

Abstract
Background
The choline‐derived metabolite trimethylamine N‐oxide (TMAO) has been demonstrated to contribute to atherosclerosis and is associated with coronary artery disease risk.
Methods and Results
We explored the impact of TMAO on endothelial and smooth muscle cell function in vivo, focusing on disease‐relevant outcomes for atherogenesis. Initially, we observed that aortas of LDLR −/− mice fed a choline diet showed elevated inflammatory gene expression compared with controls. Acute TMAO injection at physiological levels was sufficient to induce the same inflammatory markers and activate the well‐known mitogen‐activated protein kinase, extracellular signal–related kinase, and nuclear factor‐κB signaling cascade. These observations were recapitulated in primary human aortic endothelial cells and vascular smooth muscle cells. We also found that TMAO promotes recruitment of activated leukocytes to endothelial cells. Through pharmacological inhibition, we further showed that activation of nuclear factor‐κB signaling was necessary for TMAO to induce inflammatory gene expression in both of these relevant cell types as well as endothelial cell adhesion of leukocytes.
Conclusions
Our results suggest a likely contributory mechanism for TMAO‐dependent enhancement in atherosclerosis and cardiovascular risks.
Keywords: atherosclerosis, cardiovascular disease, endothelial cell, inflammation, leukocyte adhesion, nuclear factor‐κB signaling, trimethylamine N‐oxide, vascular smooth muscle cell
Subject Categories: Vascular Biology, Diet and Nutrition, Cell Signalling/Signal Transduction, Inflammation, Atherosclerosis
Introduction
Numerous recent case–control and longitudinal studies have shown a striking association between trimethylamine N‐oxide (TMAO) levels and cardiovascular disease risks in a variety of cohorts.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 TMAO is derived primarily from dietary choline and carnitine through the action of gut microbiota, which metabolize these constituents to trimethylamine (TMA).1, 11, 12 TMA is absorbed in turn and travels via the portal circulation to the liver, where it is oxidized by flavin‐containing monoxygenases, primarily FMO3, to TMAO.1, 11, 12 In studies in mice, supplementation with dietary choline, carnitine, precarnitine (γ‐butyrobetaine), or even TMAO alone was sufficient to enhance macrophage cholesterol accumulation and atherogenic plaque formation, supporting a causal relationship.1, 2, 11 TMAO also has significant effects on cholesterol metabolism in the bile acid compartments.1, 11
We report studies of the effects of elevated TMAO levels on vascular cells in vitro and in vivo. The development of atherosclerosis is preceded by functional and transcriptional changes in vascular endothelial and smooth muscle cells and by the accumulation of subendothelial low‐density lipoprotein particles.13, 14 We provide evidence that TMAO directly activates inflammatory pathways, including nuclear factor‐κB (NF‐κB) signaling in both human aortic endothelial cells (HAECs) and smooth muscle cells, providing another potential pathway by which TMAO contributes to coronary artery disease.
Methods
Animals
All animal experiments were approved by the University of California Los Angeles animal care and use committee, in accordance with public health service guidelines. For choline feeding studies, female LDLR knockout (LDLR−/−) mice aged 8 weeks and on a C57BL/6J background were purchased from the Jackson Laboratory (Bar Harbor, ME). Beginning at 9 weeks of age, the mice were given acidified water supplemented with or without 1.3% choline chloride (n=8 for each group) for 3 weeks. At the end of the treatment period, the mice were fasted for 4 hours before blood and tissues were collected for further analysis. For acute TMAO exposure studies, LDLR−/− female mice were fasted for 4 hours and then injected with vehicle or TMAO (86 μmol; n=3 for each group) for various periods of time before aorta samples were collected for examination of activation of signaling pathways or inflammatory gene expression.
Cell Culture
HAECs were cultured, as described previously.15 In brief, HAECs were isolated from anonymous discarded aorta clippings from heart transplant donors and expanded into primary cultures. The culture dishes or plates for HAECs were coated with 0.1% gelatin solution. HAECs were cultured in M199 medium supplemented with 20% FBS (Atlanta Biologicals), 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mmol/L sodium pyruvate, 65 μg/mL heparin (Sigma‐Aldrich), and 50 μg/mL endothelial cell growth supplement (BD Biosciences). Leukocytes for adhesion assay were grown in RPMI media with l‐glutamine (Corning Scientific) containing 20% heat‐inactivated FBS (Atlanta Biologicals), 100 U/mL penicillin, and 100 μg/mL streptomycin. Primary human vascular smooth muscle cells (VSMCs) were cultured in 0.1% gelatin solution using DMEM supplemented with 20% FBS (Atlanta Biologicals), 100 U/mL penicillin, 100 μg/mL streptomycin, and 1 mmol/L sodium pyruvate. All cell culture treatments were carried out in DMEM supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mmol/L sodium pyruvate, and 5% FBS for the indicated period of time. Treatments using TMA in media were prepared from a salt form of the compound TMA‐HCl, which was purchased from Sigma‐Aldrich. For signaling experiments, cells were placed in DMEM containing 5% FBS and penicillin/streptomycin for 2 hours to acclimate the cells and reduce background prior to the addition of treatments. All experiments using primary human cells were repeated in at least 3 separate donors to confirm robust function of TMAO.
RNA Extraction and Reverse Transcription
Cells or tissue were homogenized in QIAzol (Qiagen), and RNA extraction was carried out using the manufacturer's protocol. RNA samples were reverse transcribed using a high‐capacity cDNA reverse transcription kit (Applied Biosystems) with random primers.
Quantitative Polymerase Chain Reaction
Quantitative polymerase chain reaction (qPCR) was carried out using a KAPA SYBR Fast qPCR kit (Kapa Biosystems), as recommended by the manufacturer. Samples were run on a LightCycler 480 II (Roche) and analyzed using the Roche LightCycler 1.5.0 software. All qPCR targets were normalized to RPL13A expression and quantified using the ΔCt method. All qPCR primer sequences were obtained from PrimerBank and are listed in Table.
Table 1.
Quantitative Polymerase Chain Reaction Primers Used for the Study
| Primer Name | Target Species | Fwd Sequence 5′→3′ | Rev Sequence 5′→3′ |
|---|---|---|---|
| CD68 | Mouse | gacctacatcagagcccgagt | cgccatgaatgtccactg |
| COX‐2 | Mouse | TTCAACACACTCTATCACTGGC | AGAAGCGTTTGCGGTACTCAT |
| E‐Selectin | Mouse | ATGCCTCGCGCTTTCTCTC | GTAGTCCCGCTGACAGTATGC |
| ICAM1 | Mouse | cccacgctacctctgctc | gatggatacctgagcatcacc |
| KC | Mouse | CTCAAGAATGGTCGCGAGGCT | AGAGCAGTCTGTCTTCTTTCTCCGTT |
| MCP‐1 | Mouse | CTTCTGGGCCTGCTGTTCA | CCAGCCTACTCATTGGGATCA |
| MIP‐2 | Mouse | GGTTCTTCCGTTGAGGGACA | TCCAGAGCTTGAGTGTGACG |
| TNF‐α | Mouse | ctgtagcccacgtcgtagc | ttgagatccatgccgttg |
| VCAM1 | Mouse | CTGTTCCAGCGAGGGTCTAC | CACAGCCAATAGCAGCACAC |
| IL‐1b | Mouse | agttgacggaccccaaaag | agctggatgctctcatcagg |
| IL‐6 | Mouse | gctaccaaactggatataatcagga | ccaggtagctatggtactccagaa |
| COX‐2 | Human | cttcacgcatcagtttttcaag | tcaccgtaaatatgatttaagtccac |
| E‐Selectin | Human | ccccgaagggtttggtgagg | ccggaactgccaggcttgaa |
| ICAM1 | Human | tctgtcgggccaggagcact | gggaggcgtggcttgtgtgt |
| IL‐6 | Human | TTCAATGAGGAGACTTGCCTG | CTGGCATTTGTGGTTGGGTC |
COX‐2 indicates cyclooxygenase 2; IL, interleukin; MCP‐1, monocyte chemotactic protein 1; MIP‐2, macrophage inflammatory protein 2; TNF‐α, tumor necrosis factor‐α.
Nuclear and Cytosolic Fractionation
Cell lysates were fractionated using NE‐PER kits (Thermo Scientific). Approximately 5×106 cells per condition were pelleted via centrifugation and then fractionated by a series of spins, as indicated by the manufacturer's protocol. Cellular fractions were then quantified by Bradford assay (Thermo Scientific) and normalized based on total protein content to a 1‐μg/μL final solution to be assessed via immunoblot.
Immunoblot Procedure and Analysis
HAECs were grown to confluence and then treated for the indicated period of time. Cells were lysed in whole‐cell extraction buffer containing 62.5 mmol/L Tris‐HCl (pH 6.8), 2% (wt/vol) sodium dodecyl sulfate, and 10% glycerol. Samples were then sonicated and diluted 1:5 in water, and protein content was measured using a BCA protein assay kit (Pierce). Total protein concentration was normalized to 3 μg/μL, and samples were then denatured in 1× LDS loading buffer (Life Technologies) with 1× reducing agent (Life Technologies) at 95°C for 5 minutes. Samples were then loaded at 10 μL per well into 4% to 12% Bis‐Tris gels (Invitrogen) and separated out at 130 V for 2 hours. Protein was then transferred to polyvinylidene difluoride membranes (Immobilon) for 1.5 hour at 35 V. Following transfer, membranes were washed with TBST, and then blocked in 5% skim milk (Gibco) in TBST for 1 hour at room temperature. Membranes were then placed in primary antibodies (1:2000) on a shaker overnight at 4°C. The following day, membranes were washed 3× in TBST and then placed in secondary antibodies (1:2000) for 1 hour at room temperature. Blots were then washed 3× in TBST and placed in Amersham ECL detection solution (GE Health Sciences). Blots were imaged using the ChemiDoc MP system (Bio‐Rad), and bands were quantified using ImageJ software (National Institutes of Health).
Antibodies
Rabbit polyclonal antibodies that recognize β‐actin, phospho‐(Ser536) and total NF‐κB; phospho‐(Thr‐180/Tyr‐182) and total p38 mitogen‐activated protein kinase; and phospho‐(Thr‐202/Try‐204) and total extracellular signal–related kinase 1/2 were purchased from Cell Signaling Technology. Horseradish peroxidase–conjugated secondary antibodies that recognize rabbit IgG were purchased from KLP Antibodies. Antibodies recognizing β‐tubulin (rabbit) and lamin A/C were purchased from Santa Cruz Biotechnology.
Measurement of Plasma Metabolites
For plasma lipid‐, lipoprotein‐, and glucose‐level determinations, mice were fasted for 4 hours before bleeding. Total cholesterol, high‐density lipoprotein cholesterol, unesterified/free cholesterol, triglycerides, free fatty acid, and glucose levels were determined by enzymatic colorimetric assay.16 Quantification of TMAO and TMA in plasma samples was performed using stable isotope dilution high‐performance liquid chromatography with online electrospray ionization tandem mass spectrometry on an AB SCIEX 5000 triple quadrupole mass spectrometer interfaced with a Shimadzu high‐performance liquid chromatography system equipped with silica column (4.6×250 mm, 5 μm Luna Silica; Regis) at a flow rate of 0.8 mL/min, and the separation was performed, as reported previously.17
Pharmacological Inhibitors
Inhibitors of NF‐κB and G protein–coupled receptor (GPCR) signaling were treated as described in figure legends 5 and 6. NF‐κB activation inhibitor IV (InSolution NF‐κB inhibitor) and Gallein were purchased from Millipore. Pertussis toxin and BAY 11‐7082 were purchased from Sigma‐Aldrich.
Leukocyte Adhesion Assay
This assay was adapted from previously published protocols.18 HAECs were stimulated for adhesion for 5 hours in RPMI media (Corning Scientific) containing 5% FBS (Atlanta Biologicals) with indicated treatments. At 2 hours prior to assay, leukocytes were spun down at 300 g for 10 minutes and pelleted. Cells were then resuspended in PBS containing 25 μmol/L Vybrant CFDA SE Cell Tracer (Life Technologies) for 15 minutes at 37°C. Labeled leukocytes were then spun down at 1000 rpm for 10 minutes and resuspended in RPMI media (Corning Scientific) with 5% FBS (Atlanta Biologicals). The spin and resuspension was repeated twice more to remove excess dye. Leukocytes were then added to HAECs for 30 minutes to allow adhesion. The combined cell mixture was then washed 3× with PBS to remove unbound leukocytes. Finally, cells were fixed in 4% paraformaldehyde and imaged. Quantification for adhesion assays were performed by placing a 1‐mm grid over each image and counting the number of fluorescent points per 100‐μm square. Each mean value reflects 9 images taken, 3 each from separate donors.
Mouse Macrophage Isolation
Primary mouse peritoneal macrophages were harvested, as described previously.19 Briefly, at 4 days prior to euthanasia, mice were intraperitoneally injected with 1 mL of 3.85% thioglycolate. Mice were then fasted overnight (≈16 hours), and macrophages isolated by intraperitoneal lavage with sterile PBS. After overnight incubation, macrophages were treated with various agents in DMEM supplemented with 5% FBS.
Statistical Analysis
Direct comparisons for experiments using a sample size >3 were performed using 2‐tailed Student t tests. For some experiments in which n=3, nonparametric t tests using a Mann–Whitney test were performed. For these comparisons, sample median±25% of the median interquartile ranges were reported. For multiple comparisons testing, a 2‐way ANOVA with a Tukey post hoc test was used. All statistical analyses were performed using R statistical software (R Foundation for Statistical Computing). Values were considered significant at P<0.05. All data are presented as mean±SE.
Results
Chronic Choline Feeding Leads to Vascular Inflammation in Mice
Given the importance of inflammation in atherosclerosis development, we initially tested whether chronic choline supplementation could promote inflammatory gene expression in cells of the vasculature. Atherosclerosis‐prone LDLR−/− mice were fed a chemically defined diet, referred to as normal chow (0.07–0.08% total choline) or chow with 1.3% choline provided ad libitum in drinking water. After 3 weeks of diet, choline feeding did not alter serum lipid profiles or glucose levels (Figure 1A); however, a substantial increase in circulating TMAO was observed (Figure 1B). Aortas from these mice were probed using qPCR for expression of inflammatory genes. Choline‐fed mice showed significantly enhanced expression of monocyte chemotactic protein 1, macrophage inflammatory protein 2, tumor necrosis factor‐α, ICAM1, KC, cyclooxygenase 2, E‐selectin, and VCAM1, as well as induction of the macrophage marker CD68 (Figure 1C).
Figure 1.
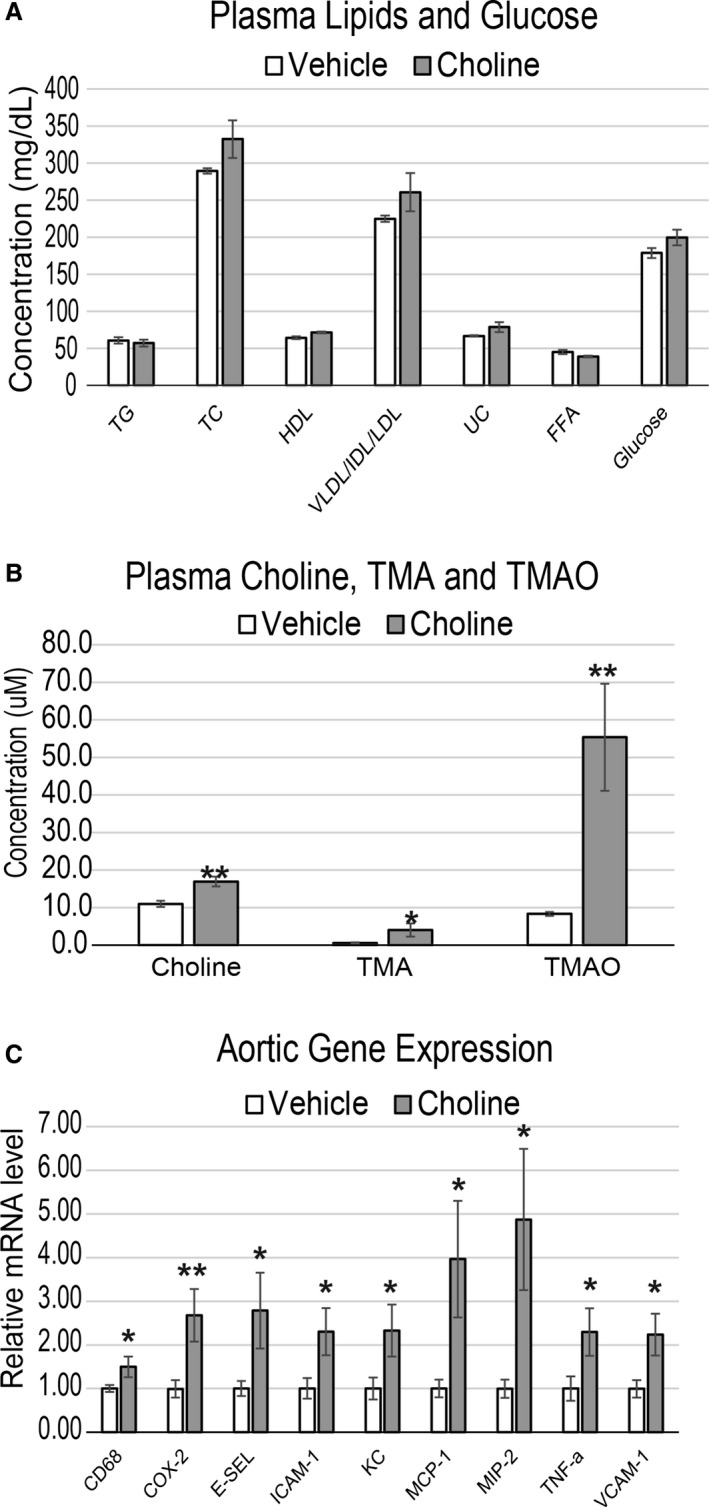
Choline feeding causes inflammation in atherosclerosis‐prone mouse aortas that is accompanied by increased plasma choline, TMA, and TMAO but not lipids. A through C, LDLR −/− female mice were fed chow or chow with either 0.07% (vehicle) or 1.3% choline for 3 weeks. Plasma was quantified for circulating levels of TG, TC, HDL, VLDL/IDL/LDL, UC, FFA, and glucose (A), as well as choline, TMA, and TMAO (B). Then aortas were harvested and quantitative polymerase chain reaction was used to quantify expression levels of inflammatory genes (C). All genes expressed as mean±SEM and normalized to RPL13A expression. n=8 mice per group; *P<0.05; **P<0.01. COX‐2 indicates cyclooxygenase 2; E‐SEL, E‐selectin; FFA, free fatty acids; HDL, high‐density lipoprotein; IDL, xxx; LDL, low‐density lipoprotein; MCP‐1, monocyte chemotactic protein 1; MIP‐2, macrophage inflammatory protein 2; TC, total cholesterol; TG, triglyceride; TMA, trimethylamine; TMAO, trimethylamine N‐oxide; TNF‐a, tumor necrosis factor α; UC, unesterified cholesterol; VLDL, very low‐density lipoprotein.
Acute TMAO Injection Activates Inflammatory Signaling and Gene Expression In Vivo
We next tested whether the observed effects of long‐term choline feeding could be recapitulated by acute TMAO administration. LDLR−/− mice were injected intraperitoneally with vehicle or a sterile solution of TMAO. The dose of TMAO used produced an initial spike in circulating levels to ≈100 μmol/L over the first hour, which returned essentially to near baseline levels ≈5 hours after injection in mice fed a normal chow diet (data not shown). Of note, the peak plasma TMAO levels with this protocol were similar to levels observed in mice chronically exposed to the choline‐supplemented diet (Figure 1C) and in some human clinical studies.9, 10, 17 At 30 minutes after intraperitoneal injection of TMAO, aortas were harvested and then assessed for changes in activation of p38 mitogen‐activated protein kinase, extracellular signal–related kinase 1/2, and p65 NF‐κB, all of which have been shown to play substantial roles in cellular inflammation contributing to the development of atherosclerosis.18, 20, 21, 22, 23, 24 Immediately following injection (30 minutes), TMAO‐exposed mice showed elevated phosphorylation of functional residues on p38 mitogen‐activated protein kinase (Thr‐180/Tyr‐182), extracellular signal–related kinase 1/2 (Thr‐202/Tyr‐204), and p65 NF‐κB (Ser536) (Figure 2A and 2B). To show that the enhanced phosphorylation of p65 NF‐κB was in fact leading to elevated nuclear abundance, the lysate was fractionated into nuclear and cytosolic regions. Consistent with the phosphorylation, TMAO increased nuclear abundance of total p65 NF‐κB in these aortas (Figure 2C). The nuclear/cytoplasmic ratios of p65 in aortas of vehicle‐ and TMAO‐treated mice were 0.58 and 1.12, respectively. In addition, in a separate cohort of mice similarly injected with vehicle versus sterile TMAO, qPCR analysis of expression of inflammatory genes in aortas harvested 5 hours after injection showed significant elevations in cyclooxygenase 2, interleukin 6, E‐selectin, and ICAM1 within the TMAO‐administered group (Figure 2D). These data confirm that acute exposure to physiologically relevant levels of TMAO is sufficient to enhance inflammatory signaling and gene expression in aortas of atheroprone LDLR−/− mice.
Figure 2.
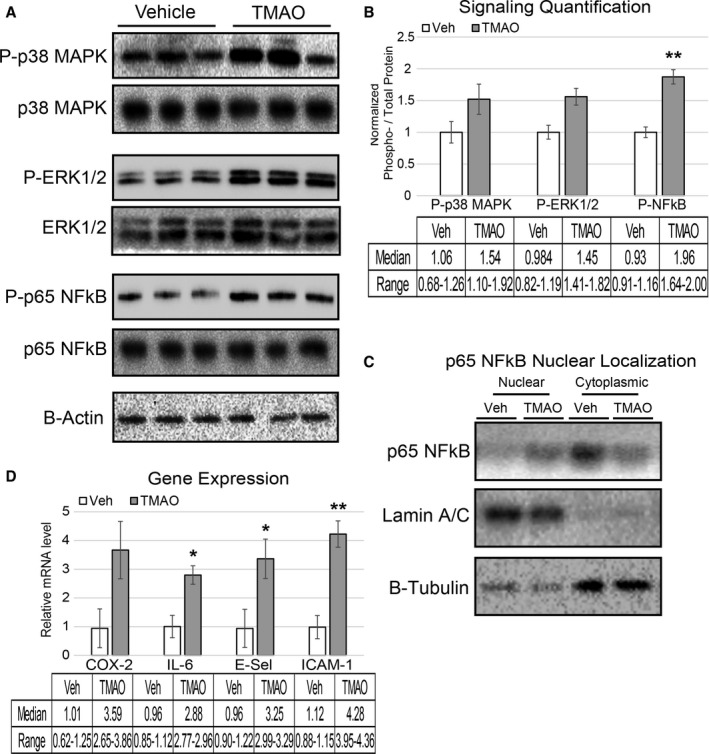
Acute TMAO injection causes activation of signaling cascades and elevated inflammatory gene expression. A and B, LDLR −/− female mice were fasted for 4 hours and then injected with vehicle or TMAO (86 μmol). Aortas were harvested 30 minutes following injection and immunoblotted (A) and quantified (B) for phosphorylation of p38 MAPK (Thr‐180/Tyr‐182), ERK1/2 (Thr‐202/Try‐204), and p65 NFkB (Ser536). n=3, nonparametric t test using a Mann–Whitney U test was performed for comparisons. Median and range are reported for each group. C, Lysate from the same aortas was fractionated into nuclear and cytosolic compartments and immune‐probed for the presence of p65 NFkB with lamin A/C and β‐tubulin, indicating the purity of the nuclear and cytosolic fractions, respectively. D, A separate cohort of LDLR −/− mice underwent the same fasting–injection procedure, and then aortas were harvested 5 hours after injection, and qPCR was used to quantify expression levels of inflammatory genes. All data were expressed as mean±SEM. Immunoblot quantification was normalized to P‐residue/total protein, with β‐actin serving as a loading control; qPCR genes normalized to RPL13A expression. n=3, nonparametric t test using a Mann–Whitney U test was performed for comparisons. Median and range are reported for each group; *P<0.05; **P<0.01. ERK1/2 indicates extracellular signal–related kinase 1/2; MAPK, mitogen‐activated protein kinase; NFkB, nuclear factor‐κB; qPCR, quantitative polymerase chain reaction; TMAO, trimethylamine N‐oxide; TNF‐a, tumor necrosis factor α; Veh, vehicle.
TMAO Induces Inflammatory Signaling and Gene Expression in Human Endothelial and Smooth Muscle Cells
We next tested whether these in vivo observations could be recapitulated in primary cultures of HAEC and VSMC lines. We found that treatment of these 2 cell lines with TMAO resulted in phosphorylation of the p38 mitogen‐activated protein kinase/extracellular signal–related kinase/NF‐κB pathway (Figure 3A, 3B, 3E, and 3F), enhanced p65 NF‐κB nuclear localization (Figure 3C and 3G), and induced inflammatory transcripts (cyclooxygenase 2, interleukin 6, E‐selectin, and ICAM1) in HAECs (Figure 3D) and similar targets (cyclooxygenase 2, interleukin 6, tumor necrosis factor α, and ICAM1) in VSMCs (Figure 3H). Given the important role of macrophages in atherosclerosis, we also subjected primary mouse peritoneal macrophages to similar treatments and conditions. We did not observe overlapping functions for TMAO in inducing inflammatory gene expression after 6 hours of treatment, indicating a specific activity in HAECs and VSMCs compared with macrophages (Figure 3I). Nevertheless, we cannot rule out the possibility that a longer treatment period of TMAO may influence inflammatory gene expression in macrophages.
Figure 3.
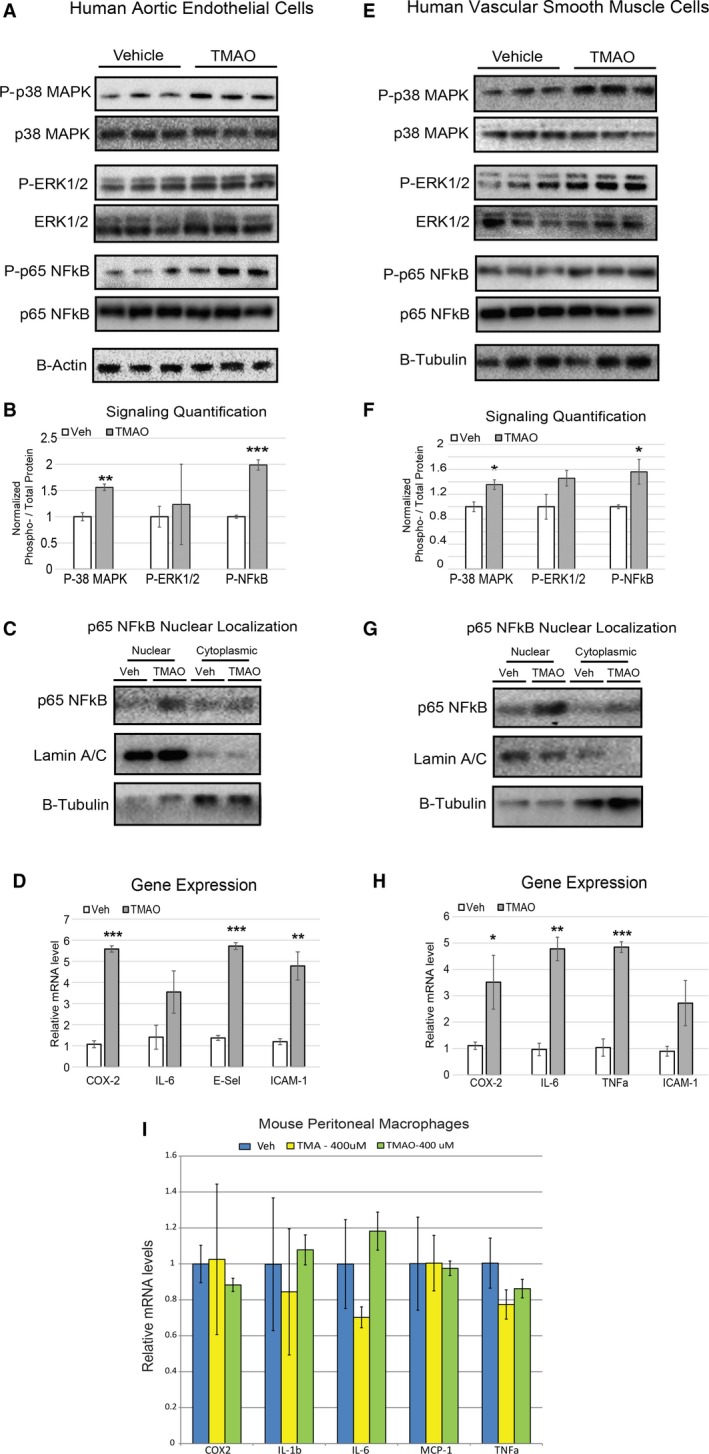
TMAO induces inflammation cascades and transcription in primary human endothelial and smooth muscle cells. A through C, Immunoblots (A), corresponding quantifications (B), and cell fractionations (C) of HAECs treated with vehicle (PBS) or TMAO (100 μmol/L) for 40 minutes (n=6). D, HAECs were treated with vehicle (PBS) or TMAO (200 μmol/L) for 6 hours and then quantified for inflammatory gene expression (n=6). E through H, The same experimental conditions were used as in panels A through D with primary VSMCs instead of HAECs. All data are expressed as mean±SEM. Immunoblot quantification was normalized to P‐residue/total protein. β‐actin and β‐tubulin were used as the loading controls for HAECs and VSMCs, respectively, and quantitative polymerase chain reaction genes normalized to RPL13A expression. I, Mouse peritoneal macrophages were treated with indicated amounts of vehicle, trimethylamine, or TMAO (n=4 for each group) and then evaluated for inflammatory gene expression. All experiments were confirmed in at least 3 separate donors to confirm robust function. *P<0.05; **P<0.01; ***P<0.001. COX‐2 indicates cyclooxygenase 2; E‐Sel, E‐selectin; ERK1/2 indicates extracellular signal–related kinase 1/2; HAEC, human aortic endothelial cell; IL‐6, interleukin 6; MAPK, mitogen‐activated protein kinase; NFkB, nuclear factor‐κB; qPCR, quantitative polymerase chain reaction; TMAO, trimethylamine N‐oxide; Veh, vehicle; VSMC, vascular smooth muscle cell.
TMAO Efficacy for Enhancing Inflammation Surpasses That of TMA at Physiological Levels
Physiological ranges of TMAO are ≈10‐ to 20‐fold higher than TMA in humans and ≈7.5‐fold higher in various inbred strains of mice (data not shown). In LDLR−/− mice, we observed that TMAO levels were 14‐fold higher than TMA in both chow and chow plus choline groups (Figure 1B). To assess the effective physiological ranges of TMA and TMAO, we tested the capacity of TMAO to exert its functions compared with equal concentrations of TMA, reflecting a supraphysiological concentration of TMA. Reaching statistical significance for only 3 observations (cyclooxygenase 2 expression in both cell types and E‐selectin in HAECs), TMAO trended toward a more robust capacity to induce phosphorylation of NF‐κB (Figure 4A, 4B, 4D and 4E) and enhance inflammatory gene expression (Figure 4C and 4F).
Figure 4.
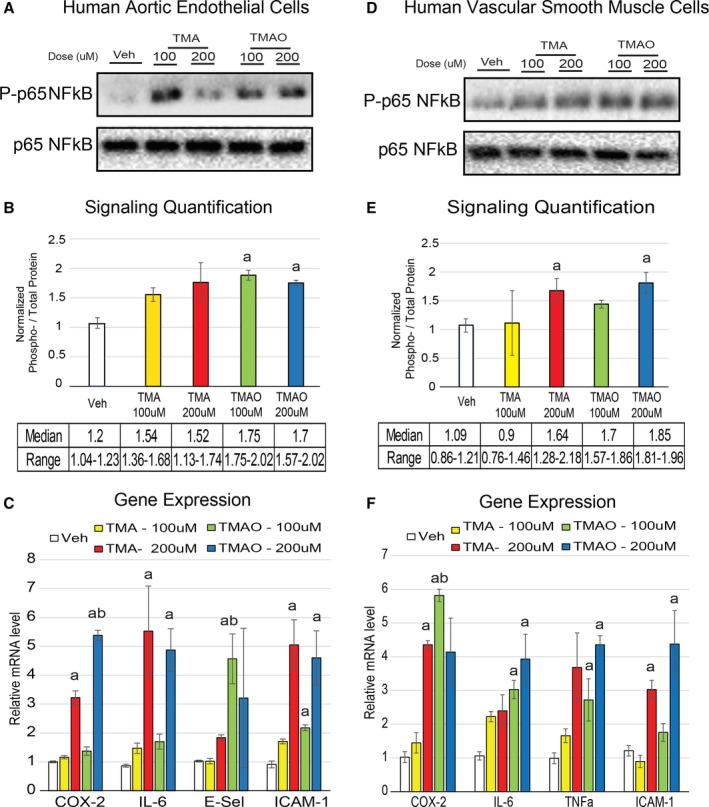
TMAO is more efficacious than TMA at affecting cell inflammation at physiological concentrations. A and B, HAECs were treated for 40 minutes with indicated concentrations of vehicle, TMA or TMAO, and then immunoblotted (A) and quantified (B) for phosphorylation of p65 NF‐κB (Ser536) (n=3). C, HAECs were treated for 6 hours in the same conditions as were used in panels A and B and then subjected to qPCR for inflammatory gene expression (n=5). D‐F, The same procedures were carried out as were used in panels A through C except with vascular smooth muscle cells instead of HAECs. All experiments were confirmed in at least 3 separate donors, and qPCR genes normalized to RPL13A expression. a P<0.05 vs vehicle treatment group; b P<0.05 of TMAO vs matched concentration of TMA. COX‐2 indicates cyclooxygenase 2; E‐Sel, E‐selectin; HAEC, human aortic endothelial cell; IL‐6, interleukin 6; NFkB, nuclear factor‐κB; qPCR, quantitative polymerase chain reaction; TMA, trimethylamine; TMAO, trimethylamine N‐oxide; TNF‐a, tumor necrosis factor α; Veh, vehicle.
Activation of NF‐κB and Gßγ Is Required for TMAO Effects on HAECs
To show that activation of signaling pathways is required for TMAO‐induced inflammatory gene expression, a pharmacological inhibitor of NF‐κB was used. A brief (30‐minute) pretreatment with 100 nmol/L NF‐κB activation inhibitor IV was sufficient to block TMAO‐induced expression of target transcripts in both cell types (Figure 5A and 5B). The same experiments were confirmed using a different NF‐κB inhibitor (BAY 11‐7082), suggesting that inhibition of TMAO‐induced target transcripts with either NF‐κB activation inhibitor was not due to off‐target pharmacological effects (data not shown). To investigate the presence of a specific receptor for TMAO using HAEC gene expression as a functional outcome, we performed similar experiments with broad‐class inhibitors of GPCR subunit signaling in HAECs. Pharmacological inhibition of Gßγ by pretreatment with 10 μmol/L Gallein, but not 200 ng/mL Gα inhibitor pertussis toxin, abolished the capacity of TMAO to enhance downstream target transcripts in HAECs (Figure 5C through 5E). These observations suggest that activation of NF‐κB and Gßγ signaling is required for TMAO to exert inflammatory effects on endothelial cells.
Figure 5.
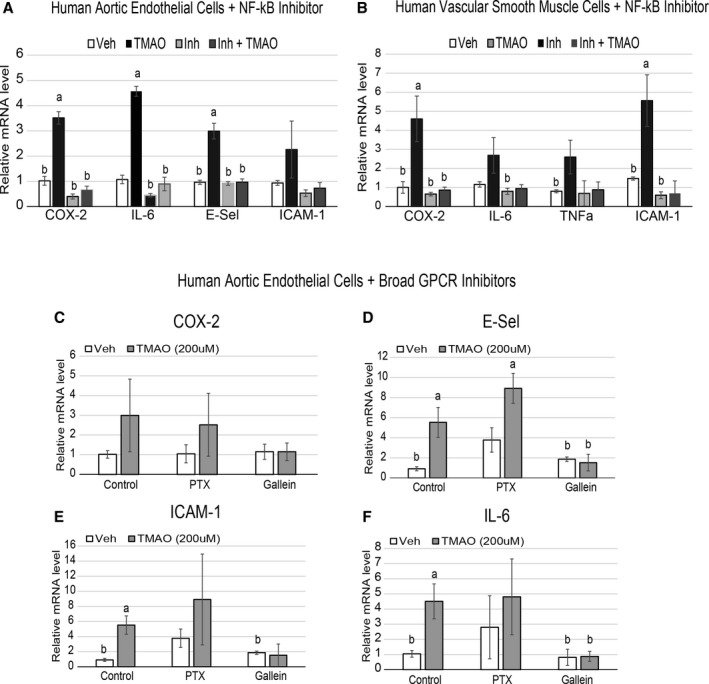
NFkB and Gßγ signaling is required for TMAO‐induced inflammatory gene expression in endothelial and smooth muscle cells. A and B, HAECs (A) or vascular smooth muscle cells (B) were pretreated for 30 minutes with control (DMSO) or 100 nmol/L NFkB activation inhibitor followed by an overnight treatment with or without 200 μmol/L TMAO and probed by qPCR for inflammatory gene expression (n=6). ANOVA with a Tukey post hoc test was used with TMAO and inhibitor as the 2 factors. We did not observe interaction between inhibitor and TMAO groups. C through F, HAECs were pretreated for 1 hour with control (DMSO), 200 ng/mL PTX, or 10 μmol/L Gallein, followed by addition of vehicle (PBS) or 200 μmol/L TMAO, then probed by qPCR for inflammatory gene expression. All experiments were confirmed in at least 3 separate donors, and qPCR genes normalized to RPL13A expression. a P<0.05 compared with vehicle‐alone group; b P<0.05 compared with TMAO‐alone treatment group. COX‐2 indicates cyclooxygenase 2; E‐Sel, E‐selectin; HAEC, human aortic endothelial cell; IL‐6, interleukin 6; NFkB, nuclear factor‐κB; PTX, pertussis toxin; qPCR, quantitative polymerase chain reaction; TMAO, trimethylamine N‐oxide; TNF‐a, tumor necrosis factor α; Veh, vehicle.
TMAO Enhances Endothelial Recruitment of Leukocytes
Increased leukocyte adhesion has been appreciated as a marker for endothelial cell activation, especially in the pathological context of atherosclerosis.23, 24 To investigate whether TMAO can play a role in this perturbation, we adapted an assay to measure the capacity of endothelia to recruit activated leukocytes.18 Primary HAECs were briefly cultured in media in the presence versus absence of TMAO and then subsequently exposed to labeled leukocytes to monitor adhesion. TMAO pretreatment significantly enhanced leukocyte adherence compared with vehicle‐ or TMA‐treated HAECs (Figure 6A and 6B). To mechanistically link the inflammatory signaling activation via TMAO to this important functional outcome, the same experiment was performed using pretreatment with a pharmacological inhibitor of NF‐κB signaling (NF‐κB activation inhibitor IV). The NF‐κB inhibitor effectively abolished the capacity of TMAO to enhance endothelial adhesion of leukocytes (Figure 6C and 6D). These observations link the inflammatory effects of TMAO to cardiovascular functional perturbations.
Figure 6.
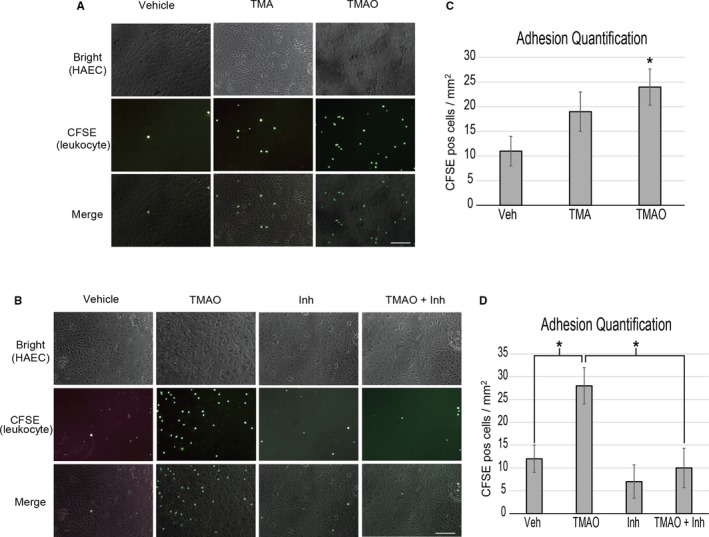
TMAO enhances HAEC recruitment of activated leukocytes through NFkB. A, HAECs were stimulated for adhesive processes for 5 hours with vehicle (PBS) or 400 μmol/L TMA or TMAO then presented with prelabeled, activated leukocytes and quantified (C). B, The same procedure as was used in panel A was followed except a 1‐hour inhibitor treatment (NFkB activation inhibitor) step was added prior to stimulation. D, Quantification of adhered leukocytes of panel B. ANOVA with a Tukey post hoc test was used with TMAO and inhibitor as the 2 factors. We did not observe interaction between inhibitor and TMAO groups. Images reflect representative examples of 9 images taken per group using 3 separate HAEC donor samples. Scale bars=100 μm. HAEC indicates human aortic endothelial cell; Inh, inhibitor; NFkB, nuclear factor‐κB; TMA, trimethylamine; TMAO, trimethylamine N‐oxide.
Discussion
Our observations support a role for TMAO in the activation of inflammatory pathways in cells of the vasculature, leading to endothelial cell leukocyte recruitment and atherosclerosis. Both in vivo studies in mice and in vitro studies in cultured HAECs and VSMCs revealed that physiological levels of TMAO could induce expression of cytokines and adhesion molecules. This activation was mediated, at least in part, by the NF‐κB signaling pathway, which has been linked previously to reduced vascular responsiveness and inflammation.
An important question that remains is how this small molecule causes cellular activation of NF‐κB signaling and subsequent events. Although a receptor for TMAO has not yet been identified, its physiological precursor TMA has been described as activating a GPCR, TAAR5.25, 26 TAAR5 is thought to have evolved as a highly conserved olfactory receptor, enabling the detection of the pungent “fishy odor” of TMA.26, 27 Genetic ablation of TAAR5 in mice removes an attraction to TMA odor.27 It has been shown that the TAAR5 receptor does not recognize TMAO,26 although given the structural similarity with TMA, a GPCR‐mediated mechanism of TMAO action is a potential scenario. Our results suggest a likely GPCR‐mediated mechanism for TMAO, specifically through activity of the Gßγ subunit complex in promoting endothelial cell inflammation.
TMAO also possesses several unique and intriguing biophysical properties. The molecule has been described to function as an organic osmolyte in shielding peptide regions from hydration, thus stabilizing structures and altering the protein folding process.28, 29, 30 Small amounts of TMAO have been predicted to cause condensed protein backbones (compared with other aqueous environments) in computational simulations.30 Furthermore, TMAO has been shown to function as a protein chaperone mimetic and to enable substantial protein stabilization. At high concentrations, it functions in freeze‐avoidance responses, particularly in cold water–dwelling organisms that also commonly live in an environment under significant amounts of pressure.31, 32, 33 Some deep sea fish and crustaceans have taken advantage of these features; abundance of the molecule can be found in the muscle and other organs of many dwellers 1000 m below sea level.33 Consequently, the ability of TMAO to alter protein conformation may facilitate its mechanism of action, perhaps due to intercalation with cellular proteins, and consequent pathway activation. It is also notable that other small molecules such as ethanol have been described as affecting membrane and lipid raft fluidity, altering localization and function of signaling components such as NF‐κB.34 It is easy to envision that the amphipathic character of the small TMAO molecule might similarly elicit such perturbations in membrane dynamics. Similarly, altering protein–protein or membrane–protein interactions could be an alternative mechanism for TMAO function.
Our studies by no means rule out other potential mechanisms by which TMAO may contribute to atherosclerosis. A previous report showed that TMAO enhanced uptake of cholesterol in peritoneal macrophages, a critical step in atherosclerosis.1 Although we did not observe TMAO inducing inflammation in macrophages, these observations could be due to differential regulation of receptor or signaling mechanisms across cell types. Varied responses to a biological compound mediated through tissue‐specific receptor coupling has been observed for lysophosphatidic acid,35 which has been demonstrated to enhance p65 NF‐κB phosphorylation in HAECs through GPCR‐mediated activation of rho kinase 2,36 a phenomenon specific to endothelial cells. Additional studies have shown that a single receptor underlies diverse cellular outcomes in response to lysophosphatidic acid treatment, such as neurite retraction,37 presumably through cell‐type specific regulation of signaling intermediates (eg, rho). A similar scenario could exist in which TMAO treatment leads to different cellular outcomes through tissue‐specific coupling of downstream events for a single GPCR.
Sources of Funding
This work was supported by NIH and Office of Dietary Supplements Grants T32HL69766 (Seldin), HL30568, NIH HL28481 and R01HL103866, P20HL113452, R01 HL103866 06A1, R01 DK106000, and NIH 2 P01 HL030568‐31A1. Hazen is also supported by an endowment from the Leonard Krieger Fund. Mass spectrometry studies were performed in a facility supported in part by an AB SCIEX Center of Innovation Award.
Disclosures
Hazen is named as the co‐inventor on issued and pending patents held by the Cleveland Clinic relating to cardiovascular diagnostics and/or therapeutics; he is also paid as a consultant for the following companies: Esperion, and Procter & Gamble; and has received research funds from Astra Zeneca, Pfizer, Procter & Gamble, Roche Diagnostics Inc, and Takeda. Hazen reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics and/or therapeutics from Cleveland Heart Laboratory, Siemens, Esperion, and Frantz Biomarkers, LLC.
Acknowledgments
We greatly thank Tiffany Wang for her help with RNA isolation and qPCR analysis and Rosa Chen for her help with manuscript preparation. We thank the laboratory of Dr Peter Tontonoz for the use of their microscopy facilities and the laboratory of Dr Luisa Iruela‐Arispe for sharing their expertise on endothelial cell protocols.
(J Am Heart Assoc. 2016;5:e002767 doi: 10.1161/JAHA.115.002767)
References
- 1. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota‐generated metabolite trimethylamine‐N‐oxide. Eur Heart J. 2014;35:904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang WH, Wang Z, Shrestha K, Borowski AG, Wu Y, Troughton RW, Klein AL, Hazen SL. Intestinal microbiota‐dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail. 2015;21:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lever M, George PM, Slow S, Bellamy D, Young JM, Ho M, McEntyre CJ, Elmslie JL, Atkinson W, Molyneux SL, Troughton RW, Frampton CM, Richards AM, Chambers ST. Betaine and trimethylamine‐N‐oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: an observational study. PLoS One. 2014;9:e114969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Troseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP, Aakhus S, Gude E, Bjorndal B, Halvorsen B, Karlsen TH, Aukrust P, Gullestad L, Berge RK, Yndestad A. Microbiota‐dependent metabolite trimethylamine‐N‐oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–726. [DOI] [PubMed] [Google Scholar]
- 8. Mente A, Chalcraft K, Ak H, Davis AD, Lonn E, Miller R, Potter MA, Yusuf S, Anand SS, McQueen MJ. The relationship between trimethylamine‐N‐oxide and prevalent cardiovascular disease in a multiethnic population living in Canada. Can J Cardiol. 2015;31:1189–1194. [DOI] [PubMed] [Google Scholar]
- 9. Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa‐Boyle B, Li XS, Levison BS, Hazen SL. Gut microbiota‐dependent trimethylamine N‐oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stubbs JR, House JA, Ocque AJ, Zhang S, Johnson C, Kimber C, Schmidt K, Gupta A, Wetmore JB, Nolin TD, Spertus JA, Yu AS. Serum trimethylamine‐N‐oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J Am Soc Nephrol. 2016;27:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, Org E, Wu Y, Li L, Smith JD, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. gamma‐Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L‐carnitine to TMAO. Cell Metab. 2014;20:799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ. Trimethylamine‐N‐oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. [DOI] [PubMed] [Google Scholar]
- 14. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. [DOI] [PubMed] [Google Scholar]
- 15. Kim JB, Xia YR, Romanoski CE, Lee S, Meng Y, Shi YS, Bourquard N, Gong KW, Port Z, Grijalva V, Reddy ST, Berliner JA, Lusis AJ, Shih DM. Paraoxonase‐2 modulates stress response of endothelial cells to oxidized phospholipids and a bacterial quorum‐sensing molecule. Arterioscler Thromb Vasc Biol. 2011;31:2624–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mehrabian M, Qiao JH, Hyman R, Ruddle D, Laughton C, Lusis AJ. Influence of the apoA‐II gene locus on HDL levels and fatty streak development in mice. Arterioscler Thromb. 1993;13:1–10. [DOI] [PubMed] [Google Scholar]
- 17. Wang Z, Levison BS, Hazen JE, Donahue L, Li XM, Hazen SL. Measurement of trimethylamine‐N‐oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem. 2014;455:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackson SM, Parhami F, Xi XP, Berliner JA, Hsueh WA, Law RE, Demer LL. Peroxisome proliferator‐activated receptor activators target human endothelial cells to inhibit leukocyte‐endothelial cell interaction. Arterioscler Thromb Vasc Biol. 1999;19:2094–2104. [DOI] [PubMed] [Google Scholar]
- 19. Orozco LD, Bennett BJ, Farber CR, Ghazalpour A, Pan C, Che N, Wen P, Qi HX, Mutukulu A, Siemers N, Neuhaus I, Yordanova R, Gargalovic P, Pellegrini M, Kirchgessner T, Lusis AJ. Unraveling inflammatory responses using systems genetics and gene‐environment interactions in macrophages. Cell. 2012;151:658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol Med Today. 1999;5:439–447. [DOI] [PubMed] [Google Scholar]
- 21. Bochkov VN, Mechtcheriakova D, Lucerna M, Huber J, Malli R, Graier WF, Hofer E, Binder BR, Leitinger N. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR‐1 and Ca(++)/NFAT. Blood. 2002;99:199–206. [DOI] [PubMed] [Google Scholar]
- 22. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. [DOI] [PubMed] [Google Scholar]
- 23. Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF‐kappa B and cytokine‐inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 24. Tak PP, Firestein GS. NF‐kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suska A, Ibanez AB, Lundstrom I, Berghard A. G protein‐coupled receptor mediated trimethylamine sensing. Biosens Bioelectron. 2009;25:715–720. [DOI] [PubMed] [Google Scholar]
- 26. Wallrabenstein I, Kuklan J, Weber L, Zborala S, Werner M, Altmuller J, Becker C, Schmidt A, Hatt H, Hummel T, Gisselmann G. Human trace amine‐associated receptor TAAR5 can be activated by trimethylamine. PLoS One. 2013;8:e54950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Q, Korzan WJ, Ferrero DM, Chang RB, Roy DS, Buchi M, Lemon JK, Kaur AW, Stowers L, Fendt M, Liberles SD. Synchronous evolution of an odor biosynthesis pathway and behavioral response. Curr Biol. 2013;23:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arakawa T, Timasheff SN. The stabilization of proteins by osmolytes. Biophys J. 1985;47:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mashino T, Fridovich I. Effects of urea and trimethylamine‐N‐oxide on enzyme activity and stability. Arch Biochem Biophys. 1987;258:356–360. [DOI] [PubMed] [Google Scholar]
- 30. Hu CY, Lynch GC, Kokubo H, Pettitt BM. Trimethylamine N‐oxide influence on the backbone of proteins: an oligoglycine model. Proteins. 2010;78:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yancey PH, Gerringer ME, Drazen JC, Rowden AA, Jamieson A. Marine fish may be biochemically constrained from inhabiting the deepest ocean depths. Proc Natl Acad Sci USA. 2014;111:4461–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yancey PH, Siebenaller JF. Trimethylamine oxide stabilizes teleost and mammalian lactate dehydrogenases against inactivation by hydrostatic pressure and trypsinolysis. J Exp Biol. 1999;202:3597–3603. [DOI] [PubMed] [Google Scholar]
- 33. Yancey PH, Fyfe‐Johnson AL, Kelly RH, Walker VP, Aunon MT. Trimethylamine oxide counteracts effects of hydrostatic pressure on proteins of deep‐sea teleosts. J Exp Zool. 2001;289:172–176. [DOI] [PubMed] [Google Scholar]
- 34. Jonsson AS, Palmblad JE. Effects of ethanol on NF‐kappaB activation, production of myeloid growth factors, and adhesive events in human endothelial cells. J Infect Dis. 2001;184:761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, Chun J. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–186. [DOI] [PubMed] [Google Scholar]
- 36. Shimada H, Rajagopalan LE. Rho kinase‐2 activation in human endothelial cells drives lysophosphatidic acid‐mediated expression of cell adhesion molecules via NF‐kappaB p65. J Biol Chem. 2010;285:12536–12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fukushima N, Kimura Y, Chun J. A single receptor encoded by vzg‐1/lpA1/edg‐2 couples to G proteins and mediates multiple cellular responses to lysophosphatidic acid. Proc Natl Acad Sci USA. 1998;95:6151–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]