generated liver-specific conditional ALR knockout (ALR-L-KO) mice. The data indicate that ALR is critical for mitochondrial function, lipid homeostasis and cell survival, and abnormality in ALR gene function may be an important determinant in the development of steatohepatitis and its complications.
MATERIALS AND METHODS
Animal protocols were approved by Institutional Animal Care and Use Committees according to NIH guidelines.
Generation of ALR-L-KO mouse
Details are described in Supplemental Material and Supplemental Figure 1. Briefly, ALR-targeting vector with LoxP sites placed between exons 1 and 2 and after exon 3 of the Gfer gene was constructed, and standard molecular techniques were employed to generate ALRfloxed/floxed mouse. To generate liver-specific ALR-knockout mouse (ALR-L-KO), hemizygous Alb-Cre transgenic mice were first crossed with ALRfloxed/floxed mice. F1 Alb-Cre;ALRfloxed/+ offsprings were then crossed with ALRfloxed/floxed mice to generate mice with following genotypes: Alb-Cre; ALRfloxed/floxed (homozygous ALR-L-KO); Alb-Cre; ALRfloxed/+ ; ALRfloxed/floxed; and ALRfloxed/null. The mice were maintained on a mixed B6.SV129 background. No difference between characteristics of the wild type ALR+/+ and ALRfloxed/floxed mice was observed from birth till over 1 year.
All of the other procedures are established standard techniques and described in the Supplemental Material Section.
Statistical analysis
All data are presented as mean ± S.D. Statistical significance was determined by Student's t-test using GraphPad Prism. A p-value of <.05 was considered significant.
RESULTS
General characteristics, hepatic histopathology and ALR expression in ALR-L-KO mice
Relative to paired WT mice, the body weights of ALR-L-KO mice were similar at 1 and 2 weeks, lower at 4 and 6 weeks, and again similar at 8 weeks (Figure 1A). Liver weights were not different between genotypes, leading to a significantly higher liver/body weight ratio in ALR-L-KO mice at 4–6 weeks (Figure 1A).
Figure 1. General characteristics of ALR-L-KO mouse.
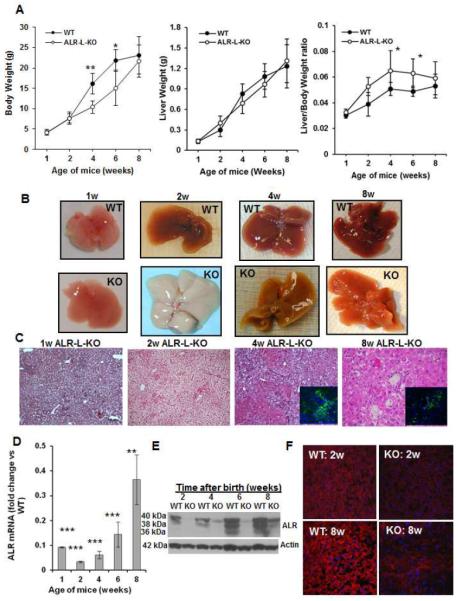
(A) Liver and body weight of the ALR-L-KO mice and their WT littermates. (B) The ALR-L-KO liver turned almost white at 2 weeks, regained normal color at 4 weeks, but developed granularity that increased further at 8 weeks. (C) H/E-stained sections of ALR-L-KO liver show development of steatosis at one week that becames excessive at 2 weeks. At 4 weeks, strong inflammation and ductular proliferation are apparent. Inset shows A6-immunostained oval/biliary epithelial cells. Persistence of inflammation, ductular proliferation with oval/biliary cell expansion (inset) continued at 8 weeks; several necrotic foci are also seen. Hepatic ALR mRNA (D) and protein (E) in ALR-L-KO mice at indicated ages. (F) Immunolabeling show almost no ALR in ALR-L-KO liver at 2 weeks and lower patchy expression at 8 weeks, while all of the hepatocytes in WT liver show strong expression. Fold change of the specific mRNA/GAPDH mRNA ratio in relation to control or WT mRNA/GAPDH ratio normalized to 1 is shown in all figures. (A,D) P*<.05, ** <.01 and *** <0.001 vs WT.
Macroscopically, ALR-L-KO livers appeared normal at one day (not shown) and one week (Figure 1B) after birth, but became milky white by 2 weeks. They regained normal color by 4 weeks, but became progressively coarsened and granular by 8 weeks (Figure 1B).
Histologically, ALR-L-KO livers had normal architecture up to 2 weeks postpartum. Lipid accumulation began at week one and progressed to profound mixed macro- and microvesicular steatosis, with hepatocyte swelling and minimal inflammation at 2 weeks (Figure 1C; Supplemental Figure 2). Steatosis was markedly reduced at 4 weeks but the livers developed scattered lobular mixed inflammation with focal hepatocyte necrosis and prominent bile ductular proliferation accompanied by accumulation of A6-positive cells (hepatic progenitor cells: HPCs or oval cells) (Figure 1C, inset). Ductular proliferation was still apparent at 8 weeks together with increasing portal/periportal and lobular mixed inflammation, hepatocyte necrosis and mitotic activity (Figure 1C). Isolated and clustered A6-positive cells, found only in the bile ducts of WT liver (not shown), were persistently present in and around the portal areas of the ALR-L-KO livers (Figure 1C inset). Atypical ductular proliferation was confirmed by keratin19 staining of biliary epithelial cells, and α-fetoprotein mRNA expression also increased strongly at 2 and 4 weeks (Supplemental Figures 3A and 3B). A6- as well as keratin19-positive cells were persistently present in the bililary areas even at 6 months and decreased somewhat at 1 year in the ALR-L-KO mice (Supplemental Figure 3C).
Hepatic ALR mRNA and protein in ALR-L-KO mice decreased strongly at 1–2 weeks, and although amounts increased from 4 weeks onward, they remained lower compared to WT mice (Figure 1D, E). In contrast, ALR in WT livers increased progressively until 6 weeks when levels stabilized. The native ALR is post-translationally modified from 22-kDa protein to 3 species with approximate mw of 36-, 38- and 40-kDa (5). Interestingly, while 38- and 40-kDa ALR was predominantly observed at 2 and 4 weeks and all 3 ALR species at 8 weeks in WT liver, only 40-kDa ALR appeared in ALR-L-KO liver at 4 weeks and 38- but not 36-kDa ALR at 8 weeks (Figure 1E). Strong immunohistochemical expression of ALR protein at 2 weeks in WT hepatocytes contrasted with almost undetectable ALR in ALR-L-KO hepatocytes; by 8 weeks, patchy ALR immunostaining in ALR-L-KO livers still was less intense than in WT liver (Figure 1F).
Cell death, regeneration, inflammation and fibrosis in ALR-L-KO mouse
Livers from WT mice showed no detectable apoptosis (TUNEL staining) between 2 and 8 weeks, and the robust proliferation (Ki67 staining) at 2 weeks was muted by 4 weeks (Figure 2A, B). In contrast, ALR-L-KO livers showed profound hepatocyte apoptosis at 2 weeks which decreased progressively, while substantial cell proliferation was seen at 4 and 8 weeks; this proliferation occurred in oval/biliary cells as well as the parenchyma (Figure 2B inset).
Figure 2. Apoptosis and proliferation of hepatocytes in ALR-L-KO mice.
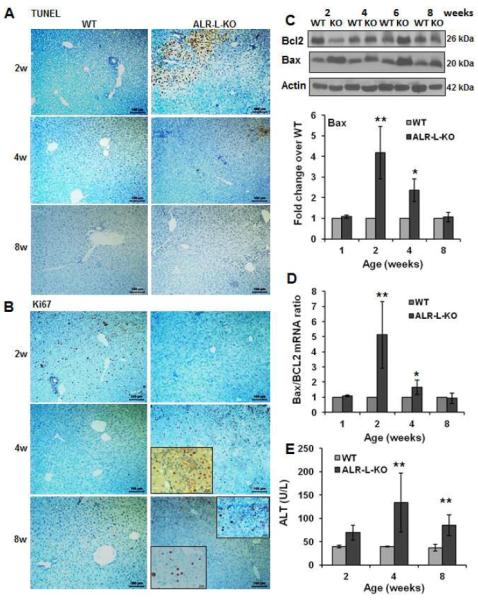
(A) TUNEL staining at 2 weeks shows profound apoptosis of hepatocytes in ALR-L-KO mice that reduces at 4 and 8 weeks. (B) Ki67 labeling of the same sections show no proliferation of hepatocytes at 2 weeks in ALR-L-KO liver in contrast to strong proliferation in the WT liver. At 4 weeks, ALR-L-KO liver shows significant Ki67 labeling in the parenchyma as well as portal area (inset), which reduces significantly at 8 weeks. (C) Western blot (representative of least 4 independent samples) shows increased expression of Bax and reduced expression of Bcl2 at 2 weeks in ALR-L-KO liver. Bax expression decreased and Bcl2 expression increased subsequently in the ALR-L-KO liver and was similar to WT levels at 8 weeks. (D) Bax/Bcl2 mRNA ratio increased strongly in ALR-L-KO liver as compared to WT liver at 2 weeks. (E) Serum ALT in ALR-L-KO mice. Bar graphs show averages of 4 independent values ± S.D. *P<.05 and **P<.01 vs corresponding values in WT liver.
Expression of both pro-apoptotic Bax and anti-apoptotic Bcl2 was stable in WT liver (Figure 2C), while at 2 weeks ALR-L-KO livers showed robust increases in Bax and moderate decreases in Bcl2. Expression of both proteins reverted first partly (4 weeks) then completely (8 weeks) to WT levels, as did the ratio of their encoding mRNAs (Figure 2D). Consistent with these data, caspase-3 activity was significantly increased in ALR-L-KO liver at 2 weeks and declined at later times (Supplemental Figure 4). Increased serum ALT levels in ALR-L-KO mice corresponded temporally to histologic evidence of hepatocyte injury (Figure 2E).
Increased CD45 staining indicated persistent portal and parenchymal inflammation in ALR-L-KO mice (Figure 3A). Hepatic mRNAs encoding the inflammatory cytokines IL1β and TNFα were significantly higher in ALR-L-KO than WT mice at 4 and 8 weeks (Figure 3B); for IL6, IFNγ and IL33 these increases were significant only at 8 weeks. NKG2D and CD8 mRNA, indicative of NKT and CD8+ T cells respectively, also were increased at 8 weeks in ALR-L-KO liver (Figure 3C). In contrast there was no significant increase in CD4 mRNA.
Figure 3. Hepatic inflammation and fibrosis in ALR-L-KO mice.
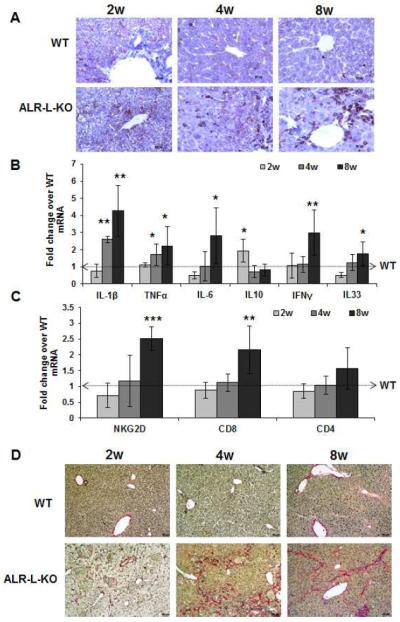
(A) CD45 staining indicates progressively increased inflammatory cells in the ALR-L-KO liver. (B) Hepatic mRNA expression of inflammatory cytokines (IL1β, TNFα, IL6, IFNγ and IL33) increased strongly at 8 weeks in ALR-L-KO relative to WT mice. Anti-inflammatory IL10 increased at 2 weeks and declines to the WT level at 4 and 8 weeks. (C) Hepatic mRNA expression of NKG2D and CD8, but not CD4, increased at 8 weeks. *P<.05, **P<.01 and ***P<.001 vs expression in WT liver. (D) Sirius Red staining shows mostly pericellular fibrosis at 2 weeks and periportal fibrosis at 4 weeks, whereas at 8 weeks there is bridging fibrosis.
Sirius Red staining (Figure 3D) showed mild pericellular fibrosis at 2 weeks in ALR-L-KO livers, with focal portal fibrosis in a background of portal mixed inflammation and bile ductular proliferation by 4 weeks. This was accompanied by accumulation of α-smooth muscle actin (α-sma)-positive cells (activated stellate cells or portal myofibroblasts) in fibrous areas (Supplemental Figure 5A). It is possible that epithelialmesenchymal transition may also contribute to fibrosis development in ALR-L-KO mice. By 8 weeks, there was focal early bridging fibrosis (Figure 3D) and a decrease in numbers of α-sma-positive cells (Supplemental Figure 5A). Results of morphometric analysis of the Sirius Red-stained sections were consistent with the above findings (Supplemental Figure 5B). Moreover, expression of the molecules associated with activation of HSCs (TIMP1, MMP13 and Collagen 1) also increased in the ALR-L-KO livers (Supplemental Figure 5C).
ALR deficiency causes mitochondrial dysfunction and injury
Ultrastructural examination at 2 weeks showed markedly fewer mitochondria per hepatocyte in ALR-L-KO mice, with the majority swollen, abnormally shaped and with increased spacing or loss of cristae (Figure 4A). There was also significant endoplasmic reticulum dilatation and robust lipid deposition. These changes persisted with lesser magnitude at later times (e.g., 8 weeks; Figure 4A).
Figure 4. Mitochondrial degeneration and reduced respiration in ALR-L-KO liver.
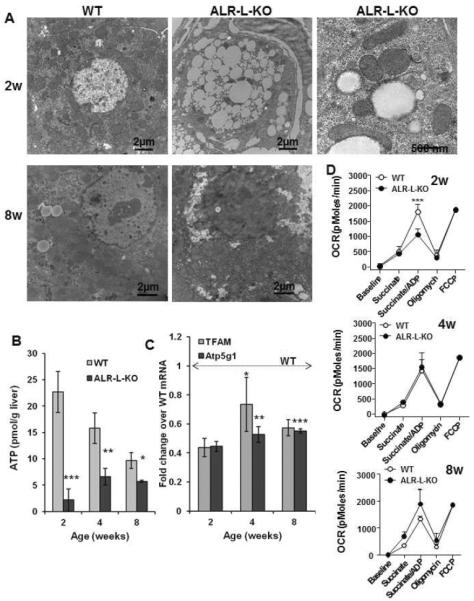
(A) TEM showing relatively few mitochondria at 2 weeks in ALR-L-KO hepatocytes, most of which are abnormally shaped with loss or abnormal spacing of cristae. Profound lipid accumulation is also apparent. Bar=500 nm. At 8 weeks, ALR-L-KO liver regains mitochondria, majority of which demonstrating normal structure. (B) ATP was strongly reduced in ALR-L-KO liver at 2 weeks and despite recovery of mitochondria, was still significantly lower in ALR-L-KO liver than in WT liver at 4 and 8 weeks. (C) mRNA expression of Atp5g1 and TFAM was significantly lower (p<0.05) in ALR-L-KO liver than WT values. *p=0.009; **p=0.006; ***p=0.002 Vs. 2 weeks ALR-L-KO. (D) ALR deficiency significantly reduces mitochondrial respiration at 2 week of age. Mitochondrial respiration, as a measure of oxygen consumption rate (OCR), was quantified in both WT and ALR-L-KO mice using Seahorse technology. *P<.05, **P<.01 and ***P<.001 vs WT values.
ATP content was greater in the WT liver at 2 weeks compared to 4–8 weeks (Figure 4B). In contrast, ALR-L-KO livers at 2 weeks had extremely low ATP, which may be due its consumption during apoptosis and mitochondrial dysfunction. Although ATP then increased by 4 weeks (Figure 4B) and remained constant at 8 weeks, it was significantly lower than in WT mice. Because of the observed mitochondrial abnormalities in ALR-L-KO mice, we analyzed expression of Atp5g1 that encodes a subunit of mitochondrial ATP synthase (13,14) and TFAM (mtTFA) that encodes mitochondrial transcription factor A, a key activator of mitochondrial transcription and genome replication (15). Expression of both Atp5g1 and TFAM was significantly lower in ALR-L-KO liver at 2 weeks relative to WT liver; expression levels of these transcripts then increased at 4 and 8 weeks without reaching the WT levels (Figure 4C).
Upon assessing further the role of ALR in mitochondrial function, as expected, mitochondria from the 2-week old ALR-L-KO liver showed reduced respiration (Figure 4D). Since the isolation procedure discards the majority of damaged mitochondria, the strongly reduced respiration rate seen in remaining mitochondria reflects the critical role of ALR in their survival and function. Further, to ensure fair comparison between functional WT and ALR-L-KO mitochondria, mitochondrial respiration was normalized to the maximal respiratory capacity, as quantified following carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) stimulation. Interestingly, the respiratory activity of ALR-L-KO mitochondria at 4 and 8 weeks was similar to the corresponding WT mitochondria (Figure 4D). These data imply that early-stage ALR deficiency significantly inhibits mitochondrial respiratory capacity (week 2); its reversal at week 4, with increase in ALR, most likely indicates a compensatory mechanism with recovery in Atp5g1 and TFAM expression and robust proliferation of hepatocytes and HPCs. It can be postulated that the 40-kDa ALR may be responsible for mitochondrial function as indicated by its predominance at 2 weeks in WT mice and appearance at 4 weeks in ALR-L-KO liver (Figure 1E).
ALR deficiency causes ROS generation, mitochondrial DNA damage and hepatocyte death in vivo and in vitro
The ALR deficiency-induced liver damage associated with increased oxidative stress is shown by marked decrease in hepatic and mitochondrial glutathione and a concomitant increase in hepatic malondialdehyde (lipid oxidation product) in 2 week old ALR-L-KO mice compared with WT mice; both these differences disappear by 8 weeks (Supplemental Figure 6).
To recapitulate in vitro the in vivo mitochondrial damage and oxidative stress, we infected ALRfloxed/floxed hepatocytes with adenovirus containing Cre recombinase (Adeno-Cre). ALR mRNA expression decreased time-dependently after Adeno-Cre transfection (Figure 5A). The DCFDA-based measurements showed significantly increased ROS production during ALR depletion (Figure 5B). Adeno-Cre-mediated ALR deletion caused a time-dependent decrease in mitochondrial mitotracker uptake and increase in the nuclear and mitochondrial 8-oxoguanine content (Figure 5C), consistent with involvement of increased mitochondrial reactive oxygen species (ROS) production in cell death (16,17). 8-Oxoguanine, a common DNA damaging lesion resulting from ROS (18), causes mismatched pairing with adenine resulting in G→T and A→C substitutions (19), and fosters abnormal gene expression/function. Even at the earliest time point (9h after Adeno-Cre infection) there was mitochondrial DNA damage indicating a crucial role for ALR in redox regulation.
Figure 5. ALR deficiency causes increased oxidative stress, mitochondrial injury and DNA damage.
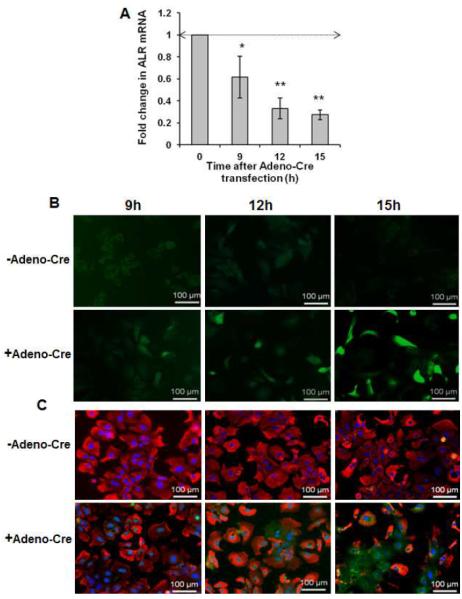
(A) Infection of ALRfloxed/floxed hepatocytes with Adeno-Cre causes time-dependent decrease in ALR mRNA expression. *P<.05 and **P<0.01 vs expression at “0” time. (B) DCFDA fluorescence assay shows increasing oxidative stress in Adeno-Cre infected ALRfloxed/floxed hepatocytes. (C) Analysis of mitochondrial and DNA damage at indicated times after Adeno-Cre infection in ALRfloxed/floxed hepatocytes using mitotracker (red) and 8 Oxoguanine (green).
ALR deficiency induces steatosis and abnormal expression of enzymes involved in lipid metabolism
The basal triglyceride content was high at 1 week in both WT and ALR-L-KO mice. Higher triglycerides at 1 week are most likely due to the transition from a previously glucose-based energy source in the fetus to fatty acids as a major source of energy in the neonate (20). A further robust increase in triglycerides occurred at 2 weeks in ALR-L-KO livers, which decreased to WT levels by 8 weeks (Figure 6A). Cholesterol content also increased at 2 weeks in ALR-L-KO liver (Figure 6B), but this was relatively modest as compared to the increase in triglycerides.
Figure 6. Neutral lipid accumulation and changes in genes associated with lipid metabolism in ALR-L-KO mice.
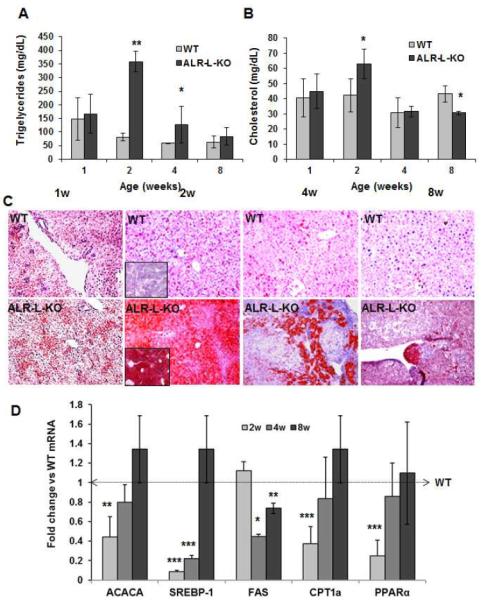
(A,B) Triglyceride accumulation increased strongly at 2 weeks in ALR-L-KO mice and declined to the basal value by 8 weeks; increase in cholesterol was modest at 2 weeks compared to WT liver. *P<.05 and **P<.001 vs WT. (C) Intense lipid (Oil Red-O) staining, seen at 2 weeks in ALR-L-KO liver, decreased at 4 and 8 weeks. (D) mRNA expressions of ACACA, SREBP1, CPT1a and PPARα all decreased at 2 weeks and recovered to the WT values by 8 weeks. FAS expression was similar to WT level at 2 weeks but reduced significantly at 4 weeks, and recovered by 8 weeks. *P<.05, **P<0.01 and ***P<.001 vs WT.
Oil red-O staining showed greater lipid distribution in WT and ALR-L-KO livers at 1 week as compared to the WT livers at 2, 4 and 8 weeks (Figure 6C). At 2 weeks, Oil Red-O staining was much more intense and evenly distributed in ALR-L-KO compared to WT livers. By 4 weeks, ALR-L-KO livers showed increased Oil Red-O staining only in focal areas, which reduced further at 8 weeks (Figure 6C).
Levels of mRNAs encoding acetyl-CoA carboxylase (ACACA) and sterol regulatory element-binding protein 1c (SREBP-1c) were decreased in ALR-L-KO livers at 2 weeks. Interestingly, expression of the SREBP-1c target gene fatty acid synthase (FAS) was similar in both ALR-L-KO and WT livers at 2 and 8 weeks (Figure 6D). However, expression (Figure 6D) and activity (Supplemental Figure 7) of carbamyl palmitoyl transferase 1a (CPT1a), which is responsible for fatty acid transport into mitochondria, were strongly reduced in ALR-L-KO livers at 2 weeks. Moreover, expression of peroxisome proliferator-activated receptor (PPAR)α, which regulates peroxisomal and mitochondrial fatty acid β-oxidation (21), was also reduced significantly at 2 weeks in ALR-L-KO livers (Figure 6D).
ALRfloxed/floxed hepatocytes depleted of ALR by Adeno-Cre infection showed increased lipid accumulation that did not increase markedly in the presence of exogenous oleic or palmitic acids (Supplemental Figure 8A). Exogenously added fatty acids increased lipid accumulation but did not cause marked lipotoxicity in control hepatocytes till 15 hours (Supplemental Figures 8A, 8B) indicating that the effects on mitochondrial damage (Figure 6) and lipid accumulation (Supplemental Figure 8A) are due to ALR depletion in Adeno-Cre-treated ALRfloxed/floxed hepatocytes. Adeno-Cre infection did not affect fatty acid synthase (FAS) mRNA expression but increased ACACA mRNA at 9h that declined to basal values by 12h and 15h, whereas expression of CPT1a decreased time-dependently (Supplemental Figure 8C).
ALR-L-KO mice develop hepatocellular carcinoma
ALR-L-KO mice did not show significant differences from WT mice in the appearance or histopathology of the liver between 8 weeks and 6 months, but developed nodular foci of high grade dysplasia after 6 months (Supplemental Figure 2). At 1 year the mice showed varying degrees of increased liver mass relative to body mass, with more than 70% developing tumors and approximately 60% demonstrated HCC (Figure 7A). Perhaps notably, ALR mRNA and protein expression were similar in WT and ALR-L-KO livers at 1 year (Figure 7B). Histologically, a clear distinction between tumor and non-tumor regions was apparent. Tumor cells were pleomorphic and often steatotic, with frequent mitotic figures and variably anaplastic nuclei (Figure 7C). Ultrastructural examination showed cytosolic and autophagosomal lipid accumulation and dilated ER (ER stress) (typical of solid organ tumors including HCC) in the neoplastic hepatocytes (Figure 7D).
Figure 7. Liver tumor development in ALR-L-KO mice and ALR in murine and human fatty liver disease.
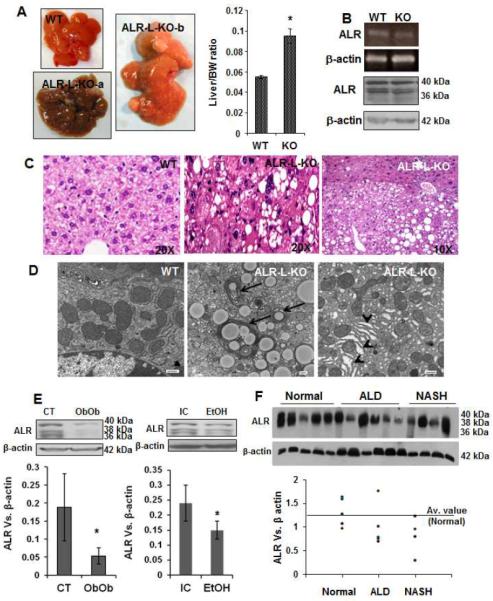
(A) Macroscopically, there were multiple tumors in ALR-L-KO livers at 1 year. Bar graph shows liver/body weight ratio in ALR-L-KO and WT mice (*P<.01). (B) ALR mRNA and protein expressions as determined by semi-quantitative RT-PCR and Western blot analysis, respectively, were similar in WT and ALR-L-KO livers. (C) The tumor (HCC) and non-tumor regions of the ALR-L-KO liver are shown. HCC is characterized by the loss of liver structure with mitotic figures and anaplastic nuclei. (D) TEM shows lipid accumulation and autophagosomes (arrows) as well as ER dilation (arrowheads) in ALR-L-KO liver at 1 year. Bar=500nm. (E) Western analysis shows hepatic ALR expression in Ob/Ob mice (8 weeks of age), mice fed Lieber-De Carlie isocaloric (IC) or alcoholic (EtOH) diet for 5 weeks that resulted in fatty liver, and (F) humans with advanced ALD or NASH. Bar graphs in (E) show ratio of ALR vs β-actin expression (n=5 each) ± SD. In the graph for (F), ALR vs β-actin ratio was plotted individually.
Association of decreased ALR with hepatic steatosis and human diseases
Significant resolution of hepatic steatosis with re-emergence of ALR in the ALR-L-KO mouse suggested a relationship between hepatic ALR deficiency and fatty liver disease. Therefore, we measured hepatic ALR in (a) alcohol-treated mice with fatty livers, (b) Ob/Ob mice that develop fatty livers spontaneously (kindly provided by Dr. Marsha Cole, Department of Pharmacology, University of Pittsburgh) and (c) humans with advanced alcoholic liver disease, nonalcoholic steatohepatitis (NASH) and HCV-induced liver disease or control livers (de-identified samples from the Health Sciences Tissue Bank, University of Pittsburgh Medical Center). The livers of Ob/Ob and alcohol-treated mice had significantly lower ALR as compared to controls (Figure 7E). More interestingly, hepatic ALR expression was also lower than the average value for control livers in humans with ALD (4/5) and NASH (4/4) (Figure 7F), but not with HCV-infection and comparable fibrosis (Supplemental Figure 9).
DISCUSSION
We report for the first time that ALR is critical for both mammalian embryonic development (failure to obtain homozygous ALRnull/null mouse) and for the postnatal survival and physiologic functions of hepatocytes in vivo. Depletion of hepatic ALR shortly after birth led to profound steatosis, mitochondrial degeneration and apoptosis of hepatocytes by 2 weeks, followed by continued cell death and regeneration, persistent inflammation, ductular proliferation, and eventually HCC. Hepatocyte loss at 2 weeks is consistent with our previous in vitro finding showing progressive loss of ALR leading to ATP depletion, release of cytochrome c and hepatocyte death (12). In this study (12), we found that hepatocyte death via apoptosis ensues with >30–40% cellular and mitochondrial ALR depletion. This calculation may not be extended to whole ALR-L-KO livers, in vivo, due to variable depletion of ALR from individual cells at any given point.
ALR plays a critical role as a component of the mitochondrial disulfide relay system that facilitates disulfide bond formation and transport of newly synthesized proteins across the outer mitochondrial membrane (22). In this system, Mia40 (mitochondrial IMS and assembly pathway 40 kDa) oxidizes incoming proteins and is reoxidized by ALR, which in turn transfers its electrons to cytochrome c which is then acted upon by cytochrome c oxidase to convert oxygen to water (23–25). The central role of ALR in this respiratory chain prevents the generation of pathological levels of ROS, shown for example by the deletion of ALR gene in ALRfloxed/floxed hepatocytes causing oxidative stress and mitochondrial damage. The critical importance of ALR in hepatic mitochondrial survival and function is also underscored by the recovery of ALR-L-KO mice upon regaining ALR. This functional in vivo role of ALR is reminiscent of an analogous situation in Saccharomyces cerevisiae, in which disruption of ERV-1, the yeast ortholog of ALR, leads to complete loss of the mitochondrial genome with subsequent cell death (26,27).
The appearance of ALR-expressing hepatocytes following excessive apoptosis and almost complete loss of ALR in ALR-L-KO mice by 2 weeks is interesting and likely reflects the complexity of the in vivo condition. Deletion of target gene expression using AlbCre-LoxP technology occurs progressively beginning at about one week post-birth and is nearly complete by 2–12 months (28–30). Liver regeneration following profound loss as seen during fulminant liver failure (in contrast to replication of existing hepatocytes after partial hepatectomy) depends upon HPCs in bile ductules and canals of Hering (31–33). The existence of robust ductular proliferation and presence of Ki67-positive hepatocytes emerging from these sites in ALR-L-KO mice are most consistent with HPC-associated regeneration. This interpretation is supported by strongly increased α-fetoprotein expression at 2 and 4 weeks in ALR-L-KO livers, whereas this protein normally declines rapidly after birth (34). It may be that the profound loss of hepatocytes due to ALR depletion is followed by preferential regeneration of progenitor cells that either lack Cre recombinase or have an inactive enzyme. We note that ALR in ALR-L-KO mice did not reach WT levels till almost 1 year, which could suggest the continued death of hepatocytes with replacement by ALR expressing cells.
Excessive accumulation of triglycerides in ALR-L-KO livers at 2 weeks could be related to mitochondrial damage, reduced ATP content, decreased mitochondrial metabolic capacity and/or impaired fatty acid β-oxidation (35,36). Hepatic expression and activity of CPT1a, that regulates free fatty acid transport into mitochondria for β-oxidation, was strongly decreased in the ALR-L-KO mice. Reduced expression of PPARα, which regulates both peroxisomal and mitochondrial fatty acid β-oxidation, provides additional support to our contention that ALR deficiency has a strong negative effect on fatty acid β-oxidation. Unaltered FAS expression and lower ACACA expression in ALR-L-KO mice than in WT mice at 2 weeks likely exclude accumulation of fatty acids due to increased synthesis. Inhibition of CPT1a activity by high levels of malonyl CoA can also be ruled out since expression of ACACA that catalyzes malonyl CoA synthesis was reduced. These results and the decrease in Atp5g1 and TFAM expression in ALR-L-KO liver suggest an interesting possibility that ALR, which is also present in the nucleus (6,37), may influence expression of genes involved in lipid and energy metabolism at the transcriptional level. It is worth noting that an ontological survey showed that the CPT1a, TFAM and ACACA gene expression is targeted by microRNAs-370, -122, and -33 raising the speculation that ALR might modulate the expression of these and/or other functional miRNAs.
Despite the incremental increase in ALR that did not reach WT levels, hepatic steatosis was reversed by 4 weeks and only a few cells with lipid accumulation were seen at 8 weeks in ALR-L-KO livers. This observation suggests that a threshold concentration of ALR, especially in mitochondria, is required for hepatocyte function. Based on the Western analysis, we predict that the high mw ALR (40 kDa) might be required for this function.
Despite reversal of steatosis and regeneration with ALR-expressing cells, there was a strong inflammatory response with neutrophil infiltration and increased expression of pro-inflammatory cytokines. Continued death of hepatocytes due to mediators released by infiltrating cells (notably neutrophils, NKT and CD8+ T cells) likely sustained the inflammatory environment throughout the life of the ALR-L-KO mice. Thus continued inflammation, hepatocyte death and regeneration (from oval cells/HPCs) precede dysplasia and tumor development in the ALR-L-KO mouse. In this regard, oval cells have been detected in human livers in conditions associated with increased risk of neoplastic development (38–40). Indeed, A6- and keratin19-positive cells continued to be found in ALR-L-KO liver till 1 year (Supplemental Figure 3C), suggesting that detrimental/carcinogenic progenitors might persist and give rise to the tumors. However, the possibility that individual ALR-deficient hepatocytes persisted in this environment and contributed to neoplasia cannot be excluded. Our results may therefore have direct implications for analysis of the complications of advanced alcoholic (ASH) and nonalcoholic steatohepatitis (NASH), as both diseases involve severe mitochondrial dysfunction and can progress to inflammation, fibrosis/cirrhosis, and liver cancer (41–43). Although hepatic ALR levels were lower in subjects with ASH and NASH, ALR levels were similar in the ALR-L-KO and WT mice at 1 year when they developed HCC. Another study reported greater levels of hepatic ALR in patients with liver cirrhosis, HCC and cholangiocarcinoma (44), and hepatic ALR expression was similar in normal and HCV livers (Supplemental Figure 9). Thus, it is also plausible that increased ALR might support survival and replication of neoplastic hepatocytes. Although a definitive conclusion cannot be drawn from the human data due to limited sample size, we found 48 sequence variants, 17 of which were novel (not in SNPdb), in a preliminary study in which the ALR gene from 100 human samples was sequenced (not shown). This raises the speculation that some of these variants may be present among the population vulnerable to ASH and/or NASH.
Our present understanding of the precise mechanisms of advanced ASH and NASH, which occur in only a subset of the population exposed to associated risk factors, is inadequate. Based on the results of these studies, we propose that the ALR-L-KO mouse may be an important model to investigate mechanisms relevant not only to the clinical development of ALD or NASH, but also to the fibroinflammatory and neoplastic consequences that arise from these diseases. The full role of this ubiquitous protein in human physiology remains to be elucidated.
Supplementary Material
Acknowledgement
We thank Dr. George Michalopoulos for his advice and comments, Mr. John Prelich and Ms. Rachel Stewart for excellent technical assistance, and Dr. Berthony Deslouches for his help during the initial stages of this work. A6 rat anti-mouse Ab was a kind gift from Dr. Valentina Factor, NIH/NCI.
Grant support: This work was supported by a VA Merit Review Award (1IO1BX001174) and grants from NIH (DK 54411, PO1AIO81678 and R21AA020846) to CRG, as well as support from Cincinnati Children's Hospital Medical Center and University of Cincinnati.
Abbreviations
- ACACA
acetyl-CoA carboxylase
- ALR
augmenter of liver regeneration
- CPT
carbamyl palmitoyl transferase
- DCFDA
dichlorofluorescin diacetate
- FAS
fatty acid synthase
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- KO
knockout
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- SREBP
sterol regulatory element-binding protein
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None of the authors have any financial conflict of interest to be disclosed.
Authors' contributions: Chandrashekhar R. Gandhi- Concept, design, study supervision and writing. The study was supported by Dr. Gandhi's funding.
J. Richard Chaillet: ALR-L-KO mouse was developed in Dr. Chaillet's laboratory under his direct supervision.
Michael Nalesnik: Performed histopathology and was involved in writing of the manuscript.
Sudhir Kumar: Postdoctoral fellow in Dr. Gandhi's laboratory, performed experiments including immunostaining and in vitro cell culture experiments and statistical analysis.
Anil Dangi: Postdoctoral fellow in Dr. Gandhi's laboratory. Performed experiments including immunostaining and RNA analysis.
A. Jake Demetris: Involved with analysis of histopathology and review of the manuscript.
Robert Ferrell: Performed analysis of human samples to determine sequence variants in the ALR gene.
Tong Wu: Analysis of histopathology and critical review of the manuscript.
Senad Divanovic: Analysis of the Sea Horse assay data and critical review of this manuscript.
Traci Stankeiwicz: Performed the Sea Horse assay.
Benjamin Schaffer: Experiments of development of the ALR-L-KO mouse.
Donna B. Stolz: Generation of histopathological images including the electron microscopy and interpretation of their results.
Stephen A.K. Harvey: Performed gene array analysis of the ALR-L-KO livers, evaluation of which was important in determining specific molecules to be analyzed.
Jiang Wang: Reviewed the histopathology of ALR-L-KO liver at later stages (6 months-1 year) and provided expert analysis.
Thomas E. Starzl: Conceptual development of this work and writing of the manuscript.
References
- 1.Francavilla A, Hagiya M, Porter KA, et al. Augmenter of liver regeneration: Its place in the universe of hepatic growth factors. Hepatology. 1994;20:747–757. [PubMed] [Google Scholar]
- 2.Gandhi CR. Augmenter of liver regeneration. Fibrogenesis Tissue Repair. 2012;5:10. doi: 10.1186/1755-1536-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagiya M, Francavilla A, Polimeno L, et al. Cloning and sequence analysis of the rat augmenter of liver regeneration (ALR) gene: Expression of biologically active recombinant ALR and demonstration of tissue distribution. Proc Natl Acad Sci. 1994;91:8142–8146. doi: 10.1073/pnas.91.17.8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giorda R, Hagiya M, Seki T, et al. Analysis of the structure and expression of the augmenter of liver regeneration (ALR) gene. Molecular Medicine. 1996;2:97–108. [PMC free article] [PubMed] [Google Scholar]
- 5.Gandhi CR, Kuddus R, Subbotin VM, et al. A fresh look at augmenter of liver regeneration in rats. Hepatology. 1999;29:1435–1445. doi: 10.1002/hep.510290522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tury A, Mairet-Coello G, Lisowsky T, et al. Expression of the sulfhydryl oxidase ALR (Augmenter of Liver Regeneration) in adult rat brain. Brain Res. 2005;1048:87–97. doi: 10.1016/j.brainres.2005.04.050. [DOI] [PubMed] [Google Scholar]
- 7.Farrell SR, Thorpe C. Augmenter of liver regeneration: a flavin-dependent sulfhydryl oxidase with cytochrome c reductase activity. Biochemistry. 2005;44:1532–1541. doi: 10.1021/bi0479555. [DOI] [PubMed] [Google Scholar]
- 8.Allen S, Balabanidou V, Sideris DP, et al. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J Mol Biol. 2005;353:937–944. doi: 10.1016/j.jmb.2005.08.049. [DOI] [PubMed] [Google Scholar]
- 9.Lange H, Lisowsky T, Gerber J, et al. An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep. 2001;2:715–720. doi: 10.1093/embo-reports/kve161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francavilla A, Vujanovic NL, Polimeno L, et al. The in vivo effect of hepatotrophic factors augmenter of liver regeneration, hepatocyte growth factor, and insulin-like growth factor-II on liver natural killer cell functions. Hepatology. 1997;25:411–415. doi: 10.1002/hep.510250225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandhi CR, Murase N, Starzl TE. Cholera toxin-sensitive GTP-binding protein-coupled activation of augmenter of liver regeneration (ALR) receptor and its function in rat Kupffer cells. J Cell Physiol. 2010;222:365–373. doi: 10.1002/jcp.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thirunavukkarasu C, Wang LF, Harvey SA, et al. Augmenter of liver regeneration: an important intracellular survival factor for hepatocytes. J Hepatol. 2008;48:578–88. doi: 10.1016/j.jhep.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dyer MR, Walker JE. Sequences of members of the human gene family for the c subunit of mitochondrial ATP synthase. Biochem J. 1993;293:51–64. doi: 10.1042/bj2930051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li HS, Zhang JY, Thompson BS, et al. Rat mitochondrial ATP synthase ATP5G3: cloning and upregulation in pancreas after chronic ethanol feeding. Physiol Genomics. 2001;6:91–98. doi: 10.1152/physiolgenomics.2001.6.2.91. [DOI] [PubMed] [Google Scholar]
- 15.Tiranti V, Rossi E, Ruiz-Carrillo A, et al. Chromosomal localization of mitochondrial transcription factor A (TCF6), single-stranded DNA-binding protein (SSBP), and endonuclease G (ENDOG), three human housekeeping genes involved in mitochondrial biogenesis. Genomics. 1995;25:559–564. doi: 10.1016/0888-7543(95)80058-t. [DOI] [PubMed] [Google Scholar]
- 16.Lemasters JJ, Thurman RG. Hypoxia and reperfusion injury to liver. Prog Liver Dis. 1993;11:85–114. [PubMed] [Google Scholar]
- 17.Kaplowitz N, Tsukamoto H. Oxidative stress and liver disease. Prog Liver Dis. 1996;14:131–159. [PubMed] [Google Scholar]
- 18.Kanvah S, Joseph J, Schuster GB, et al. Oxidation of DNA: Damage to Nucleobases. Acc Chem Res. 2010;43:280–287. doi: 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- 19.Cheng KC, Cahill DS, Kasai H, et al. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and C→A substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 20.Chalmers RA, Stanley CA, English N, et al. Mitochondrial carnitine-acylcarnitine translocase deficiency presenting as sudden neonatal death. J Pediatr. 1997;131:220–225. doi: 10.1016/s0022-3476(97)70157-4. [DOI] [PubMed] [Google Scholar]
- 21.Rakhshandehroo M, Knoch B, Müller M, et al. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010;2010:ID612089. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riemer J, Bulleid N, Herrmann JM. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science. 2009;324:1284–1287. doi: 10.1126/science.1170653. [DOI] [PubMed] [Google Scholar]
- 23.Bihlmaier K, Mesecke N, Terziyska N, et al. The disulfide relay system of mitochondria is connected to the respiratory chain. J Cell Biol. 2007;179:389–395. doi: 10.1083/jcb.200707123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banci L, Bertini I, Calderone V, et al. Molecular recognition and substrate mimicry drive the electron-transfer process between MIA40 and ALR. Proc Natl Acad Sci USA. 2011;108:4811–4816. doi: 10.1073/pnas.1014542108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dabir DV, Hasson SA, Setoguchi K, et al. A small molecule inhibitor of redox-regulated protein translocation into mitochondria. Dev Cell. 2013;25:81–92. doi: 10.1016/j.devcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lisowsky T. Dual function of a new nuclear gene for oxidative phosphorylation and vegetative growth in yeast. Mol Gen Genet. 1992;232:58–64. doi: 10.1007/BF00299137. [DOI] [PubMed] [Google Scholar]
- 27.Lisowsky T. ERV1 is involved in the cell-division cycle and the maintenance of mitochondrial genomes in Saccharomyces cerevisiae. Curr Genet. 1994;26:15–20. doi: 10.1007/BF00326299. [DOI] [PubMed] [Google Scholar]
- 28.Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knockouts using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 29.Gu J, Weng Y, Zhang QY, et al. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–25901. doi: 10.1074/jbc.M303125200. [DOI] [PubMed] [Google Scholar]
- 30.Xu Z, Chen L, Leung L, et al. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci USA. 2005;102:4120–4125. doi: 10.1073/pnas.0500660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michalopoulos GK. Liver regeneration: alternative epithelial pathways. Int J Biochem Cell Biol. 2011;43:173–179. doi: 10.1016/j.biocel.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Best J, Dollé L, Manka P, et al. Role of liver progenitors in acute liver injury. Front Physiol. 2013;4:258. doi: 10.3389/fphys.2013.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riehle KJ, Dan YY, Campbell JS, et al. New concepts in liver regeneration. J Gastroenterol Hepatol. 2011;26(Suppl 1):203–212. doi: 10.1111/j.1440-1746.2010.06539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tilghman SM, Belayew A. Transcriptional control of the murine albumin/alpha-fetoprotein locus during development. Proc Natl Acad Sci USA. 1982;79:5254–5257. doi: 10.1073/pnas.79.17.5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boison D, Scheurer L, Zumsteg V, et al. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA. 2002;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fromenty B, Pessayre D. Inhibition of mitochondrial β-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 37.Lu C, Li Y, Zhao Y, et al. Intracrine hepatopoietin potentiates AP-1 activity through JAB1 independent of MAPK pathway. FASEB J. 2002;16:90–92. doi: 10.1096/fj.01-0506fje. [DOI] [PubMed] [Google Scholar]
- 38.Libbrecht L, Roskams T. Hepatic progenitor cells (HPCs) in human liver tumor development. Semin Cell Dev Biol. 2002;13:389–396. doi: 10.1016/s1084952102001258. [DOI] [PubMed] [Google Scholar]
- 39.Lowes KN, Brennan BA, Yeoh GC, et al. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am J Pathol. 1999;154:537–541. doi: 10.1016/S0002-9440(10)65299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roskams TA, Libbrecht L, Desmet VJ. Progenitor cells in diseased human liver. Semin Liver Dis. 2003;23:385–396. doi: 10.1055/s-2004-815564. [DOI] [PubMed] [Google Scholar]
- 41.Powell EE, Cooksley WG, Hanson R, et al. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 42.Day C, Saksena S. Non-alcoholic steatohepatitis: definitions and pathogenesis. J Gastroenterol Hepatol. 2002;17:S377–S384. doi: 10.1046/j.1440-1746.17.s3.31.x. [DOI] [PubMed] [Google Scholar]
- 43.Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24:3–20. doi: 10.1055/s-2004-823098. [DOI] [PubMed] [Google Scholar]
- 44.Thasler WE, Schlott T, Thelen P, et al. Expression of augmenter of liver regeneration (ALR) in human liver cirrhosis and carcinoma. Histopathology. 2005;47:57–66. doi: 10.1111/j.1365-2559.2005.02172.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.