

Abstract
Background
Diabetes is associated with prolongation of the QT interval of the electrocardiogram and enhanced dispersion of ventricular repolarization, factors that, together with atherosclerosis and myocardial ischemia, may promote the occurrence of electrical disorders. Thus, we tested the possibility that alterations in transmembrane ionic currents reduce the repolarization reserve of myocytes, leading to action potential (AP) prolongation and enhanced beat‐to‐beat variability of repolarization.
Methods and Results
Diabetes was induced in mice with streptozotocin (STZ), and effects of hyperglycemia on electrical properties of whole heart and myocytes were studied with respect to an untreated control group (Ctrl) using electrocardiographic recordings in vivo, ex vivo perfused hearts, and single‐cell patch‐clamp analysis. Additionally, a newly developed algorithm was introduced to obtain detailed information of the impact of high glucose on AP profile. Compared to Ctrl, hyperglycemia in STZ‐treated animals was coupled with prolongation of the QT interval, enhanced temporal dispersion of electrical recovery, and susceptibility to ventricular arrhythmias, defects observed, in part, in the Akita mutant mouse model of type I diabetes. AP was prolonged and beat‐to‐beat variability of repolarization was enhanced in diabetic myocytes, with respect to Ctrl cells. Density of Kv K+ and L‐type Ca2+ currents were decreased in STZ myocytes, in comparison to cells from normoglycemic mice. Pharmacological reduction of Kv currents in Ctrl cells lengthened AP duration and increased temporal dispersion of repolarization, reiterating features identified in diabetic myocytes.
Conclusions
Reductions in the repolarizing K+ currents may contribute to electrical disturbances of the diabetic heart.
Keywords: diabetes, electrophysiology, repolarization, variability
Subject Categories: Electrophysiology, Arrhythmias, Animal Models of Human Disease, Basic Science Research
Introduction
Diabetes is a major risk factor for cardiovascular diseases, including ventricular arrhythmia and sudden death.1, 2, 3 Prolongation of the QT interval and increased dispersion of repolarization have been observed in the diabetic population,4, 5, 6, 7, 8, 9, 10 and these variables, together with intervening comorbidities,11 represent critical substrates for the occurrence of electrical disturbances.4, 12, 13, 14, 15, 16, 17 But whether alterations at the cellular level contribute to protracted electrical recovery and repolarization variability with diabetes remain to be determined.
Prolongation of the action potential (AP) in cardiomyocytes is typically observed in the hypertrophied and failing heart,18 with major consequences on intracellular Ca2+ homeostasis and contractile force developed by the myocardium.18, 19, 20 Moreover, the protracted AP predisposes myocytes to the occurrence of early afterdepolarizations, which may trigger extrasystoles and tachyarrhythmias and/or aggravate the dispersion of repolarization of the myocardium in the presence of local nonpropagating responses.21 Similarly, beat‐to‐beat variability of repolarization, which is observed at the single‐cell level and in the whole heart,22, 23 has detrimental consequences on myocardial electrical stability by facilitating the development of reentrant arrhythmias.15, 16, 21, 24 Importantly, these defects may be equally operative in the diabetic heart, representing important variables in the propensity to develop ventricular arrhythmias and sudden death in the presence of this disease.
In the present study, we have evaluated the properties and temporal dispersion of cardiac electrical recovery in diabetic mice and their propensity to develop arrhythmias together with the implementation of a novel analysis of AP to establish the duration and degree of variability of myocyte repolarization. Kv and L‐type Ca2+ current densities were reduced in diabetic cells, and Kv current inhibition in control myocytes prolonged the AP profile and enhances its variability. These results suggest that defective repolarizing K+ current contributes to electrical disturbances of the diabetic heart.
Materials and Methods
In Vivo Studies
Female C57Bl/6 mice (n=170) or male C57BL/6‐Ins2Akita/J (Akita; #003548; n=8; The Jackson Laboratory, Bar Harbor, ME) and male wild‐type (WT; n=7) mice were studied in accord with the Guide for Care and Use of Laboratory Animals; experiments were approved by the local animal care committee. When needed, isoflurane (1–1.5%, inhalation) was employed as a methodology of anesthesia.
To induce hyperglycemia, female C57Bl/6 mice at 3 to 6 months of age (n=112) were treated with streptozotocin (STZ; Sigma‐Aldrich, St. Louis, MO) for 4 to 6 consecutive days (≈100 mg/kg body weight per day, intraperitoneally). Female naïve C57Bl/6 mice at 3 to 6 months of age (n=58) served as control animals. STZ was dissolved in 0.9% saline solution containing 20 mmol/L of sodium citrate tribasic dihydrate (Sigma‐Aldrich). Final STZ concentration was 5 mg/L. Glucose levels were assessed using a TRUEtrack meter (Home Diagnostics, Inc., Fort Lauderdale, FL) and test strips. Animals with blood glucose level >300 mg/dL were included in the study. Glucose levels and their changes over time in STZ‐treated and Akita mice are reported in Figure S1.
Echocardiography was performed in mice using a VisualSonics Vevo 2100 System equipped with a MS550D high‐frequency (22–55 MHz) linear transducer (VisualSonics Inc., Toronto, Ontario, Canada). Short‐axis views of the left ventricle (LV) were obtained in conscious mice20, 25 to compute LV volume and ejection fraction (EF) by the Teichholz formula. Diastolic function was assessed using pulsed‐wave Doppler imaging of the transmitral filling pattern in the apical 4‐chamber view of the heart in mice anesthetized with isoflurane. Early transmitral filling wave (E‐wave) and late filling wave attributable to atrial contraction (A‐wave) were obtained. Deceleration time of the E‐wave was determined by measuring the time needed for the down‐slope of the peak of the E‐wave to reach baseline.20
Electrocardiograms (ECGs) were recorded under isoflurane anesthesia by inserting needle electrodes subcutaneously into mouse limbs (lead II). Electrical signals were amplified (Animal Bio Amp; ADInstruments, Houston, TX), digitized using a 4‐kHz A/D converter (MPVS‐400; Millar Instruments, Houston, TX) and recorded using LabChart software (ADInstruments), with low‐ and high‐pass filtering at 100 and 3 Hz, respectively. Surface ECG intervals were measured using LabChart (version 7 or 8; ADInstruments).20 To reduce electrical noise and motion artifacts, 50 consecutive beats were averaged by the software and employed to calculate heart rate and electrocardiographic parameters. PR interval, QRS duration, and QT interval were measured by determining the earliest onset and latest offset of atrial and ventricular deflections from the averaged cycles.20 Rate‐corrected QT interval (QTc) was calculated using the Bazett formula (QTc=QT/√RR). This analysis was extended to a total of 500 beats to determine average interval durations and variability.
To record ECGs in freely roaming, unanesthetized animals, telemetric biopotential transmitters (ETA‐F10; Data Science International, New Brighton, MN) were implanted subcutaneously in mice anesthetized with isoflurane (1–1.5%, inhalation).20 Leads were secured subcutaneously corresponding to position II. Subsequently, signals were transmitted from implants to RPC‐1 receivers and Data Exchange Matrix (Data Science International) and stored in a computer. The telemetric ECG was analyzed during normal activity and for 90 minutes after pharmacological treatments. All recordings were digitized at 2 kHz and analyzed offline with Ponemah software (version 5.10).
LV hemodynamics and pressure‐volume (PV) loops were obtained in anesthetized mice (isoflurane, 1.5%) in the closed‐chest preparation with a MPVS‐400 system for small animals (Millar Instruments) equipped with a PVR‐1045 catheter.20, 25 The mouse was intubated and ventilated (MiniVent Type 845; Hugo Sachs Elektronik‐Harvard Apparatus, GmbH, March, Germany) with isoflurane anesthesia; the right carotid artery was then exposed and the pressure transducer was inserted and advanced in the LV cavity. Data were acquired with Chart 5 or LabChart8 (ADInstruments) software.
For combined autonomic blockade, atropine (0.5 mg/kg body weight, intraperitoneally; Hospira , Lake Forest, IL) plus propranolol (1 mg/kg body weight, intraperitoneally; Fluka Chemie GmbH, Buchs, Switzerland) were administered to animals.20, 26 Compounds were dissolved in USP saline solution.
To establish the propensity to develop arrhythmias, mice were treated with caffeine (120 mg/kg body weight, intraperitoneally; Sigma‐Aldrich) and epinephrine (2 mg/kg body weight, intraperitoneally; Sigma‐Aldrich).27, 28, 29 Compounds were dissolved in USP saline solution. Occurrence of arrhythmia was monitored in anesthetized control (Ctrl) and STZ mice for 19.6±5.3 (n=7) and 19.6±3.4 minutes (n=7) after administration of drugs, respectively. By telemetry, occurrence of arrhythmias in conscious mice was monitored for 90 minutes after treatment with caffeine and epinephrine.
Ex Vivo Properties of the Mouse Heart
To assess ex vivo electrical properties, hearts were perfused through the aorta in a Langendorff apparatus (Radnoti LLC, Monrovia, CA) at a constant pressure of 80 mm Hg with Krebs–Henseleit buffer (KHB; Sigma‐Aldrich), containing (in mmol/L): 118 NaCl, 4.7 KCl, 11 glucose, 1.2 MgSO4, 1.2 KH2PO4, 1.8 CaCl2 and 25 NaHCO3, gassed with 95% O2 and 5% CO2 (pH 7.4) at 37°C.19, 20 Temperature was maintained by immersing the heart in a water‐heated glassware reservoir (Radnoti LLC), containing preheated KHB. Hearts were stimulated at 125‐ms basic cycle length with a 2‐ms square pulse at 1.5‐fold its threshold level (4‐channel stimulator, BMS 414; Crescent Electronics, Sandy, UT), using a minicoaxial electrode (Harvard Apparatus, Cambridge, MA). Monophasic action potentials (MAPs) were recorded using a micro‐MAP‐Tip electrode (Harvard Apparatus) initially positioned on the apical and then on the basal region of the epicardium of the LV free wall. Subsequently, the LV was opened and the MAP electrode was positioned on the endocardial aspect of the LV free wall. MAP signal was amplified (Animal Bio Amp; ADInstruments), digitized by a 4‐kHz A/D converter (Power Lab 8/30; ADInstruments), and recorded with LabChart software, with low‐ and high‐pass filtering at 1 kHz and 0.3 Hz, respectively. Data were analyzed with LabChart software.
Myocyte Isolation and Patch‐Clamp Studies
With the animals under deep anesthesia (isoflurane), hearts were excised and LV myocytes were enzymatically dissociated as reported previously.19, 20, 25, 30 Briefly, the heart was connected to a plastic cannula for retrograde perfusion through the aorta in a Langendorff system (Radnoti LLC) at 37°C at constant flow. Perfusate consisted of a Ca2+‐free solution gassed with 85% O2 and 15% N2. After 5 minutes, 0.1 mmol/L of CaCl2, 274 units/mL of collagenase (type 2; Worthington Biochemical Corporation, Lakewood, NJ), and 0.57 units/mL of protease (Type XIV; Sigma‐Aldrich) were added to the solution, which contained (in mmol/L): NaCl 126, KCl 4.4, MgCl2 5, HEPES 5, glucose 22, taurine 20, creatine 5, Na pyruvate 5, and NaH2PO4 5 (pH 7.4). At completion of digestion, atria and right ventricle were dissected and free wall of the LV was cut in small pieces, and these fragments were shaken in resuspension solution and filtered using a 200‐μm nylon mesh (Spectrum Laboratories Inc, Piscataway, NJ). For electrophysiological studies, only rod‐shaped myocytes exhibiting cross‐striations and showing no spontaneous contractions or contractures were selected; cells were used within 8 hours after enzymatic digestion.
Isolated LV myocytes were placed in a bath on the stage of an IX51, IX53, or IX71 (Olympus, Tokyo, Japan) microscopes for patch‐clamp measurements.19, 25, 30, 31 Experiments were conducted at 37°C. Data were acquired by means of the whole‐cell patch‐clamp technique in voltage‐ and current‐clamp modes using Multiclamp 700A, 700B, and Axoclamp 900A amplifiers (Molecular Devices, Sunnyvale, CA). Electrical signals were digitized using 250‐kHz 16‐bit resolution A/D converters (Digidata 1322, 1440A, and 1550; Molecular Devices) and recorded using pCLAMP 9.0 and 10 software (Molecular Devices) with low‐pass filtering at 2 kHz. Membrane capacitance (Cm) was measured in voltage‐clamp mode using a 5‐mV voltage step and pCLAMP software algorithm; this parameter was employed to normalize transmembrane currents.19, 20, 25, 30 Pipettes were pulled by means of a vertical (PB‐7; Narishige International Inc., East Meadow, NY), or horizontal (P‐1000; Sutter Instrument Co, Novato, CA) glass microelectrode pullers.
For AP measurements, current‐clamp mode was employed.19, 20, 30, 31 Cells were stimulated with current pulses ≈1.5 times threshold. Myocytes were bathed with Tyrode solution containing (in mmol/L): NaCl 140, KCl 5.4, MgCl2 1, HEPES 5, glucose 5.5, and CaCl2 1 (pH 7.4, adjusted with NaOH). The composition of the pipette solution was (in mmol/L): NaCl 10, KCl 113, MgCl2 0.5, K2‐ATP 5, glucose 5.5, HEPES 10, EGTA 10, and CaCl2 1 (pH 7.2 with KOH). To test the effects of inhibition of Kv currents, APs were continuously recorded for the same cells before and after exposure to 4‐aminopyridine (4‐AP; 0.1 mmol/L).30, 32, 33, 34, 35 At this concentration level, 4‐AP has been previously reported to abolish the large, slowly inactivating phase of the Kv‐based IK,slow 32, 34, 35 leading to prolongation of AP.33 To test the acute effects of high glucose, APs were continuously recorded for the same cells before and after exposure to a Tyrode solution containing 20 mmol/L of glucose. Experiments testing the effects of high glucose were performed at room temperature.
Voltage‐gated outward K+ Kv currents were assessed in voltage‐clamp mode with Tyrode solution containing 0.3 mmol/L of CdCl2 to block L‐type Ca2+ current.30 Current‐voltage (I‐V) relations were determined applying depolarizing steps in the range from −50 to +80 mV, in 10‐mV increments, 500 ms in duration. Test steps were preceded by a prestep to −30 mV, 20 ms in duration, from holding potential (Vh) of −70 mV. Amplitude of Kv currents was measured as the difference between the peak outward current at the beginning of the test step and Iss, the current at the end of the 500‐ms step.30 Currents were normalized by Cm.
L‐type Ca2+ current (ICaL) was measured in voltage‐clamp mode.20, 25, 30 Myocytes were bathed with a modified Na+‐K+–free Tyrode solution of the following composition (in mmol/L): N‐methyl‐d‐glucamine (NMDG) 140, CsCl 4, MgCl2 1, HEPES 5, glucose 5.5, CaCl2 1, and 4‐AP 2 (pH 7.4 with CsOH). Composition of the pipette solution was (in mmol/L): NMDG 10, CsCl 113, MgCl2 0.5, Tris‐ATP 5, glucose 5.5, HEPES 10, EGTA 5, and TEA‐Cl 20 (pH 7.2 with CsOH). ICaL current‐voltage (I‐V) relation and activation properties were determined applying depolarizing steps 300 ms in duration from Vh −70 mV in 10‐mV increments. ICaL amplitude was measured as the current difference between the peak inward current at the beginning of the step and the current at the end of the 300‐ms pulse. ICaL was normalized by Cm.
Equilibrium potential for Ca+ was defined by the intercept of the I‐V relation to the zero‐current axis. A linear fitting of the positive potential portion of the I‐V relation was employed for this determination.20 At each potential tested (Vm), Ca2+ conductance (g) was calculated as
where ICaL is the amplitude of the L‐type Ca2+ current at Vm.
Values of normalized Ca2+ conductance (G=g/gmax) were plotted to obtain activation curves; half maximal activation potential (V1/2G) and slope of the activation curve (kG) were derived by fitting the data with the Boltzmann equation:
A 2‐pulse protocol was utilized to assess the voltage dependence of steady‐state inactivation of ICaL. Prepulses were introduced to depolarize the cell to different membrane voltages starting from −80 mV for 300 ms, in 10‐mV increments. Each prepulse was followed by a single 300‐ms test pulse, which depolarized the cell to 0 mV. Values of normalized Ca2+ conductance (G=g/gmax) were plotted with Vm relative to the preconditioning steps to determine steady‐state inactivation curves, fitted with a Boltzmann equation, as indicated above.
Inactivation time course of ICaL was studied with a 1000‐ms depolarizing pulse at 0 mV.30 Traces were fitted between the inward peak and to 500 ms from the beginning of the pulse to 0 mV with a biexponential function20, 31:
where Af and As are, respectively, the fraction of the fast and slow inactivating components, τf and τs are the time constants of the fast and slow inactivating component, and C is the offset constant.
ICaL reactivation was studied using a protocol consisting of 4 preconditioning pulses at 1‐Hz depolarizing cells from Vh −70 to 0 mV for 50 ms. The test pulse was elicited with progressive delay, ranging from 50 to 500 ms, in increaments of 50 ms. With each test pulse, ICaL was activated by depolarizing cells from Vh −70 to 0 mV for 200 ms. For each interpulse duration (t), ICaL reactivation was calculated by dividing the amplitude of the current measured during the test pulse (I(t)) by the maximal amplitude of the current measured with prolonged interpulse duration (Imax). Values of ICa reactivation were plotted to obtain reactivation curves and fitted with a monoexponential function:
Quantification of Temporal Variability
Variability of electrocardiographic parameters was computed using SD and RSD (RSD=(SD/mean)×100)22, 36, 37 on 10 consecutive series of 50 beats that were averaged to reduce electrical noise and motion artifacts. To minimize the confounding impact of severe functional defects and structural remodeling of the myocardium, the analysis of temporal variability was initially restricted to STZ‐treated animals up to 2 months after onset of diabetes. Similarly, 7‐month‐old Akita mice were studied because EF was preserved at this age.
Variability of AP repolarization was computed using SD, RSD, and short‐term variability (STV)22, 36, 37 on 70 consecutive waveforms of the 100 APs obtained from each cell in the control and diabetic groups. STV was calculated using the following formula:
where APD is the duration of the duration of the AP at a given repolarization level. For experiments in which myocytes were exposed to 4‐AP to inhibit Kv currents, 10 consecutive waveforms at steady state after drug application were used for the analysis of variability.
Newly Developed Repolarization Analysis
A newly developed algorithm for assessment of the repolarization phase was employed in this study. This method, based on fitting of the AP decay, was introduced to obtain detailed information on the repolarization properties of myocytes. By this strategy, the attempt was made to identify recovery abnormalities and temporal dispersion throughout the entire repolarization phase of the AP, with the exception of the final portion, from 95% of repolarization to return to resting membrane potential. A signal‐processing method previously used for ECG recordings38 was adapted for analysis of APs. Briefly, Axon binary files containing a sequence of APs in the form of sequential sweeps were converted into text files. Subsequently, data were imported in Matlab (MathWorks, Inc., Natick, MA) software as matrices with size b x S, where b is the number of sequential sweeps (APs) and S is the number of samples collected during each sweep. Each repolarization phase of the AP was modeled using the following steps: (1) segmentation of the repolarization phase of the AP and normalization of the AP amplitude in the interval [0–1]; (2) fitting of repolarization intervals based on least square approximation, subjected to monotonically decreasing behavior constraints38; and (3) interpolation of the reciprocal functions of the obtained fitting over a fixed range of the AP repolarization. Details are reported in Data S1.
For each myocyte, the obtained values of duration of repolarization segments for sequential APs computed with the algorithm were then employed to calculated average AP profile and degree of beat‐to‐beat variability. Parameters obtained in each cells were then used to compute average AP profile for control myocytes, STZ‐myocytes and for cells before and after exposure to 4‐AP or high glucose. Similarly, fold change and statistical comparison between 2 experimental groups were calculated. Repolarization properties were graphically visualized using Prism software, with repolarization levels on the y axis and repolarization time on the x axis.
Data Analysis
Data are presented as median and interquartile ranges (IQRs) or mean±SEM. Linear regressions and Pearson's correlation coefficient were calculated with Prism software. Statistical analysis was performed using SigmaPlot 11.0. Data were initially tested for normality (Shapiro–Wilk) and equal variance for assignment to parametric or nonparametric analysis. Parametric tests included Student t test or paired t test for nonpaired or paired comparison between 2 sets of data, respectively. When normality or equal variance were not met, nonparametric analysis was performed using the Mann–Whitney rank‐sum test or the Wilcoxon signed‐rank test for nonpaired or paired comparison between 2 sets of data, respectively. For categorical data analysis, Fisher's exact test was used.20 P<0.05 was considered significant.
Results
Hyperglycemia and Diabetic Cardiomyopathy
To define the effects of elevated glucose levels on cardiac functions, Ctrl and STZ‐injected diabetic (STZ) female mice were monitored by echocardiography at ≈2 weeks, ≈1 month, and ≈2 months from onset of the hyperglycemic state. EF was initially preserved in STZ mice, but decreased by 12% at 2 months, together with an increase in diastolic LV volume (Figure 1A). By transmitral flow Doppler echocardiography, passive LV filling (E wave) was reduced, whereas deceleration time was prolonged in STZ mice, at 2 weeks and 1 month, with respect to Ctrl (Figure 1B and 1C). Thus, impairment of diastolic function with diabetes precedes the reduction in EF. Additionally, by hemodynamics and PV measurements in the closed‐chest preparation at 2 to 3 months after onset of hyperglycemia, developed pressure was comparable in STZ and Ctrl mice. However, +dP/dt was attenuated and the time constant of pressure decay (tau) was prolonged in the STZ group. Despite these alterations, stroke volume and cardiac output were preserved in diabetic animals (Figure 1D).
Figure 1.
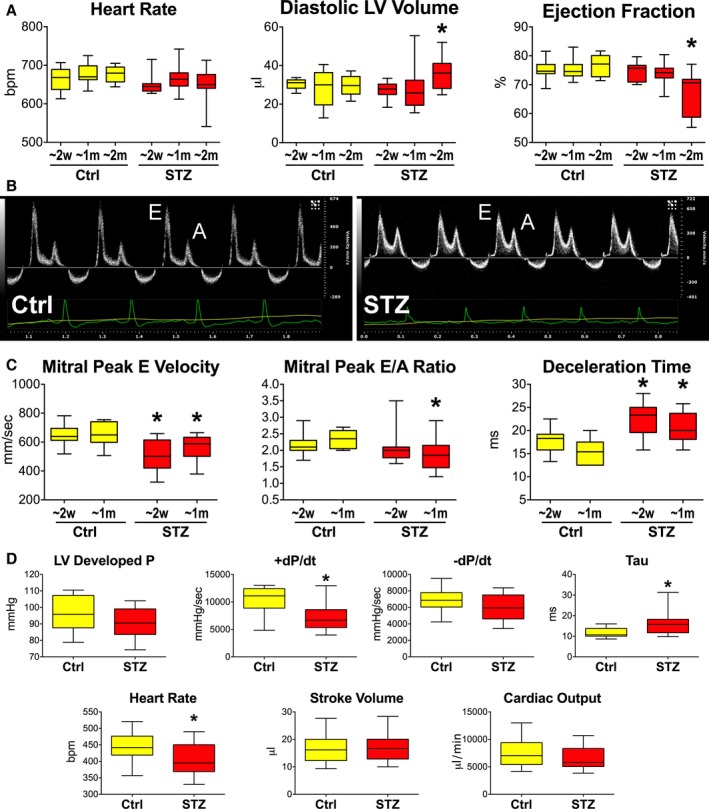
Hyperglycemia is accompanied by a progressive deterioration of cardiac function. A, Echocardiographic parameters in female streptozotocin (STZ) mice (STZ) at ≈2 weeks (≈2 weeks, n=11), ≈1 month (≈1 month; n=20), and ≈2 months (≈2 months; n=14) after onset of hyperglycemia and in control (Ctrl) mice (≈2 weeks, n=11; ≈1 month, n=20; ≈2 months, n=10). Quantitative data are shown as median and interquartile ranges (IQRs). *P<0.05 vs corresponding time point in Ctrl. B, Transmitral flow Doppler echocardiograms from Ctrl and STZ female mice. E, Early passive filling wave; A, active filling wave. C, Quantitative data for Ctrl (≈2 weeks, n=11; ≈1 month, n=8) and STZ female mice at ≈2 weeks (≈2 weeks; n=10) and ≈1 month (≈1 month; n=10) after onset of hyperglycemia. Results are shown as median and IQRs. *P<0.05 vs corresponding time point in Ctrl. D, Hemodynamic and volumetric parameters obtained by PV catheterization in Ctrl (n=13) and STZ female mice 60 to 100 days (mean, 83.7±3.1 days) after onset of hyperglycemia (n=13). Data are shown as median and IQR. *P<0.05 vs Ctrl. bpm indicates beats per minute; LV, left ventricular; P, pressure.
To establish whether hyperglycemia affected electrical activation and recovery of the myocardium before changes in EF become apparent, surface ECGs were acquired. In diabetic hearts at 2 weeks, atrioventricular conduction (PR interval) and ventricular activation (QRS interval) were similar to Ctrl. In contrast, indices of electrical recovery (QT interval and heart‐rate–adjusted QTc) were prolonged in diabetic mice by 16.9% and 14.4%, respectively (Figure 2A and 2B). These alterations were maintained at the later time point (≈1 month; Figure 2C).
Figure 2.
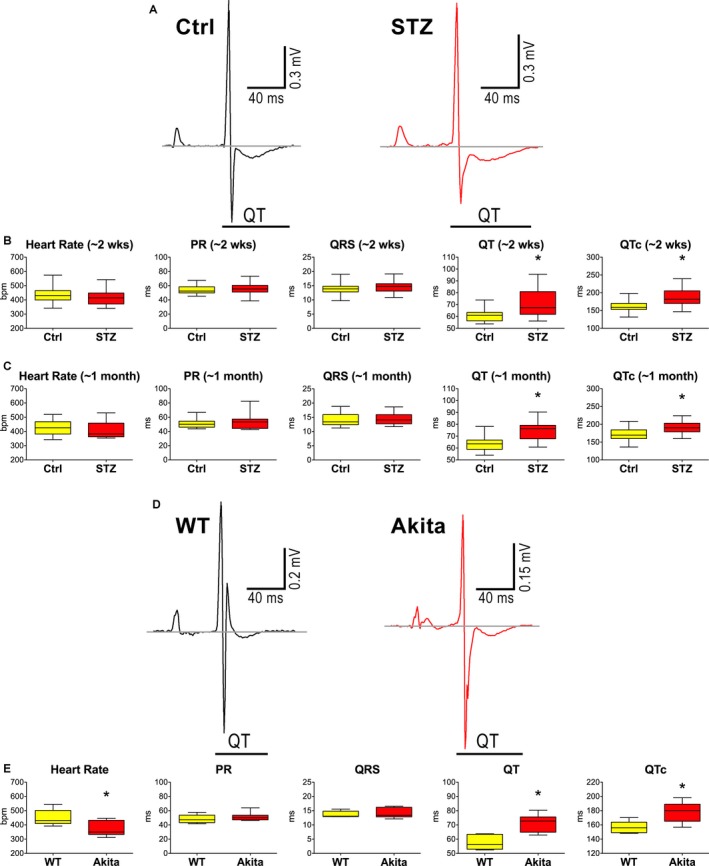
Hyperglycemia leads to protracted electrical recovery of the heart in vivo. A, Electrocardiograms obtained in control (Ctrl) and streptozotocin (STZ)‐treated (STZ) female mice. B, Electrocardiographic parameters in Ctrl (n=23) and STZ (n=22) female mice at 14 to 18 days (≈2 weeks) after onset of hyperglycemia are shown as median and interquartile ranges (IQRs). *P<0.05 vs Ctrl. C, Electrocardiographic parameters in Ctrl (n=11) and STZ (n=27) female mice at 31 to 38 days (≈1 month) after onset of hyperglycemic condition are shown as median and IQRs. *P<0.05 vs Ctrl. D, ECGs obtained in wild‐type (WT) and male Akita mice. E, Electrocardiographic parameters in wild‐type (WT, n=6) and male Akita (n=7) mice at 7 months of age are shown as median and IQRs. *P<0.05 vs WT. wks indicates weeks.
To corroborate findings observed in mice with diabetes induced by STZ administration and to reduce the potential confounding off‐target effects of pharmacological treatment, we studied male Akita mice, a rodent model of naturally occurring diabetes type I attributable to a spontaneous mutation of the insulin 2 gene that causes misfolding of the insulin protein and hyperglycemia (see Figure S1).39 With respect to age‐ and sex‐matched wild‐type (WT) animals, Akita mice displayed prolonged QT interval and QTc (Figure 2D and 2E), whereas cardiac function, evaluated by echocardiography, was relatively preserved (Figure S2). Thus, hyperglycemia is characterized early by delays in electrical recovery of the myocardium and diastolic dysfunction, whereas systolic defects occur later in the process.
Hyperglycemia and Electrical Disturbances
Clinically, there are several inherited and acquired conditions, including diabetes,4, 5, 9, 10, 12, 13 that are characterized by prolongation of the QT interval, a factor that enhances the incidence of ventricular arrhythmia and sudden cardiac death.14 Moreover, increased dispersion of ventricular repolarization occurs in diabetic patients5, 6, 7, 8, 10 with an important implication on propensity to develop electrical disturbances.4, 13, 15, 16 To test the possibility that prolonged electrical recovery with hyperglycemia was associated with increased repolarization variability and rhythm disturbances, the degree of temporal dispersion of myocardial repolarization and susceptibility to develop arrhythmias were quantified. In both STZ‐treated and Akita mice, hyperglycemia was coupled with an increase in QT and QTc variability, evaluated by SD and RSD22, 36, 37 (Figure 3A through 3C). Additionally, because diabetes is associated with autonomic imbalance,40 an important determinant of QT variability,41 ECGs were obtained in control and chronic diabetic mice after complete autonomic blockade with atropine and propranolol.26 In spite of this intervention, QT interval and QTc and respective temporal variability remained elevated in diabetic animals (Figure 3D and 3E and Figure S3).
Figure 3.
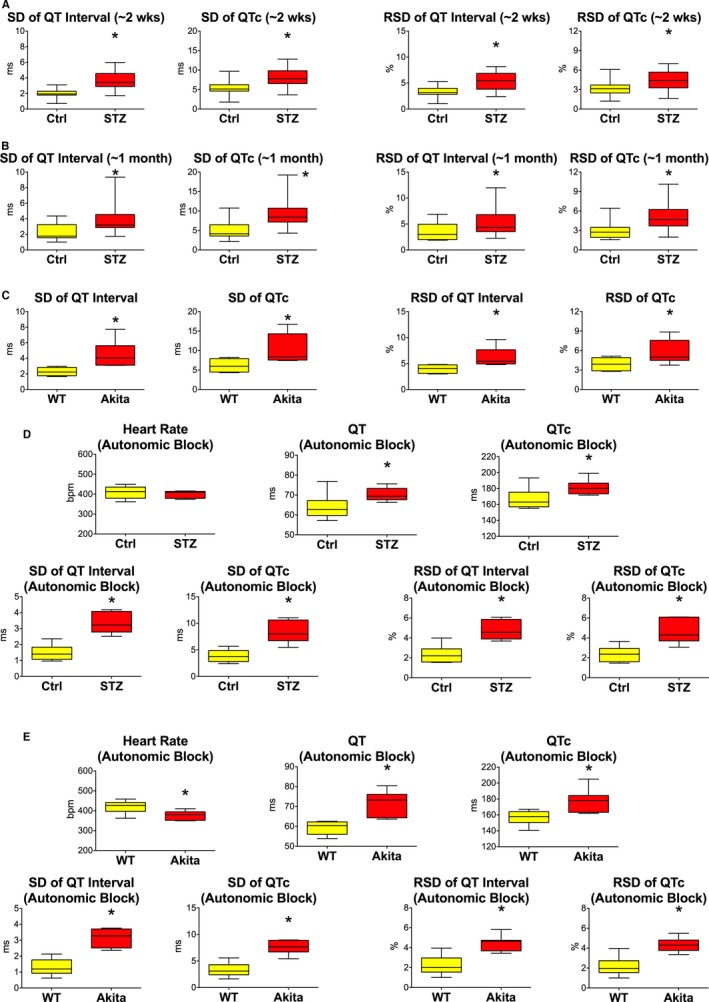
Hyperglycemia enhances temporal dispersion of electrical recovery. A and B, Variability of the electrical recovery evaluated by SD and RSD in control (Ctrl) and streptozotocin (STZ) female mice at ≈2 weeks (A, same animals as in Figure 2B) and ≈1 month (B, same animals as in Figure 2C) after onset of the hyperglycemia is shown as median and interquartile ranges (IQRs). *P<0.05 vs Ctrl. C, Variability of the electrical recovery evaluated by SD and RSD in WT and Akita male mice (same animals as in Figure 2E) is shown as median and IQRs. *P<0.05 vs WT. D, Electrocardiographic parameters and variability of repolarization assessed after complete autonomic block in Ctrl (n=10) and STZ (n=6) female mice at 168 to 183 days after onset of hyperglycemic conditions are shown as median and IQRs. *P<0.05 vs Ctrl. E, Electrocardiographic parameters and variability of repolarization assessed after complete autonomic block in WT (n=6) and Akita (n=7) male mice at 9 months of age are shown as median and IQRs. *P<0.05 vs WT. bpm indicates beats per minute.
To establish the propensity of diabetic mice to develop ventricular arrhythmias, ECGs were evaluated in anesthetized Ctrl and STZ mice before and after administration of caffeine and epinephrine.27, 28, 29 This intervention induced ventricular tachycardia (VT) in 1 of the 7 Ctrl mice, whereas in the majority of diabetic animals (6 of 7), it elicited a bidirectional pattern of ventricular activation (Figure 4A through 4C), previously identified as bidirectional VT (BVT).28, 29, 42 BVT was associated with a reduction in RR interval (Figure 4D) coupled with enhanced sinus discharge. Occurrence of BVT in STZ animals after administration of caffeine and epinephrine was also confirmed in conscious mice by telemetry (Figure 4E). Collectively, these data indicate that hyperglycemia leads to prolongation of the electrical recovery, enhances dispersion of ventricular repolarization, and increases susceptibility to develop ventricular arrhythmia.
Figure 4.
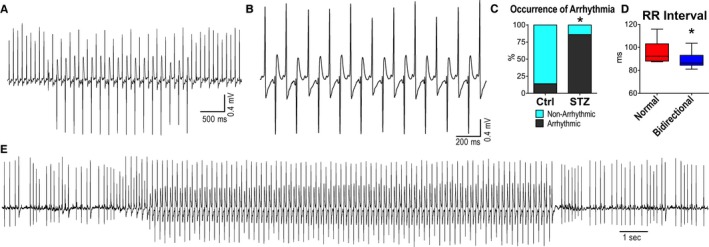
Hyperglycemia enhances the propensity to develop arrhythmias. A and B, Electrocardiographic recordings of sustained bidirectional ventricular tachycardia (VT) in a streptozotocin (STZ)‐treated female mouse after administration of caffeine and epinephrine, under anesthesia. C, Occurrence of ventricular arrhythmias in control (Ctrl; n=7) and STZ (n=7; 13–24 days after STZ administration) anesthetized female mice. *P<0.05 vs Ctrl. D, RR interval duration before (normal) and during the appearance of bidirectional VT (bidirectional) in STZ female mice (n=6; 13–24 days after STZ administration). *P<0.05 vs normal. E, Telemetric ECG recording of sustained bidirectional VT in a conscious diabetic female mouse, 28 days after STZ administration. The mouse was treated with caffeine and epinephrine. After drug administration, bidirectional VT was observed in 1 of the 2 diabetic female mice implanted with telemetry. Arrhythmia was not observed after treatment in 4 Ctrl female mice.
Hyperglycemia and Myocyte Electrical Properties
To determine whether alterations in electrophysiological properties of cardiomyocytes mirrored the electrical defects observed in vivo, isolated cell preparations from the LV free wall of STZ and Ctrl mice were studied by patch‐clamp. The standard analysis of AP characteristics19, 30 was complemented with a newly developed algorithm for assessment of the repolarization phase.
At a 2‐Hz stimulation rate, resting membrane potential, AP amplitude, and dV/dtmax were comparable in control and diabetic myocytes. In contrast, dV/dtmin was reduced by 63% whereas the duration of AP at 10%, 30%, 50%, and 70% were significantly prolonged in diabetic cells (Figure 5A and 5B). Using the newly developed algorithm, an uninterrupted prolongation of AP duration was found in diabetic myocytes, in the range comprised from 0.19%, which corresponds to the first analyzed data point of the repolarization, to 79.4% of the repolarization phase (Figure 5C). Prolongation of the AP varied from 1.2‐ to 2.8‐fold, with respect to Ctrl cells, reaching a maximum at 23.2% of the repolarization phase. Importantly, values of duration of repolarization computed with the newly developed approach were similar to results obtained at discrete repolarization levels, determined with standard analysis (see Figure 5B); this correspondence was documented by linear regression and Pearson's correlation test between the 2 approaches (Figure S4). Additionally, exposure of Ctrl myocytes to 20 mmol/L of glucose, a concentration of the sugar comparable to levels found in diabetes, had minimal consequences on duration of AP (Figure S5), suggesting that chronic exposure to high‐glucose in vivo induces electrical remodeling in cardiomyocytes.
Figure 5.
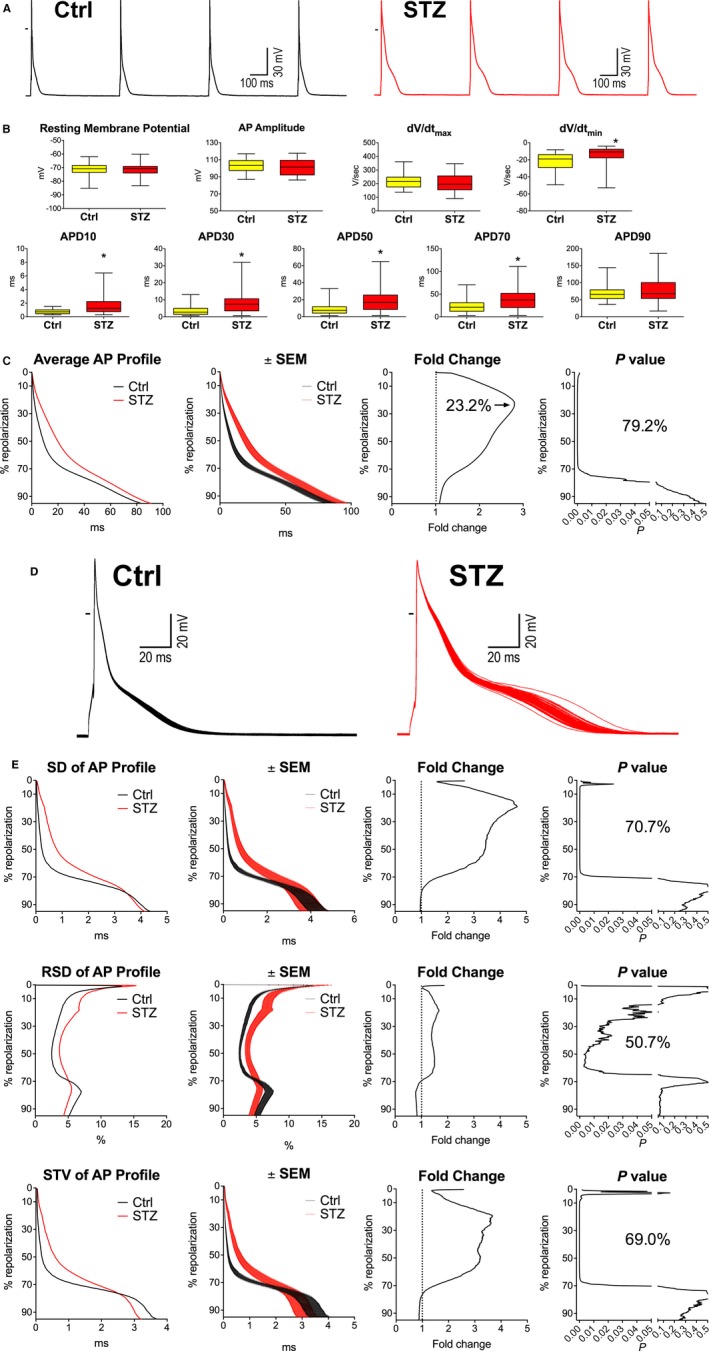
Hyperglycemia alters electrical properties of cardiomyocytes. A, APs recorded at 2‐Hz pacing rate in cardiomyocytes isolated from control (Ctrl) and streptozotocin (STZ)‐treated female mice. B, AP properties of Ctrl myocytes (n=41; from 20 mice) and myocytes obtained from STZ‐treated female animals (n=39; from 14 mice at 12–60 days after onset of hyperglycemia) stimulated at 2 Hz are shown as median and interquartile ranges. *P<0.05 vs Ctrl. C, Analysis and comparison of repolarization properties of APs reported in (B) using the novel algorithm. Graph of average AP profile represents the calculated average repolarization phase of the AP for Ctrl and STZ myocytes. Corresponding error bars, fold changes for the duration of the AP in STZ vs Ctrl cells, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively. D, Superimposed sequential APs collected in Ctrl and STZ myocytes. E, Variability of repolarization of the AP evaluated by SD, RSD, and STV in Ctrl and STZ myocytes reported in (B) using the novel algorithm. Graphs of SD, RSD, and STV of the AP profile represent the calculated average variability of the repolarization phase of the AP for Ctrl and STZ myocytes. Corresponding error bars, fold changes for the variability of the AP in STZ vs Ctrl cells, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively. AP indicates action potential; STV, short‐term variability.
To establish whether the enhanced variability of electrical recovery in diabetic mice was associated with increased temporal dispersion of the AP in cardiomyocytes, SD, RSD, and short‐term variability (STV)22, 36, 37 were calculated for 70 consecutive waveforms of the 100 APs obtained from each cell (Figure 5D). The initial 30 excitation waveforms were excluded from the analysis to allow the AP profile to reach a steady‐state configuration in response to commencement of the stimulation protocol. Using the 3 parameters of variability, repolarization was found to be more heterogeneous in diabetic myocytes; however, dissimilarities were detected in progressively broader repolarization ranges when RSD, STV, and SD were employed, respectively (Figure 5E). The enhanced temporal dispersion of the AP in diabetic cardiomyocytes covered a range comprising 50.7% to 70.7% of the whole repolarization.
In a subset of cells, APs were acquired at 2, 4, and 6 Hz to establish whether the mechanisms of rate adaptation of AP in Ctrl and diabetic myocytes enhanced or diminished differences between the electrical behaviors of the 2 cell types. At faster pacing rates, prolongation of AP of diabetic myocytes covered a broader range of repolarization (Figure 6). Similarly, the enhanced temporal dispersion of AP of diabetic cells, evaluated with SD and STV, involved a larger fraction of the repolarization, which, however, was not observed with RSD (Figure S6).
Figure 6.
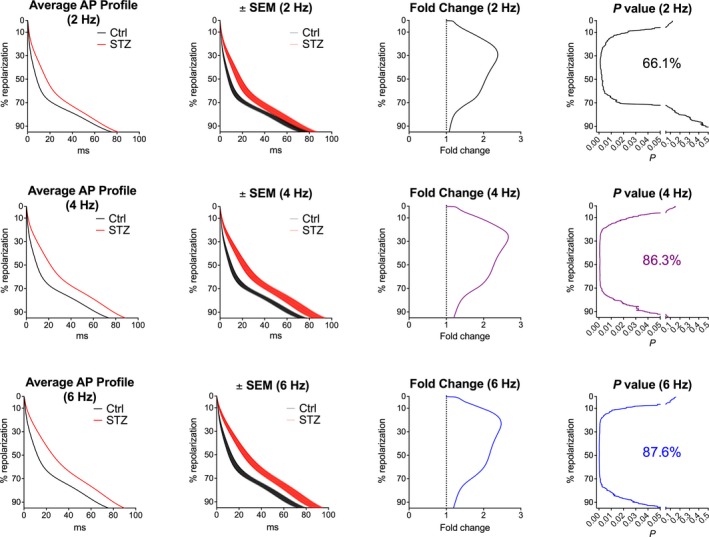
Hyperglycemia and AP properties at different pacing rates. Analysis and comparison of repolarization properties of AP using the novel algorithm. Control (Ctrl) myocytes (n=22; from 12 mice) and myocytes obtained from streptozotocin (STZ) ‐treated female mice (n=27; from 12 mice at 18–45 days after onset of hyperglycemia) were stimulated at 2‐, 4‐, and 6‐Hz pacing rates. Graphs of average AP profile represent the calculated average repolarization phase of the AP for Ctrl and STZ myocytes. Corresponding error bars, fold changes for the duration of the AP in STZ vs Ctrl cells, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively. AP indicates action potential
In the attempt to define the impact of the protracted duration of AP on the electrical recovery of the intact myocardium, local MAPs were evaluated in a small cohort of perfused hearts in the apical and basal regions of the epicardial LV free wall and in the endocardium. MAP duration at 50% repolarization was increased in the apex of hearts from STZ mice, with respect to Ctrl, whereas electrical signals from other anatomical regions did not differ significantly (Figure 7). These initial results (1) link the alterations of the repolarization properties of cardiomyocyte to the defective electrical recovery observed in vivo and (2) may be indicative of a reduced spatial electrical gradient in diabetic hearts.
Figure 7.
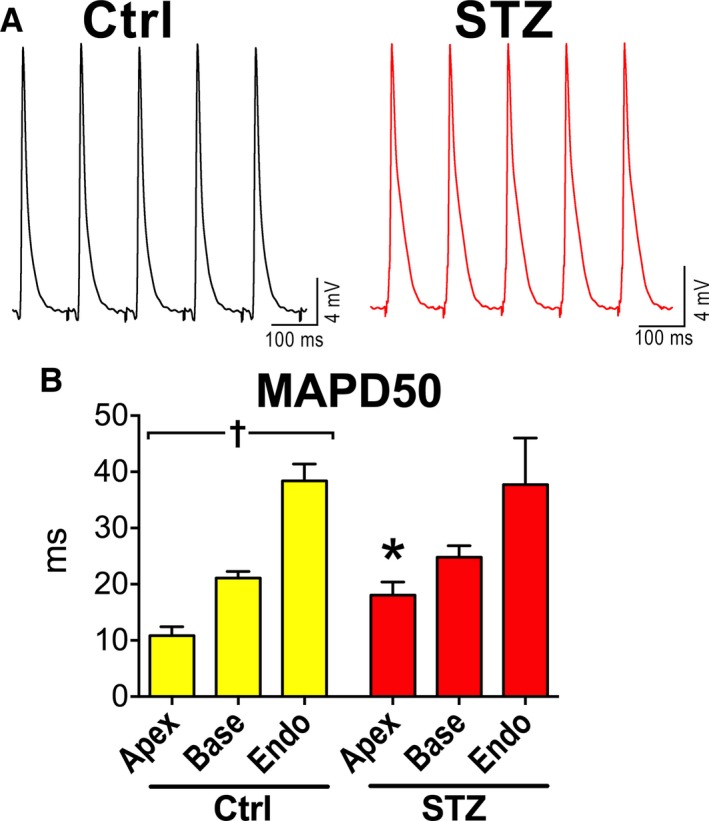
Hyperglycemia and myocardial electrical properties. A, Monophasic action potentials obtained in the apical region of the epicardial left ventricular (LV) free wall of hearts from control (Ctrl) and streptozotocin (STZ) female mice. B, Duration of monophasic action potentials at 50% repolarization (MAPD50) in the apical (Apex) and basal (Base) regions of the epicardial aspects and endocaridal (Endo) aspect of the LV free wall in hearts from Ctrl (n=10; Apex n=9; Base, n=10; Endo, n=9) and STZ female mice (n=5; 12–30 days after induction of diabetes; Apex, n=5; Base, n=4; Endo, n=4). Data are reported as mean±SEM. *P<0.05 vs Ctrl; † P<0.05 within the group.
Hyperglycemia and Ionic Current Remodeling
To define the ionic basis for the observed prominent prolongation of the early and intermediate repolarization phase of the AP of diabetic myocytes, the density of Kv K+ and L‐type Ca2+ (ICaL) currents, operative in this range of potentials, were tested. In agreement with previous findings with other experimental models of diabetes,43, 44, 45, 46, 47, 48 outward Kv currents and ICaL were significantly reduced in STZ myocytes (Figure 8A through 8D and Figure S7). These alterations may respectively promote and oppose prolongation of AP duration. Thus, these findings point to Kv currents as an important determinant for the delayed repolarization phase of AP occurring with hyperglycemia.
Figure 8.
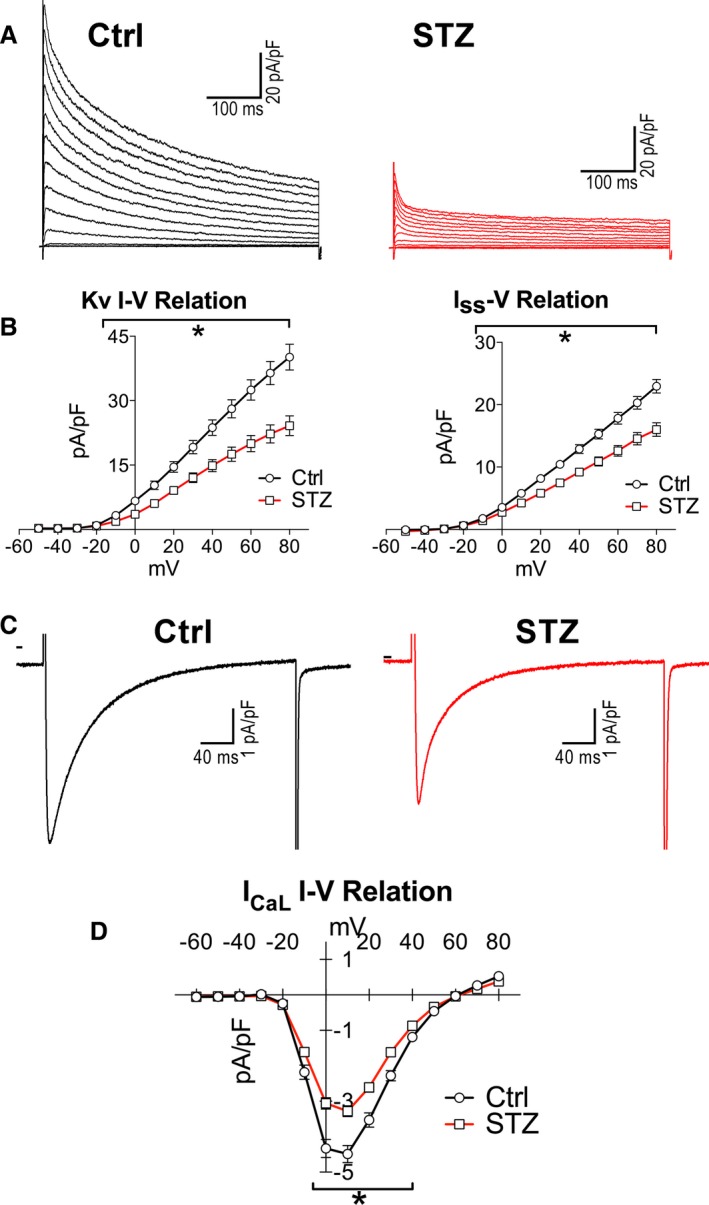
Hyperglycemia reduces Kv currents. A, Whole‐cell voltage‐gated K+ current recorded in voltage clamp in 1 control (Ctrl) and 1 streptozotocin (STZ) myocyte. B, I‐V relation for Kv currents and Iss for Ctrl myocytes (n=25; from 4 mice) and myocytes obtained from STZ‐treated female mice (n=27; from 10 mice at 12–30 days after onset of hyperglycemia) are shown as mean±SEM. *P<0.05 vs Ctrl. C, Whole‐cell voltage‐gated Ca2+ current recorded in voltage clamp at +10 mV in 1 Ctrl and 1 STZ myocyte. D, I‐V relation for IC aL for Ctrl myocytes (n=21; from 3 mice) and myocytes obtained from STZ‐treated female mice (n=22; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. *P<0.05 vs Ctrl. Additional properties of IC aL are reported in Figure S7.
In the attempt to define the role of diminished outward Kv currents on the prolongation of AP and enhanced beat‐to‐beat variability observed in diabetic myocytes, outward K+ currents in control mouse myocytes were inhibited with 4‐AP (0.1 mmol/L).32, 33, 34, 35 This pharmacological intervention led to a prolongation of AP comprising 95% of the whole repolarization, with a maximum of 5.3‐fold increase in duration at 15.6% of the repolarization (Figure 9A and 9B). Interestingly, 4‐AP enhanced repolarization variability (Figure 9C), paralleling the behavior of diabetic myocytes. Thus, the remodeled AP profile and temporal dispersion of the repolarization of diabetic myocytes is partly mimicked by exposure to 4‐AP, supporting the notion that the reduction of Kv currents in myocytes contributes to the altered electrical properties of the diabetic heart.
Figure 9.
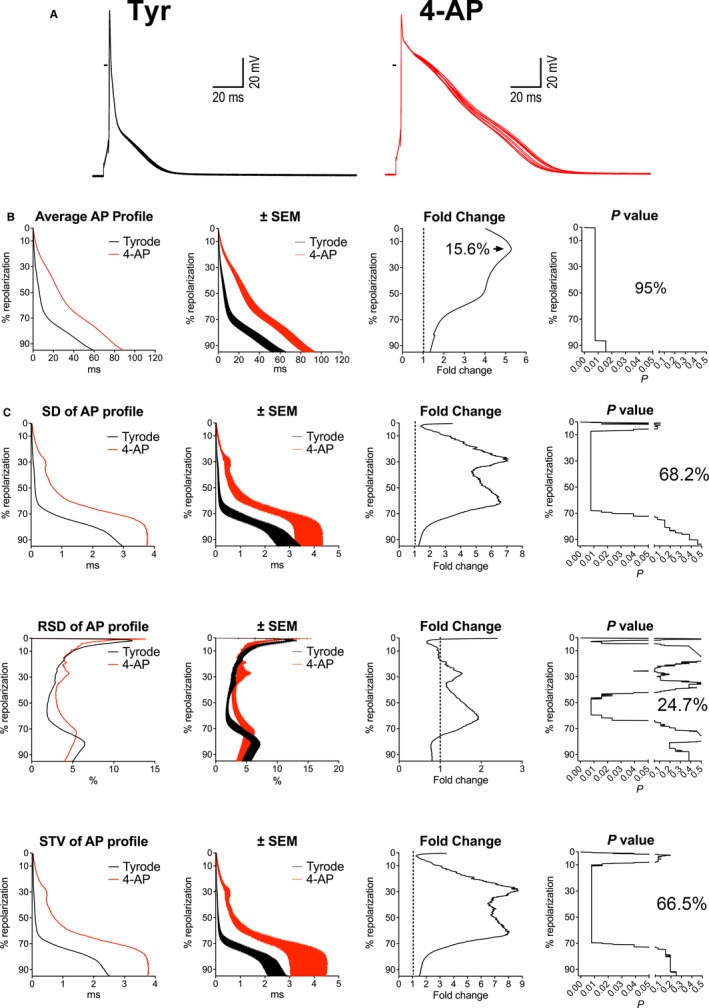
Inhibition of Kv currents prolongs the AP and enhances beat‐to‐beat variability. A, Superimposed sequential APs of a nondiabetic myocyte before and after exposure to 0.1 mmol/L of 4‐AP. B, Analysis and comparison of repolarization properties of the AP using the novel algorithm, for myocytes obtained from female mice before and after exposure to 4‐AP (n=8; from 5 mice). Graph of average AP profile represents the calculated average repolarization phase of the AP in myocytes before and after exposure to 4‐AP. Corresponding error bars, fold changes for the duration of the AP in 4‐AP vs Tyrode (Tyr), and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively. C, Variability of repolarization of the AP before and after exposure of 4‐AP evaluated by SD, RSD, and STV in cells reported in (B), using the novel algorithm. Graphs of SD, RSD, and STV of AP profile represent the calculated average variability of the repolarization phase of the AP for myocytes in Tyrode and after exposure to 4‐AP. Corresponding error bars, fold changes for the variability of the AP collected in 4‐AP vs Tyrode, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively. 4‐AP indicates 4‐aminopyridine; AP, action potential; STV, short‐term variability.
Discussion
The results of the current study indicate that hyperglycemia leads to prolongation and enhanced temporal dispersion of the repolarization phase of AP in myocytes. These defects at the cellular level are paralleled, in vivo, by delayed ventricular repolarization, increased variability of electrical recovery, and enhanced propensity to develop arrhythmias, features also observed in diabetic patients.3, 4, 5, 6, 7, 8, 9, 10, 12, 13 Densities of Kv currents are reduced in diabetic myocytes contributing, at least in part, to the alteration in repolarization dynamics, modality of rate adaptation, and beat‐to‐beat variability of AP. Thus, changes in Kv currents may constitute an important mediator of electrical remodeling of the diabetic heart.
The STZ mouse model employed here is characterized by an initial decline of diastolic function, which is commonly found in the diabetic population.1, 49, 50, 51, 52 LV filling patterns in STZ‐treated mice are altered in the first month after onset of hyperglycemia, whereas EF is still preserved. The electrical characteristics of the myocardium and cardiomyocytes were determined at this early stage of the disease to avoid the confounding variables of the advanced myopathy,52, 53 where depressed myocyte function, cell death, and myocardial scarring would have interfered with recognition of the consequences of hyperglycemia per se on these physiological parameters. By this approach, we have documented that, together with depressed diastolic performance, reduction in Kv K+ and L‐type Ca2+ current densities, protracted electrical recovery, and enhanced temporal dispersion of repolarization are initial functional defects of the myocardium dictated by diabetes. The process linking reduced ionic currents with the enhanced beat‐to‐beat variability of electrical recovery of the diabetic heart may involve complex multiscale mechanisms, from ion channels to the whole organ.22, 23, 54, 55 The reduced repolarizing effect of Kv currents has attenuated the repolarization reserve of diabetic myocytes, potentially unmasking fluctuations of ionic currents caused by stochastic behavior of ion channel gating, normally concealed by K+ efflux.
Alterations of the spatial heterogeneity of ion channel expression and AP duration have been previously reported in rodent models of diabetes,45, 56, 57 which are consistent, at least in part, with our observation of a preferential prolongation of duration of MAPs in the epicardial/apical region of STZ hearts. The altered regional properties of cardiac tissue may have affected locally the refractoriness of the myocardium, negatively interfering with electrical conduction and pattern of impulse propagation. Although further experiments are warranted, these initial findings suggests that altered spatial heterogeneity of the myocardium with diabetes represents the basis for the alternation in the polarity of the QRS axis observed during episodes of BVT.29, 58
Reductions of the K+ channel subunit, Kv4.2, interacting protein KChIP2, and Ca2+ channel α‐1C subunit have been proposed as molecular substrates for the decreased Kv currents and ICaL in myocytes from rodent models of diabetes.43, 45, 46, 47, 57, 59 Our electrophysiological data support the possibility that similar alterations occur in the STZ‐hyperglycemic mouse model employed here. Importantly, reduction in the outward K+ Kv currents and prolongation of AP duration in mammals appear to be a conserved response to a variety of physio‐ and pathological conditions dictated by an increase in workload of the myocardium, including ischemic cardiomyopathy,60, 61 hypertension,62 pressure overload,63 aging,20, 64 and heart failure.18, 65, 66 Thus, the enhanced beat‐to‐beat variability observed in diabetic myocytes may represent a feature shared with other diseased conditions.
A limitation of our study is related to the use of intracellular Ca2+‐buffered conditions19 in the analysis of AP characteristics. This approach has abrogated the modulatory action of Ca2+ transients on AP profile at different rates and has prevented the manifestation of destabilizing effects of spontaneous Ca2+ releases on transmembrane potential. Enhanced diastolic Ca2+ releases from the sarcoplasmic reticulum have been previously found in myocytes obtained from diabetic rats,67 suggesting that these spontaneous events may have contributed to the heterogeneity of electrical recovery observed in heart of STZ‐treated mice. Although these aspects represent important constraints to our study, we elected to initially dissect the contribution of ionic current remodeling on the electrical behavior of diabetic myocytes. Thus, the reported prolongation of AP and enhanced temporal dispersion of the repolarization phase of diabetic myocytes may represent a conservative evaluation of the destabilizing events occurring in diabetic cells and their implication in occurrence of ventricular arrhythmias. Additionally, because of the large number of tests performed, we cannot exclude that statistical significance by chance alone may have occurred in our analysis.
Alterations of the structural properties and intercellular coupling of the myocardium may have occurred with diabetes,52, 68, 69 weakening the electrotonic interaction between myocytes, which acts as a mechanism of suppression of repolarization variability in situ.54, 70 Thus, intervening structural remodeling may have cooperated with reduced Kv currents in promoting the repolarization heterogeneity found in the diabetic heart. Also, we cannot exclude with certainty that electrical defects observed in vivo are the consequences of local ischemic events secondary to atherosclerosis, a typical feature of the diabetic heart.11
The novel algorithm employed here has allowed us to obtain detailed information on the AP profile of control and diabetic myocytes and to define repolarization ranges in which duration and degree of heterogeneity of AP were critically altered by the hyperglycemic condition. Additionally, the changes in AP profile induced by modulation of Kv currents were outlined and the relationships with the electrical behavior of diabetic myocytes were identified, providing proof of concept that reduction in Kv currents is a critical determinant of the remodeled AP shape occurring with diabetes. The novel analytical tool reported in this study offers a comprehensive assessment of the repolarization dynamics of AP, which may facilitate recognition of ionic mechanisms underlying the electrical behavior of cardiomyocytes under various experimental conditions.
Sources of Funding
This work was supported by National Institutes of Health grants R01 HL091021, R01 HL114346, P01 AG043353, P01 HL092868, R01 AG037495, R01 HL111183, R01 AG037490, R01 HL105532, and R37 HL081737.
Disclosures
Anversa is a member of Autologous LLP and Anversa and Leri are members of AAL Scientifics Corp.
Supporting information
Data S1. Supplemental Materials and Methods.
Figure S1. Blood glucose in control and diabetic mice. A, Circulating glucose levels in nontreated (0 days) and streptozotocin (STZ)‐treated female mice as a function of time from treatment. Data are shown as mean±SEM (n=7–30 mice for each time point). B, Circulating glucose levels in wild‐type (WT) and Akita male mice as a function of age. Data are shown as mean±SEM (n=4–7 mice for each time point).
Figure S2. Cardiac function in Akita mice. Quantitative data obtained by echocardiography in male wild‐type (WT; n=7) and Akita (n=8) mice at ≈7 months of age are shown as median and interquartile ranges (IQRs).
Figure S3. Hyperglycemia and electrocardiographic parameters under complete autonomic block. A and B, Additional electrocardiographic parameters in control (Ctrl) and streptozotocin (STZ) female mice (A) and WT and Akita (B) male mice reported in Figure 3D and 3E are shown as median and interquartile ranges (IQRs).
Figure S4. Comparison of action potential (AP) parameters obtained by standard analysis and the novel algorithm. Relation between AP duration at discrete repolarization levels computed with conventional (Standard) and newly developed (Automatic) approaches. Data obtained in control (Ctrl) and streptozotocin (STZ) myocytes shown in Figure 5B and 5C are reported as mean±SEM and are fitted with linear regression.
Figure S5. High glucose and action potential (AP) profile. A, APs obtained in a myocytes from a control (Ctrl) female mouse heart before and after exposure to 20 mmol/L of glucose (high glucose). B, AP properties of Ctrl myocytes (n=6 from 3 female mice) stimulated at 1 Hz before (Tyrode; Tyr) and after exposure to 20 mmol/L of glucose (high glucose) are shown as median and interquartile ranges (IQRs). *P<0.05 vs Tyr. C, Analysis and comparison of repolarization properties of APs reported in (B) using the novel algorithm. Graph of average AP profile represents the calculated average repolarization phase of the AP in myocytes before and after exposure to 20 mmol/L glucose. Corresponding error bars, fold changes for the duration of the AP in high glucose vs Tyrode, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively.
Figure S6. Hyperglycemia and action potential (AP) properties at different pacing rates. A through C, Variability of repolarization of the AP at 2‐, 4‐, and 6‐Hz pacing rates evaluated by SD (A), RSD (B), and short‐term variability(STV) (C) using the novel algorithm for cells reported in Figure 6. Graphs of SD, RSD, and STV of the AP profile represent calculated average variability of the repolarization phase of AP for control (Ctrl) and streptozotocin (STZ) myocytes. Corresponding error bars, fold changes for the variability of AP in STZ vs Ctrl cells, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively.
Figure S7. Hyperglycemia and ICaL properties. A, Activation curves of ICaL for control (Ctrl) myocytes (n=21, from 3 mice) and myocytes from streptozotocin (STZ)‐treated female mice (n=22; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. Fitting parameters: Ctrl: V1/2G −6.8±0.3 mV, kG 4.6±0.3 mV, R 2 0.96; STZ: V1/2G −6.6±0.3 mV, kG 5.2±0.3 mV, R 2 0.96. B, Steady‐state inactivation curves of ICaL for Ctrl myocytes (n=21; from 3 mice) and myocytes from STZ‐treated mice (n=19; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. Fitting parameters: Ctrl: V1/2G −19.2±0.2 mV, kG 5.4±0.2 mV, R 2 0.99; STZ: V1/2G −18.8±0.2 mV, kG 5.6±0.2 mV, R 2 0.99. C, Recovery from inactivation curves of ICaL for Ctrl myocytes (n=15; from 3 mice) and myocytes from STZ‐treated mice (n=16; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. Fitting parameters: Ctrl: τ 45.4±1 ms, R 2 0.88; STZ: τ 51.9±1 ms, R 2 0.91. D, Inactivation time constants of ICaL for Ctrl myocytes (n=20; from 3 mice) and myocytes from STZ‐treated mice (n=17; from 4 mice at 12–19 days after induction of diabetes) are shown as median and interquartile ranges (IQRs).
Figure S8. Novel algorithm. Example of a collection of v1(n) functions when considering N=19, thus leading to L=10. Vectors are represented by small circles.
Figure S9. Novel algorithm. Simulated 19‐sample signal of the repolarization phase of the action potential (AP) and resulting fitted curve. Fitted curve is smooth with respect to original signal and exhibit monotonic decay.
(J Am Heart Assoc. 2016;5:e003078 doi: 10.1161/JAHA.115.003078)
Accompanying Data S1 and Figures S1 through S9 are available at http://jaha.ahajournals.org/content/5/2/e003078/suppl/DC1
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics C and Stroke Statistics S . Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 2. Jouven X, Lemaitre RN, Rea TD, Sotoodehnia N, Empana JP, Siscovick DS. Diabetes, glucose level, and risk of sudden cardiac death. Eur Heart J. 2005;26:2142–2147. [DOI] [PubMed] [Google Scholar]
- 3. McNally PG, Lawrence IG, Panerai RB, Weston PJ, Thurston H. Sudden death in type 1 diabetes. Diabetes Obes Metab. 1999;1:151–158. [DOI] [PubMed] [Google Scholar]
- 4. Rossing P, Breum L, Major‐Pedersen A, Sato A, Winding H, Pietersen A, Kastrup J, Parving HH. Prolonged QTc interval predicts mortality in patients with type 1 diabetes mellitus. Diabet Med. 2001;18:199–205. [DOI] [PubMed] [Google Scholar]
- 5. Okin PM, Devereux RB, Lee ET, Galloway JM, Howard BV; Strong Heart S . Electrocardiographic repolarization complexity and abnormality predict all‐cause and cardiovascular mortality in diabetes: the Strong Heart Study. Diabetes. 2004;53:434–440. [DOI] [PubMed] [Google Scholar]
- 6. Cardoso CR, Salles GF, Deccache W. Prognostic value of QT interval parameters in type 2 diabetes mellitus: results of a long‐term follow‐up prospective study. J Diabetes Complications. 2003;17:169–178. [DOI] [PubMed] [Google Scholar]
- 7. Cox AJ, Azeem A, Yeboah J, Soliman EZ, Aggarwal SR, Bertoni AG, Carr JJ, Freedman BI, Herrington DM, Bowden DW. Heart rate‐corrected QT interval is an independent predictor of all‐cause and cardiovascular mortality in individuals with type 2 diabetes: the Diabetes Heart Study. Diabetes Care. 2014;37:1454–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miki T, Tobisawa T, Sato T, Tanno M, Yano T, Akasaka H, Kuno A, Ogasawara M, Murase H, Saitoh S, Miura T. Does glycemic control reverse dispersion of ventricular repolarization in type 2 diabetes? Cardiovasc Diabetol. 2014;13:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Veglio M, Giunti S, Stevens LK, Fuller JH, Perin PC; Group EICS . Prevalence of Q‐T interval dispersion in type 1 diabetes and its relation with cardiac ischemia: the EURODIAB IDDM Complications Study Group. Diabetes Care. 2002;25:702–707. [DOI] [PubMed] [Google Scholar]
- 10. Suys BE, Huybrechts SJ, De Wolf D, Op De Beeck L, Matthys D, Van Overmeire B, Du Caju MV, Rooman RP. QTc interval prolongation and QTc dispersion in children and adolescents with type 1 diabetes. J Pediatr. 2002;141:59–63. [DOI] [PubMed] [Google Scholar]
- 11. Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. [DOI] [PubMed] [Google Scholar]
- 12. Veglio M, Sivieri R, Chinaglia A, Scaglione L, Cavallo‐Perin P. QT interval prolongation and mortality in type 1 diabetic patients: a 5‐year cohort prospective study. Neuropathy Study Group of the Italian Society of the Study of Diabetes, Piemonte Affiliate. Diabetes Care. 2000;23:1381–1383. [DOI] [PubMed] [Google Scholar]
- 13. Whitsel EA, Boyko EJ, Rautaharju PM, Raghunathan TE, Lin D, Pearce RM, Weinmann SA, Siscovick DS. Electrocardiographic QT interval prolongation and risk of primary cardiac arrest in diabetic patients. Diabetes Care. 2005;28:2045–2047. [DOI] [PubMed] [Google Scholar]
- 14. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berger RD, Kasper EK, Baughman KL, Marban E, Calkins H, Tomaselli GF. Beat‐to‐beat QT interval variability: novel evidence for repolarization lability in ischemic and nonischemic dilated cardiomyopathy. Circulation. 1997;96:1557–1565. [DOI] [PubMed] [Google Scholar]
- 16. Hinterseer M, Beckmann BM, Thomsen MB, Pfeufer A, Ulbrich M, Sinner MF, Perz S, Wichmann HE, Lengyel C, Schimpf R, Maier SK, Varro A, Vos MA, Steinbeck G, Kaab S. Usefulness of short‐term variability of QT intervals as a predictor for electrical remodeling and proarrhythmia in patients with nonischemic heart failure. Am J Cardiol. 2010;106:216–220. [DOI] [PubMed] [Google Scholar]
- 17. de Bruyne MC, Hoes AW, Kors JA, Hofman A, van Bemmel JH, Grobbee DE. Prolonged QT interval predicts cardiac and all‐cause mortality in the elderly. The Rotterdam Study. Eur Heart J. 1999;20:278–284. [DOI] [PubMed] [Google Scholar]
- 18. Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. [DOI] [PubMed] [Google Scholar]
- 19. Signore S, Sorrentino A, Ferreira‐Martins J, Kannappan R, Shafaie M, Del Ben F, Isobe K, Arranto C, Wybieralska E, Webster A, Sanada F, Ogorek B, Zheng H, Liu X, Del Monte F, D'Alessandro DA, Wunimenghe O, Michler RE, Hosoda T, Goichberg P, Leri A, Kajstura J, Anversa P, Rota M. Inositol 1, 4, 5‐trisphosphate receptors and human left ventricular myocytes. Circulation. 2013;128:1286–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Signore S, Sorrentino A, Borghetti G, Cannata A, Meo M, Zhou Y, Kannappan R, Pasqualini F, O'Malley H, Sundman M, Tsigkas N, Zhang E, Arranto C, Mangiaracina C, Isobe K, Sena BF, Kim J, Goichberg P, Nahrendorf M, Isom LL, Leri A, Anversa P, Rota M. Late Na(+) current and protracted electrical recovery are critical determinants of the aging myopathy. Nat Commun. 2015;6:8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burashnikov A, Antzelevitch C. Late‐phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin Electrophysiol. 2006;29:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaniboni M, Pollard AE, Yang L, Spitzer KW. Beat‐to‐beat repolarization variability in ventricular myocytes and its suppression by electrical coupling. Am J Physiol Heart Circ Physiol. 2000;278:H677–H687. [DOI] [PubMed] [Google Scholar]
- 23. Johnson DM, Heijman J, Bode EF, Greensmith DJ, van der Linde H, Abi‐Gerges N, Eisner DA, Trafford AW, Volders PG. Diastolic spontaneous calcium release from the sarcoplasmic reticulum increases beat‐to‐beat variability of repolarization in canine ventricular myocytes after beta‐adrenergic stimulation. Circ Res. 2013;112:246–256. [DOI] [PubMed] [Google Scholar]
- 24. Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, Volders PG, Vos MA. Increased short‐term variability of repolarization predicts d‐sotalol‐induced torsades de pointes in dogs. Circulation. 2004;110:2453–2459. [DOI] [PubMed] [Google Scholar]
- 25. Rota M, Boni A, Urbanek K, Padin‐Iruegas ME, Kajstura TJ, Fiore G, Kubo H, Sonnenblick EH, Musso E, Houser SR, Leri A, Sussman MA, Anversa P. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res. 2005;97:1332–1341. [DOI] [PubMed] [Google Scholar]
- 26. D'Souza A, Bucchi A, Johnsen AB, Logantha SJ, Monfredi O, Yanni J, Prehar S, Hart G, Cartwright E, Wisloff U, Dobryznski H, DiFrancesco D, Morris GM, Boyett MR. Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat Commun. 2014;5:3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin‐dependent protein kinase II promotes life‐threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock‐in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–e82. [DOI] [PubMed] [Google Scholar]
- 29. Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rota M, Hosoda T, De Angelis A, Arcarese ML, Esposito G, Rizzi R, Tillmanns J, Tugal D, Musso E, Rimoldi O, Bearzi C, Urbanek K, Anversa P, Leri A, Kajstura J. The young mouse heart is composed of myocytes heterogeneous in age and function. Circ Res. 2007;101:387–399. [DOI] [PubMed] [Google Scholar]
- 31. Rota M, Vassalle M. Patch‐clamp analysis in canine cardiac Purkinje cells of a novel sodium component in the pacemaker range. J Physiol. 2003;548:147–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brouillette J, Clark RB, Giles WR, Fiset C. Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol. 2004;559:777–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DuBell WH, Lederer WJ, Rogers TB. K(+) currents responsible for repolarization in mouse ventricle and their modulation by FK‐506 and rapamycin. Am J Physiol Heart Circ Physiol. 2000;278:H886–H897. [DOI] [PubMed] [Google Scholar]
- 34. Liu J, Kim KH, London B, Morales MJ, Backx PH. Dissection of the voltage‐activated potassium outward currents in adult mouse ventricular myocytes: I(to, f), I(to, s), I(K, slow1), I(K, slow2), and I(ss). Basic Res Cardiol. 2011;106:189–204. [DOI] [PubMed] [Google Scholar]
- 35. London B, Guo W, Pan X, Lee JS, Shusterman V, Rocco CJ, Logothetis DA, Nerbonne JM, Hill JA. Targeted replacement of KV1.5 in the mouse leads to loss of the 4‐aminopyridine‐sensitive component of I(K, slow) and resistance to drug‐induced QT prolongation. Circ Res. 2001;88:940–946. [DOI] [PubMed] [Google Scholar]
- 36. Priori SG, Napolitano C, Diehl L, Schwartz PJ. Dispersion of the QT interval. A marker of therapeutic efficacy in the idiopathic long QT syndrome. Circulation. 1994;89:1681–1689. [DOI] [PubMed] [Google Scholar]
- 37. Niemeijer MN, van den Berg ME, Eijgelsheim M, van Herpen G, Stricker BH, Kors JA, Rijnbeek PR. Short‐term QT variability markers for the prediction of ventricular arrhythmias and sudden cardiac death: a systematic review. Heart. 2014;100:1831–1836. [DOI] [PubMed] [Google Scholar]
- 38. Cabasson A, Meste O, Blain G, Bermon S. Quantifying the PR interval pattern during dynamic exercise and recovery. IEEE Trans Biomed Eng. 2009;56:2675–2683. [DOI] [PubMed] [Google Scholar]
- 39. Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early‐onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. [DOI] [PubMed] [Google Scholar]
- 40. Kuehl M, Stevens MJ. Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat Rev Endocrinol. 2012;8:405–416. [DOI] [PubMed] [Google Scholar]
- 41. Nayyar S, Roberts‐Thomson KC, Hasan MA, Sullivan T, Harrington J, Sanders P, Baumert M. Autonomic modulation of repolarization instability in patients with heart failure prone to ventricular tachycardia. Am J Physiol Heart Circ Physiol. 2013;305:H1181–H1188. [DOI] [PubMed] [Google Scholar]
- 42. Levy S, Aliot E. Bidirectional tachycardia: a new look on the mechanism. Pacing Clin Electrophysiol. 1989;12:827–834. [DOI] [PubMed] [Google Scholar]
- 43. Lopez‐Izquierdo A, Pereira RO, Wende AR, Punske BB, Abel ED, Tristani‐Firouzi M. The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am J Physiol Heart Circ Physiol. 2014;306:H747–H754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shimoni Y, Firek L, Severson D, Giles W. Short‐term diabetes alters K+ currents in rat ventricular myocytes. Circ Res. 1994;74:620–628. [DOI] [PubMed] [Google Scholar]
- 45. Sato T, Kobayashi T, Kuno A, Miki T, Tanno M, Kouzu H, Itoh T, Ishikawa S, Kojima T, Miura T, Tohse N. Type 2 diabetes induces subendocardium‐predominant reduction in transient outward K+ current with downregulation of Kv4.2 and KChIP2. Am J Physiol Heart Circ Physiol. 2014;306:H1054–H1065. [DOI] [PubMed] [Google Scholar]
- 46. Lu Z, Jiang YP, Xu XH, Ballou LM, Cohen IS, Lin RZ. Decreased L‐type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3‐kinase signaling. Diabetes. 2007;56:2780–2789. [DOI] [PubMed] [Google Scholar]
- 47. Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, Gomez AM. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–615. [DOI] [PubMed] [Google Scholar]
- 48. Wang DW, Kiyosue T, Shigematsu S, Arita M. Abnormalities of K+ and Ca2+ currents in ventricular myocytes from rats with chronic diabetes. Am J Physiol. 1995;269:H1288–H1296. [DOI] [PubMed] [Google Scholar]
- 49. Liu JE, Palmieri V, Roman MJ, Bella JN, Fabsitz R, Howard BV, Welty TK, Lee ET, Devereux RB. The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol. 2001;37:1943–1949. [DOI] [PubMed] [Google Scholar]
- 50. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188. [DOI] [PubMed] [Google Scholar]
- 51. Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology. 2002;98:33–39. [DOI] [PubMed] [Google Scholar]
- 52. Bell DS. Diabetic cardiomyopathy. Diabetes Care. 2003;26:2949–2951. [DOI] [PubMed] [Google Scholar]
- 53. Bell DS. Diabetic cardiomyopathy. A unique entity or a complication of coronary artery disease? Diabetes Care. 1995;18:708–714. [DOI] [PubMed] [Google Scholar]
- 54. Pueyo E, Corrias A, Virag L, Jost N, Szel T, Varro A, Szentandrassy N, Nanasi PP, Burrage K, Rodriguez B. A multiscale investigation of repolarization variability and its role in cardiac arrhythmogenesis. Biophys J. 2011;101:2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lemay M, de Lange E, Kucera JP. Effects of stochastic channel gating and distribution on the cardiac action potential. J Theor Biol. 2011;281:84–96. [DOI] [PubMed] [Google Scholar]
- 56. Shimoni Y, Severson D, Giles W. Thyroid status and diabetes modulate regional differences in potassium currents in rat ventricle. J Physiol. 1995;488(Pt 3):673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Panguluri SK, Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Tipparaju SM. MicroRNA‐301a mediated regulation of Kv4.2 in diabetes: identification of key modulators. PLoS One. 2013;8:e60545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nishiyama A, Ishii DN, Backx PH, Pulford BE, Birks BR, Tamkun MM. Altered K(+) channel gene expression in diabetic rat ventricle: isoform switching between Kv4.2 and Kv1.4. Am J Physiol Heart Circ Physiol. 2001;281:H1800–H1807. [DOI] [PubMed] [Google Scholar]
- 60. Kaprielian R, Wickenden AD, Kassiri Z, Parker TG, Liu PP, Backx PH. Relationship between K+ channel down‐regulation and [Ca2+]i in rat ventricular myocytes following myocardial infarction. J Physiol. 1999;517(Pt 1):229–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rozanski GJ, Xu Z, Zhang K, Patel KP. Altered K+ current of ventricular myocytes in rats with chronic myocardial infarction. Am J Physiol. 1998;274:H259–H265. [DOI] [PubMed] [Google Scholar]
- 62. Cerbai E, Barbieri M, Li Q, Mugelli A. Ionic basis of action potential prolongation of hypertrophied cardiac myocytes isolated from hypertensive rats of different ages. Cardiovasc Res. 1994;28:1180–1187. [DOI] [PubMed] [Google Scholar]
- 63. Volk T, Nguyen TH, Schultz JH, Faulhaber J, Ehmke H. Regional alterations of repolarizing K+ currents among the left ventricular free wall of rats with ascending aortic stenosis. J Physiol. 2001;530:443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walker KE, Lakatta EG, Houser SR. Age associated changes in membrane currents in rat ventricular myocytes. Cardiovasc Res. 1993;27:1968–1977. [DOI] [PubMed] [Google Scholar]
- 65. Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing‐induced heart failure. Circ Res. 1996;78:262–273. [DOI] [PubMed] [Google Scholar]
- 66. Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–385. [DOI] [PubMed] [Google Scholar]
- 67. Yaras N, Ugur M, Ozdemir S, Gurdal H, Purali N, Lacampagne A, Vassort G, Turan B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54:3082–3088. [DOI] [PubMed] [Google Scholar]
- 68. Nygren A, Olson ML, Chen KY, Emmett T, Kargacin G, Shimoni Y. Propagation of the cardiac impulse in the diabetic rat heart: reduced conduction reserve. J Physiol. 2007;580:543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wong TC, Piehler KM, Kang IA, Kadakkal A, Kellman P, Schwartzman DS, Mulukutla SR, Simon MA, Shroff SG, Kuller LH, Schelbert EB. Myocardial extracellular volume fraction quantified by cardiovascular magnetic resonance is increased in diabetes and associated with mortality and incident heart failure admission. Eur Heart J. 2014;35:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lesh MD, Pring M, Spear JF. Cellular uncoupling can unmask dispersion of action potential duration in ventricular myocardium. A computer modeling study. Circ Res. 1989;65:1426–1440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Materials and Methods.
Figure S1. Blood glucose in control and diabetic mice. A, Circulating glucose levels in nontreated (0 days) and streptozotocin (STZ)‐treated female mice as a function of time from treatment. Data are shown as mean±SEM (n=7–30 mice for each time point). B, Circulating glucose levels in wild‐type (WT) and Akita male mice as a function of age. Data are shown as mean±SEM (n=4–7 mice for each time point).
Figure S2. Cardiac function in Akita mice. Quantitative data obtained by echocardiography in male wild‐type (WT; n=7) and Akita (n=8) mice at ≈7 months of age are shown as median and interquartile ranges (IQRs).
Figure S3. Hyperglycemia and electrocardiographic parameters under complete autonomic block. A and B, Additional electrocardiographic parameters in control (Ctrl) and streptozotocin (STZ) female mice (A) and WT and Akita (B) male mice reported in Figure 3D and 3E are shown as median and interquartile ranges (IQRs).
Figure S4. Comparison of action potential (AP) parameters obtained by standard analysis and the novel algorithm. Relation between AP duration at discrete repolarization levels computed with conventional (Standard) and newly developed (Automatic) approaches. Data obtained in control (Ctrl) and streptozotocin (STZ) myocytes shown in Figure 5B and 5C are reported as mean±SEM and are fitted with linear regression.
Figure S5. High glucose and action potential (AP) profile. A, APs obtained in a myocytes from a control (Ctrl) female mouse heart before and after exposure to 20 mmol/L of glucose (high glucose). B, AP properties of Ctrl myocytes (n=6 from 3 female mice) stimulated at 1 Hz before (Tyrode; Tyr) and after exposure to 20 mmol/L of glucose (high glucose) are shown as median and interquartile ranges (IQRs). *P<0.05 vs Tyr. C, Analysis and comparison of repolarization properties of APs reported in (B) using the novel algorithm. Graph of average AP profile represents the calculated average repolarization phase of the AP in myocytes before and after exposure to 20 mmol/L glucose. Corresponding error bars, fold changes for the duration of the AP in high glucose vs Tyrode, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively.
Figure S6. Hyperglycemia and action potential (AP) properties at different pacing rates. A through C, Variability of repolarization of the AP at 2‐, 4‐, and 6‐Hz pacing rates evaluated by SD (A), RSD (B), and short‐term variability(STV) (C) using the novel algorithm for cells reported in Figure 6. Graphs of SD, RSD, and STV of the AP profile represent calculated average variability of the repolarization phase of AP for control (Ctrl) and streptozotocin (STZ) myocytes. Corresponding error bars, fold changes for the variability of AP in STZ vs Ctrl cells, and statistical difference are reported in the ±SEM, fold change, and P‐value graphs, respectively.
Figure S7. Hyperglycemia and ICaL properties. A, Activation curves of ICaL for control (Ctrl) myocytes (n=21, from 3 mice) and myocytes from streptozotocin (STZ)‐treated female mice (n=22; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. Fitting parameters: Ctrl: V1/2G −6.8±0.3 mV, kG 4.6±0.3 mV, R 2 0.96; STZ: V1/2G −6.6±0.3 mV, kG 5.2±0.3 mV, R 2 0.96. B, Steady‐state inactivation curves of ICaL for Ctrl myocytes (n=21; from 3 mice) and myocytes from STZ‐treated mice (n=19; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. Fitting parameters: Ctrl: V1/2G −19.2±0.2 mV, kG 5.4±0.2 mV, R 2 0.99; STZ: V1/2G −18.8±0.2 mV, kG 5.6±0.2 mV, R 2 0.99. C, Recovery from inactivation curves of ICaL for Ctrl myocytes (n=15; from 3 mice) and myocytes from STZ‐treated mice (n=16; from 4 mice at 12–19 days after induction of diabetes) are shown as mean±SEM. Fitting parameters: Ctrl: τ 45.4±1 ms, R 2 0.88; STZ: τ 51.9±1 ms, R 2 0.91. D, Inactivation time constants of ICaL for Ctrl myocytes (n=20; from 3 mice) and myocytes from STZ‐treated mice (n=17; from 4 mice at 12–19 days after induction of diabetes) are shown as median and interquartile ranges (IQRs).
Figure S8. Novel algorithm. Example of a collection of v1(n) functions when considering N=19, thus leading to L=10. Vectors are represented by small circles.
Figure S9. Novel algorithm. Simulated 19‐sample signal of the repolarization phase of the action potential (AP) and resulting fitted curve. Fitted curve is smooth with respect to original signal and exhibit monotonic decay.