

Abstract
Background
γ‐Glutamyltransferase (GGT) has been linked to an increased risk of several cardiovascular outcomes; however, the relationship of GGT with sudden cardiac death (SCD) has not been investigated previously. We aimed to assess the association of GGT with risk of SCD.
Methods and Results
Serum GGT activity was assessed at baseline in the Kuopio Ischemic Heart Disease prospective cohort of 1780 men, and 136 SCDs were recorded during 22 years of follow‐up. Correction for within‐person variability was made using data from repeated measurements taken several years apart. The regression dilution ratio of loge GGT adjusted for age was 0.68 (95% CI 0.61–0.74). Serum GGT was log‐linearly associated with risk of SCD. The hazard ratio for SCD per 1 SD higher baseline loge GGT values (2‐fold higher) was 1.30 (95% CI 1.10–1.54; P=0.002) after adjustment for several established risk factors and remained consistent with further adjustment for alcohol consumption, resting heart rate, lipids, and C‐reactive protein (hazard ratio 1.26, 95% CI 1.05–1.50; P=0.014). The corresponding hazard ratios were 1.48 (95% CI 1.15–1.89; P=0.002) and 1.40 (95% CI 1.07–1.82; P=0.014) after correction for within‐person variability. Hazard ratios remained unchanged after accounting for incident coronary events and did not vary importantly by levels or categories of prespecified conventional risk factors.
Conclusions
GGT is positively, log‐linearly, and independently associated with future risk of SCD in the general male population. Further research is needed to replicate these findings.
Keywords: γ‐glutamyltranferase, risk factor, sudden cardiac death
Subject Categories: Risk Factors, Primary Prevention, Epidemiology
Introduction
The majority of sudden cardiac deaths (SCDs) occur in the general population and outside of the hospital, with few or no early warning signs.1 Coronary heart disease (CHD) with its sequelae (eg, scarring from previous myocardial infarction, heart failure) is the most common pathology underlying SCD, especially for persons aged >40 years.2, 3 Despite a progressive decline in CHD over past decades due to major advances in treatment and prevention, SCD remains a vast public health problem accounting for 15% to 20% of all deaths.4, 5 In a large percentage of persons, SCD is usually the first manifestation of cardiac disease.6 Out‐of‐hospital SCD is an unrecognized but major contributor to overall SCD. Risk factors for SCD include atherosclerotic cardiovascular disease (CVD) risk factors, especially among middle‐aged and older persons.7 Although these established risk factors explain a large proportion of the risk of SCD,8 its pathogenesis is still not fully established because multiple factors appear to be involved. These established risk factors are often absent in a large proportion of those who have SCD,9 making identification of persons at increased risk a difficult undertaking.
There is a need to further assess potential risk factors that may have causal or predictive relevance to SCD (in‐ and out‐of‐hospital SCD) and that will help tailor preventive and therapeutic interventions. γ‐Glutamyltransferase (GGT), a biomarker that is routinely used in clinical practice to help indicate potential hepatic or biliary disease, has been shown to be positively and independently associated with CHD,10, 11 stroke,10, 11 CVD,11, 12 cardiac arrhythmias,13 and congestive heart failure.14, 15 The proposed mechanistic pathways underpinning these associations include the pro‐oxidant and proinflammatory activities16 of GGT. GGT may be linked to the risk of SCD; however, there has been no previous prospective evaluation of its association with SCD. We aimed to evaluate, in detail, the nature and magnitude of the prospective association of GGT with risk of SCD in a population‐based cohort of 1780 apparently healthy men from eastern Finland. Repeated measurements of GGT were performed in a subset of men to help quantify within‐person variability in GGT values. In subsidiary analysis, we assessed the associations with out‐of‐hospital SCD and non‐SCD.
Methods
Study Population
The study population consisted of a representative sample of men living in the city of Kuopio and its surrounding rural communities in eastern Finland. These men were participants in the Kuopio Ischemic Heart Disease (KIHD) risk factor study, a longitudinal population‐based study designed to investigate risk factors for CVD and related outcomes.17 Men were aged 42 to 61 years during baseline examinations performed between March 1984 and December 1989. Of 3433 potentially randomly eligible and randomly selected men, 2682 (78%) volunteered to participate; 186 did not respond to the invitation, and 367 declined to give informed consent. The final cohort for the present analysis included 1780 men who were not missing information on serum GGT, covariates, and outcomes. The research ethics committee of the University of Eastern Finland approved the study, and each participant gave written informed consent.
Ascertainment of Outcomes
All SCDs that occurred from study enrollment through 2012 were included. There were no losses to follow‐up. In the KIHD study, participants are under continuous surveillance for the development of new CVD events, including incident cases and deaths.18 The sources of information on outcomes were based on a comprehensive review of national hospital discharge records, inpatient physician claims data, study ECGs, death certificate registers, and medical–legal reports. Deaths were coded according to the International Classification of Diseases, 9th or 10th revision codes. The diagnostic classification of events was based on symptoms, ECG findings, cardiac enzyme elevations, autopsy findings (80% of all cardiac deaths), and history of CHD. A death was determined to be an SCD when it occurred within 1 hour of the onset of an abrupt change in symptoms or within 24 hours after the onset of symptoms, including unwitnessed cases when clinical and autopsy findings did not reveal a noncardiac cause of sudden death.19 The witnessed participant was to have been alive and symptom free within 1 hour before the event. SCDs that occurred under out‐of‐hospital conditions were also defined as events that occurred in places that had been reported accurately in hospital documents. The deaths due to aortic aneurysm rupture; cardiac rupture or tamponade; and pulmonary embolism, cancer, or other noncardiac comorbidities were not included as SCDs. Documents were cross‐checked in detail by 2 physicians. Non‐SCDs were also carefully documented using standardized criteria. Cardiac deaths that did not lead to death during the 24 hours following the onset of symptoms were considered non‐SCDs. The independent events committee of the KIHD study, masked to clinical data, performed classification of outcomes.
Measurement of Risk Factors
Collection of blood specimens and measurement of serum lipids, lipoproteins, and glucose have been described previously.20, 21 Blood samples were taken between 8 and 10 am. In addition to fasting, participants were instructed to abstain from drinking alcohol for at least 3 days and from smoking for at least 12 hours before assessment. The serum samples were stored frozen at −80°C for 0.2 to 2.5 years. The cholesterol content of lipoprotein fractions and serum triglycerides were measured enzymatically (Boehringer Mannheim).22 Fasting plasma glucose was measured by the glucose dehydrogenase method (Merck) after protein precipitation by trichloroacetic acid. Serum GGT activity was measured using the kinetic method (Thermo Fisher Scientific) and C‐reactive protein (CRP) with an immunometric assay (Immulite High Sensitivity C‐Reactive Protein Assay; Diagnostic Products Corp). Serial measurements of GGT were performed at 4 and 11 years after baseline measurements over a 22‐year period in a random subset of participants. Smoking, alcohol consumption, and blood pressure were assessed, as described previously.20 Body mass index was computed as the ratio of weight in kilograms to the square of height in meters. Standard resting 12‐lead ECG was also recorded. The ECG criterion for left ventricular hypertrophy was based on either the Sokolow–Lyon or Romhilt–Estes point score.23, 24, 25
Statistical Analysis
Values of skewed variables (GGT, CRP, and triglycerides) were log‐transformed to achieve approximately symmetrical distributions. We performed descriptive analyses summarizing the baseline characteristics of the participants. Cross‐sectional associations of GGT with various risk markers were assessed using linear regression models adjusted for age. Time‐to‐event analyses were conducted using Cox proportional hazards models after confirming proportional hazards assumptions using Schoenfeld residuals.26 To quantify and correct for within‐person variability in levels of GGT, that is, the extent to which an individual person's GGT measurements vary around a long‐term average level of exposure (“usual levels”),27 adjusted regression dilution ratios were estimated by regressing available repeated measurements on baseline values.28 To characterize the shape of the association between GGT and SCD risk, hazard ratios (HRs) were calculated within quartiles of baseline GGT values and plotted against mean GGT values within each quartile. Floating variances were used to calculate 95% CIs for the log HR in each group, including the reference group, to allow for comparisons across the groups, regardless of the arbitrarily chosen reference category (bottom quartile).26 Because the association showed a log‐linear shape, HRs were calculated per 1 SD higher loge GGT values. The SD of baseline loge GGT was 0.63 (equivalent to ≈2‐fold higher circulating GGT, e0.63=1.88). HRs were adjusted for age, body mass index, systolic blood pressure, prevalent CHD, smoking status, history of diabetes mellitus, left ventricular hypertrophy, history of hypertension, use of medications (antihypertensive agents and lipid‐lowering drugs), alcohol consumption, resting heart rate, triglycerides, total cholesterol, high‐density lipoprotein cholesterol, and CRP. We performed subgroup analyses using interaction tests to assess statistical evidence of any differences in hazards across levels or categories of prespecified individual‐level characteristics, including age at survey, smoking status, history of diabetes mellitus, history of hypertension, left ventricular hypertrophy, body mass index, systolic blood pressure, and CRP. To minimize biases as a result of including cases with prevalent but undetected CHD, heart failure, and cardiac arrhythmias, sensitivity analysis involved excluding the first 5 years of follow‐up. All statistical analyses were conducted using Stata version 14 (Stata Corp).
Results
Baseline Characteristics and Correlates of GGT
Table 1 summarizes baseline characteristics of the 1780 men in the current study. The mean age was 53 years (SD 5 years). Median GGT value was 20 U/L (interquartile range 15–32 U/L). During a mean follow‐up of 22 years, there were 136 SCDs (annual rate 3.50 per 1000 person‐years at risk; 95% CI 2.96–4.14). Of the total SCDs, 108 occurred out of the hospital. There were 66 non‐SCDs. Serum GGT values were weakly to moderately and positively correlated with physical measures (body mass index, blood pressure, and resting heart rate) and with several lipid, metabolic, and inflammation (CRP) markers. Weak inverse correlations were observed for age (r=−0.03) and high‐density lipoprotein cholesterol (r=−0.04). Baseline GGT values were higher in men with diabetes compared with men without diabetes, in men with a history of hypertension compared with men without a history of hypertension, and in current smokers compared with nonsmokers (Table 2).
Table 1.
Baseline Participant Characteristics
| Overall (N=1780) Mean (SD) or n (%) | Without SCD (n=1644) Mean (SD) or n (%) | With SCD (n=136) Mean (SD) or % | P Value | |
|---|---|---|---|---|
| Loge GGT, U/L | 3.11 (0.63) | 3.09 (0.61) | 3.39 (0.76) | <0.0001 |
| Questionnaire/prevalent conditions | ||||
| Age at survey, y | 52.6 (5.0) | 52.5 (5.1) | 54.0 (3.8) | 0.002 |
| Alcohol consumption, g/week | 76.1 (140.2) | 74.0 (140.1) | 103.9 (140.1) | 0.023 |
| History of diabetes | 82 (4.6) | 68 (4.1) | 14 (10.3) | 0.001 |
| Current smokers | 577 (32.4) | 515 (31.3) | 62 (45.6) | 0.001 |
| Left ventricular hypertrophy | 19 (1.1) | 17 (1.0) | 2 (1.5) | 0.634 |
| History of hypertension | 517 (29) | 473 (28.8) | 44 (32.4) | 0.377 |
| History of CHD | 391 (22.0) | 334 (20.3) | 57 (41.9) | <0.001 |
| Use of antihypertensives | 325 (18.3) | 287 (17.5) | 38 (27.9) | 0.002 |
| Medication for dyslipidemia | 11 (0.6) | 11 (0.7) | 0 (0.0) | 0.339 |
| Physical measurements | ||||
| BMI, kg/m2 | 26.8 (3.5) | 26.7 (3.5) | 28.1 (4.2) | <0.0001 |
| SBP, mm Hg | 133.9 (16.6) | 133.6 (16.5) | 138.6 (18.0) | 0.001 |
| DBP, mm Hg | 89.0 (10.5) | 88.9 (10.5) | 91.0 (10.3) | 0.031 |
| Resting heart rate, bpm | 62.5 (10.7) | 62.4 (10.6) | 63.9 (11.1) | 0.122 |
| Lipid markers | ||||
| Total cholesterol, mmol/L | 5.93 (1.10) | 5.91 (1.09) | 6.21 (1.13 | 0.004 |
| HDL‐C, mmol/L | 1.29 (0.30) | 1.30 (0.30) | 1.23 (0.26) | 0.015 |
| Loge triglycerides, mmol/L | 0.09 (0.50) | 0.08 (0.49) | 0.19 (0.55) | 0.020 |
| Metabolic and inflammatory markers | ||||
| Fasting plasma glucose, mmol/L | 5.33 (1.21) | 5.30 (1.14) | 5.77 (1.94) | <0.0001 |
| Serum creatinine, μmol/L | 89.4 (22.5) | 89.2 (13.4) | 91.7 (70.7) | 0.251 |
| Loge CRP, mg/L | 0.29 (0.96) | 0.26 (0.95) | 0.69 (1.01) | <0.0001 |
BMI indicates body mass index; CHD, coronary heart disease; CRP, C‐reactive protein; DBP, diastolic blood pressure; GGT, γ‐glutamyltransferase; HDL‐C, high‐density lipoprotein cholesterol; SBP, systolic blood pressure; SCD, sudden cardiac death.
Table 2.
Cross‐Sectional Correlates of GGT
| Pearson Correlation r (95% CI)a | Percentage Difference (95% CI) in GGT Levels Per 1 SD Higher or Compared With Reference Category of Correlateb | |
|---|---|---|
| Loge GGT, U/L | — | — |
| Questionnaire/prevalent conditions | ||
| Age at survey, y | −0.03 (−0.08 to 0.02) | −2% (−5 to 1) |
| Alcohol consumption, g/week | 0.27 (0.23–0.31)*** | 19% (15–22)*** |
| History of diabetes | ||
| No | — | Ref |
| Yes | — | 47% (28–69)*** |
| Smoking status | ||
| Other | — | Ref |
| Current | — | 11% (4–18)** |
| Left ventricular hypertrophy | ||
| No | — | Ref |
| Yes | — | −4% (−28 to 28) |
| History of hypertension | ||
| No | — | Ref |
| Yes | — | 27% (20–36)*** |
| History of CHD | ||
| No | — | Ref |
| Yes | — | 17% (9–25)*** |
| Use of antihypertensives | ||
| No | — | Ref |
| Yes | — | 24% (15–34)*** |
| Medication for dyslipidemia | ||
| No | — | Ref |
| Yes | — | 25% (−14 to 81) |
| Physical measurements | ||
| BMI, kg/m2 | 0.34 (0.30–0.38)*** | 24% (21–27)*** |
| SBP, mm Hg | 0.21 (0.17–0.26)*** | 14% (11–18)*** |
| DBP, mm Hg | ||
| Resting heart rate, bpm | 0.14 (0.10–0.19)*** | 9% (6–13)*** |
| Lipid markers | ||
| Total cholesterol, mmol/L | 0.10 (0.05–0.14)*** | 6% (3–9)*** |
| HDL‐C, mmol/L | −0.04 (−0.09 to 0.01) | −2% (−5 to 0) |
| Loge triglycerides, mmol/L | 0.26 (0.21–0.30)*** | 18% (14–21)*** |
| Metabolic and inflammatory markers | ||
| Fasting plasma glucose, mmol/L | 0.21 (0.16–0.25)*** | 14% (11–17)*** |
| Serum creatinine, μmol/L | 0.00 (−0.04 to 0.05) | 0% (−3 to 3) |
| Loge CRP, mg/L | 0.27 (0.22–0.31)*** | 18% (15–21)*** |
BMI indicates body mass index; CHD, coronary heart disease; CRP, C‐reactive protein; DBP, diastolic blood pressure; HDL‐C, high‐density lipoprotein cholesterol; GGT, γ‐glutamyltransferase; Ref, reference.
Asterisks indicate the level of statistical significance: **P<0.01; ***P<0.001.
Pearson correlation coefficients between loge GGT and the row variables.
Percentage change in GGT levels per 1‐SD increase in the row variable (or for categorical variables, the percentage difference in mean GGT levels for the category vs the reference) adjusted for age.
Correction for Within‐Person Variability
Serial measurements of GGT taken at 4 and 11 years after baseline over 22 years were available in a random sample of 624 men, providing a total of 1143 repeated measurements of GGT. The regression dilution ratio of loge GGT, adjusted for age, was 0.68 (95% CI 0.61–0.74), suggesting that the associations using single or baseline measurements of GGT with SCD could underestimate the association by [(1/0.68)−1]×100=47%.
GGT and Risk of SCD
SCD rates per 1000 person‐years of follow‐up across quartiles of GGT were 5.67 (95% CI 4.31–7.46) for the fourth quartile, 3.54 (95% CI: 2.52–4.98) for the third quartile, 3.09 (95% CI 2.10–4.354) for the second quartile, and 2.14 (95% CI: 1.46–3.15) for the first quartile. Cumulative hazard curves demonstrated a greater risk of SCDs among men in the top quartile of GGT levels compared with those in the bottom quartile (P=0.0001 for log‐rank test) (Figure 1). Baseline and usual GGT values were log‐linearly associated with risk of SCD in analyses adjusted for conventional risk factors (age, body mass index, systolic blood pressure, prevalent CHD, smoking status, history of diabetes mellitus, left ventricular hypertrophy, history of hypertension, use of antihypertensive agents and lipid‐lowering drugs) (Figure 2). The age‐adjusted HR per 1‐SD change in baseline loge GGT value was 1.56 (95% CI 1.34–1.82; P<0.001), which was somewhat attenuated following further adjustment for established risk factors (HR 1.30, 95% CI 1.10–1.54; P=0.002). The results remained consistent on further adjustment for alcohol consumption, resting heart rate, triglycerides, total cholesterol, and high‐density lipoprotein cholesterol (HR 1.26, 95% CI 1.05–1.50; P=0.014) and further for CRP (HR 1.23, 95% CI 1.02–1.47; P=0.028). The corresponding HRs per 1‐SD change in usual loge GGT values were 1.93 (95% CI 1.55–2.41; P<0.001), 1.48 (95% CI 1.15–1.89; P=0.002), 1.40 (95% CI 1.07–1.82; P=0.014), and 1.35 (95% CI 1.03–1.77; P=0.028), respectively. The HRs remained materially unchanged after further adjusting for incident coronary events (Table 3). HRs did not vary importantly by levels or categories of prespecified conventional risk factors (P for interaction >0.10 for each) (Figure 3), and the main results remained the same in analyses that excluded the first 5 years of follow‐up (data not shown). In separate analyses for out‐of‐hospital SCD and non‐SCD, the initial positive associations of GGT with out‐of‐hospital SCD and non‐SCD in analyses adjusted for several established risk factors were less robust on further adjustment for potential confounders (Table 3). To put the strength of the association of GGT with risk of SCD into context, comparisons were made to the associations of GGT with the combined outcome of SCDs plus resuscitated sudden cardiac arrests. The associations were similar compared with those observed for GGT and SCD (Table 4).
Figure 1.
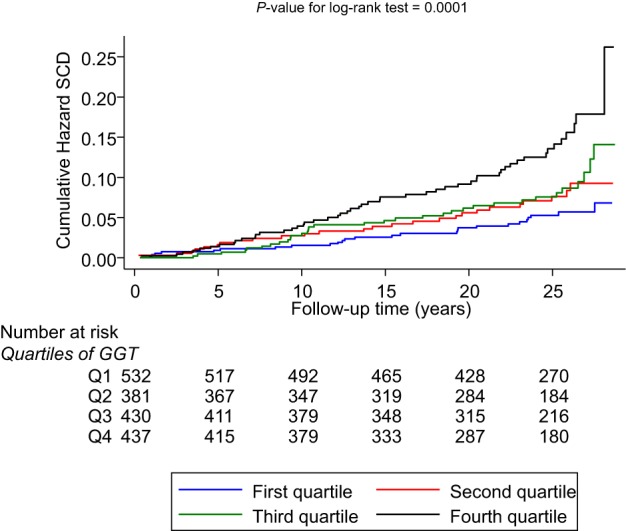
Cumulative hazard curves for sudden cardiac death by quartiles of GGT. The median GGT level was 12.0 IU/L (range 11–14 IU/L) for the lowest quartile; 18.0 IU/L (range 16–19 IU/L) for the second quartile; 25.0 IU/L (range 23–29 IU/L) for the third quartile; and 46.0 IU/L (range 38–68 IU/L) for the top quartile. GGT indicates γ‐glutamyltransferase; Q, quartile; SCD, sudden cardiac death.
Figure 2.
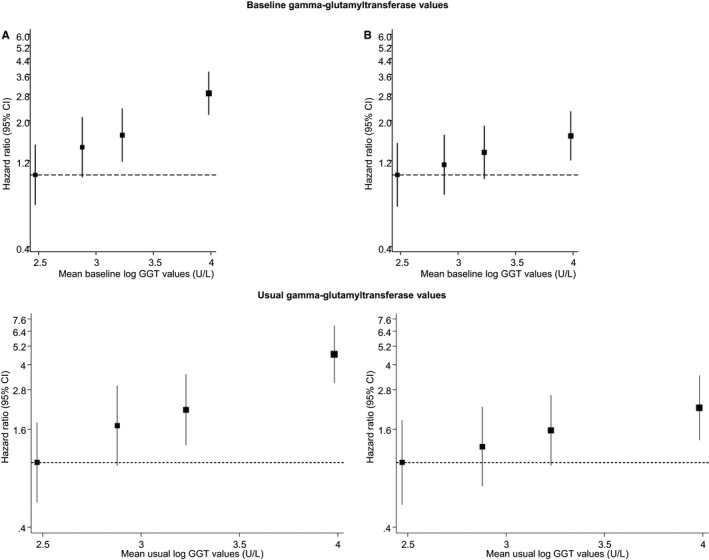
Hazard ratios for sudden cardiac death, by quartiles of baseline and usual values of GGT. A, Adjusted for age. B, Adjusted for age, body mass index, systolic blood pressure, prevalent coronary heart disease, smoking status, history of diabetes, left ventricular hypertrophy, history of hypertension, and use of medications (antihypertensive agents and lipid‐lowering drugs). GGT indicates γ‐glutamyltransferase.
Table 3.
Associations of Baseline and Usual Levels of GGT With SCDs, Out‐of‐Hospital SCDs, and Non‐SCDs
| Models | SCD | P Value | Out‐of‐Hospital SCD | P Value | Non‐SCD | P Value |
|---|---|---|---|---|---|---|
| HR (95% CI) | HR (95% CI) | HR (95% CI) | ||||
| n=136/1780 | n=108/1780 | n=66/1780 | ||||
| Baseline GGT | ||||||
| Model 1 | 1.56 (1.34–1.82) | <0.001 | 1.47 (1.24–1.74) | <0.001 | 1.46 (1.16–1.83) | 0.001 |
| Model 2 | 1.30 (1.10–1.54) | 0.002 | 1.23 (1.01–1.49) | 0.038 | 1.34 (1.04–1.72) | 0.022 |
| Model 3 | 1.26 (1.05–1.50) | 0.014 | 1.17 (0.95–1.44) | 0.139 | 1.29 (0.99–1.68) | 0.059 |
| Model 4 | 1.23 (1.02–1.47) | 0.028 | 1.16 (0.95–1.43) | 0.152 | 1.27 (0.97–1.67) | 0.077 |
| Model 5 | 1.23 (1.03–1.48) | 0.025 | 1.17 (0.95–1.44) | 0.149 | 1.29 (0.99–1.68) | 0.065 |
| Usual GGT | ||||||
| Model 1 | 1.93 (1.55–2.41) | <0.001 | 1.76 (1.37–2.27) | <0.001 | 1.74 (1.24–2.43) | 0.001 |
| Model 2 | 1.48 (1.15–1.89) | 0.002 | 1.35 (1.02–1.79) | 0.038 | 1.53 (1.06–2.21) | 0.022 |
| Model 3 | 1.40 (1.07–1.82) | 0.014 | 1.26 (0.93–1.70) | 0.139 | 1.46 (0.99–2.15) | 0.059 |
| Model 4 | 1.35 (1.03–1.77) | 0.028 | 1.25 (0.92–1.70) | 0.152 | 1.43 (0.96–2.12) | 0.077 |
| Model 5 | 1.36 (1.04–1.78) | 0.025 | 1.25 (0.92–1.70) | 0.149 | 1.45 (0.98–2.14) | 0.065 |
Model 1: Adjusted for age. Model 2: Model 1 plus body mass index, systolic blood pressure, prevalent coronary heart disease, smoking status, history of diabetes, left ventricular hypertrophy, history of hypertension, and use of medications (antihypertensive agents and lipid‐lowering drugs). Model 3: Model 2 plus alcohol consumption, resting heart rate, triglycerides, total cholesterol, and high‐density lipoprotein cholesterol. Model 4: Model 3 plus C‐reactive protein. Model 5: Model 4 plus incident coronary heart disease as a time‐dependent covariate. HRs are reported per 1‐SD increase in loge GGT levels; 1 SD higher loge GGT was approximately equivalent to 2‐fold higher GGT levels. GGT indicates γ‐glutamyltransferase; HR, hazard ratio; SCD, sudden cardiac death.
Figure 3.
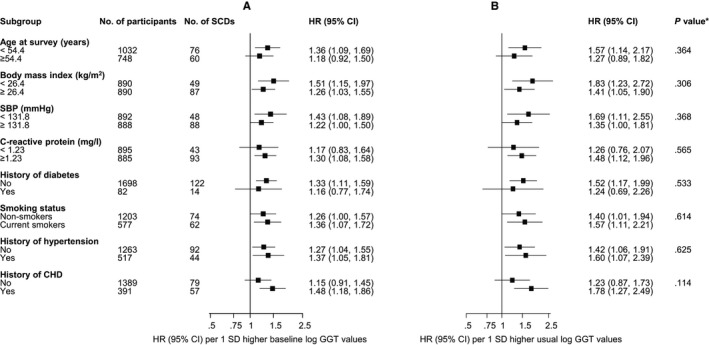
HRs for baseline and usual levels of GGT and SCD risk by several participant‐level characteristics. A, Baseline levels of GGT. B, Usual levels of GGT. HRs were adjusted for age, body mass index, SBP, prevalent CHD, smoking status, history of diabetes, left ventricular hypertrophy, history of hypertension, and use of medications (antihypertensive agents and lipid‐lowering drugs). *, P value for interaction. CHD indicates coronary heart disease; GGT, γ‐glutamyltransferase; HR, hazard ratio; SCD, sudden cardiac death; SBP, systolic blood pressure.
Table 4.
Associations of Baseline and Usual Levels of GGT With the Combined Outcome of Sudden Cardiac Deaths and Resuscitated Sudden Cardiac Arrests
| HR (95% CI) | P Value | |
|---|---|---|
| n=158/1780 | ||
| Baseline GGT levels | ||
| Model 1 | 1.53 (1.33–1.77) | <0.001 |
| Model 2 | 1.30 (1.11–1.52) | 0.001 |
| Model 3 | 1.26 (1.06–1.49) | 0.008 |
| Model 4 | 1.23 (1.03–1.46) | 0.019 |
| Model 5 | 1.24 (1.04–1.46) | 0.016 |
| Usual GGT levels | ||
| Model 1 | 1.87 (1.52–2.31) | <0.001 |
| Model 2 | 1.47 (1.16–1.85) | 0.001 |
| Model 3 | 1.40 (1.09–1.80) | 0.008 |
| Model 4 | 1.35 (1.05–1.74) | 0.019 |
| Model 5 | 1.36 (1.06–1.75) | 0.016 |
Model 1: Adjusted for age. Model 2: Model 1 plus body mass index, systolic blood pressure, prevalent coronary heart disease, smoking status, history of diabetes, left ventricular hypertrophy, history of hypertension, and use of medications (antihypertensive agents and lipid‐lowering drugs). Model 3: Model 2 plus alcohol consumption, resting heart rate, triglycerides, total cholesterol, and high‐density lipoprotein cholesterol. Model 4: Model 3 plus C‐reactive protein. Model 5: Model 4 plus incident coronary heart disease as a time‐dependent covariate. HRs are reported per 1‐SD increase in loge GGT levels; 1 SD higher loge GGT was approximately equivalent to 2‐fold higher GGT levels. GGT indicates γ‐glutamyltransferase; HR, hazard ratio.
Discussion
In this population of middle‐aged and older men without a history of heart failure and cardiac arrhythmias at baseline, we have assessed—for the first time—the shape, magnitude, and independence of the prospective association of both baseline and usual values of GGT with risk of SCD. There were weak to modest correlations of GGT values with several conventional cardiovascular risk factors. In analyses adjusted for age and established risk factors, we observed an approximately log‐linear association of GGT with risk of SCD. The association remained persistent on further adjustment for a comprehensive panel of potential confounders and on accounting for incident coronary events during follow‐up. The overall findings remained consistent across several subgroups and levels of cardiovascular risk markers. Significant associations with out‐of‐hospital SCD and non‐SCD were observed in analyses adjusted for several established risk factors but were less robust after further adjustment for potential confounders.
Possible Explanations for Findings
Several studies have demonstrated GGT levels to be associated with a variety of CVD end points; however, no study has evaluated the specific end point of SCD. The finding of a positive and log‐linear association of GGT with SCD is consistent with that observed for GGT and CVD,11, 12 thereby highlighting important clues regarding the mechanistic pathways underlying the association between GGT and SCD. The most likely culprits are the proinflammatory activities of GGT16 and the direct involvement of GGT in atheromatous plaque formation.29, 30 Given the length of time between baseline measurements of GGT and subsequent occurrence of SCD in our study (>20 years), there is a likelihood that the effects of inflammation on SCD are mediated through a chronic process, which is most likely to be the development and progression of coronary atherosclerosis.7 Indeed, pathological signs of inflammation are evident in the coronary atherosclerotic lesions and vulnerable plaques that are found in the majority of cases of SCD.31, 32 The GGT–SCD association observed in our study remained robust after adjusting for CRP. This was unexpected, given that elevated levels of GGT may reflect chronic subclinical inflammation, a state characterized by elevated levels of CRP, which is also secreted by the liver and is directly and strongly correlated with levels of GGT12 (as demonstrated in the current study). The persistent association on further adjustment for CRP reflects the possibility that other pathways apart from inflammatory processes may also play a role in the etiology between GGT and SCD. Our data showed that the joint association of elevated GGT and CRP levels was associated with a stronger risk of SCD, but the estimate was imprecise and was not statistically significant because of the small number of SCDs in each category (data not shown). Other likely pathways include the involvement of GGT in impairing antioxidant defense systems and promoting oxidative stress16 and underlying fatty liver,16 which is also associated with insulin resistance and oxidative stress,33, 34 all known to be associated with increased risk of CVD.35, 36, 37 Through its involvement in atheromatous plaque formation,29, 30 GGT may also cause acute myocardial ischemia, which is the most common trigger for fatal arrhythmias.38 It is difficult to place our findings in the context of previous population‐based prospective studies because we were unable to locate any previously published articles exploring the prospective association between serum GGT and the specific cardiovascular end point of SCD. Further research is needed to help unravel the mechanistic pathways of GGT in the pathogenesis of SCD.
Implications of Findings
Our findings highlight a clear and independent link between serum GGT and the risk of SCD and may have clinical implications. Given the long‐term association between baseline levels of GGT and risk of SCD, assays for GGT (which are sensitive, well standardized, simple, quick, inexpensive, commonly measured as part of routine liver function panels, and do not require a fasting state prior to venipuncture) may have the potential to be used in the identification of persons at high risk of SCD. In addition, early intervention in the development of the disease process will be important for prevention of SCD events. Although the causal nature of this association remains to be investigated, there is a possibility that lowering levels of GGT may decrease the risk of future SCD. Those with elevated GGT levels might benefit from nutritional and lifestyle modifications (eg, weight loss and physical activity39) that decrease GGT levels. Formal risk assessment analyses are warranted to assess the role of GGT in the prediction of SCD.
Strengths and Limitations
The notable strengths of this study include a large sample that was selected to be a nationally representative population‐based sample of middle‐aged men, that was well characterized, and that involved high response, and there were no losses during follow‐up, reducing the risk of selection bias. Participants were monitored prospectively using established databases for hospital admissions, including out‐of‐hospital outcome events, supplemented with reliable data on a comprehensive panel of lifestyle and biological markers to allow adequate adjustment for potential confounding, enabling reliable assessments of the associations. The mean follow‐up period in this study was sufficiently long to ascertain the risk for SCD in the general‐population setting. Serial measurements of GGT made within a subset of men over time were available, enabling quantification of the extent of within‐person variability over the long period of follow‐up. The KIHD study made such a correction on the basis of 2 repeated measurements, indicating an independent association of GGT with SCD. Nevertheless, studies are needed with repeated measurements of GGT in larger numbers of participants to assess GGT variability in greater detail. The reliability of the data was also confirmed by our ability to replicate the independent association of GGT with other cardiovascular outcomes.11 Our study had a number of limitations in addition to the several strengths listed. We included a relatively small number of SCDs; therefore, these findings need to be confirmed in larger scale studies with more SCD events. Measurements of other liver function enzymes were not made in the KIHD study, preventing comparison of the separate and joint associations of different liver function enzymes with SCD risk. Participants in the current study were not suitable for risk prediction analysis because we did not have complete measurements of risk factors used in standard risk‐prediction algorithms for SCD. The KIHD study involved prolonged blood storage, which could cause underestimation of the associations; however, GGT is not known to be influenced by prolonged storage or repeated freeze–thaw cycles.40 The KIHD study included only middle‐aged white men from eastern Finland, an area known for its high prevalence and incidence of atherosclerotic vascular diseases. The annual rate of SCDs of 3.50 per 1000 person‐years at risk reflects the high rate of CHD mortality in the study region, which has the highest rate in the world.41, 42 Consequently, our findings cannot necessarily be extrapolated to women, young persons, elderly persons, and other populations. Although a comprehensive panel of confounders was taken into account to ensure the validity of our results, potential residual confounding due to errors in risk‐marker measurements and other unmeasured confounders cannot be entirely ruled out.
Conclusions
GGT activity is positively and log‐linearly associated with future risk of SCD in the general male population. Further research is needed to replicate these findings and to assess the potential clinical utility of GGT as a risk assessment tool or validated causal therapeutic target for SCD.
Sources of Funding
The Academy of Finland, Helsinki, Finland; Finnish Foundation for Cardiovascular Research, Helsinki, Finland, and Finnish Cultural Foundation, Helsinki, Finland, supported the Kuopio Ischemic Heart Disease Study. These sources had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Disclosures
None.
Acknowledgments
We thank the staff of the Kuopio Research Institute of Exercise Medicine and the Research Institute of Public Health and University of Eastern Finland, Kuopio, Finland, for the data collection in the study.
(J Am Heart Assoc. 2016;5:e002858 doi: 10.1161/JAHA.115.002858)
References
- 1. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. [DOI] [PubMed] [Google Scholar]
- 2. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116:1887–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Authors/Task Force M , Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–2867. pii: ehv316. [DOI] [PubMed] [Google Scholar]
- 4. Myerburg RJ, Castellanos A. Sudden cardiac death In: Zipes DP, Jalife J, eds. Cardiac Electrophysiology: From Cell to Bedside. 5th ed Philadelphia, PA: Saunders Elsevier; 2009:797–808. [Google Scholar]
- 5. Gillum RF. Geographic variation in sudden coronary death. Am Heart J. 1990;119:380–389. [DOI] [PubMed] [Google Scholar]
- 6. Myerburg RJ, Junttila MJ. Sudden cardiac death caused by coronary heart disease. Circulation. 2012;125:1043–1052. [DOI] [PubMed] [Google Scholar]
- 7. Albert CM, Ma J, Rifai N, Stampfer MJ, Ridker PM. Prospective study of C‐reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death. Circulation. 2002;105:2595–2599. [DOI] [PubMed] [Google Scholar]
- 8. Grundy SM, D'Agostino RB Sr, Mosca L, Burke GL, Wilson PW, Rader DJ, Cleeman JI, Roccella EJ, Cutler JA, Friedman LM. Cardiovascular risk assessment based on US cohort studies: findings from a National Heart, Lung, and Blood Institute Workshop. Circulation. 2001;104:491–496. [DOI] [PubMed] [Google Scholar]
- 9. Spooner PM, Zipes DP. Sudden death predictors: an inflammatory association. Circulation. 2002;105:2574–2576. [DOI] [PubMed] [Google Scholar]
- 10. Fraser A, Harris R, Sattar N, Ebrahim S, Smith GD, Lawlor DA. Gamma‐glutamyltransferase is associated with incident vascular events independently of alcohol intake: analysis of the British Women's Heart and Health Study and Meta‐Analysis. Arterioscler Thromb Vasc Biol. 2007;27:2729–2735. [DOI] [PubMed] [Google Scholar]
- 11. Kunutsor SK, Apekey TA, Khan H. Liver enzymes and risk of cardiovascular disease in the general population: a meta‐analysis of prospective cohort studies. Atherosclerosis. 2014;236:7–17. [DOI] [PubMed] [Google Scholar]
- 12. Kunutsor SK, Bakker SJ, Kootstra‐Ros JE, Gansevoort RT, Dullaart RP. Circulating gamma glutamyltransferase and prediction of cardiovascular disease. Atherosclerosis. 2014;238:356–364. [DOI] [PubMed] [Google Scholar]
- 13. Alonso A, Misialek JR, Amiin MA, Hoogeveen RC, Chen LY, Agarwal SK, Loehr LR, Soliman EZ, Selvin E. Circulating levels of liver enzymes and incidence of atrial fibrillation: the Atherosclerosis Risk in Communities cohort. Heart. 2014;100:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wannamethee SG, Whincup PH, Shaper AG, Lennon L, Sattar N. Gamma‐glutamyltransferase, hepatic enzymes, and risk of incident heart failure in older men. Arterioscler Thromb Vasc Biol. 2012;32:830–835. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y, Tuomilehto J, Jousilahti P, Salomaa V, Li B, Antikainen R, Mahonen M, Katzmarzyk PT, Hu G. Serum gamma‐glutamyltransferase and the risk of heart failure in men and women in Finland. Heart. 2013;99:163–167. [DOI] [PubMed] [Google Scholar]
- 16. Emdin M, Pompella A, Paolicchi A. Gamma‐glutamyltransferase, atherosclerosis, and cardiovascular disease: triggering oxidative stress within the plaque. Circulation. 2005;112:2078–2080. [DOI] [PubMed] [Google Scholar]
- 17. Salonen JT. Is there a continuing need for longitudinal epidemiologic research? The Kuopio Ischaemic Heart Disease Risk Factor Study. Ann Clin Res. 1988;20:46–50. [PubMed] [Google Scholar]
- 18. Karppi J, Kurl S, Makikallio TH, Ronkainen K, Laukkanen JA. Serum beta‐carotene concentrations and the risk of congestive heart failure in men: a population‐based study. Int J Cardiol. 2013;168:1841–1846. pii: S0167‐5273(12)01701‐9. [DOI] [PubMed] [Google Scholar]
- 19. Laukkanen JA, Makikallio TH, Rauramaa R, Kiviniemi V, Ronkainen K, Kurl S. Cardiorespiratory fitness is related to the risk of sudden cardiac death: a population‐based follow‐up study. J Am Coll Cardiol. 2010;56:1476–1483. [DOI] [PubMed] [Google Scholar]
- 20. Salonen JT, Nyyssonen K, Korpela H, Tuomilehto J, Seppanen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86:803–811. [DOI] [PubMed] [Google Scholar]
- 21. Kunutsor SK, Khan H, Laukkanen JA. Serum albumin concentration and incident type 2 diabetes risk: new findings from a population‐based cohort study. Diabetologia. 2015;58:961–967. [DOI] [PubMed] [Google Scholar]
- 22. Salonen JT, Salonen R, Seppanen K, Rauramaa R, Tuomilehto J. HDL, HDL2, and HDL3 subfractions, and the risk of acute myocardial infarction. A prospective population study in eastern Finnish men. Circulation. 1991;84:129–139. [DOI] [PubMed] [Google Scholar]
- 23. Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol. 2001;6:343–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okin PM, Roman MJ, Devereux RB, Pickering TG, Borer JS, Kligfield P. Time‐voltage QRS area of the 12‐lead electrocardiogram: detection of left ventricular hypertrophy. Hypertension. 1998;31:937–942. [DOI] [PubMed] [Google Scholar]
- 25. Oikarinen L, Karvonen M, Viitasalo M, Takala P, Kaartinen M, Rossinen J, Tierala I, Hanninen H, Katila T, Nieminen MS, Toivonen L. Electrocardiographic assessment of left ventricular hypertrophy with time‐voltage QRS and QRST‐wave areas. J Hum Hypertens. 2004;18:33–40. [DOI] [PubMed] [Google Scholar]
- 26. Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York: Springer; 2000. [Google Scholar]
- 27. Fibrinogen Studies C , Wood AM, White I, Thompson SG, Lewington S, Danesh J. Regression dilution methods for meta‐analysis: assessing long‐term variability in plasma fibrinogen among 27,247 adults in 15 prospective studies. Int J Epidemiol. 2006;35:1570–1578. [DOI] [PubMed] [Google Scholar]
- 28. Rosner B, Willett WC, Spiegelman D. Correction of logistic regression relative risk estimates and confidence intervals for systematic within‐person measurement error. Stat Med. 1989;8:1051–1069; discussion 1071‐3. [DOI] [PubMed] [Google Scholar]
- 29. Franzini M, Corti A, Martinelli B, Del Corso A, Emdin M, Parenti GF, Glauber M, Pompella A, Paolicchi A. Gamma‐glutamyltransferase activity in human atherosclerotic plaques–biochemical similarities with the circulating enzyme. Atherosclerosis. 2009;202:119–127. [DOI] [PubMed] [Google Scholar]
- 30. Paolicchi A, Emdin M, Ghliozeni E, Ciancia E, Passino C, Popoff G, Pompella A. Human atherosclerotic plaques contain gamma‐glutamyl transpeptidase enzyme activity. Circulation. 2004;109:1440. [DOI] [PubMed] [Google Scholar]
- 31. Davies MJ, Thomas A. Thrombosis and acute coronary‐artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137–1140. [DOI] [PubMed] [Google Scholar]
- 32. Spaulding CM, Joly LM, Rosenberg A, Monchi M, Weber SN, Dhainaut JF, Carli P. Immediate coronary angiography in survivors of out‐of‐hospital cardiac arrest. N Engl J Med. 1997;336:1629–1633. [DOI] [PubMed] [Google Scholar]
- 33. Targher G, Bertolini L, Rodella S, Lippi G, Franchini M, Zoppini G, Muggeo M, Day CP. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring). 2008;16:1394–1399. [DOI] [PubMed] [Google Scholar]
- 34. Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. [DOI] [PubMed] [Google Scholar]
- 35. Chamberlain AM, Agarwal SK, Ambrose M, Folsom AR, Soliman EZ, Alonso A. Metabolic syndrome and incidence of atrial fibrillation among blacks and whites in the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 2010;159:850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Youn JY, Zhang J, Zhang Y, Chen H, Liu D, Ping P, Weiss JN, Cai H. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol. 2013;62:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo Y, Lip GY, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012;60:2263–2270. [DOI] [PubMed] [Google Scholar]
- 38. Davies MJ. Anatomic features in victims of sudden coronary death. Coronary artery pathology. Circulation. 1992;85:I19–I24. [PubMed] [Google Scholar]
- 39. Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non‐alcoholic fatty liver disease in adults: a systematic review. J Hepatol. 2012;56:255–266. [DOI] [PubMed] [Google Scholar]
- 40. Rhone DP, White FM. Effects of storage in the cold on activity of gamma‐glutamyltransferase in serum. Clin Chem. 1976;22:103–104. [PubMed] [Google Scholar]
- 41. Tunstall‐Pedoe H, Kuulasmaa K, Amouyel P, Arveiler D, Rajakangas AM, Pajak A. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case‐fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90:583–612. [DOI] [PubMed] [Google Scholar]
- 42. Keys A. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Cambridge (MA): Harvard University Press; 1980. [Google Scholar]