

Abstract
Background
Despite the high prevalence of chronic venous insufficiency and varicose veins in the Western world, suitable pharmaceutical therapies for these venous diseases have not been explored to date. In this context, we recently reported that a chronic increase in venous wall stress or biomechanical stretch is sufficient to cause development of varicose veins through the activation of the transcription factor activator protein 1.
Methods and Results
We investigated whether deleterious venous remodeling is suppressed by the pleiotropic effects of statins. In vitro, activator protein 1 activity was inhibited by two 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors, rosuvastatin and atorvastatin, in stretch‐stimulated human venous smooth muscle cells. Correspondingly, both statins inhibited venous smooth muscle cell proliferation as well as mRNA expression of the activator protein 1 target gene monocyte chemotactic protein 1 (MCP1). In isolated mouse veins exposed to an increased level of intraluminal pressure, statin treatment diminished proliferation of venous smooth muscle cells and protein abundance of MCP1 while suppressing the development of varicose veins in a corresponding animal model by almost 80%. Further analyses of human varicose vein samples from patients chronically treated with statins indicated a decrease in venous smooth muscle cell proliferation and MCP1 abundance compared with samples from untreated patients.
Conclusions
Our findings imply that both atorvastatin and rosuvastatin effectively inhibit the development of varicose veins, at least partially, by interfering with wall stress–mediated activator protein 1 activity in venous smooth muscle cells. For the first time, this study reveals a potential pharmacological treatment option that may be suitable to prevent growth of varicose veins and to limit formation of recurrence after varicose vein surgery.
Keywords: activator protein 1, 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors, smooth muscle cells, vascular remodeling, veins
Subject Categories: Animal Models of Human Disease, Hypertension, Vascular Disease, Pathophysiology, Remodeling
Introduction
The term chronic venous insufficiency (CVI) is often used to functionally describe the full spectrum of venous diseases such as development of varicose veins. Depending on the population and the study design, the prevalence of CVI has been reported in up to 40% of women and 17% of men,1 whereas the mean prevalence of varicose veins in the adult population varies between 5% and 30%, with female predominance.2, 3 As one of the most common medical conditions in the United States, health care costs for the treatment of CVI‐associated diseases such as venous ulcers exceed $1 billion annually. Aside from surgical intervention, no causative treatment options are available to inhibit the maladaptive venous remodeling that leads to formation of varicose veins or varicose recurrences after surgical interventions. This is mainly related to the lack of mechanistic knowledge delineating molecular targets that are rate‐limiting for the onset or progression of venous insufficiency or the development of varicose veins.
Clinically, insufficient varicose veins are characterized by impaired venous return and thus reflux of venous blood. Morphologically, the appearance of these veins is swollen and bulged and frequently accompanied by incompetent valves. Consequently, the increase in venous volume results in venous hypertension and thus a chronic increase in venous wall stress.2, 4 In this context, we showed that these environmental conditions activate the transcription factor activator protein 1 (AP‐1), which controls expression of genes with products that promote cellular activity and proliferation in the venous wall. Among them, monocyte chemoattractant protein 1 (MCP1) contributes to the recruitment of circulating monocytes and proliferation of vascular smooth muscle cells (SMCs),5, 6 whereas matrix metalloproteinase (MMP) 2 promotes degradation and thus structural rearrangement of the extracellular matrix in the vessel wall.7, 8 Consequently, an increase in wall stress stimulates expression of MMP2 and elevates gelatinase activity in the media of affected veins and human venous SMCs (HUVSMCs).9, 10 Likewise, MMP9 activity is increased in varicose veins of human patients11 and in rat veins on exposure to higher pressure levels.12 Blocking the activity of AP‐1 by specific decoy oligodeoxynucleotides that mimic the DNA binding site of AP‐1 in an animal model was successful in preventing pressure‐induced varicose‐like venous remodeling.9 Although the outcome of this study delineated AP‐1 as a valuable clinical target to interfere with varicose vein development, so far it has not been applicable to humans. This study aimed at transferring these findings from bench to bedside by validating that 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors, which are known to block AP‐1 activity as part of their pleiotropic effects,13, 14 have the capacity to interfere with activation of venous cells and/or varicose vein development.
Material and Methods
Antibodies
The anti–human/mouse MMP2 antibody (DLN‐12481) was purchased from Dianova. The anti–human/mouse myocardin antibodies (sc‐21561 and sc‐33766) were obtained from Santa Cruz Biotechnology, as was the monoclonal anti–mouse CD31 antibody (clone: MEC 13.3; sc‐18916). The anti–human proliferating cell nuclear antigen (PCNA) (ab2426), anti–mouse MCP1 (clone ECE.2; ab8101), anti–human MCP1 (ab9669), anti–mouse Ki67 (ab16667), anti–human c‐Fos (ab7963), anti–phosphorylated c‐Fos (ab192442), anti–c‐Fos (ab7963), and anti–human MMP9 (ab38898) antibodies were obtained from Abcam.
Cell Culture
HUVSMCs were isolated from human umbilical cord veins and cultured in DMEM (Invitrogen) supplemented with 15% FCS. The phenotype of these cells was confirmed by positive immunofluorescence for smooth muscle actin and desmin. Only cells cultured up to passage 4 were used throughout the study. The isolation of HUVSMCs was approved by the local ethics committee (Heidelberg, Germany; reference 336/2005) and conformed to the principles outlined in the Declaration of Helsinki (1997). HUVSMC proliferation on stimulation with statin treatment was assessed by counting the number of cells in randomized microscopic fields of view of the culture. To expose HUVSMCs to biomechanical stretch, cells were cultured on plastic dishes or BioFlex Collagen type I 6‐well plates (Flexcell). Stretching was performed by using a Flexcell FX‐5000 Tension System with 13% cyclic elongation at 0.5 Hz. Cyclic elongation is needed to prevent the cells from evading the biomechanical stimulus through rearranging their focal contacts. Cultured cells were fixed with methanol at 0°C to 4°C and blocked with 1% BSA/PBS for 30 minutes prior to immunofluorescence analysis.
Morphological Analyses of Tissue Samples and Cells
Mouse and human tissue specimens were fixed for 24 hours in zinc fixative, dehydrated, and embedded in paraffin. Specimens of human great saphenous veins from varicose vein excision (CEAP [clinical, etiological, anatomical, pathological] classification C2–C3) and nondilated veins (eg, from bypass surgeries) were collected in agreement with the local ethics committee and conformed to the principles outlined in the Declaration of Helsinki (1997). In general, 37% of the samples were from male patients, and 63% were from female patients (patients’ mean age: 52 years). Patients treated with statins (preferentially simvastatin and pravastatin) for other reasons were ≈20% older and more often suffered from cardiovascular diseases.
Immunofluorescence detection of MMP2, MMP9, PCNA, Ki67, MCP1, c‐Fos, phosphorylated c‐Fos, and CD31 was performed on 4‐ to 5‐μm‐thick paraffin sections or fixed cells by using the corresponding primary antibodies in combination with Cy5‐, Cy3‐, or Cy2‐labeled secondary antibodies (Dianova). Nuclei were visualized by counterstaining with DAPI. Immunofluorescence staining intensity was quantified by using the Cell^R software (Olympus) analyzing at least 3 different regions of the specimen per experimental group and animal. Immunofluorescence images of human tissue sections were automatically fused by multiple image alignment to obtain a high‐resolution overview of the whole sample. Exposure times for digital imaging of individual molecular targets were kept constant. Because of the size of human tissue sections, evaluation of the overall MMP2, MCP1, MMP9, and myocardin abundance in the vessel wall was determined by using a scoring system and was independently performed by 2 investigators who were blinded with regard to the identities of the specimens. Specimens were analyzed and rated according to the staining intensity and the number of cells stained within the intima/media of the vessel wall (the endothelial cell monolayer was lost in most specimens because of the surgical procedure). Specimens showing no staining were rated as 0, and with strong staining intensity, most of the cells were rated as 5.
Animal Models
All animal studies were performed with permission of the Regional Council Karlsruhe and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996). NMRI mice (aged 8–12 weeks) were fed with animal food (R/M‐H; ssniff GmbH) supplemented with simvastatin (SimvaHEXAL; HEXAL AG), atorvastatin (Sortis; Pfizer), or rosuvastatin (Crestor; AstraZenica) or with control diet for up to 15 days. Based on animal weight and ingestion, the applied dose of the corresponding statins was 10 to 20 mg/kg per day. Ligation of mouse auricle veins was performed as described previously.9 In brief, mice were anesthetized with isoflurane, and 1 of the 3 first‐order veins was ligated using a surgical thread (silk, 7.0; Ethicon). Because of the highly collateralized auricle vasculature, this procedure does not lead to ischemia or necrosis. Remodeling of collateral veins was documented on a daily basis by using a high‐resolution digital camera (Olympus). Images were morphometrically analyzed using the software ImageJ (US National Institutes of Health). Mouse auricles were dissected and processed for paraffin embedding and histological examination.
Perfusion of Isolated Mouse Veins
NMRI mice (aged 8–12 weeks) were sacrificed, and the facial veins were extracted and inserted into a perfusion chamber (Culture Myograph; DMT). The chambers were placed in an incubator at 37°C and 5% CO2, and the veins were continuously perfused for 18 hours with DMEM (Invitrogen) containing 15% FCS at a transmural pressure of 4 or 16 mm Hg. Histological integrity of the segments was routinely checked by immunohistochemical staining for the endothelial cell marker CD31. After perfusion, vessel segments were fixed in zinc fixative (4°C for 24 hours) and subsequently dehydrated in a series of ethanol and isopropanol, embedded in paraffin, and processed for sectioning.
Electrophoretic Mobility Shift Assay
Preparation of nuclear extracts from the cultured HUVSMCs and subsequent nondenaturing 4% polyacrylamide gel electrophoresis were carried out, as described previously.15 The double‐stranded gel shift oligodeoxynucleotide for AP‐1 (5′‐GTGCTGACTCAGCAC‐3′) was end‐labeled with [γ‐32P]ATP by using the 5′‐end labeling kit from Amersham GE Healthcare. The double‐stranded AP‐1 oligodeoxynucleotide was prepared from complementary single‐stranded bonded oligodeoxynucleotides obtained from Biomers, as described prevously.16 To ascertain the binding specificity, 2.0 μL of a c‐jun gel supershift antibody (2 mg/mL, sc‐1694; Santa Cruz Biotechnology) per 6.0 μL of nuclear extract (10 μg protein) was added 30 minutes before addition of the labeled oligonucleotide probe and incubated for another 30 minutes at room temperature.
Quantitative Polymerase Chain Reaction
Total RNA was isolated from cultured cells by solid phase extraction with the RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions. cDNA was synthesized using the Omniscript RT Kit (Qiagen), according to the manufacturer's protocol. Quantitative reverse‐transcription polymerase chain reaction was performed in a Rotor‐Gene Q (Qiagen). Amplification of the 60S ribosomal protein L32 (RPL32) cDNA served as an internal standard. Primers based on the following sequences were used for amplification: human MCP1, forward AGCAAGTGTCCCAAAGAAGC, reverse CCTGAACCCACTTCTGCTTGG (annealing temperature 55°C); human RPL32, forward AGGCATTGACAACAGGGTTC, reverse GTTGCACATCAGCAGCACTT (annealing temperature 56°C). Cycle threshold was set within the exponential phase of the polymerase chain reaction. Quantification of the polymerase chain reaction product was performed using the ΔCt method.
Statistical Analysis
Results are expressed as mean±SD. Differences between 2 matched experimental groups were analyzed by unpaired Student t test (if the corresponding data had passed the Kolmogorov–Smirnov normality analyses) or Wilcoxon rank sum test (if the corresponding data had not passed the Kolmogorov–Smirnov normality analyses), with a probability value of P<0.05 considered statistically significant. Differences among ≥3 experimental groups were analyzed by ANOVA, followed by the Tukey‐Kramer multiple comparisons test (if the corresponding data had passed the Kolmogorov–Smirnov normality analyses) or the Kruskal–Wallis test followed by Dunn's multiple comparisons test (if the corresponding data had not passed the Kolmogorov–Smirnov normality analyses), with a probability value of P<0.05 considered statistically significant.
Results
HMG‐CoA Reductase Inhibitors Attenuate AP‐1 Activity in Biomechanically Stimulated Venous SMCs
Recently, we reported that an increase in venous pressure is sufficient to promote the formation of varicose‐like veins in mice.9, 17 In this context, we showed that the transcriptional activity of AP‐1 appeared to be rate‐limiting for the development of varicose‐like veins and thus may represent a druggable target. To translate our findings into a therapeutic approach, we tested the capacity of HMG‐CoA reductase inhibitors to interfere with venous remodeling because these drugs have been reported to attenuate AP‐1 activity.13 Subsequent in vitro experiments revealed that treatment with atorvastatin or rosuvastatin, but not simvastatin, inhibited the activity of AP‐1 in HUVSMCs that were exposed to biomechanical stretch (Figure 1A). Real‐time polymerase chain reaction analyses showed that mRNA expression of MCP1, a prototypic transcriptional target of AP‐1, was significantly downregulated on treatment with atorvastatin or rosuvastatin (Figures 1B and 2). Both statins also decreased the nuclear abundance of phosphorylated (activated) c‐Fos, a subunit of AP‐1 (Figure 1C). Apart from AP‐1–related effects, we were able to confirm that these statins also inhibit the proliferation of HUVSMCs (Figure 3A and B)—another relevant process linked to venous wall remodeling.
Figure 1.
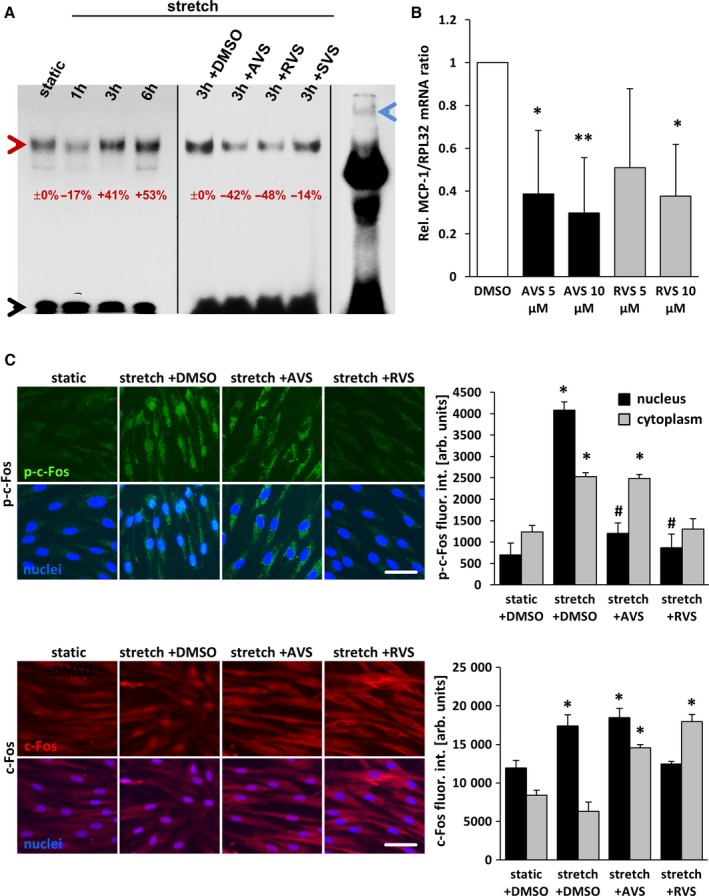
Statins inhibit stretch‐induced activation of AP‐1 in HUVSMCs. A, Nuclear extracts from HUVSMCs, stretched for 1 to 6 hours and/or treated or pretreated with 10 μmol/L AVS, RVS, or SVS, were exposed to 32P‐labeled double‐stranded gel shift ODNs mimicking a consensus AP‐1 DNA binding site, separated by nondenaturing polyacrylamide gel electrophoresis and subjected to autoradiography (red arrowhead: AP‐1–ODN/protein complex; black arrowhead: unbound AP‐1–ODN; blue arrowhead: band of the AP‐1–ODN/protein complex supershifted by using an anti–c‐Jun antibody). The percentaged alterations in the band intensities are indicated with static or stretch set to 0%. B, mRNA expression ratios of MCP1 and RPL32 (housekeeping gene) in statin‐treated HUVSMCs were determined by real‐time polymerase chain reaction. Control conditions (DMSO) were set to 1 (*P<0.05/**P<0.01 vs DMSO, n=3). C, HUVSMCs were exposed to biomechanical stretch for 24 hours with or without statin pretreatment (1 hour at 10 μmol/L). Abundance of the AP‐1 subunit c‐Fos (red fluorescence) and activated (phosphorylated) c‐Fos (green fluorescence) in the nuclei (blue fluorescence, black bars) and cytoplasm (gray bars) was determined by quantitative immunofluorescence analyses (*P<0.05 vs static plus DMSO, # P<0.05 vs stretch plus DMSO; bars represent the mean±SD of values obtained from 5 microscopic fields of view of 1 of 2 experiments with comparable results; scale bars = 50 μm). arb. unit indicates arbitrary unit; AP‐1, activator protein 1; AVS, atorvastatin; DMSO, dimethyl sulfoxide; fluor. int., fluorescence intensity; h, hour; HUVSMC, human umbilical vein smooth muscle cell; ODN, oligodeoxynucleotide; p‐c‐Fos, phosphorylated c‐Fos; Rel., relative; RVS, rosuvastatin; SVS, simvastatin.
Figure 2.
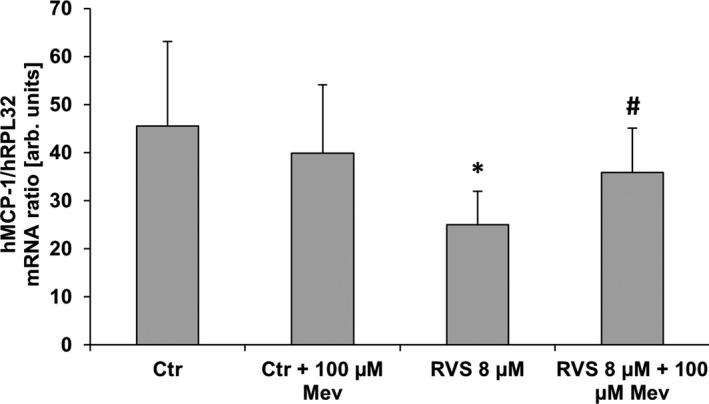
MCP1 mRNA expression in stretch‐stimulated HUVSMCs. mRNA expression ratio of MCP1 and RPL32 (house‐keeping gene) was determined by real‐time polymerase chain reaction from samples of HUVSMCs exposed to biomechanical stretch for 6 hours (0.5 Hz, 13% elongation) and treated with RVS and/or Mev to compensate for 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase–specific effects (control: DMSO‐treated cells). RVS significantly decreased MCP1 mRNA expression, which is blocked by mevalonate treatment. *P<0.05 vs control, # P<0.05 vs RVS, n=8; unpaired nonparametric 2‐tailed Mann–Whitney test was applied. arb. unit indicates arbitrary unit; Ctr, control; h, human; HUVSMC, human umbilical vein smooth muscle cell; Mev, mevalonic acid; RVS, rosuvastatin.
Figure 3.
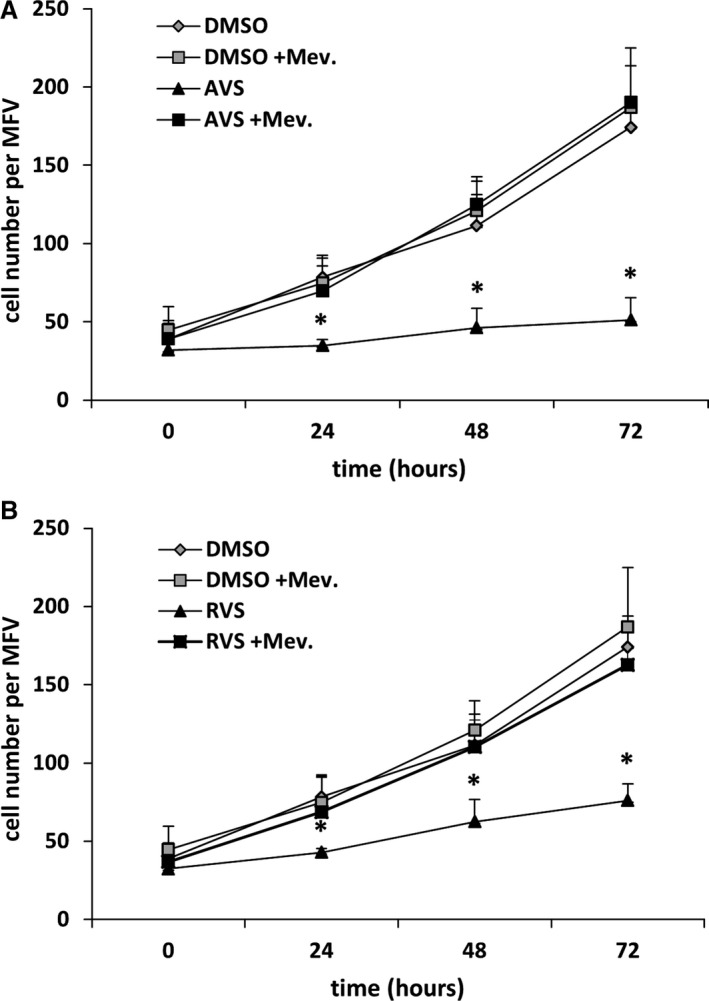
AVS and RVS inhibit human VSMC proliferation. Proliferation of VSMCs treated with AVS (A, 1 μmol/L) or RVS (B, 5 μmol/L) and/or Mev (100 μmol/L) to counteract 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibition was determined for 72 hours by counting the number of cells within defined MFV. *P<0.05 vs corresponding DMSO control, the curves represent the mean±SD for 1 of 3 experiments, with comparable results performed in pentaplicates. AVS indicates atorvastatin; DMSO, dimethyl sulfoxide; Mev, mevalonic acid; MFV, microscopic fields of view; RVS, rosuvastatin; VSMC, venous smooth muscle cell.
Atorvastatin and Rosuvastatin Inhibit Pressure‐Induced Activation of Endothelial Cells and SMCs in Mouse Veins
To scrutinize the relevance of the aforementioned findings, we used small murine vein segments as a more physiological model. The chronic increase in venous pressure as an initial determinant of varicose vein remodeling was mimicked by raising the intraluminal pressure from 4 to 16 mm Hg for 18 hours. Although the abundance of c‐Fos was not altered under these conditions (Figure 4B), an increase in venous pressure significantly elevated the level of phosphorylated c‐Fos, which was diminished on treatment with atorvastatin and rosuvastatin (Figure 4A). Likewise, cellular proliferation and protein abundance of MCP1 and MMP2 as additional transcriptional targets of AP‐1 were increased on raising the venous pressure and decreased on treatment with HMG‐CoA reductase inhibitors (Figure 5). In contrast, abundance of myocardin—a marker of the quiescent vascular SMC phenotype that is decreased in stretch‐stimulated SMCs17, 18—was stabilized in statin‐treated veins (Figure 6).
Figure 4.
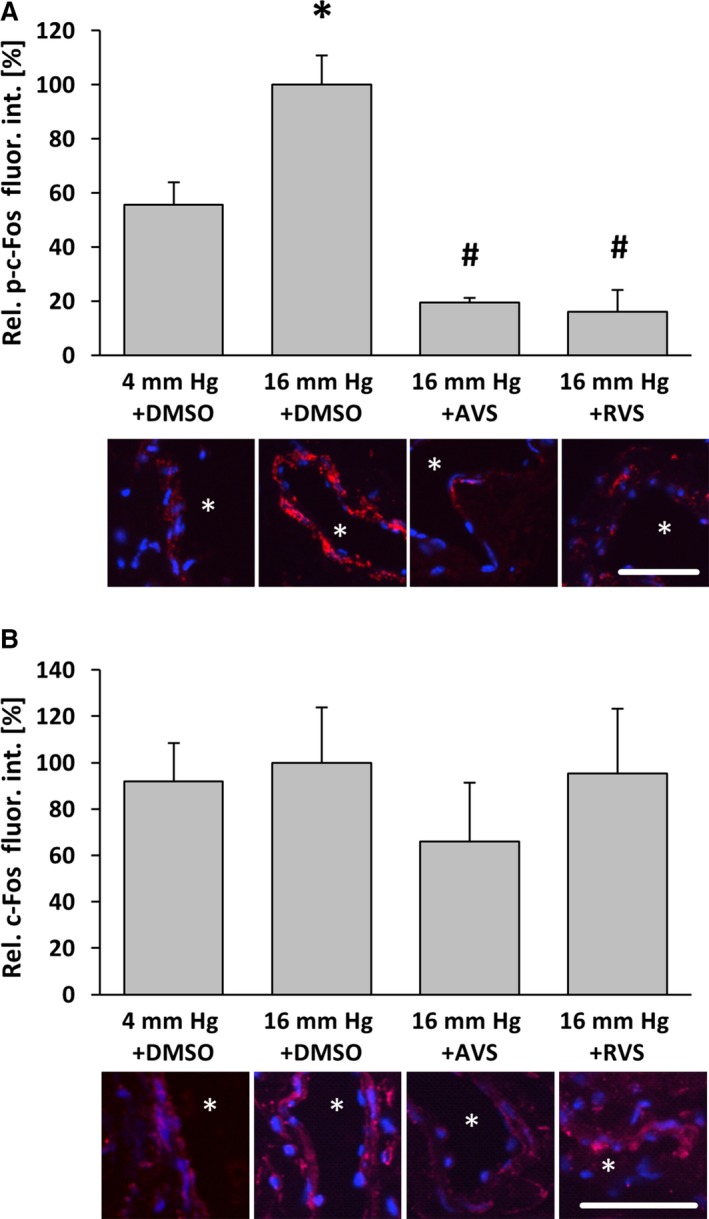
Atorvastatin and Rosuvastatin attenuate pressure‐induced phosphorylation of c‐Fos in venous wall cells. Exposure of mouse facial vein segments to a supraphysiological intraluminal pressure of 16 mm Hg increased the level of phosphorylated c‐Fos, whereas total c‐Fos abundance remained unaltered. A, *P<0.05 vs 4 mm Hg plus DMSO, n=3–4, 16 mm Hg values were set to 100%. B, No significant changes, n=5–6. This effect was blocked by pretreatment with atorvastatin (10 μmol/L) 1 hour prior to biomechanical stimulation. A, # P<0.05 vs 16 mm Hg plus DMSO, n=5; asterisks in the representative images mark the lumen, scale bars = 50 μm. AVS indicates atorvastatin; DMSO, dimethyl sulfoxide; fluor. int., fluorescence intensity; p‐c‐Fos, phosphorylated c‐Fos; Rel., relative; RVS, rosuvastatin.
Figure 5.
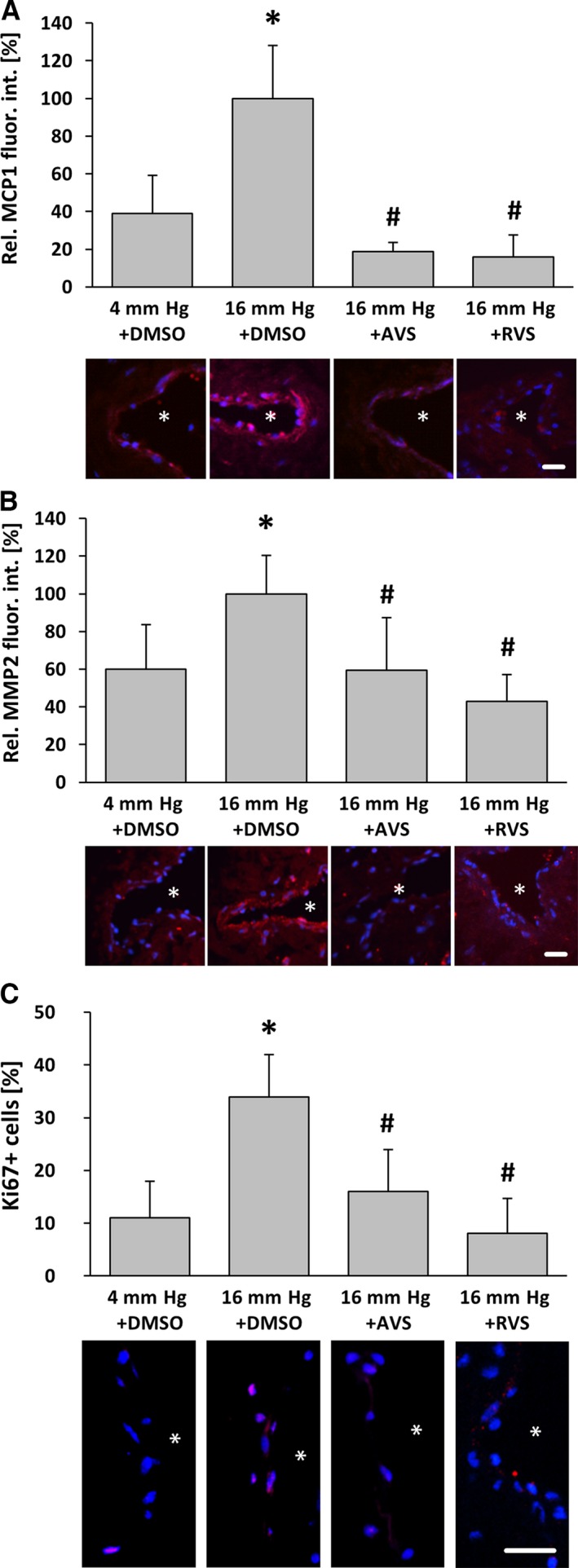
AVS counteracts pressure‐induced activation of venous wall cells. Exposure of mouse facial vein segments to a supraphysiological intraluminal pressure of 16 mm Hg increased protein abundance of MCP1 and MMP2 (A, B) as well as proliferation (C) of both endothelial and smooth muscle cells in the venous vessel wall. A and B, *P<0.05 vs 4 mm Hg plus DMSO, n=4–5. C, *P<0.05 vs 4 mm Hg plus DMSO, n=4–5; proliferation marker Ki67. This effect was blocked by pretreatment with AVS (10 μmol/L) 1 hour prior to biomechanical stimulation. A–C, # P<0.05 vs 16 mm Hg plus DMSO, n=4–5, 16 mm Hg values were set to 100%; asterisks in the representative images mark the lumen, scale bars = 20 μm. AVS indicates atorvastatin; DMSO, dimethyl sulfoxide; fluor. int., fluorescence intensity; Rel., relative; RVS, rosuvastatin.
Figure 6.
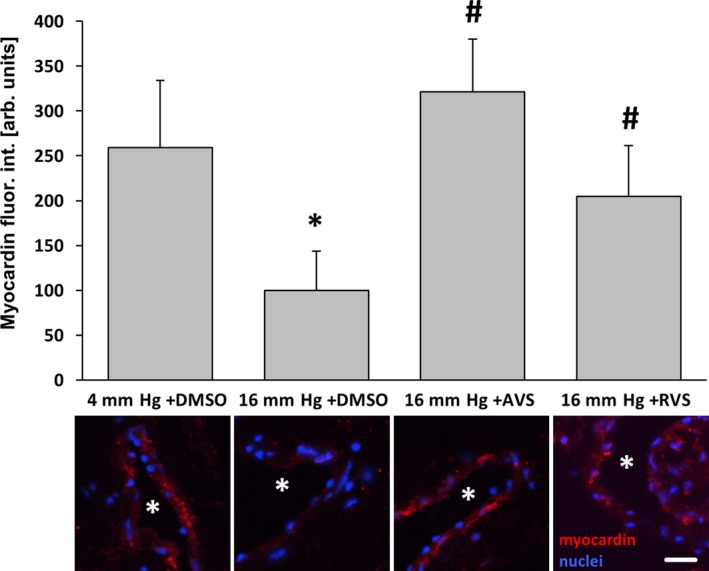
AVS and RVS attenuate the pressure‐induced decrease in myocardin abundance in veins. Exposure of mouse facial vein segments to a supraphysiological intraluminal pressure of 16 mm Hg decreased myocardin abundance of smooth muscle cells in the vessel wall (*P<0.05 vs 4 mm Hg plus DMSO, # P<0.05 vs 16 mm Hg, n=5–9), which was blocked by administration (10 μmol/L) of AVS or RVS 1 hour prior and during biomechanical stimulation. Asterisks in stain panels mark the lumen, scale bar = 20 μm. arb. unit indicates arbitrary unit; AVS, atorvastatin; DMSO, dimethyl sulfoxide; fluor. int., fluorescence intensity; Rel., relative; RVS, rosuvastatin.
Atorvastatin and Rosuvastatin, But Not Simvastatin, Inhibit Venous Remodeling
We next investigated the capacity of HMG‐CoA reductase inhibitors to attenuate venous remodeling in vivo. To this end, mice were fed with statins for 11 days and underwent local auricle vein ligation, thus elevating pressure in the connected venous network and inducing its rapid enlargement and corkscrew‐like appearance. Although only rosuvastatin treatment significantly changed total cholesterol plasma levels in these mice (Figure 7), both atorvastatin and rosuvastatin, but not simvastatin, attenuated venous remodeling under these conditions (Figures 8A, 8B and 9). As has been shown previously,9 this process was associated with an increase in proliferation and MMP2 protein abundance both in endothelial cells and SMCs of individually dissected veins (Figures 8C and 10). Moreover, phosphorylated c‐Fos was detected in nuclei of remodeling veins but not in veins from statin‐treated mice (Figure 8C). Although MCP1 abundance was also investigated by immunofluorescence, it was not detected in this phase of remodeling (data not shown).
Figure 7.
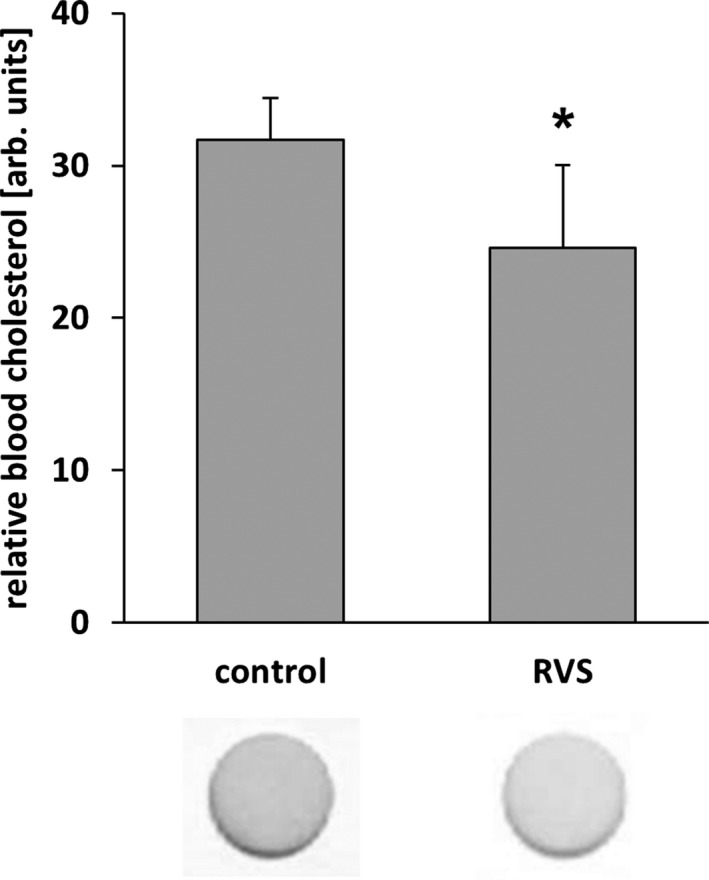
RVS lowers blood cholesterol levels in NMRI mice. Blood from mice fed with a statin‐supplemented diet (15 days) was analyzed with a commercially available colorimetric test stick whereby the cholesterol concentration was quantified through densitometric assessment of the color intensity (*P<0.05 vs control diet, n=4–5). arb. unit indicates arbitrary unit; RVS, rosuvastatin.
Figure 8.
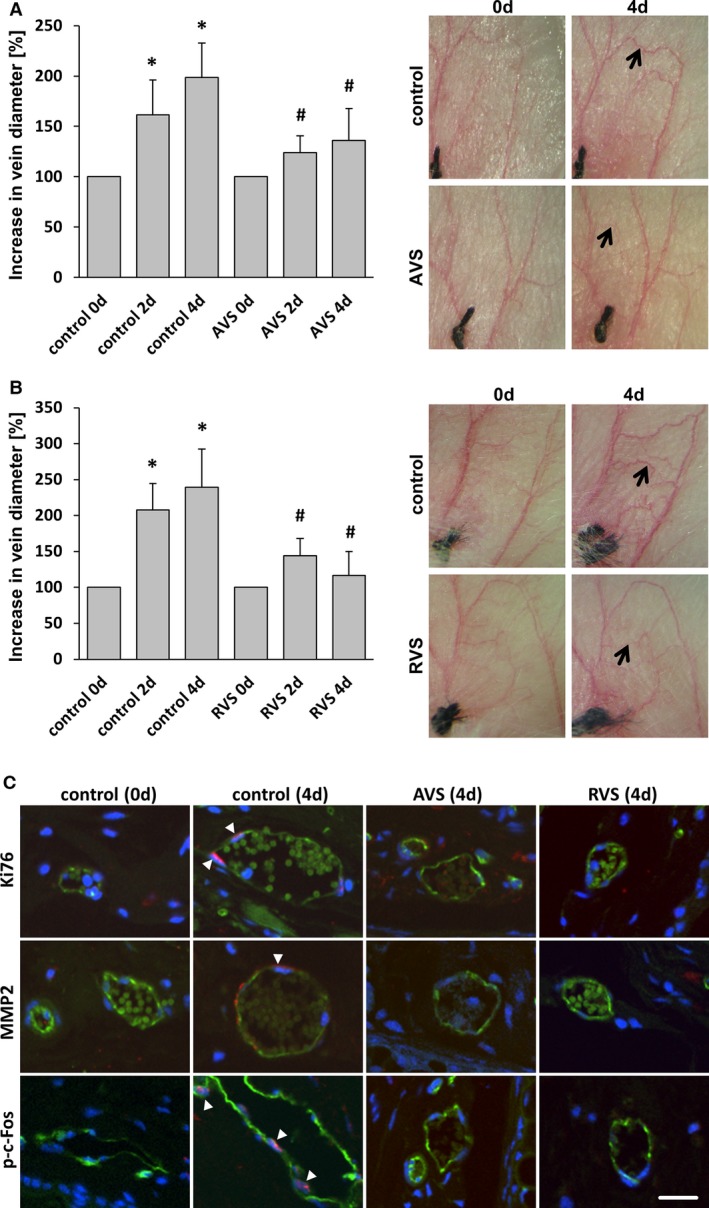
Treatment with AVS or RVS blocks varicose‐like venous remodeling in the mouse auricle model. Daily imaging of the vasculature of the mouse auricle for 4 days after ligation of a central vein revealed increased tortuosity and the growth (A and B, arrows) of connected small collateral veins; this was significantly inhibited by systemic administration of AVS or RVS. A and B, *P<0.05 vs control day 0, # P<0.05 vs corresponding control day 2/4, n=5–7. Representative images show the venous network of mouse auricles (A and B) and cross‐sections of corresponding blood vessels (C) (confocal images; green fluorescence: CD31) showing Ki67‐positive nuclei (arrowheads), MMP2 accumulation (arrowhead), and p‐c‐Fos–positive nuclei (arrowheads) in remodeling veins (red fluorescence). Corresponding activity was not observed in veins from statin‐treated mice (scale bars = 20 μm). AVS indicates atorvastatin; d, days; p‐c‐Fos, phosphorylated c‐Fos; RVS, rosuvastatin.
Figure 9.
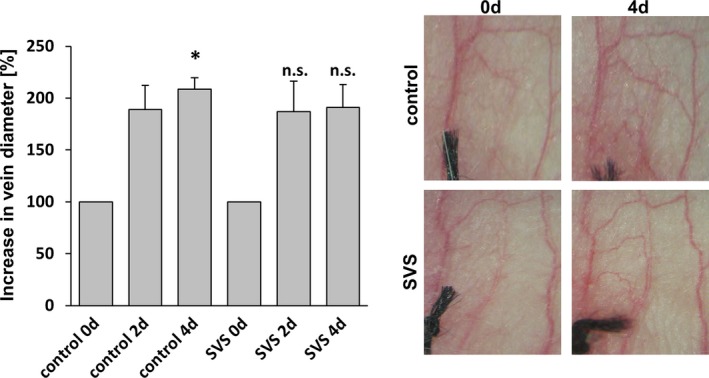
Treatment with SVS does not block varicose‐like venous remodeling in the mouse auricle model. Daily imaging of the vasculature of the mouse auricle for 4 days after ligation of a central vein revealed increased tortuosity and the growth of connected small collateral veins; was not affected by SVS treatment. *P<0.05 vs control day 0, n.s. vs corresponding control day 2/4, n=5). Representative images show the venous network of mouse auricles. d indicates days; n.s., not significant; SVS, simvastatin.
Figure 10.
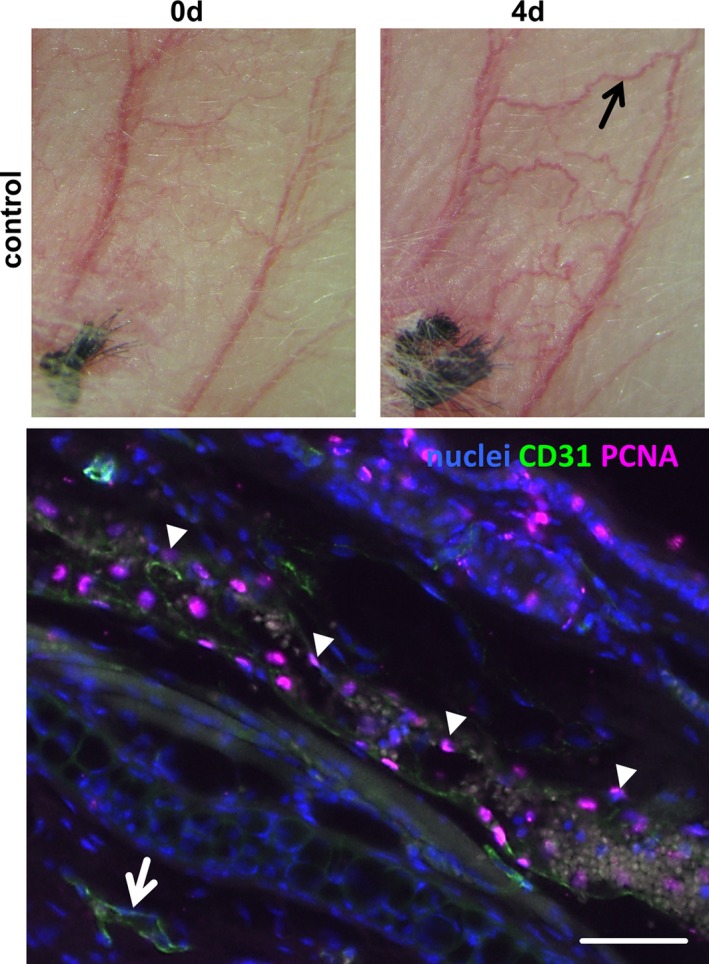
Proliferation in an auricle vein on induction of venous hypertension. At 4 days after ligation of a central auricle vein, the marked vein (black arrow) was dissected and processed for longitudinal sectioning. The representative image below shows the immunofluorescence detection of Ki67 (pink fluorescence, arrowheads) in the nuclei (blue fluorescence) of this blood vessel. No proliferating cells were detected in adjacent nonremodeling veins (arrow, scale bar = 50 μm). d indicates days.
Statin Treatment Decreases Cellular Proliferation and Expression of AP‐1 Target Genes in Human Varicose Veins
Long‐term statin‐based therapies are often administered to hypercholesteremic patients who sometimes also have varicose veins. We collected saphenous vein samples from such patients (see Material and Methods) for subsequent immunofluorescence analysis. The results were compared with human varicose and nondilated (healthy) veins. In line with the observations made earlier, the total protein abundance of c‐Fos was only marginally altered among the different groups (Figure 11A and 11B); however, although often heterogeneously distributed, especially in varicose veins, c‐Fos was partially localized in the nuclei of vascular SMCs (Figure 11A, arrows). Phosphorylated (active) c‐Fos was rarely detected in vascular SMCs but always traceable in endothelial cells, although absent from their nuclei (Figure 11A). In varicose veins, the overall abundance of phosphorylated c‐Fos in vascular SMCs was increased in varicose veins (Figure 11C) and accumulated preferentially in the nuclei of these cells (Figure 11A, arrows). According to these findings, proliferation and protein abundance of MCP1 as well as MMP2 was increased in varicose veins and was attenuated in vein specimens from statin‐treated patients (Figure 12). The highest levels of MCP1 were partially associated with phosphorylated c‐Fos–positive nuclei (compare Figure 11D and 11E). Further analyses revealed a decrease in MMP9 and an attenuated decrease in myocardin abundance in vein samples from statin‐treated patients (Figure 13).
Figure 11.
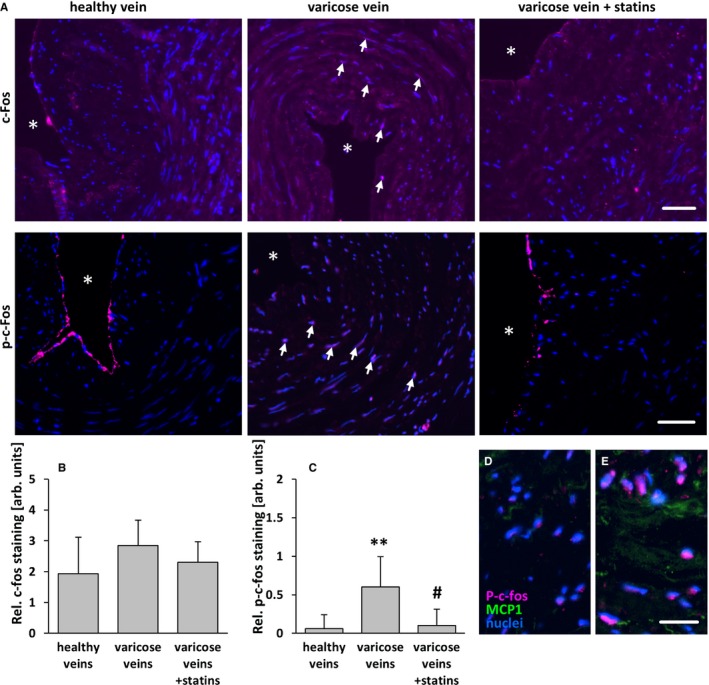
Statin treatment diminishes phosphorylation of c‐Fos of SMCs in human varicose veins. c‐Fos and p‐c‐Fos (activated) were detected by immunofluorescence analyses in cross‐sections of excised human varicose saphenous vein segments from patients chronically treated or not treated with statins and from nondilated (healthy) human veins. A, In representative images, asterisks mark the lumen, and arrows mark nuclei positive for c‐Fos and p‐c‐Fos in varicose veins). Although the c‐Fos–specific fluorescence intensity was not significantly altered (B), the intensity of the p‐c‐Fos–specific fluorescence (often detectable in the nuclei of SMCs in varicose veins, arrows) was significantly increased in the walls of varicose veins but not in those from statin‐treated patients. C, **P<0.01 vs healthy veins, # P<0.01 vs varicose veins, n=8–10, scale bars = 50 μm. Highest levels of MCP1 were in detected in areas with p‐c‐Fos–positive nuclei (D and E) (scale bar = 20 μm). AU indicate arbitrary unit; p‐c‐Fos, phosphorylated c‐Fos; Rel., relative; SMC, smooth muscle cell.
Figure 12.
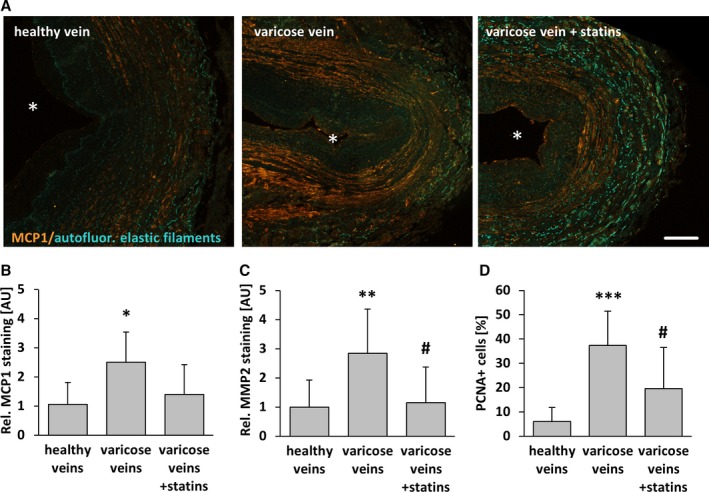
Statin treatment diminishes SMC activation in human varicose veins. MCP1, MMP2, and PCNA (proliferation marker) were detected by immunofluorescence analyses in cross‐sections of human excised varicose saphenous vein segments from patients chronically treated or not treated with statins and from nondilated (healthy) human veins. A, In representative images of MCP1 staining, asterisks mark the lumen. Compared with healthy veins, the abundance of the aforementioned markers was significantly increased in the walls of varicose veins but not in those from statin‐treated patients. B–D, *P<0.05, **P<0.01, *** P<0.01 vs healthy veins, # P<0.05 vs varicose veins, n=9–10, scale bar = 200 μm. AU indicate arbitrary unit; autofluor., autofluorescent; Rel., relative.
Figure 13.
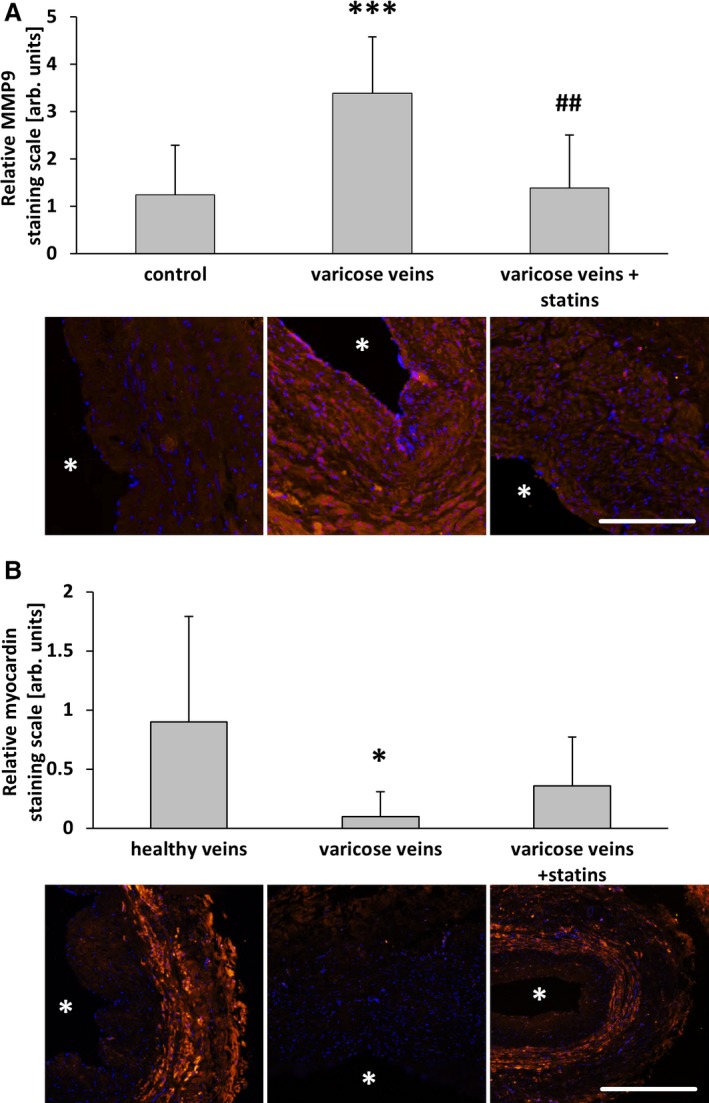
Statin treatment diminished smooth muscle cell activation in human varicose veins. MMP9 and myocardin were detected by immunofluorescence staining (asterisks mark the lumen) in cross‐sections of human great saphenous veins from varicose vein excision, those from patients chronically treated with statins and nondilated (healthy) veins. Compared with healthy veins, the abundance of MMP9 was significantly increased in the walls of varicose veins but not in those from statin‐treated patients. A, *** P<0.001 vs healthy, ## P<0.01 vs varicose veins, n=9–10. The abundance of myocardin was significantly decreased in the walls of varicose veins but not in those from statin‐treated patients. B, *P<0.05 vs healthy, n=9–10, scale bars = 200 μm. AU indicate arbitrary unit; Rel., relative.
Discussion
Insufficient venous return may evolve from dysfunctional venous valves, increased body mass index, or pregnancy. Other risk factors for CVI include sex, age, and prolonged standing at work.2, 3, 19 Even ablation of veins—however insufficient—through stripping, sclerotherapy, radiofrequency ablation, or endovenous laser treatment may restrict overall venous return and often results in development of varicose recurrences within 5 years after intervention.20 Although underlying causes may differ, all of the aforementioned conditions ultimately contribute to an increase in intraluminal pressure, namely, venous hypertension, and thus increase in venous wall stress.4 This biomechanical stimulus is sufficient to elicit varicose‐like remodeling of the venous vessel wall.9 In line with this effect, lowering venous pressure through collateral bypasses prevents formation of bulged and tortuous veins. Likewise, surgical techniques referred to as Cure Conservatrice et Hémodynamique de l'Insuffisance Veineuse en Ambulatoire, or CHIVA, correct venous insufficiency by establishing bypasses and avoiding obstruction of venous drainage wherever possible. This surgical strategy, which indirectly lowers venous wall stress, has been reported to result in a lower rate of recurrences.21, 22
Although a plethora of surgical therapies are available to remove varicose and thus insufficient veins, studies of pharmacological therapies are scarce. Considering arterial diseases, statins have repeatedly been reported to counteract progression of arterial diseases and reduce the 5‐year incidence of major coronary events or stroke,23, 24 effects that may be exerted independently from their HMG‐CoA reductase inhibitory capacity. A pleiotropic effect mediated by this class of drugs is inhibition of the activity of transcription factors such as AP‐1 or nuclear factor‐κB,13 which control expression of genes that have been associated with pathological vascular remodeling processes25, 26, 27 and responses of vascular cells to elevated biomechanical stress.28 Accordingly, atorvastatin and especially rosuvastatin, which is a more potent and water‐soluble inhibitor of HMG‐CoA reductase, inhibited proliferation of AP‐1 activity in stretch‐stimulated venous SMCs as well as expression of its target genes MCP1 and MMP2 in isolated perfused veins exposed to supraphysiological pressure levels. By applying the mouse auricle vein ligation model,9, 16 we simply mimicked a local increase in venous pressure as it may occur with venous valve failure or prolonged standing without provoking necrosis or severe inflammatory responses. Based on that model, we showed for the first time that progressive remodeling leading to venous dilatation and tortuosity is effectively inhibited by atorvastatin or rosuvastatin. Treatment with these statins was further accompanied by a decrease of MMP2 expression in and proliferation of both venous endothelial cells and SMCs. The same changes were observed in varicose vein samples from patients receiving HMG‐CoA reductase inhibitors. In line with the current literature,29 MMP9 abundance was also diminished in these samples.
The putative beneficial impact of statins on the SMC phenotype in varicose veins may also be concluded from the higher level of myocardin—the coactivator of serum response factor—present in these cells, considered a marker for the contractile and quiescent SMCl phenotype.16, 17, 29 Collectively, our data suggest that statin treatment (1) may slow down the progression of primary varicose vein development and (2) may reduce the rate of varicose recurrences after surgical removal of insufficient veins by blocking AP‐1 activity and limiting the wall stress–dependent activation of venous SMCs.
Because of the pleiotropic nature of statin‐mediated effects, we cannot fully exclude the possibility that mechanisms other than those related to AP‐1 activity may have directly or indirectly contributed to the observed effects. Statins have been shown, for instance, to inhibit activation of the Rho/Rho‐kinase pathway in biomechanically activated venous SMCs; this signaling cascade is critical for adequate responses to elevated wall stress.31 Nevertheless, our earlier studies9 clearly indicated that interfering with the activity of AP‐1 is sufficient to attenuate venous remodeling and thus will significantly contribute to the impact of an antivaricose treatment. In this context, our findings may spur clinical studies to scrutinize the overall relevance of corresponding therapeutic approaches, for instance, by assessing the progression of varicose vein development in patients chronically treated with statins or the rate of varicose recurrences after surgical removal in this group of patients. This research appears to be even more important considering that the effective concentrations of statins in human blood (1–10 nmol/L, 1–2 mg/kg per day) are much lower than those usually applied in cell culture (1–10 μmol/L) or animal models (10–500 mg/kg per day)32 to compensate for a possible resistance to the main pharmacological effect (eg, statins rarely lower cholesterol serum levels in rodents33). Nevertheless, conclusions drawn from those experimental studies have spurred many novel therapeutic options, and in vitro observations are often compatible with statin‐related effects in human patients. Although we cannot exclude the possibility that lower statin concentrations in human blood may limit their potential therapeutic efficacy, our findings delineate a novel pharmacological treatment option suitable to delay the development of severe CVI or to support existing surgical strategies by preventing varicose recurrences.
Sources of Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB TR23, projects C5 and C6), the German Cardiac Society (DGK) and the German Society of Phlebology (DGP).
Disclosures
None.
Acknowledgments
The authors would like to acknowledge the excellent technical assistance of Gudrun Scheib and Maria Harlacher.
(J Am Heart Assoc. 2016;5:e002405 doi: 10.1161/JAHA.115.002405)
References
- 1. Beebe‐Dimmer JL, Pfeifer JR, Engle JS, Schottenfeld D. The epidemiology of chronic venous insufficiency and varicose veins. Ann Epidemiol. 2005;15:175–184. [DOI] [PubMed] [Google Scholar]
- 2. Eberhardt RT, Raffetto JD. Chronic venous insufficiency. Circulation. 2005;111:2398–2409. [DOI] [PubMed] [Google Scholar]
- 3. Eberhardt RT, Raffetto JD. Chronic venous insufficiency. Circulation. 2014;130:333–346. [DOI] [PubMed] [Google Scholar]
- 4. Pfisterer L, Konig G, Hecker M, Korff T. Pathogenesis of varicose veins—lessons from biomechanics. Vasa. 2014;43:88–99. [DOI] [PubMed] [Google Scholar]
- 5. Schepers A, Eefting D, Bonta PI, Grimbergen JM, de Vries MR, van Weel V, de Vries CJ, Egashira K, van Bockel JH, Quax PH. Anti‐MCP‐1 gene therapy inhibits vascular smooth muscle cells proliferation and attenuates vein graft thickening both in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2006;26:2063–2069. [DOI] [PubMed] [Google Scholar]
- 6. Spinetti G, Wang M, Monticone R, Zhang J, Zhao D, Lakatta EG. Rat aortic MCP‐1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2004;24:1397–1402. [DOI] [PubMed] [Google Scholar]
- 7. Zhang HW, Wang X, Zong ZH, Huo X, Zhang Q. AP‐1 inhibits expression of MMP‐2/9 and its effects on rat smooth muscle cells. J Surg Res. 2009;157:e31–e37. [DOI] [PubMed] [Google Scholar]
- 8. Lim CS, Davies AH. Pathogenesis of primary varicose veins. Br J Surg. 2009;96:1231–1242. [DOI] [PubMed] [Google Scholar]
- 9. Feldner A, Otto H, Rewerk S, Hecker M, Korff T. Experimental hypertension triggers varicosis‐like maladaptive venous remodeling through activator protein‐1. FASEB J. 2011;25:3613–3621. [DOI] [PubMed] [Google Scholar]
- 10. Meng X, Mavromatis K, Galis ZS. Mechanical stretching of human saphenous vein grafts induces expression and activation of matrix‐degrading enzymes associated with vascular tissue injury and repair. Exp Mol Pathol. 1999;66:227–237. [DOI] [PubMed] [Google Scholar]
- 11. Jacob MP, Cazaubon M, Scemama A, Prie D, Blanchet F, Guillin MC, Michel JB. Plasma matrix metalloproteinase‐9 as a marker of blood stasis in varicose veins. Circulation. 2002;106:535–538. [DOI] [PubMed] [Google Scholar]
- 12. Raffetto JD, Barros YV, Wells AK, Khalil RA. MMP‐2 induced vein relaxation via inhibition of [Ca2+]e‐dependent mechanisms of venous smooth muscle contraction. Role of RGD peptides. J Surg Res. 2010;159:755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MP, Nilsson J, Pachinger O, Weidinger F. HMG‐CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:58–63. [DOI] [PubMed] [Google Scholar]
- 14. Dechend R, Fiebler A, Lindschau C, Bischoff H, Muller D, Park JK, Dietz R, Haller H, Luft FC. Modulating angiotensin II‐induced inflammation by HMG Co‐A reductase inhibition. Am J Hypertens. 2001;14:55S–61S. [DOI] [PubMed] [Google Scholar]
- 15. Wagner AH, Gebauer M, Pollok‐Kopp B, Hecker M. Cytokine‐inducible CD40 expression in human endothelial cells is mediated by interferon regulatory factor‐1. Blood. 2002;99:520–525. [DOI] [PubMed] [Google Scholar]
- 16. Millet A, Curet P. [Value of digital angiography in the preoperative evaluation of arteritis of the leg. Results of a national survey]. J Radiol. 1989;70:581–583. [PubMed] [Google Scholar]
- 17. Pfisterer L, Meyer R, Feldner A, Drews O, Hecker M, Korff T. Bortezomib protects from varicose‐like venous remodeling. FASEB J. 2014;28:3518–3527. [DOI] [PubMed] [Google Scholar]
- 18. Pfisterer L, Feldner A, Hecker M, Korff T. Hypertension impairs myocardin function—a novel mechanism facilitating arterial remodeling. Cardiovasc Res. 2012;96:120–129. [DOI] [PubMed] [Google Scholar]
- 19. Scott TE, LaMorte WW, Gorin DR, Menzoian JO. Risk factors for chronic venous insufficiency: a dual case‐control study. J Vasc Surg. 1995;22:622–628. [DOI] [PubMed] [Google Scholar]
- 20. Flessenkamper I, Hartmann M, Hartmann K, Stenger D, Roll S. Endovenous laser ablation with and without high ligation compared to high ligation and stripping for treatment of great saphenous varicose veins: results of a multicentre randomised controlled trial with up to 6 years follow‐up. Phlebology. 2016;31:23–33. [DOI] [PubMed] [Google Scholar]
- 21. Pares JO, Juan J, Tellez R, Mata A, Moreno C, Quer FX, Suarez D, Codony I, Roca J. Varicose vein surgery: stripping versus the CHIVA method: a randomized controlled trial. Ann Surg. 2010;251:624–631. [DOI] [PubMed] [Google Scholar]
- 22. Carandina S, Mari C, De Palma M, Marcellino MG, Cisno C, Legnaro A, Liboni A, Zamboni P. Varicose vein stripping vs haemodynamic correction (CHIVA): a long term randomised trial. Eur J Vasc Endovasc Surg. 2008;35:230–237. [DOI] [PubMed] [Google Scholar]
- 23. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 24. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high‐risk individuals: a randomised placebo‐controlled trial. Lancet. 2002;360:7–22. [DOI] [PubMed] [Google Scholar]
- 25. Ahn JD, Morishita R, Kaneda Y, Lee SJ, Kwon KY, Choi SY, Lee KU, Park JY, Moon IJ, Park JG, Yoshizumi M, Ouchi Y, Lee IK. Inhibitory effects of novel AP‐1 decoy oligodeoxynucleotides on vascular smooth muscle cell proliferation in vitro and neointimal formation in vivo. Circ Res. 2002;90:1325–1332. [DOI] [PubMed] [Google Scholar]
- 26. Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. Low doses of lipopolysaccharide and minimally oxidized low‐density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein‐1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stadlbauer TH, Wagner AH, Holschermann H, Fiedel S, Fingerhuth H, Tillmanns H, Bohle RM, Hecker M. AP‐1 and STAT‐1 decoy oligodeoxynucleotides attenuate transplant vasculopathy in rat cardiac allografts. Cardiovasc Res. 2008;79:698–705. [DOI] [PubMed] [Google Scholar]
- 28. Li C, Xu Q. Mechanical stress‐initiated signal transduction in vascular smooth muscle cells in vitro and in vivo. Cell Signal. 2007;19:881–891. [DOI] [PubMed] [Google Scholar]
- 29. Nomura S, Yoshimura K, Akiyama N, Mikamo A, Furutani A, Aoki H, Matsuzaki M, Hamano K. HMG‐CoA reductase inhibitors reduce matrix metalloproteinase‐9 activity in human varicose veins. Eur Surg Res. 2005;37:370–378. [DOI] [PubMed] [Google Scholar]
- 30. Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428:185–189. [DOI] [PubMed] [Google Scholar]
- 31. Kozai T, Eto M, Yang Z, Shimokawa H, Luscher TF. Statins prevent pulsatile stretch‐induced proliferation of human saphenous vein smooth muscle cells via inhibition of Rho/Rho‐kinase pathway. Cardiovasc Res. 2005;68:475–482. [DOI] [PubMed] [Google Scholar]
- 32. Bjorkhem‐Bergman L, Lindh JD, Bergman P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br J Clin Pharmacol. 2011;72:164–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bjorkhem‐Bergman L, Acimovic J, Torndal UB, Parini P, Eriksson LC. Lovastatin prevents carcinogenesis in a rat model for liver cancer. Effects of ubiquinone supplementation. Anticancer Res. 2010;30:1105–1112. [PubMed] [Google Scholar]