

Abstract
Background
The genetic basis of stroke susceptibility remains to be elucidated. STR1 quantitative trait locus (STR1/QTL) was identified on rat chromosome 1 of stroke‐prone spontaneously hypertensive rat (SHRSP) upon Japanese‐style stroke‐permissive diet (JD), and it contributes to 20% of the stroke phenotype variance.
Methods and Results
Nine hundred eighty‐six probe sets mapping on STR1 were selected from the Rat RAE230A array and screened through a microarray differential expression analysis in brains of SHRSP and stroke‐resistant SHR (SHRSR) fed with either regular diet or JD. The gene encoding Ndufc2 (NADH dehydrogenase [ubiquinone] 1 subunit), mapping 8 Mb apart from STR1/QTL Lod score peak, was found significantly down‐regulated under JD in SHRSP compared to SHRSR. Ndufc2 disruption altered complex I assembly and activity, reduced mitochondrial membrane potential and ATP levels, and increased reactive oxygen species production and inflammation both in vitro and in vivo. SHRSR carrying heterozygous Ndufc2 deletion showed renal abnormalities and stroke occurrence under JD, similarly to SHRSP. In humans, T allele variant at NDUFC2/rs11237379 was associated with significant reduction in gene expression and with increased occurrence of early‐onset ischemic stroke by recessive mode of transmission (odds ratio [OR], 1.39; CI, 1.07–1.80; P=0.012). Subjects carrying TT/rs11237379 and A allele variant at NDUFC2/rs641836 had further increased risk of stroke (OR=1.56; CI, 1.14–2.13; P=0.006).
Conclusions
A significant reduction of Ndufc2 expression causes complex I dysfunction and contributes to stroke susceptibility in SHRSP. Moreover, our current evidence may suggest that Ndufc2 can contribute to an increased occurrence of early‐onset ischemic stroke in humans.
Keywords: complex I, early‐onset ischemic stroke, knockout rat model, mitochondria, Ndufc2, stroke‐prone spontaneously hypertensive rat
Subject Categories: Basic Science Research, Gene Expression & Regulation, Cerebrovascular Disease/Stroke
Introduction
It is known that both environmental and genetic factors contribute to increased stroke predisposition in humans.1, 2 Unraveling the genetic causes of stroke may take significant advantage of the availability of an inbred animal model, the stroke‐prone spontaneously hypertensive rat (SHRSP).3 The latter represents a suitable model for studies on the human disease4 and, based on previous investigations, carries at least 3 quantitative trait loci (QTL) for stroke susceptibility.5 One of them (STR1) maps on rat chromosome 1; it is included between Klk and Mt1pa markers, extending for an overall 80 cM length, and shows a 6.7 Lod score peak in correspondence of the anonymous marker, D1Mit3. STR1 contributes to 20% of overall stroke phenotype variance.5 Notably, its effect was confirmed by congenic experimentation.6 In fact, congenic animals carrying SHRSP/STR1 within the stroke‐resistant (SHRSR) genomic background had increased stroke occurrence. However, no apparent suitable candidate genes were initially discovered near the Lod score peak area. In the attempt to identify the gene(s) responsible for stroke phenotype within STR1, we performed, based on previous literature,7 a microarray differential expression analysis of all sequences contained inside STR1 in brains of both SHRSP and SHRSR fed with either regular or stroke permissive diet. One gene was found to be differentially modulated (ie, down‐regulated) in brains of SHRSP. This gene was Ndufc2 (NADH dehydrogenase [ubiquinone] 1, subcomplex unknown, 2), mapping 8 Mb apart from the Lod score peak.
Here, we report both in vitro and in vivo evidence of the significant functional impact of Ndufc2‐reduced expression on mitochondrial function, oxidative stress accumulation, and cell viability. We describe the stroke phenotype of SHRSR carrying heterozygous Ndufc2 deletion. Moreover, we report results of a genetic screening carried out in a human cohort of subjects with early‐onset ischemic stroke by testing available NDUFC2 single nucleotide polymorphisms (SNPs), which may generate the hypothesis that Ndufc2 is a genetic risk factor for stroke also in humans.
Methods
Brain Expression Profiling of Potential Candidate Genes Within STR1/QTL on Rat Chromosome 1 in SHRSP
Ten 6‐week‐old SHR/molBbb (SHRSR) and ten 6‐week‐old SHRSP/Bbb (SHRSP) rats, both inbred for 20 years at the MDC center in Berlin and derived from the original colonies established in Japan, were fed with either regular (RD; n=5) or Japanese‐style diet (JD; n=5) for 4 weeks and were then sacrificed. Brains were removed, immediately frozen, and subsequently extracted for total RNA by using the QIAGEN's RNeasy Total RNA Isolation kit (Qiagen GmbH, Hilden, Germany). Preparation quality was assessed by agarose‐formaldehyde gel electrophoresis. For synthesis of double‐stranded cDNA from 15 μg of total RNA, the cDNA Synthesis System kit (Roche Diagnostics, Indianapolis, IN) was used. Biotinylated cRNA was synthesized with PerkinElmer's (Waltham, MA) nucleotide analogs using Ambion's MEGAScript T7 kit (Ambion, Austin, TX). After fragmenting the cRNA for target preparation using the standard Affymetrix protocol, 15 μg of fragmented cRNA was hybridized for 16 hours at 45°C to Rat Genome 230 A Array. After hybridization, the arrays were washed and stained with streptavidin‐phycoerythrin in the Affymetrix Fluidics Station 400 and further scanned using the AFFYMETRIX GeneChip Scanner 3000 7G (Affymetrix Inc, Santa Clara, CA). Image data were analyzed with GCOS 1.4 using Affymetrix default analysis settings and global scaling as a normalization method. All arrays passed our formal quality criteria (3′–5′ ratio of Gapdh <3; RawQ <3) and were quantile‐normalized using Robust Multiarray Average (RMA).8 Outliers in replicates were removed according to a Nalimov test P‐value threshold of 10−3.9 After removal of probe sets located outside the major stroke QTL interval on rat chromosome 1 (92–220 Mbps), the remaining 986 probe sets were analyzed using ANOVA statistic followed by a false discovery rate (FDR) multiple testing correction.10 Probes that underwent 0.1% FDR (N=87) were k‐means clustered. The cluster analysis was done after applying probe‐set standardization using an average Euclidean distance function. K was selected according to the absolute minimum in the Davies Bouldin cluster estimation procedure.11 Furthermore, to estimate contribution of treatment and strain differences and the interaction to the general ANOVA effect, a 2‐way ANOVA was performed on probe sets representing Ndufc2.
Data of microarray analysis are available online (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-1102; accession numbers: ArrayExpress acc#: E‐MTAB‐1102).
Sequencing of Ndufc2 in the 2 Parental Strains
As previously reported,12 genomes of SHR/NHsd (Harlan Laboratories, Bethesda, MD) and of SHRSP/Gla (Western Infirmary, Glasgow, UK) were sequenced on an Illumina platform to a depth of at least 23× (Illumina, San Diego, CA). In brief, 5 μg of DNA was used to construct paired‐end whole‐genome libraries with 300‐ to 600‐base‐pair (bp) insert size. Genomic DNA was prepared from liver or spleen and quality checked before sequencing on an Illumina HiSeq2000, following the manufacturer's instructions. Quality‐filtered Illumina paired‐end and/or mate pair reads were mapped to the Brown Norway reference genome, RGSC‐3.4,13 using the read alignment software Burrows‐Wheeler Aligner (BWA‐0.5.8c).14 Genomic variants (single‐nucleotide variant and short indels [1–15 bp]) were detected using the Genome Analysis Toolkit (GATK version 1.0.6001).15, 16 Sequence differences between strains SHR/NHsd and SHRSP/Gla, compared to other rat strains, were accessed using custom Perl scripts.
Determination of Ndufc2 Expression by Reverse‐Transcription Polymerase Chain Reaction in Brain Tissue and in Cellular Extracts
Two micrograms of total RNA were used for cDNA synthesis using Superscript III First‐Strand (Invitrogen, Carlsbad, CA) and random examer primers according to manufacturer's instructions. The following oligonucleotides were used: Ndufc2: forward 5′‐GGCTTGTCTACATCGGCTTC‐3′; reverse 5′‐ TGATGGTCCCTCACAGCATA‐3′; ß‐actin: forward 5′‐AGATGACCCAGATCATGTTTGAGA‐3′; reverse 5′‐ATAGGGACATGCGGAGACCG‐3′. Real‐time quantitative polymerase chain reaction (RT‐PCR) was performed using a 2× SYBR Green PCR Master Mix (Applied Biosystems, Forster City, CA) containing the double‐stranded DNA‐binding fluorescent probe, Sybr Green, and all necessary components except primers. Quantitative PCR conditions included an initial denaturation step of 94°C/10 minutes followed by 40 cycles of 94°C/15 seconds and 60°C/15 seconds. Standards, samples, and negative controls (no‐template) were analyzed in triplicate. Concentrations of mRNA were calculated from serially diluted standard curves simultaneously amplified with the unknown samples and corrected for β‐actin mRNA levels. Levels of Ndufc2 mRNA in JD‐fed rats were compared to levels of RD‐fed rats. Levels of Ndufc2 mRNA in Ndufc2‐silenced cells were compared to nonsilenced cells.
Western Blotting of Total Proteins Extracted From Brain Tissue and Cellular Extracts
Brain total proteins were extracted as previously reported for tissues.17 For cellular protein extraction, cells were washed twice with ice‐cold PBS and lysed with lysis buffer. Protein concentrations were determined by the Bradford method.18 Then, 40 μg of total proteins were separated on 12% SDS‐PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Amersham, Piscataway, NJ). Nonspecific binding sites were blocked with 5% nonfat dried milk for 2 hours at room temperature (RT). Membranes were then incubated overnight with the following primary antibodies: anti‐Ndufc2 (1:200) rabbit polyclonal (Catalog No.: NBP1‐59610; Novus Biologicals, Littleton, CO); anti‐Nf‐κBp65 (nuclear factor kappa B; 1:200; Catalog No.: SC‐8008; Santa Cruz Biotechnology, Santa Cruz, CA); anti‐SOD2 (superoxide dismutase 2, mitochondrial; 1:200; Catalog No.: S5569; Sigma‐Aldrich, Milan, Italy), anti‐phospho‐MAPKp38 (mitogen‐activated protein kinase; 1:200) mouse monoclonal (Catalog No.: SC‐7973; Santa Cruz Biotechnology); anti‐phospho‐JNK1 (c‐Jun N‐terminal kinase 1; 1:200) mouse monoclonal (Catalog No.: SC‐6254; Santa Cruz Biotechnology); anti‐MAPKp38 (1:200) rabbit polyclonal (Catalog No.: SC‐535; Santa Cruz Biotechnology); anti‐JNK1 (1:200) rabbit polyclonal (Catalog No.: SC‐474; Santa Cruz Biotechnology); anti‐IκB (nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor; 1:200) rabbit polyclonal (Catalog No.: SC‐945; Santa Cruz Biotechnology); anti‐c‐Jun mouse monoclonal antibody (1:200; Catalog No.: L70B11; Cell Signaling Technology, Milan, Italy); anti‐β‐actin (1:5000; Catalog No.: A5441; Sigma‐Aldrich). Secondary antibodies were: 1:5000 antirabbit (Catalog No.: SC‐2004; Santa Cruz Biotechnology) and 1:5000 antimouse (Catalog No.: SC‐2005; Santa Cruz Biotechnology).
Signals were revealed with an enhanced chemiluminescence detection system (ECL; Amersham) and visualized by a ChemiDoc XRS+imaging system (Bio‐Rad, Richmond, CA). Finally, protein levels were normalized using β‐actin levels. Protein bands were scanned and quantified densitometrically.
Detection of Brain‐Oxidized Proteins
To detect levels of brain‐oxidized proteins from total proteins, the Oxyblot Detection kit (Millipore, Milan, Italy) was used as previously reported.17 Five selected protein bands from immunoblots were scanned and quantified densitometrically. Experiments were performed in triplicate.
In Vitro Ndufc2 Silencing in A10 Cells
A vascular smooth muscle cell line (A10) obtained from rat embryonic thoracic aorta (purchased from LGC Promochem, Milan, Italy) was cultured in DMEM containing normal glucose (5.5 mmol/L), 10% FBS, and supplemented with penicillin (100 U/mL)/streptomycin (100 μg/mL) at 37°C in 95% O2 and 5% CO2. Cells were plated in 100‐mm‐diameter dishes (4×105) and 60‐mm‐diameter dishes (1.5×105), passaged upon reaching confluence with 2 mL of trypsin and used at the 12th passage and 70% confluence. Cells were washed with PBS, and then OPTI‐MEM–reduced serum medium (Invitrogen, Carlsbad, CA) was added to the cells. Ndufc2‐specific siRNA (Mission siRNA; Sigma‐Aldrich) and a nucleic acid transferring agent, Lipofectamine 2000 (Invitrogen) were incubated in OPTI‐MEM–reduced serum medium for 20 minutes at RT to form a siRNA‐Lipofectamine complex. The siRNA‐Lipofectamine complex‐containing medium was added to cells to a final siRNA concentration of 33 nmol/L. Five hours later, the complex‐containing medium was replaced with DMEM supplemented with 10% FBS. Cells transfected with Lipofectamine and no‐target siRNA (Sigma‐Aldrich) were used as controls. Seventy‐two hours later, both silenced and nonsilenced cells were used for all analyses described below. Six experiments were performed.
Determination of Reactive Oxygen Species Production in Ndufc2‐Silenced A10 Cells
Intracellular production of superoxide was evaluated by dichlorodihydrofluorescein ester (DCHF), as previously reported.17 For this purpose, A10 cells were incubated in 1 mL of 0.05 mmol/L of PBS solution of the permeable agent, DCHF (Sigma‐Aldrich), for 30 minutes. Cells were lyzed with 2% Triton‐X. Lysates were collected on ice, centrifuged at 13 800 g at 4°C for 10 minutes, and then fluorescence intensity was read. In this assay, oxidative stress is quantified by monitoring intracellular DCF content by a fluorometer (Mithras LB 940; Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany) with excitation at 495 nm and emission at 525 nm. Fluorescence intensity measurements are expressed as arbitrary units.
Characterization of Inflammatory Pathway in Ndufc2‐Silenced A10 Cells
RNA extracted from both silenced and nonsilenced cells was analyzed by quantitative RT‐PCR using an RT2Profiler PCR array (Rat Inflammation array; SuperArray Bioscience Corporation, Frederick, MD). Procedure and data analysis have been previously described.19 Results are expressed as relative levels of each gene mRNA under condition of either presence or absence of Ndufc2 referred to the expression of this gene in control cells (that were chosen to represent 1× expression of each gene). Results were considered significant when mRNA expression was 2.5‐fold higher or lower than that of control cells. Experiments were performed in triplicate.
Assessment of Necrosis by Flow Cytometry With Propidium Iodide Staining
Cells were harvested by incubation with 2 mL of trypsin for 5 minutes. After addition of 10% FBS DMEM medium, the suspension was centrifuged at low speed at RT. Each pellet was washed with PBS. After washing, 5×105 cells were resuspended in 2 mL of cold PBS and centrifuged at 4°C at 500g for 5 minutes. The supernatant was removed, pellets energically vortexed, and then gently resuspended in 400 μL of hypotonic solution of propidium iodide (PI; 50 mg/mL in 0.1% sodium citrate plus 0.1% Triton X‐100; Sigma‐Aldrich). Cells were incubated in the dark at 4°C for 20 minutes. Then, fluorescence‐activated cell sorting (FACS) was performed by a FACS Calibur flow cytometer (BD Biosciences, San Jose CA). For this assay, red fluorescence was measured corresponding to the red color of PI (FL‐3 detector). To exclude doublets and cell aggregates from the analysis, a doublet discrimination module was used. Peak width was estimated using the coefficient of variation of the signals within each peak. Cells treated with a high dose of H2O2 were used as positive control.
Analysis of Mitochondrial Function
Complex I assembly evaluation
Mitochondria were isolated from whole rat brains using the Mitochondria Isolation Kit for Rodent Tissue (Abcam, Cambridge, MA). Preparation of total mitochondrial extracts for Blue Native (BN)–PAGE (BNG) analysis was performed as described previously.20, 21 Mitochondrial pellet was resuspended at a protein concentration of ≈1 mg/mL in a solubilization buffer containing 50 mmol/L of Bis‐Tris (pH 7.0), 1% (v/v) n‐dodecyl‐β‐d‐maltoside, and 750 mmol/L of ε‐amino‐N‐caproic acid. The suspension was kept on ice for 1 hour with occasional vortexing and was then clarified by centrifugation at 14 000g for 30 minutes. Then, 0.9 mL of the resulting supernatant, containing both membrane and water‐soluble proteins, was mixed with 0.1 mL of concentrated BN‐PAGE loading buffer (10×) containing 0.75 mol/L of ε‐amino‐N‐caproic acid and 3% (w/v) Serva blue G‐250. Samples were stored at −20°C until analysis. Protein concentrations were determined by bicinchoninic acid protein assay using BSA as a standard. Intact mitochondrial complexes were then separated by electrophoresis using a minigel system (Bio‐Rad Mini‐Protean 3) and a 4% to 15% gradient acrylamide gel (Mini‐Protean Precast GelS; Bio‐Rad). Mitochondrial protein complexes were transferred to a methanol‐activated PVDF membrane (Bio‐Rad) using the BioRad Mini Trans‐blot System. Membranes were then blocked overnight at 4°C in 5% milk/PBS solution, washed for 10 minutes in PBS 0.05% Tween‐20, and incubated for 3 hours with the primary antibodies (MitoProfile Total OXPHOS Rodent WB Antibody Cocktail; Abcam) to a final concentration of 6.0 μg/mL in 1% milk/PBS solution. Membranes were washed in PBS 0.05% Tween‐20 and incubated for 1 hour with the secondary antibody at RT (goat anti‐mouse, 1:5000, HRP‐linked). Rabbit polyclonal Hsp60 was used as loading control (Anti‐Hsp60 antibody‐Loading Control; Abcam) at a concentration of 1 μg/mL in 1% milk/PBS solution at 1:3000 dilution. Bands were detected using ECL Luminata Crescendo (Millipore), and images were acquired using the ChemiDoc XRS+imaging system. Experiments were performed in triplicate.
Ndufc2‐silenced cells were harvested using trypsin and washed twice with PBS. After removal of residual PBS, cells were immediately treated with the Mithochondria Isolation Kit for Cultured Cells (Abcam). Samples were then treated as reported above for brain tissues.
Complex I activity
Brains were dissected out, weighted, wetted from blood, and placed in ice‐cold respiration medium (sucrose 250 mmol/L, MgCl2 5 mmol/L, EDTA 1 mmol/L [pH 7.4], HEPES 20 mmol/L [pH 7.4], and KH2PO4 2 mmol/L). Tissue was chopped finely with a pair of scissors and rinsed in new respiration medium to remove any blood. Disrupted tissue was then homogenized on ice by 20 to 30 passes in a tight‐fitting glass/Teflon power‐driven Potter‐Elvejhem homogenizer in ≈25 mL of respiration buffer. Homogenate was centrifuged at 600 rpm for 10 minutes at 4°C. The supernatant, rich with mitochondria, was then centrifuged at 4°C at 12 000 rpm for 10 minutes. The external part of the pellet, which is the cleaner part, was resuspended in a small volume of respiration buffer by a miniglass/Teflon power‐driven Potter‐Elvejhem. Mitochondrial protein concentrations were determined as stated above.18
Silenced cells were washed once with cold PBS 1× and treated with lysis buffer.
Activity of mitochondrial complex I was analyzed using an ELISA microplate assay kit (Abcam). Capture antibody for complex I was precoated in the wells of the microplate, and samples containing 50 μg of brain mitochondrial extracts and 40 μg of cell lysates were added to wells. In this assay, complex I activity is measured by oxidation of NADH to NAD+, which leads to increased absorbance at 450 nm. Absorbance was read by a Benchmark microplate reader (Bio‐Rad). All tests were done in triplicate.
Mitochondrial membrane potential assessment
Brain mitochondrial membrane potential was determined by a fluorescent probe, TMRE (Abcam). Briefly, TMRE reagent was warmed to room temperature and diluted with PBS 1× 0.2% BSA to obtain a final TMRE staining working solution at a concentration of 500 nmol/L. Isolated mitochondria (100 μg) were incubated with 80 μL of TMRE staining working solution for 10 minutes at 37°C. Fluorescence was determined using a fluorometer (Mithras LB 940; Berthold) microplate reader at 549 nm excitation/575 nm emission. Data are expressed as fluorescent units per μg protein. Mitochondrial membrane potential of silenced cells was determined by use of the TMRE Mitochondrial Membrane Potential Assay Kit (Abcam). Fluorescence was determined by a FACS Calibur flow cytometer (FL‐2 detector). Data are expressed as fluorescent intensity.
ATP‐level measurement
Brain mitochondria (250–500 μg) were plated in a dark 96‐multiwell plate on ice and incubated, always on ice, for 5 minutes with respiration buffer (sucrose 250 mmol/L, MgCl2 5 mmol/L, EDTA 1 mmol/L, HEPES 20 mmol/L, KH2PO4 2 mmol/L, ADP 1 mmol/L, D‐Luciferin, and Luciferase firefly ready from ATP Determination Kit; Invitrogen, Milan, Italy) in the presence of both glutammate (2.5 mmol/L) and malate (2.5 mmol/L; complex I substrates) or succinate (5 mmol/L; complex II substrate). In this assay, quantitative determination of ATP with recombinant firefly luciferase and its substrate, D‐luciferin, is based on luciferase's requirement for ATP in producing light (emission ≈560 nm at pH 7.8) from the reaction. Absorbance was quantified by a fluorometer (Mithras LB 940; Berthold). The reaction was repeated after the addition of oligomycin (2 μmol/L) as a mitochondrial ATPase inhibitor and of valinomycin (2.5 μmol/L) as an oxidative phosphorylation inhibitor.22
Nduc2‐silenced cells were washed once with PBS 1× and then incubated at 37°C in 1 mL of respiration buffer (as reported above for brain tissues) in the presence of both glutammate (2.5 mmol/L) and malate (2.5 mmol/L) or succinate (5 mmol/L) for 60 minutes. Cells were washed once with ice‐cold PBS and then lyzed with 90 μL of ATP‐lysis buffer (Tris HCl [pH 7.5] 200 mmol/L, NaCl 2 mol/L, EDTA [pH 7.5] 20 mmol/L, and Triton X‐100 0.2%). Lysates were collected on ice and centrifuged at 4°C at 12 000 rpm for 15 minutes. Ten microliters of supernatant from each sample was plated in a dark 96‐multiwell plate; 100 μL of total ATP‐kit buffer from the kit were added to each well. Chemiluminescent signal was read with the fluorometer. Five microliters of cell lysate were used for protein concentration and ATP‐level normalization. The reaction was repeated after the addition of oligomycin (2 μmol/L) and valinomycin (2.5 μmol/L), as described above. Experiments were performed in triplicate.
Generation of a Genetically Manipulated Animal Model
Generation of SHR‐Ndufc2 mutant strains (SHR‐Ndufc2 em1Mcwi and SHR‐Ndufc2 em2Mcwi) was performed at the Medical College of Wisconsin (Milwaukee, WI) under protocols approved by the institutional animal care and use committee. Generation of ZFN mutants was performed as previously described.23, 24 ZFN constructs specific for rat Ndufc2 were designed, assembled, and validated by Sigma‐Aldrich to the exon 1 sequence GGCTTCCTGGGCTACTGCacgggcCTGATGGACAACATG where each ZFN monomer binds the underlined sequence on opposite strands. In vitro–transcribed ZFN mRNA SHR/NCrl (SHR) rat embryos were transferred to pseudopregnant Sprague‐Dawley female rats to be carried to parturition. The resulting pups were ear punched and DNA was extracted and screened for ZFN‐induced mutations by the Surveyor Nuclease assay (Transgenomic, Inc., Omaha, NE), as described previously,23 using primers flanking the target sequence (Ndufc2_F: 5′‐CGCATCAATATGATGAACGG‐3′ and Ndufc2_R: 5′‐CGCTGAAAACTCTAGACGGG‐3′). Two positive mutant founders were identified and confirmed to have ZFN‐induced mutations. Sanger sequencing confirmed mutant alleles within exon 1 in each founder, a 9‐bp deletion (del‐9 ACGGGCCTG; Ndufc2‐m1) and a net 107‐bp deletion (del‐111 GCCCGTGCAGTAGCCCAGGAAGCCGATGTAGACAAGCCGCGGGTCGTTCAGCTTGGGCGGGGGCAGTCTCCGGGCCTCATCCGGCAAGAATCTTAAGGGCTCATGGCCCGG plus ins‐4 TTGT; Ndufc2‐m2). The 2 founders were back‐crossed to the SHR/NCrl strain to establish the SHR‐Ndufc2 em1Mcwi and SHR‐Ndufc2 em2Mcwi breeding colonies, respectively.
Both newborn rats and embryos obtained from colonies breeding were genotyped to assess allele carrier status for the corresponding deletion. Embryos were obtained at 7 to 8 days of gestation (SHR‐Ndufc2 em2Mcwi, n=47 from 6 pregnant rats; SHR‐Ndufc2 em1Mcwi, n=20 from 2 pregnant rats), and at 10 to 12 days of gestation (SHR‐Ndufc2 em2Mcwi, n=8 from 2 pregnant rats; SHR‐Ndufc2 em1Mcwi, n=8 from 2 pregnant rats).
For genotyping of SHR‐Ndufc2 mutant strains, genomic DNA was prepared from tail clipping of each animal by salt precipitation5and subsequently used for PCR reactions.
For detection of the large 107‐bp deletion (SHR‐Ndufc2 em2Mcwi), PCR reactions were performed with 50 ng of genomic DNA in a total volume of 20 μL containing 250 nmol/L of dNTP, 10 nmol/L of each primer (forward 5′‐TCTTTGCCCACTGTGGGTTAT 3′ and reverse 5′CCCCTCCCCAGACCTTATGA‐3′), and 0.3 U of Taq Polymerase (Life Technologies, Carlsbad, CA). They were processed on a thermal cycler system (T100 Thermal Cycler; Bio‐Rad, Hercules, CA) by using “touchdown PCR” conditions (to increase specificity and reduce background amplification). Briefly, PCR was carried out at 95°C for 3 minutes followed by 14 cycles of denaturation at 95°C for 15 seconds, annealing at 63.5°C for 30 seconds with subsequent decreases of 0.5°C per cycle, and elongation at 72°C for 70 seconds, with subsequent 19 cycles at 95°C for 15 seconds, 56°C for 30 seconds, 72°C for 70 seconds, and a final extension of 72°C for 5 minutes. The resultant PCR product was a fragment of 318 bp in the wild‐type strain and a fragment of 211 bp in the SHR‐Ndufc2em2Mcwi strain. They were resolved on 2.5% agarose gels and visualized using the ChemiDoc XRS+Imaging System (Bio‐Rad).
For detection of the minor 9‐bp deletion (SHR‐Ndufc2 em1Mcwi), the PCR/single‐strand conformational polymorphism analysis was used. Briefly, genotyping was carried out using 50 ng of DNA and the same set of oligonucleotides mentioned above. In this case, the forward primer was previously labeled with ATP‐ɣ32P (PerkinElmer) by using T4 polynucleotide kinase (New England Biolabs, Boston, MA). PCR reactions were carried out with the same protocol described above, and products were loaded onto a 7% polyacrylamide gel run on a Gel Electrophoresis System (Thermo Fisher Scientific, Waltham, MA) for 4 hours. Images were acquired using the PMI Personal Molecular Imager System (Bio‐Rad).
Because only heterozygous rats were obtained for both mutant lines, the molecular analyses and phenotypic studies were performed in heterozygous, compared to wild‐type, rats of both SHR‐Ndufc2 em1Mcwi and SHR‐Ndufc2 em2Mcwi lines.
Phenotyping of SHR‐Ndufc2 em1Mcwi and SHR‐Ndufc2 em2Mcwi Lines
Feeding with JD was started at 6 weeks of age in both heterozygous and wild‐type SHR‐Ndufc2 em1Mcwi and SHR‐Ndufc2 em2Mcwi rats, along with parental rats (for number of animals used, see Table 1). A group of rats (n=6) was sacrificed both at 6 weeks of age and at the end of 4 weeks of either RD or JD for analysis of: Ndufc2 by western blot and RT‐PCR, SOD2, and Nf‐κbp65 by western blot, complex I assembly and activity, mitochondrial membrane potential, ATP levels, and tissue oxidative stress. Remaining rats were subsequently monitored up to 3 months for signs of renal damage (detected by 24‐hour proteinuria measurement in metabolic cages) and of stroke (by evidence of paresis, paralysis, hemiplegia, epilepsia, or sudden death). Animal studies were approved by the Neuromed Institutional review committee. Procedures involving animals and their care were carried out in accord with our institution's guidelines, which comply with national and international laws and policies.
Table 1.
Modulation of Inflammatory Genes in Ndufc2‐Silenced A10 Cells
| Gene Name | Control | RNAsi1 | RNAsi2 |
|---|---|---|---|
| Ccl2 | −0.83±0.77 | 1.05±0.83 | 0.73±0.98 |
| Ccl6 | −6.70±15.4 | 1.80±11.1 | 10.61±2.6 |
| Ccr5a | 86.30±17a | −42.24±21a | −8.31±15a |
| Ccr6a | 62.44±1.3a | −1.40±2.5a | −1.23±1.9a |
| Cxcl10 | 0.86±3.4 | −8.26±6.5 | −3.25±5 |
| Il10 | −0.78±20 | 0.63±14 | 13.86±3.9 |
| Il10ra | 3.46±21 | 9.49±18 | 16.40±2.5 |
| Il13a | 1.81±51a | 8.27±38a | 36.22±8.5a |
| Il13ra1a | 3.67±22a | 42.77±26a | 22.81±2a |
| Il15a | −28.93±1.9a | 3.04±2.1a | 1.89±0.15a |
| Lta | 3.17±74 | 37.17±64 | 58.72±2.1 |
| Tnfa | 2.58±12.9a | 9.37±11.9a | 10.81±1.0a |
| Tgfb1a | −531±3.85a | 3.57±3.8a | 3.38±0.25a |
| Tnfrsf1ba | −11.04±3a | 3.73±3.1a | 2.77±0.18a |
Ccl2 inidcates chemokine ligand 2; Ccl6, chemokine ligand 6; Ccr5, chemokine receptor type 5; Ccr6, chemokine receptor type 6; Cxcl10, chemokine ligand 10; Il10, interleukin‐10; Il10ra, interleukin‐10 receptor alpha; Il13, interleukin‐13; Il13ra1, interleukin‐13 receptor alpha 1; Il15, interleukin‐15; Lta, lymphotoxin‐alpha; Tgfb1, transforming growth factor beta 1; Tnf, tumor necrosis factor; Tnfrsf1b, tumor necrosis factor receptor superfamily, member 1B.
Genes markedly modulated by Ndufc2 silencing (number of experiments=3).
Genotyping of NDUFC2 Markers in an Italian Cohort of Early‐Onset Ischemic Strokes and Controls
Study population
Four hundred eighty‐four unrelated Caucasian young adults (<65 years) who were referred to the (1) Angelo Bianchi Bonomi Hemophilia and Thrombosis Center of the University of Milan and (2) to Department of Neurology, Sapienza University, Sant'Andrea Hospital, Rome, for thrombophilia screening after a first ischemic stroke were enrolled in this study. Patient and control populations were in part or in whole previously investigated in relation to polymorphisms of genes encoding procoagulant and inflammatory factors and of methionine metabolism components, chromosome 12p13, and NPR3 gene promoter polymorphisms.25, 26, 27, 28 Median time interval between ischemic stroke and blood sampling was 6 months (range, 1 month–10 years); 75% of patients had blood sampled within 2 years and 64% within 1 year after the event. Clinical records were reviewed, and when the type of stroke was not specified, neurologists who took care of patients during the acute phase were contacted. Clinical diagnosis was objectively confirmed by computed tomography scans or magnetic resonance or magnetic resonance angiography and intra‐arterial angiography. According to Trial of ORG 10172 in Acute Stroke Treatment (TOAST) classification, 14.7% of enrolled patients were classified as large artery stroke, 5.2% as cardioembolic, 16.8% as small artery (lacunar), 51.8% as undetermined etiology, and 11.6% as other determined etiology stroke. All patients underwent a cardiologic evaluation, transthoracic echocardiography, and Doppler examination of neck vessels.
One thousand one hundred sixty‐five healthy unrelated Caucasian subjects comparable for age and sex were chosen from the whole population of controls made of partners and friends who accompanied patients to the Center in the same period as patients and agreed to be investigated. Previous thrombosis in the controls was excluded using a validated structured questionnaire.29 The presence, at the time of stroke for patients and at the time of blood sampling for controls, of hypertension, hypercholesterolemia, diabetes mellitus, and smoking habit (at least 5 cigarettes daily) was recorded.
The study was approved by the institutional review board of the University of Milan, University of Rome, and University of Florence, and all subjects gave their informed consent to the study.
Genomic DNA extraction
Genomic DNA was isolated from venous peripheral blood by using the FlexiGene kit (Qiagen). Quality and concentration of DNA were determined by a NanoDrop1000 spectrophotometer (Thermo Fisher Scientific).
Genotyping
We studied 3 tagSNPs in NDUFC2: rs641836, rs584981, and rs11237379 by 7900HT real‐time PCR and TaqMan technology assays (c__26529993_10, c___614632_10 and c__2999825_10, respectively; Life Technologies). The 3 NDUFC2 tagSNPs were selected for analysis by using the algorithm‐Tagger‐pairwise Tagging (HapMap database and software, http://hapmap.ncbi.nlm.nih.gov) on NDUFC2 gene for CEU population. They captured 100% of alleles with a mean r 2 of 0.961. Polymorphisms information were assessed in the Single Nucleotide Polymorphism (dbSNP) NCBI (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp&cmd=search&term=), and ENSEMBL (http://www.ensembl.org/index.html) databases.
To assess functional relevance of the rs11237379 variant allele, we characterized NDUFC2 expression by RT‐PCR in a cohort of young healthy subjects (mean age, 35±6; females, 52%), free from cardiovascular risk factors, carrying wild‐type (n=6), heterozygous (n=12), and mutant homozygous (n=6) genotypes. Lymphocyte extraction was performed by following standard procedures with Ficoll.30 RNA extraction and cDNA were obtained as described above. Finally, RT‐PCR of human NDUFC2 was performed with the following set of oligonucleotides: forward 5′‐CCTGTATGCTGTGAGGGACC‐3′ and reverse 5′‐CGACGTTCAGCTCCACAACA‐3′. β‐actin: forward 5′‐GCAAGAGATGGCCACGGCTG‐3′ and reverse 5′‐CCACAGGACTCCATGCCCAG‐3′.
Statistical Analysis
In vitro and in vivo experiments
Data of Ndufc2 expression levels, western blot densitometric analyses, complex I activity and ATP levels, results of FACS, rat body weight, systolic blood pressure, and urinary protein levels are provided as means±SD. Comparisons between 2 groups were performed by using t test analyses. When more than 2 groups were compared at the same time, 1‐way ANOVA followed by the Bonferroni post‐hoc test was performed. Survivor function in rats monitored over JD feeding was estimated by the life‐table method. Log‐rank and Wilcoxon statistics were used for testing equality of survivor functions.
SPSS statistical software (version 12.0; SPSS, Inc., Chicago, IL) was used for statistical analysis. Statistical significance was stated at the P<0.05 level.
Human genetic analyses
Statistical analysis was performed using the SPSS package (v20). Hardy–Weinberg equilibrium (HWE) was evaluated by the chi‐square (χ2) test. Genotype distributions were compared between patients and controls or between TOAST types by χ2 analysis. Categorical variables are expressed as frequencies and percentages. Unless otherwise indicated, continuous data are given as median and interquartile range (IQR). Comparisons of continuous variables between patients and controls or among genotypes were performed by the nonparametric Mann–Whitney or Kruskal–Wallis test.
Multiple logistic regression analysis was used to estimate odds ratios (ORs) and 95% CIs for risk of stroke under the assumption of an additive, as well as a dominant and a recessive effect of each allele. To evaluate whether polymorphisms were independently associated with stroke, multiple logistic regression analyses were adjusted for traditional cardiovascular risk factors (age, sex, hypertension, diabetes mellitus, dyslipidemia, and smoking habit). A value of P<0.05 was chosen as the cut‐off level for statistical significance.
Results
Identification of Ndufc2 as a Sequence Down‐regulated in Brains of SHRSP Under JD and Related Molecular Analyses
Eight‐six probe sets were revealed significantly differentially modulated inside the STR1/QTL. After applying a K‐mean clustering with K=6 and the 2‐way ANOVA statistical analysis, as specified in the Methods section, we detected the gene encoding Ndufc2 (NADH dehydrogenase [ubiquinone] 1, subcomplex unknown, 2), mapping 8 Mb apart from the Lod score peak of the QTL, as the sequence differentially modulated, significantly down‐regulated, in the brain of SHRSP versus SHRSR when fed with JD (P values for significant Ndufc2 effects in strain [P=6.96E−05], diet [P=8.29E−10], and interaction [P=2.79E−03]; n=5 for each strain at each diet; Figure 1A and 1B).
Figure 1.
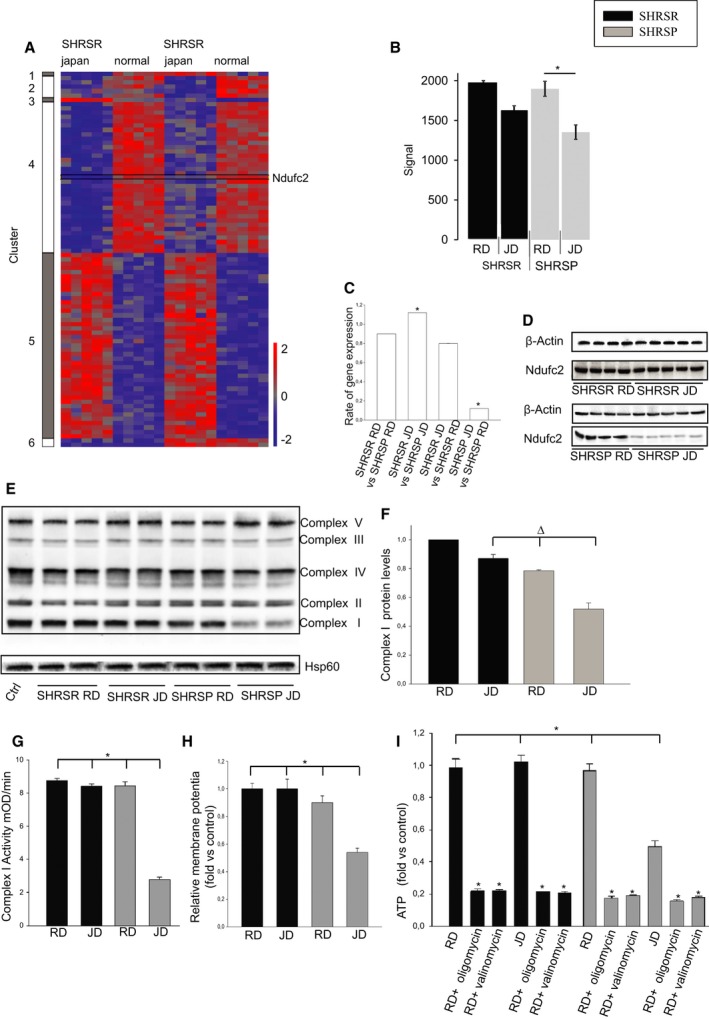
Ndufc2 is differentially modulated in brains of SHRSP under stroke‐permissive diet. Evidence of complex I dysfunction in brains of SHRSP. A, Heatmap represents expression differences between samples in a color scale ranging from red (up‐regulated) to blue (down‐regulated). Probe sets representing Ndufc2 are marked. Two‐way ANOVA analysis revealed significant Ndufc2 effects in strain (P=6.96E−05), diet (P=8.29E−10), and interaction (P=2.79E−03; n=5 for each strain at each diet). Corresponding densitometric analysis for Ndufc2 expression levels in brains of SHRSP and SHRSR under either RD or JD is shown in (B). C, Confirmation of Ndufc2 differential expression by RT‐PCR in the same tissue samples; *P<0.0001 for each ratio indicated in the figure (n=6 for each group). D, Representative western blot of Ndufc2 expression levels, confirming reduced expression in brains of SHRSP under JD. E, Representative western blot of BNG (n=3) and corresponding densitometric analysis (F). Δ P<0.01 for SHRSP JD versus both SHRSP RD and SHRSR JD. G, Complex I activity measurement; (H) mitochondrial membrane potential assessment. *P<0.0001 for SHRSP JD versus all other samples (I). ATP‐level measurement in the same tissue samples. Oligomycin was used to confirm functionality of ATP assay as an inhibitor of oxidative phosphorylation. Valinomycin was used as a mitochondrial ATPase inhibitor. *P<0.0001 for SHRSP JD versus all other samples and for each inhibited experimental point versus the noninhibited corresponding sample (n=6 for each group). Values are expressed as means±SD. Ctrl indicates control; Hsp60, heat shock protein 60; JD, Japanese‐style stroke‐permissive diet; RD, regular diet; RT‐PCR, reverse‐transcriptase polymerase chain reaction; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR.
Analysis of Ndufc2 mRNA expression and protein levels confirmed the microarray differential expression results (P<0.0001; n=6; Figure 1C and 1D).
Ndufc2 is a subunit of OXPHOS complex I. Therefore, we evaluated mitochondrial function in brains of SHRSP and SHRSR under JD. Brains of JD‐fed SHRSP showed significant reduction of both complex I assembly (P<0.01; n=3) and activity (P<0.0001; n=6; Figure 1E through 1G), mitochondrial membrane potential (P<0.0001; n=6; Figure 1H), and ATP production (P<0.0001; n=6; Figure 1I), as well as remarkable increases of both inflammation (P<0.0001; n=3) and oxidative stress (P<0.0001; n=3; Figure 2), likely as a consequence of mitochondrial uncoupling.
Figure 2.
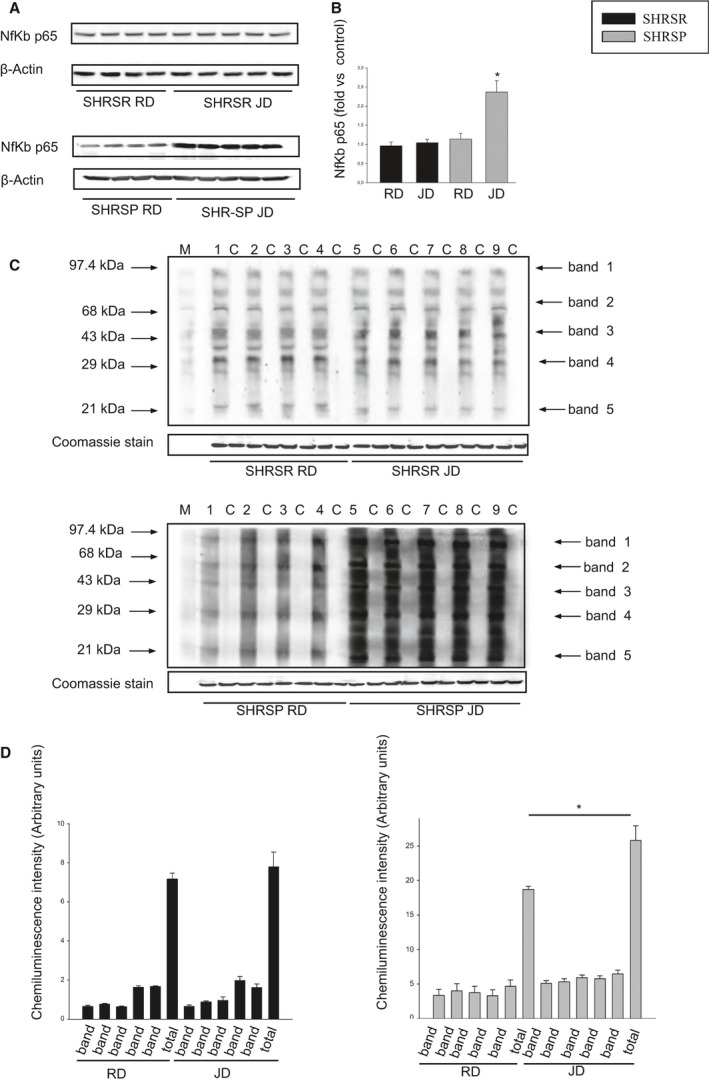
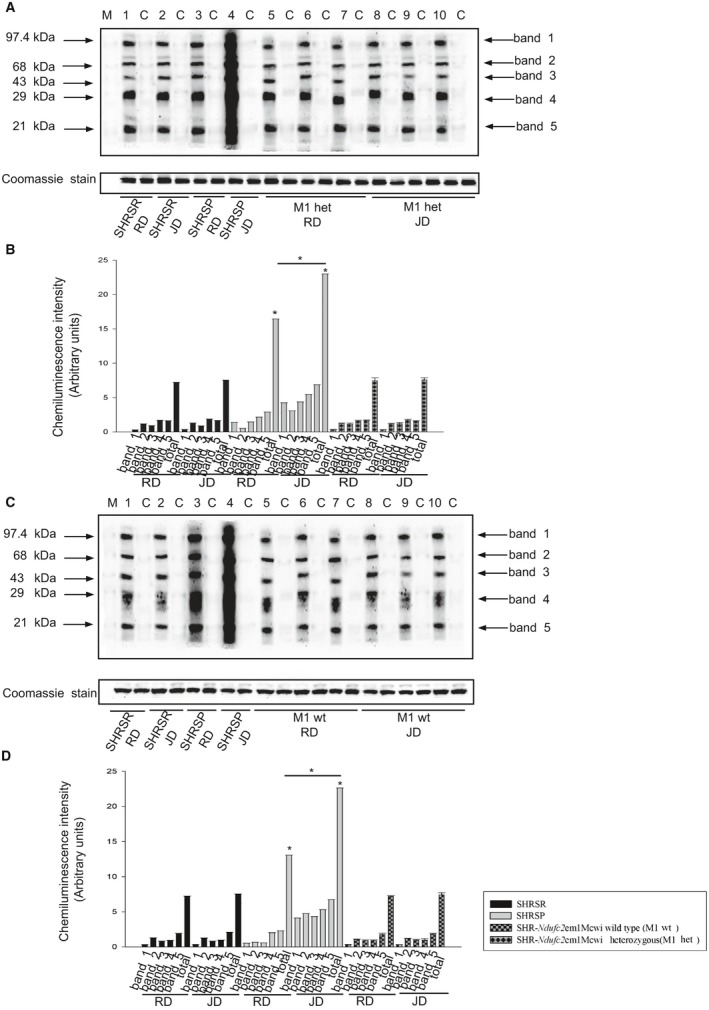
Molecular analyses in brains of SHRSP as compared to SHRSR under stroke‐permissive diet. A, Representative western blot (n=3) of Nf‐κBp65 in SHRSR and SHRSP upon either RD or JD with corresponding densitometric analysis (B). *P<0.0001 for SHRSP JD versus all other samples. D, Western blot of intracellular protein extracts immunostained for carbonylated proteins using the Oxyblot Protein Oxidation Detection kit (Millipore, Milan, Italy) in brains of SHRSR and SHRSP under either RD or JD. Each lane was loaded with 50 μg of total proteins. Lane M, DNP marker. Each sample is run with its own untreated control (C). Normalization for lane protein loading was performed using Coomassie staining. D, Densitometric analysis of Oxiblot gels. Bar graphs represent chemiluminescence intensity relative to the gel loading band. Bands 1 to 5 refer to the most prominent bands on the blots (identified by arrows), whereas total refers to the total chemiluminescence intensity from all bands. *P<0.0001 for SHRSP JD versus SHRSP RD (total band). Densitometric values are expressed as means±SD. JD indicates Japanese‐style stroke‐permissive diet; Nf‐κB, nuclear factor kappa B; RD, regular diet; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR.
Next‐generation sequencing, covering full exons, introns, and 10 kb upstream and downstream on either side of Ndufc2, did not show variations between SHRSP and SHRSR.
In Vitro Effects of Ndufc2 Silencing
We then tested whether Ndufc2 knockdown in vascular cells could recapitulate the cellular abnormalities observed in SHRSP under JD. Ndufc2 silencing was confirmed by both RT‐PCR and western blotting (P<0.0001; n=6; Figure 3A). In this experimental condition, complex I assembly (P<0.0001; n=3) and activity (P<0.0001; n=6), mitochondrial membrane potential (P<0.0001; n=6), and ATP production (P<0.0001; n=6) were significantly decreased (Figure 3B through 3F). Reactive oxygen species (ROS) production (P<0.0001; n=6), Nf‐κB signaling, and other markers of inflammation increased in Ndufc2‐silenced cells (Figure 3G and 3H; Table 1). Fluorescence‐activated cell sorting (FACS) analysis revealed a significant decrease of cell viability and a significant increase of cell necrosis (P<0.0001; n=5; Figure 3I and 3J). Expression levels of proteins underlying necrosis (mitogen‐activated protein kinase [MAPK]p38, phosphor‐JNK [c‐Jun N‐terminal kinase], and c‐Jun) were significantly augmented (P<0.001; n=3; Figure 4).
Figure 3.
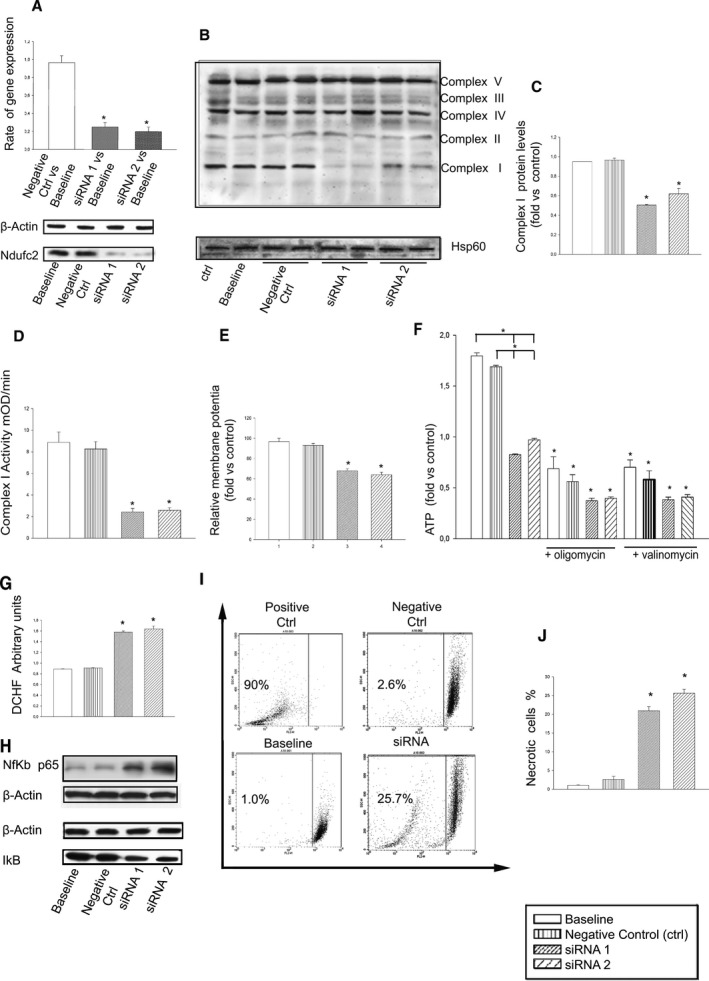
In vitro Ndufc2 silencing in A10 cells. A, Confirmation of Ndufc2 silencing by RT‐PCR and western blot; *P<0.0001 for each siRNA versus baseline (n=6). B, Representative western blot of BNG (n=3) in silenced versus nonsilenced A10 cells. All complexes belonging to OXPHOS are identified on the right side; Hsp60 was used to normalize protein expression level. C, Densitometric analysis of BNG. *P<0.0001 for each siRNA versus both baseline and negative CTRL. D, Assessment of complex I activity. E, Mitochondrial membrane potential assessment. *P<0.0001 for each siRNA versus both baseline and negative CTRL (n=6). F, Measurement of ATP levels in silenced versus nonsilenced cells and both in the absence and in the presence of oligomycin and valinomycin (to confirm functionality of the ATP assay as specified above). *P<0.0001 for each siRNA versus baseline and versus negative CTRL, and for each inhibited point versus the noninhibited corresponding sample (n=6 for each group). G, Assessment of resctive oxygen species levels; *P<0.0001 for each siRNA versus both baseline and negative CTRL (n=6 for each group). H, Representative western blot (n=3) of Nf‐κBp65 and of its inhibitor, IκB, in silenced versus nonsilenced cells, showing that increases of Nf‐κBp65 expression are accompanied by decreases of IκB expression levels. I, FACS analysis and corresponding percent values of necrotic cells (J). *P<0.0001 for each siRNA versus both baseline and negative CTRL (n=5). Values are expressed as means±SD. Ctrl indicates control; DCHF, 2′,7′‐dichlorodihydrofluorescein; FACS, fluorescence‐activated cell sorting; Hsp60, heat shock protein 60; IκB, nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor; Nf‐κB, nuclear factor kappa B; RT‐PCR, reverse‐transcriptase polymerase chain reaction.
Figure 4.
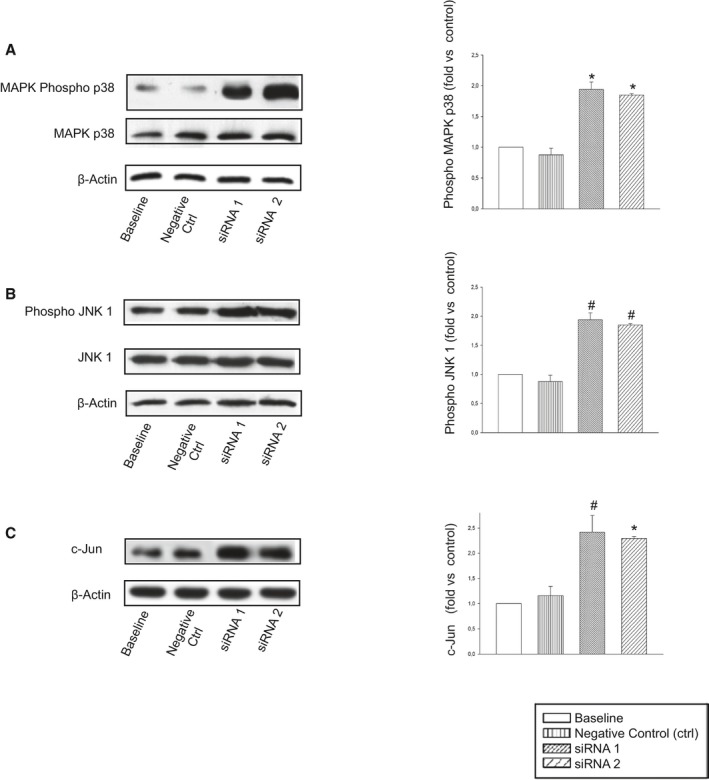
Expression levels of proteins involved in necrosis in Ndufc2‐silenced as compared to nonsilenced A10 cells. A, MAPKp38; (B) JNK1; and (C) c‐Jun. Number of western blots for each protein=3. Densitometric analyses are reported on the right side. *P<0.0001 and # P<0.001 for each siRNA versus baseline and negative CTRL. Ctrl indicates control; JNK1, c‐Jun N‐terminal kinase 1; MAPK, mitogen‐activated protein kinase.
Phenotype and Brain Molecular Analyses of SHR‐Derived Lines Carrying Ndufc2 Deletion
We generated a rat model carrying Ndufc2 deletion, as described in the Methods section, and characterized its phenotype. No homozygous knockout rat was obtained from either line (SHR‐Ndufc2 em1Mcwi [carrying 9‐bp gene deletion] and SHR‐Ndufc2 em2Mcwi [carrying 107‐bp gene deletion]). Furthermore, genotype assessment excluded the presence of homozygous knockout embryos even at early stages of pregnancy, suggesting that Ndufc2 full deletion is lethal before embryo implantation. Thus, only heterozygous and wild‐type rats of the 2 novel rat lines were characterized and compared to the 2 parental strains.
At 6 weeks of age, brains of heterozygous SHR‐Ndufc2 em2Mcwi showed a significantly lower amount of Ndufc2 mRNA (P<0.01; n=6) and protein expression (P<0.0001; n=6), as compared to both parental SHRSR and heterozygous SHR‐Ndufc2 em1Mcwi (Figure 5A through 5C). A defect in complex 1 assembly (P<0.0001; n=3) was present only in heterozygous SHR‐Ndufc2 em2Mcwi rats, whereas both complex 1 activity and ATP levels were comparable among all strains (Figure 5D through 5I). Level of oxidative stress was also unchanged among strains (data not shown).
Figure 5.
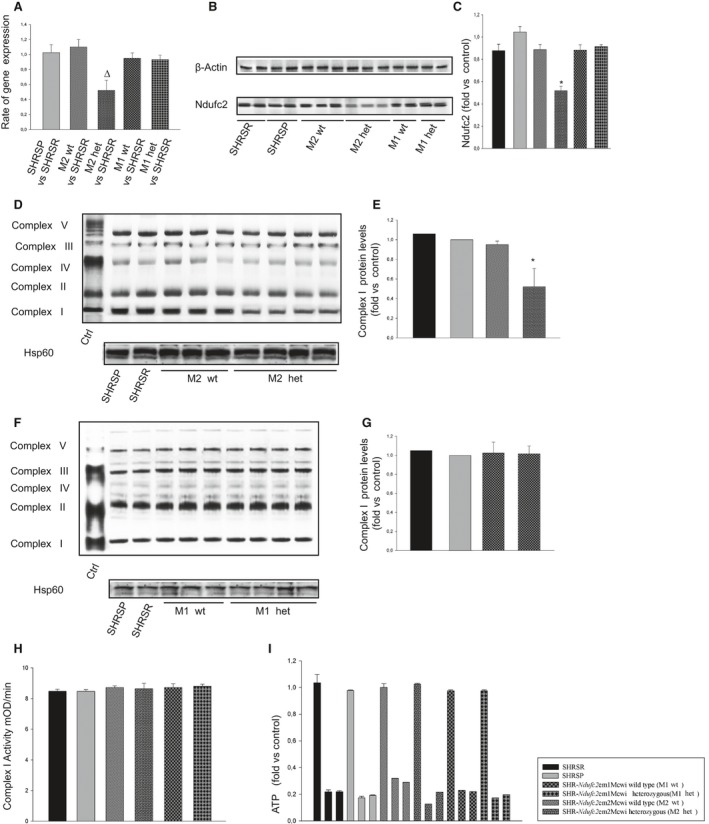
Molecular analyses of brains of all strains at 6 weeks of age. A through C, Brains of heterozygous SHR‐Ndufc2 em2Mcwi showed significantly lower amount of Ndufc2 mRNA and protein expression, as compared to both parental SHRSR and SHR‐Ndufc2 em1Mcwi. Δ P<0.01 versus SHRSR; *P<0.0001 versus all other samples (n=6 for each group). D through G, Representative western blot of BNG (n=3) showing defective complex I assembly only in heterozygous SHR‐Ndufc2 em2Mcwi rats. Corresponding densitometric analyses are shown on the right side of each blot. *P<0.0001 versus all other samples. H, Complex I activity and (I) ATP levels were comparable among all strains at this age (n=3). Values are expressed as means±SD. Ctrl indicates control; het, heterozygous; Hsp60, heat shock protein 60; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR; wt, wild type.
JD feeding was started at 6 weeks of age and maintained for 3 months. Systolic blood pressure (SBP), body weight (BW), and proteinuria levels during the dietary treatment are reported in Table 2. SBP levels were comparable among parentals, heterozygous SHR‐Ndufc2 em1Mcwi and SHR‐Ndufc2 em2Mcwi, and their wild‐type controls. Baseline BW was lower in SHRSP (P<0.001; n=18), as compared to all other lines. Levels of 24‐hour proteinuria upon JD feeding increased dramatically in SHRSP and significantly in heterozygous SHR‐Ndufc2 em2Mcwi, although to a lesser extent as compared to SHRSP (for all significance values of comparisons at different experimental time points and between strains, see Table 2).
Table 2.
SBP, BW, and Proteinuria Levels in Parental and Genetically Manipulated Rat Lines Upon JD Feeding
| BW (g) | SBP (mm Hg) | Proteinuria (mg/24 h) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 Weeks | 7 Weeks | 8 Weeks | 12 Weeks | 4 Weeks | 7 Weeks | 8 Weeks | 12 Weeks | 4 Weeks | 7 Weeks | 8 Weeks | 12 Weeks | |
| SHRSR | 220±13 (n=18) | — | 260±16 (n=18) | 289±8a (n=18) | 180±4 (n=18) | — | 189±6 (n=18) | 188±4 (n=18) | 33±5.5 (n=18) | — | 68±14 (n=18) | 85±16a (n=18) |
| SHRSP | 177±6b (n=18) | 192±6c (n=5) | — | — | 181±14 (n=18) | 193±14 (n=5) | — | — | 188±12b (n=18) | 240±30c (n=5) | — | — |
| SHR Ndufc2 em1Mcwi w.t. | 235±15 (n=10) | — | 287±12 (n=10) | 309±27a (n=10 | 177±7 (n=10) | — | 183±2 (n=10) | 188±2 (n=10) | 27±12 (n=10) | — | 56±16 (n=10) | 67±6a (n=10) |
| SHR Ndufc2 em1Mcwi hetero | 235±14 (n=17) | — | 281±18 (n=17) | 308±21a (n=17) | 174±6 (n=17) | — | 178±6 (n=17) | 182±7 (n=17) | 24±7 (n=17) | — | 43±6 (n=17) | 71±19a (n=17) |
| SHR Ndufc2 em2Mcwi w.t. | 239±13 (n=12) | — | 299±16 (n=12) | 314±49a (n=12) | 181±3 (n=12) | — | 181±9 (n=12) | 190±3 (n=12) | 31±5 (n=12) | — | 47±26 (n=12) | 72±22 ∆ (n=12) |
| SHR Ndufc2 em2Mcwi hetero | 237±27 (n=26) | — | 293±30 (n=24) | 312±36a (n=16 | 177±7 (n=26) | — | 189±2 (n=24) | 186±6 (n=16) | 84±35d (n=26) | — | 146±58e, f (n=24) | 126±27 (n=16) |
SHRSP survived up to 7 weeks of JD feeding. BW indicates body weight; JD, Japanese‐style stroke‐permissive diet; SBP, systolic blood pressure; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR.
P<0.0001 for 12 weeks vs 4 weeks for each strain.
P<0.001 vs other strains at 4 weeks.
P<0.0001 for SHRSP/7 weeks vs SHRSP/4 weeks.
P<0.05 for heterozygous M2/4 weeks vs wild‐type M2, heterozygous, and wild‐type M1/4 weeks.
P<0.0001 for heterozygous M2/8 weeks vs all other strains/8 weeks.
P<0.05 for heterozygous M2/8 weeks vs heterozygous M2/4 weeks.
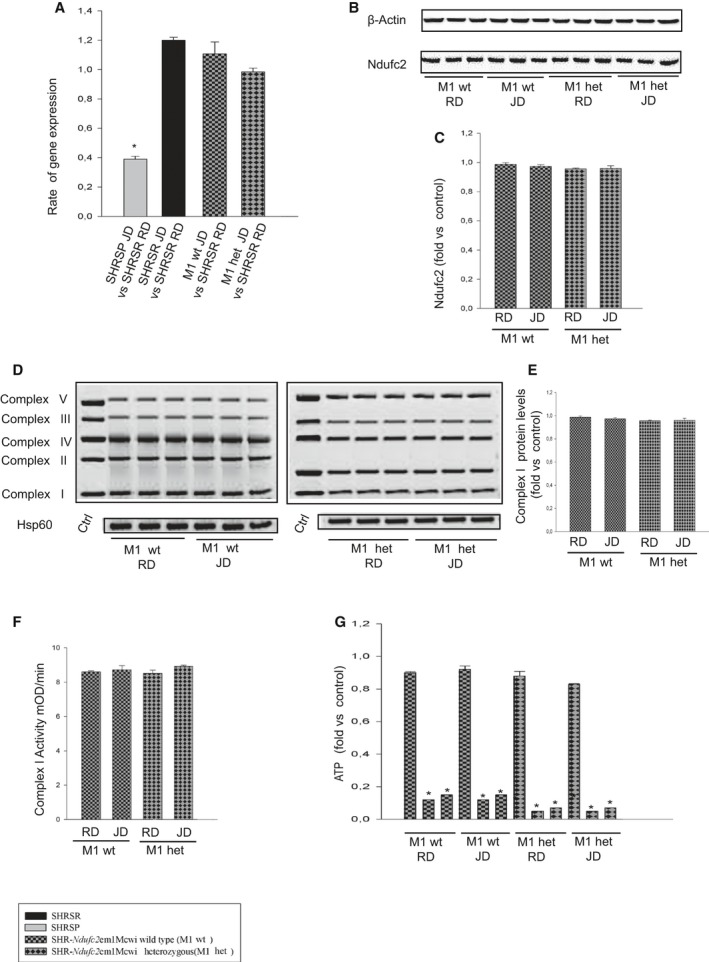
Figure 6 shows results obtained after 4 weeks of either RD or JD feeding in heterozygous and wild‐type SHR‐Ndufc2 em2Mcwi. At the end of 4 weeks of RD, Ndufc2 mRNA was significantly decreased in heterozygous SHR‐Ndufc2 em2Mcwi as compared to age‐matched SHRSR (P<0.0001; n=5; Figure 6A). JD further reduced Ndufc2 mRNA (P<0.0001; n=5; Figure 6A). Ndufc2 protein expression level was consistent with gene expression data (P<0.0001; n=3; Figure 6B and 6C). Complex I assembly was significantly reduced in both RD‐ and JD‐fed heterozygous SHR‐Ndufc2 em2Mcwi (n=3; P<0.0001; Figure 6D and 6E). Complex 1 activity (P<0.0001; n=5), mitochondrial membrane potential (P<0.0001; n=5), and ATP levels (P<0.0001; n=5) were markedly decreased only in JD‐fed heterozygous SHR‐Ndufc2 em2Mcwi (Figure 6F through 6H). Oxidative stress was also higher in heterozygous, as compared to wild‐type, SHR‐Ndufc2 em2Mcwi and to parental SHRSR (P<0.0001; n=3; Figure 7). Nf‐kB protein expression levels were higher (P<0.01; n=3), whereas those of SOD2 were lower (P<0.0001; n=3) in heterozygous SHR‐Ndufc2 em2Mcwi as compared to SHRSR, similarly to SHRSP (Figure 8). In contrast, all parameters of mitochondrial function and oxidative stress were unchanged in both wild‐type and heterozygous SHR‐Ndufc2 em1Mcwi (Figures 9 and 10).
Figure 6.
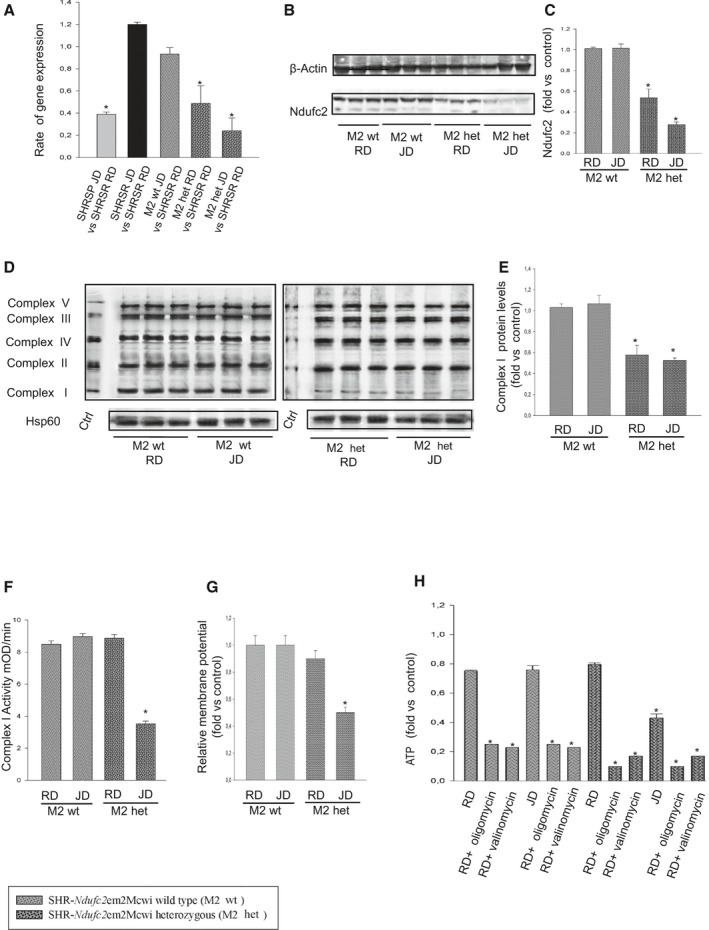
Molecular analyses in brains of both wild‐type and heterozygous SHR ‐Ndufc2 em2Mcwi lines at the end of 4 weeks exposure to either RD or JD. A, Brain Ndufc2 expression in both wild‐type and heterozygous SHR‐Ndufc2em2Mcwi is compared to that of RD‐fed parental SHRSR; *P<0.0001 for each indicated ratio (n=5). B, Ndufc2 protein expression levels in brains of wild‐type and heterozygous SHR‐Ndufc2 em2Mcwi (number of western blots=3) with corresponding densitometric analysis (C); *P<0.0001 for heterozygous SHR‐Ndufc2 em2Mcwi upon RD and JD versus wild type. D and E, Representative western blots of BNG in wild‐type and heterozygous SHR‐Ndufc2 em2Mcwi (n=3 each) with corresponding densitometric analysis. *P<0.0001 for heterozygous SHR‐Ndufc2 em2Mcwi upon RD and JD versus wild type. F, Complex I activity measurement; (G) mitochondrial membrane potential assessment; *P<0.0001 for heterozygous SHR‐Ndufc2 em2Mcwi upon JD versus RD and versus wild‐type line at both RD and JD (n=5 each). H, ATP levels measurement in both wild‐type and heterozygous rat lines upon RD or JD (n=5 each). *P<0.0001 for heterozygous SHR‐Ndufc2 em2Mcwi versus wild‐type line and for each inhibited point versus the not inhibited corresponding sample. Values are expressed as means±SD. BNG indicates Blue Native–PAGE; Ctrl, control; het, heterozygous; Hsp60, heat shock protein 60; JD, Japanese‐style stroke‐permissive diet; RD, geular diet; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR; wt, wild type.
Figure 7.
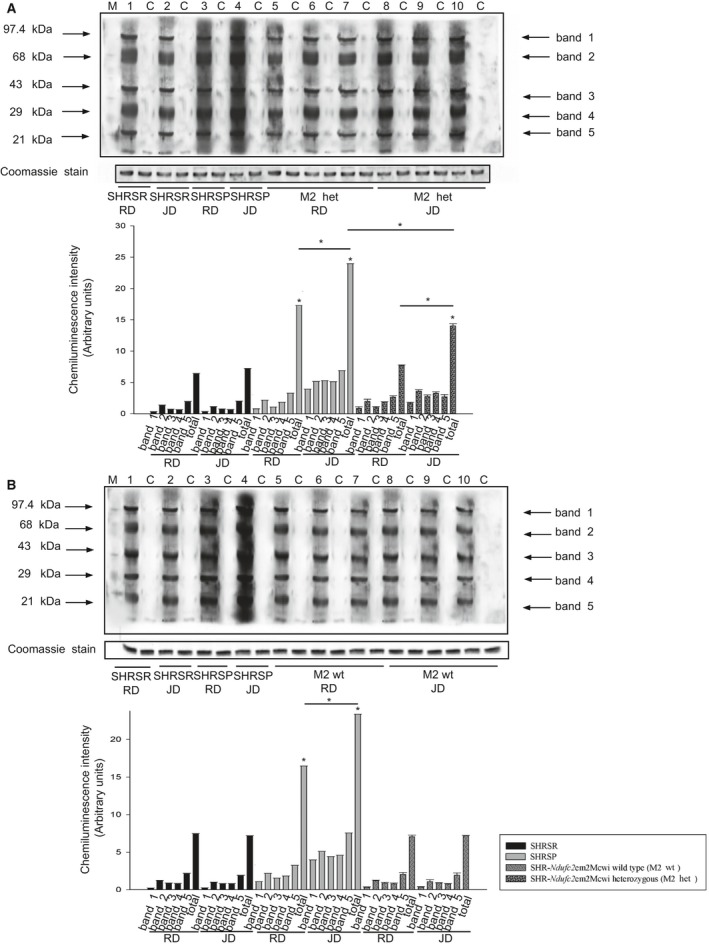
Detection of oxidative stress in brains of SHR ‐Ndufc2 em2Mcwi under either RD or JD. A, Detection of carbonylated proteins in brains of heterozygous SHR‐Ndufc2 em2Mcwi at the end of 4 weeks of either RD or JD feeding with corresponding densitometric analysis (graph below). *P<0.0001 for SHRSP versus RD and for heterozygous SHR‐Ndufc2 em2Mcwi JD versus RD, as well as versus SHRSP JD. B, Detection of carbonylated proteins in brains of wild‐type SHR‐Ndufc2 em2Mcwi at the end of 4 weeks of either RD or JD feeding with corresponding densitometric analysis (graph below); *P<0.0001 for SHRSP JD versus RD and for both versus all other strains (total band). Het indicates heterozygous; JD, Japanese‐style stroke‐permissive diet; RD, geular diet; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR; wt, wild type.
Figure 8.
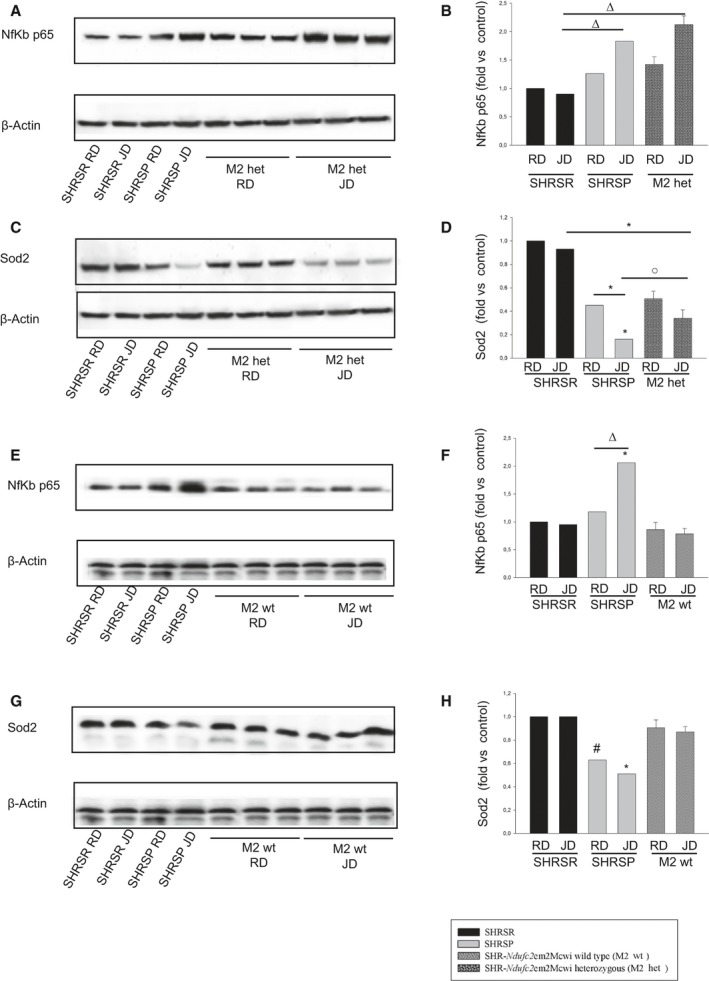
Molecular analyses in brains of wild‐type and heterozygous SHR ‐Ndufc2 em2Mcwi at the end of either RD or JD exposure. A through D, Representative western blots of Nf‐kBp65 and of SOD2 (n=3) with corresponding densitometric analysis in brains of heterozygous SHR‐Ndufc2 em2Mcwi, as compared to parental, lines at the end of 4 weeks of either RD or JD feeding. E through H, Same analyses (n=3) were performed in brains of wild‐type SHR‐Ndufc2 em2Mcwi. Densitometric values are expressed as means±SD. Significance values: (B) Δ P<0.01 for SHRSP JD and heterozygous SHR‐Ndufc2 em2Mcwi JD versus SHRSR JD. C, *P<0.0001 for heterozygous SHR‐Ndufc2 em2Mcwi JD versus SHRSR JD; for SHRSP JD versus SHRSP RD and versus all other samples; ο P<0.05 for SHRSP JD versus heterozygous SHR‐Ndufc2 em2Mcwi JD. F, *P<0.0001 for SHRSP JD versus all other samples; Δ P<0.01 for SHRSP JD versus SHRSP RD. H, # P<0.001 and *P<0.0001 for SHRSP JD versus all other samples. Het indicates heterozygous; JD, Japanese‐style stroke‐permissive diet; Nf‐κB, nuclear factor kappa B; RD, geular diet; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR; Sod2, superoxide dismutase 2, mitochondrial; wt, wild type.
Figure 9.
Molecular analyses in the brains of wild‐type and heterozygous SHR ‐Ndufc2 em1Mcwi at the end of either RD or JD exposure. A, Ndufc2 expression levels as assessed by RT‐PCR. *P<0.0001 for the indicated ratio (n=4 each). B, Representative western blot of Ndufc2 in the 2 lines upon either RD or JD with corresponding densitometric analysis (C), number of western blots=3 for each line. D, Representative western blots of BNG (n=3) with corresponding densitometric analysis (E). (F) Complex I activity assessment (n=3 for each group); (G) ATP levels measurement (n=3 for each group). Densitometric values are expressed as means±SD. Ctrl indicates control; het, heterozygous; Hsp60, heat shock protein 60; JD, Japanese‐style stroke‐permissive diet; RD, geular diet; RT‐PCR, reverse‐transcriptase polymerase chain reaction; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR; wt, wild type.
Figure 10.
Detection of oxidative stress in the brains of wild‐type and heterozygous SHR ‐Ndufc2 em1Mcwi at the end of either RD or JD exposure. A, Detection of carbonylated proteins using the Oxyblot Protein Oxidation Detection kit (n=3; Millipore, Milan, Italy) in brains of heterozygous SHR‐Ndufc2 em1Mcwi versus parental lines at the end of 4 weeks of exposure to either RD or JD. B, Corresponding densitometric analysis with values shown as means±SD. *P<0.0001 for SHRSP JD versus SHRSP RD and for both versus all other samples. Densitometric values are expressed as means±SD. C, Detection of carbonylated proteins using the Oxyblot Protein Oxidation Detection kit (n=3) in brains of wild‐type SHR‐Ndufc2 em1Mcwi versus parental lines at the end of 4 weeks of exposure to either RD or JD. D, Corresponding densitometric analysis with values shown as means±SD. *P<0.0001 for SHRSP JD versus SHRSP RD and for both versus all other samples. Het indicates heterozygous; JD, Japanese‐style stroke‐permissive diet; RD, geular diet; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR; wt, wild type.
Most important, occurrence of stroke reached 100% in SHRSP by 7 weeks of JD and 40% in heterozygous SHR‐Ndufc2 em2Mcwi by 3 months of JD feeding. In contrast, stroke did not occur in all other lines (Figure 11A). Comparison of heterozygous SHR‐Ndufc2 em2Mcwi versus SHRSR and all SHRSR‐derived lines was significantly different (P<0.002). Comparison of heterozygous SHR‐Ndufc2 em2Mcwi versus SHRSP was significantly different (P<0.001).
Figure 11.
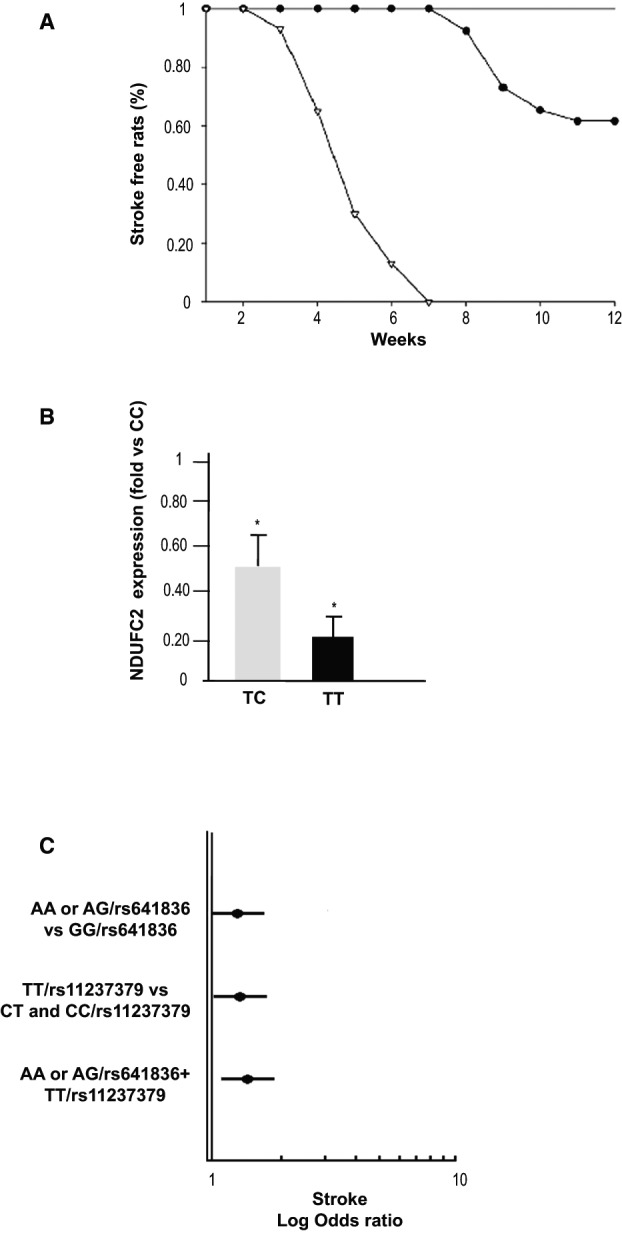
Ndufc2‐related stroke phenotype in both animal models and in humans. A, Stroke occurrence in heterozygous SHR‐Ndufc2 em2Mcwi line as compared to SHRSP and to all other lines (SHRSR, wild‐type SHR‐Ndufc2 em2Mcwi, heterozygous, and wild‐type SHR‐Ndufc2 em1Mcwi). Comparison of heterozygous SHR‐Ndufc2 em2Mcwi versus SHRSR and all SHRSR‐derived lines was significantly different (P<0.002). Comparison of heterozygous SHR‐Ndufc2 em2Mcwi versus SHRSP was significantly different (P<0.001). For numbers of animals included in these studies, see Table 1. Symbols: open triangles=SHRSP; closed circles=heterozygous SHR‐Ndufc2 em2Mcwi; straight line includes: SHRSR; wild‐type and heterozygous SHR‐Ndufc2 em1Mcwi, wild‐type SHR‐Ndufc2 em2Mcwi. B, NDUFC2 expression assessed by RT‐PCR in peripheral blood lymphocytes in a cohort of young healthy subjects carrying wild‐type (n=6), heterozygous (n=12), and double‐mutant (n=6) genotypes for NDUFC2/rs11237379. *P<0.0001 for both CT and TT versus CC wild‐type genotype (t test analysis). Values in the figure are expressed as means±SD. C, Plot showing the logarithmic ORs for stroke in A allele carriers at rs641836, TT genotype carriers at rs11237379, and A/rs641836+TT/rs11237379 carriers with respect to the other genotypes of NDUFC2 tagSNPs (at multivariable logistic regression analysis). OD indicates odds ratio; RT‐PCR, reverse‐transcriptase polymerase chain reaction; SHRSP, stroke‐prone spontaneously hypertensive rat; SHRSR, stroke‐resistant SHR.
Analysis of Human Cohort
In the attempt to translate our findings to a clinical setting, we evaluated allelic and genotypic distributions of 3 NDUFC2 tagSNPs in 484 unrelated Caucasian young adults affected by ischemic stroke and in 1165 control subjects. Table 3 shows demographic and clinical characteristics of the study subjects. No statistically significant differences were observed in age and sex between patients and controls. Instead, we observed significant differences in the prevalence of traditional cardiovascular risk factors.
Table 3.
Demographic and Clinical Characteristics of Patients With Early‐Onset Ischemic Stroke and of Related Controls
| Controls (N=1165) | Stroke Patients (N=484) | P Value | |
|---|---|---|---|
| Age median, IQR | 42 (32–52) | 44 (35–52) | 0.078 |
| Sex (male), N (%) | 522 (44.8) | 222 (45.9) | 0.668 |
| Smoking habit, N (%) | 297 (25.5) | 193 (39.9) | <0.0001 |
| Diabetes, N (%) | 4 (0.3) | 13 (2.7) | <0.0001 |
| Hypertension, N (%) | 85 (7.3) | 140 (28.9) | <0.0001 |
| Dyslipidemia, N (%) | 39 (3.3) | 106 (21.9) | <0.0001 |
IQR indicates interquartile range.
Table 4 shows both genotype distributions and allele frequencies in patients and controls of the 3 studied NDUFC2 SNPs (rs584981, rs641836, and rs11237379). Genotype distributions respected HWE in patients and controls. Prevalence of TT homozygous subjects for rs11237379 SNP was significantly higher in patients than in controls (P=0.017; Table 4). The T allele conferred increased occurrence of early‐onset ischemic stroke by recessive mode of transmission (OR, 1.39; CI, 1.07–1.80; P=0.012). Remarkably, NDUFC2 expression in peripheral blood lymphocytes was significantly and progressively lower in TC/rs11237379 (n=12) and in TT/rs11237379 (n=6) versus CC/rs11237379 (n=6) subjects (P<0.0001 for both CT and TT vs CC; Figure 11B).
Table 4.
Genotypes and Alleles Frequency Distribution for rs584981, rs641836, and rs11237379 NDUFC2 Polymorphisms in Early‐Onset Ischemic Stroke Patients and in Controls
| SNP | Controls (n=1165) n (%) | Stroke Patients (n=484) n (%) | P Value |
|---|---|---|---|
| rs584981 | |||
| TT | 696 (59.7) | 294 (60.7) | 0.846a |
| TC | 414 (35.5) | 170 (35.1) | 0.705b |
| CC | 55 (4.7) | 20 (4.1) | 0.601c |
| C frequency | 0.225 | 0.217 | |
| H‐W | 0.450 | 0.594 | |
| rs641836 | |||
| GG | 799 (68.6) | 309 (63.8) | 0.167a |
| GA | 321 (27.6) | 155 (32.0) | 0.062b |
| AA | 45 (3.9) | 20 (4.1) | 0.798c |
| A frequency | 0.176 | 0.201 | |
| H‐W | 2.752 | 0.010 | |
| rs11237379 | |||
| CC | 251 (21.5) | 94 (19.4) | 0.058a |
| CT | 590 (50.6) | 227 (46.9) | 0.334b |
| TT | 324 (27.8) | 163 (33.7) | 0.017c |
| C frequency | 0.469 | 0.429 | |
| H‐W | 0.332 | 0.874 | |
H‐W indicates Hardy‐Weinberg; SNP, single‐nucleotide polymorphism.
According to the additive model.
According to the dominant model.
According to the recessive model.
Even if the difference did not reach statistical significance (P=0.062), carriers of the A allele (AA homozygotes and GA heterozygotes) at the rs641836 SNP were more prevalent in patients than in controls (Table 4). We did not observe any difference between patients and controls with regard to the genotype distribution of the rs584981 SNP (Table 4).
Multivariable logistic regression analysis, with early‐onset ischemic stroke as the dependent variable and SNPs and traditional cardiovascular risk factors as independent variables, demonstrated that carrier status of the A allele at rs641836 SNP or of TT genotype at rs11237379 SNP was associated with increased risk of stroke (OR=1.32 [95% CI 1.03–1.70]; P=0.029 and OR=1.39 [95% CI 1.07–1.79]; P=0.013, respectively).
At multivariable logistic regression analysis, patients with the combined presence of A rs641836 allele and TT/rs11237379 genotype had the highest early‐onset ischemic stroke risk (OR=1.57 [95% CI 1.15–2.14]; P=0.004; Figure 11C).
Finally, we observed that the combined presence of A rs641836 allele and TT/rs11237379 genotype was significantly higher in large artery TOAST subtype patients (32.4%) and cardioembolic TOAST subtype patients (40%) with respect to small artery (16%), other cause (19.6%), and undetermined cause (16.0%) TOAST subtypes (P=0.003; Figure 12).
Figure 12.
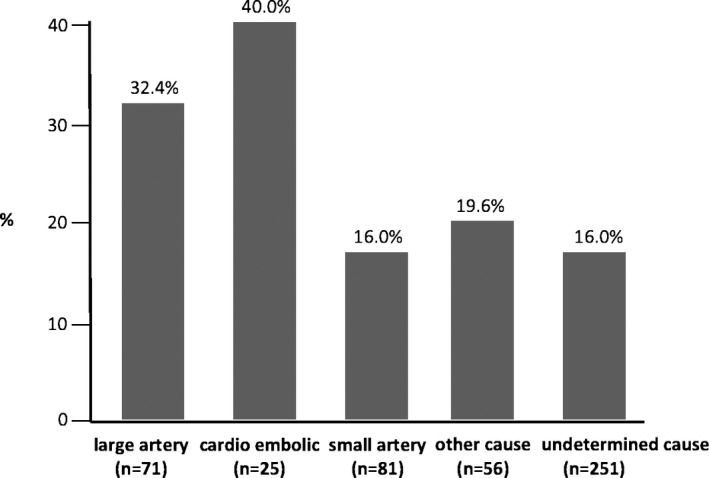
Distribution of the genetic risk condition, derived from combined presence of A/rs641836 allele+TT/rs11237379 genotype for NDUFC2, according to TOAST classification, in a case‐control cohort of Italian early‐onset ischemic strokes. Prevalence was statistically significantly different (overall P=0.003). TOAST indicates Trial of ORG 10172 in Acute Stroke Treatment.
Discussion
In this study, we demonstrate that a subunit of NADH dehydrogenase (NADH dehydrogenase [ubiquinone] 1, subcomplex unknown, 2 [Ndufc2]), located 8 Mb apart from the Lod score peak of a stroke/QTL on rat chromosome 1, is significantly suppressed by stroke‐permissive diet only in brains of SHRSP.
A constellation of cellular abnormalities, including altered complex I assembly and activity, reduced ATP production, increased inflammation, and oxidative stress, was found in brains of SHRSP upon stroke‐permissive diet. We demonstrated that Ndufc2 down‐regulation is sufficient to induce several cellular abnormalities and it predisposes to increased occurrence of stroke. By performing in vitro Ndufc2 silencing in a murine vascular cell line, we provided evidence that Ndufc2 is a fundamental component of NADH dehydrogenase to allow regular complex I assembly and activity. In fact, its down‐regulation led to decreased complex I integrity and function, decreased mitochondrial membrane potential and ATP production, enhanced ROS accumulation, and inflammation. This is consistent with reduced NADH oxidation, which is mediated primarily by complex I. As a consequence, cell viability decreased whereas necrosis significantly increased.
A novel animal model, derived from parental SHRSR, carrying heterozygous deletion of Ndufc2, showed increased predisposition to develop both renal and cerebrovascular damage. Hence, we found that brain Ndufc2 transcription decreased in the presence of 107‐bp deletion, as documented by gene expression already at 6 weeks of age. Upon JD, proteinuria preceded stroke occurrence similarly to what happens in SHRSP.31 Notably, JD further reduced brain Ndufc2 expression in this strain and decreased both complex I activity and ATP levels. Interestingly, RD feeding did not compromise complex I activity, despite significantly lower complex I assembly. This evidence suggests that, despite altered complex I assembly in the presence of reduced Ndufc2 expression, some intrinsic mitochondrial mechanisms can maintain complex I activity and regular ATP levels under RD.32 In addition, this evidence suggest that a high‐salt dietary regimen is critical for stroke development in the presence of Ndfuc2 deletion.
Notably, we did not detect any Ndufc2 mutations in SHRSP, thus suggesting that either epigenetic mechanisms (such as miRNA differential expression) or other gene mutations within the STR1 region may be responsible for Ndufc2 down‐regulation under JD.
Mitochondrion is a major source of energy for cells and its function is fundamental for life of all organisms.33 NADH dehydrogenase plays important functions in the inner mitochondrial membrane. In fact, it belongs to the oxidative phosphorylation process involved in energy production.34 Electrons are donated from NADH to complex I and then to other components of the chain in order to reduce molecular oxygen (O2) to form H2O. Flow of electrons through the OXPHOS chain is accompanied by pumping of protons across the mitochondrial inner membrane, thus creating a transmembrane electrochemical gradient. Complex I leaks electrons in the intermembrane space to generate superoxide anion (O2 −). Re‐entry of protons into the matrix is used by complex V to synthetize ATP from ADP.34
Complex I is considered the major ROS‐generating site within mitochondria. Subunits of complex I are encoded by both nuclear and mitochondria DNA. Mutations in complex I genes lead to increased mitochondrial ROS production.33, 34, 35 Importantly, the pathological relevance of NDUFC2 has been recently explored with regard to type 2 diabetes mellitus, providing mechanistic results that are strongly consistent with ours.36 NDUFC2 appears down‐regulated in skeletal muscle cells of subjects affected by insulin resistance37 and is associated with insulin secretion in vivo.38 Furthermore, NDUFC2 may represent a novel oncogene involved in breast cancer and intestinal adenocarcinoma, predicting poor prognosis.39, 40 Notably, the pathological relevance of NDUFC2/rs11237379 has been also highlighted through genome‐wide association studies in pulmonary disease.41, 42
Although it is plausible that mitochondrial dysfunction in general and, particularly, dysfunction of complex I may predispose to cardiovascular diseases and events, no information is available yet with regard to the relationship between Ndufc2 and cardiovascular disease risk. To our knowledge, the present study is the first demonstration that reduced transcription of Ndufc2 contributes to stroke.
In summary, Ndufc2, encoding a subunit of OXPHOS complex I, mapping close to the load score peak of a stroke QTL in SHRSP, exerts a key role on stroke susceptibility in this model. A SHRSR‐derived line carrying heterozygosity for Ndufc2 deletion showed increased renal and cerebrovascular phenotypes compared to the resistant strain of origin. The relevance of these experimental findings was extended to a human cohort of early‐onset ischemic stroke cases. This population was previously investigated for polymorphisms of candidate genes encoding procoagulant and inflammatory factors and methionine metabolism components: for a polymorphism of type C natriuretic peptide receptor (NPR3) promoter gene and for polymorphisms of chromosome 12p13.25, 26, 27, 28 In this population, an SNP study revealed a significant association between an intronic marker (rs11237379) of NDUFC2 and early‐onset ischemic stroke. Interestingly, a functional significance of rs11237379 was documented by its direct relationship with gene expression level, suggesting that reduced NDUFC2 expression may predispose to stroke susceptibility also in humans. Finally, its pathogenic relevance was enhanced by concomitant presence of a second allele variant at rs641836. Both SNPs may be in linkage disequilibrium with still unknown variants in the regulatory or coding regions of the gene. The different distribution of the genetic risk condition, derived from the combined presence of A rs641836 allele and TT/rs11237379 genotype, observed across TOAST stroke subtypes may suggest a prominent role of NDUFC2 in the atherosclerotic and prothrombotic mechanisms underlying stroke.
We are aware that the extension of the experimental results to the human stroke genetic analysis presented in the current work should be considered hypothesis generating rather than definitive, attributable to intrinsic study limitations. In particular, lack of an extensive genotyping for ancestry informative markers in both the autosomal and mitochondrial genomes of our human subjects did not allow a correct population stratification. Additional studies are certainly required to test the hypothesis generated by our experimental data that Ndufc2 may play a contributory role to ischemic stroke predisposition in humans.
Altogether, our novel findings, while pointing to mitochondrial dysfunction as a cause of vascular disease, provide new important insights on the genetic basis of stroke and may have great relevance for prevention and treatment of this common pathological condition.
Sources of Funding
The present work was supported by a grant (Ricerca Corrente) from the Italian Ministry of Health to Volpe and Rubattu; by the 5‰ grant to Volpe and Rubattu; by the Award grant from the Sapienza University to Volpe; and by National Institutes of Health grants RC2 HL101681 (Dwinell) and DP2‐OD008396 (Geurts).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e002701 doi: 10.1161/JAHA.115.002701)
References
- 1. Rubattu S, Gigante B, Stanzione R, De Paolis P, Tarasi D, Volpe M. In the search for stroke genes: a long and winding road. Am J Hypertens. 2004;17:197–202. [DOI] [PubMed] [Google Scholar]
- 2. Lindgren A. Stroke genetics: a review and update. J Stroke. 2014;16:114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okamoto K, Yamori Y, Nagaoka A. Establishment of the stroke prone spontaneously hypertensive rat (SHR). Circ Res. 33/34: I143–I153. [Google Scholar]
- 4. Bailey EL, Smith C, Sudlow CLM, Wardlaw JM. Is the spontaneously hypertensive stroke prone rat a pertinent model of subcortical ischemic stroke? A systematic review Int J Stroke. 2011;6:434–444. [DOI] [PubMed] [Google Scholar]
- 5. Rubattu S, Volpe M, Kreutz R, Ganten U, Ganten D, Lindpaintner K. Chromosomal mapping of genetic loci contributing to stroke in an animal model of a complex human disease. Nat Genet. 1996;13:429–434. [DOI] [PubMed] [Google Scholar]
- 6. Rubattu S, Hubner N, Ganten U, Evangelista A, Stanzione R, Di Angelantonio E, Plehm R, Langanki R, Gianazza E, Sironi L, D'Amati G, Volpe M. Reciprocal congenic lines for a major stroke‐QTL on rat chromosome 1. Physiol Genomics. 2006;27:108–113. [DOI] [PubMed] [Google Scholar]
- 7. Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, Al‐Majali KM, Trembling PM, Mann CJ, Shoulders CC, Graf D, St Lezin E, Kurtz TW, Kren V, Pravenec M, Ibrahimi A, Abumrad NA, Stanton LW, Scott J. Identification of Cd36 (Fat) as an insulin‐resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. [DOI] [PubMed] [Google Scholar]
- 8. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix Gene Chip probe level data. Nucleic Acids Res. 2003;31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, Maciver F, Mueller M, Hummel O, Monti J, Zidek V, Musilova A, Kren V, Causton H, Game L, Born G, Schmidt S, Müller A, Cook SA, Kurtz TW, Whittaker J, Pravenec M, Aitman TJ. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet. 2005;37:243–253. [DOI] [PubMed] [Google Scholar]
- 10. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series. 1995;57:289–300. [Google Scholar]
- 11. Davies DL, Bouldin DW. A cluster separation measure. IEEE Trans Pattern Anal Mach Intell. 1979;1:224–227. [PubMed] [Google Scholar]
- 12. Atanur SS, Diaz AG, Maratou K, Sarkis A, Rotival M, Game L, Tschannen MR, Kaisaki PJ, Otto GW, Ma MC, Keane TM, Hummel O, Saar K, Chen W, Guryev V, Gopalakrishnan K, Garrett MR, Joe B, Citterio L, Bianchi G, McBride M, Dominiczak A, Adams DJ, Serikawa T, Flicek P, Cuppen E, Hubner N, Petretto E, Gauguier D, Kwitek A, Jacob H, Aitman TJ. Genome sequencing reveals loci under artificial selection that underlie disease phenotypes in the laboratory rat. Cell. 2013;154:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan‐Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera V, Holt RA, Adams MD, Amanatides PG, Baden‐Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C, Glodek A, Gu Z, Jennings D, Kraft CL, Nguyen T, Pfannkoch CM, Sitter C, Sutton GG, Venter JC, Woodage T, Smith D, Lee HM, Gustafson E, Cahill P, Kana A, Doucette‐Stamm L, Weinstock K, Fechtel K, Weiss RB, Dunn DM, Green ED, Blakesley RW, Bouffard GG, De Jong PJ, Osoegawa K, Zhu B, Marra M, Schein J, Bosdet I, Fjell C, Jones S, Krzywinski M, Mathewson C, Siddiqui A, Wye N, McPherson J, Zhao S, Fraser CM, Shetty J, Shatsman S, Geer K, Chen Y, Abramzon S, Nierman WC, Havlak PH, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Li B, Liu Y, Qin X, Cawley S, Worley KC, Cooney AJ, D'Souza LM, Martin K, Wu JQ, Gonzalez‐Garay ML, Jackson AR, Kalafus KJ, McLeod MP, Milosavljevic A, Virk D, Volkov A, Wheeler DA, Zhang Z, Bailey JA, Eichler EE, Tuzun E, Birney E, Mongin E, Ureta‐Vidal A, Woodwark C, Zdobnov E, Bork P, Suyama M, Torrents D, Alexandersson M, Trask BJ, Young JM, Huang H, Wang H, Xing H, Daniels S, Gietzen D, Schmidt J, Stevens K, Vitt U, Wingrove J, Camara F, Mar Albà M, Abril JF, Guigo R, Smit A, Dubchak I, Rubin EM, Couronne O, Poliakov A, Hübner N, Ganten D, Goesele C, Hummel O, Kreitler T, Lee YA, Monti J, Schulz H, Zimdahl H, Himmelbauer H, Lehrach H, Jacob HJ, Bromberg S, Gullings‐Handley J, Jensen‐Seaman MI, Kwitek AE, Lazar J, Pasko D, Tonellato PJ, Twigger S, Ponting CP, Duarte JM, Rice S, Goodstadt L, Beatson SA, Emes RD, Winter EE, Webber C, Brandt P, Nyakatura G, Adetobi M, Chiaromonte F, Elnitski L, Eswara P, Hardison RC, Hou M, Kolbe D, Makova K, Miller W, Nekrutenko A, Riemer C, Schwartz S, Taylor J, Yang S, Zhang Y, Lindpaintner K, Andrews TD, Caccamo M, Clamp M, Clarke L, Curwen V, Durbin R, Eyras E, Searle SM, Cooper GM, Batzoglou S, Brudno M, Sidow A, Stone EA, Venter JC, Payseur BA, Bourque G, López‐Otín C, Puente XS, Chakrabarti K, Chatterji S, Dewey C, Pachter L, Bray N, Yap VB, Caspi A, Tesler G, Pevzner PA, Haussler D, Roskin KM, Baertsch R, Clawson H, Furey TS, Hinrichs AS, Karolchik D, Kent WJ, Rosenbloom KR, Trumbower H, Weirauch M, Cooper DN, Stenson PD, Ma B, Brent M, Arumugam M, Shteynberg D, Copley RR, Taylor MS, Riethman H, Mudunuri U, Peterson J, Guyer M, Felsenfeld A, Old S, Mockrin S, Collins F; Rat Genome Sequencing Project Consortium . Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. [DOI] [PubMed] [Google Scholar]
- 14. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A frame‐work for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di Castro S, Scarpino S, Marchitti S, Bianchi F, Stanzione R, Cotugno M, Sironi L, Gelosa P, Duranti E, Ruco L, Volpe M, Rubattu S. Differential modulation of UCP2 in kidneys of stroke‐prone spontaneously hypertensive rats under high salt/low potassium diet. Hypertension. 2013;61:534–541. [DOI] [PubMed] [Google Scholar]
- 18. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 19. Scarpino S, Marchitti S, Stanzione R, Evangelista A, Di Castro S, Savoia C, Quarta G, Sciarretta S, Ruco L, Volpe M, Rubattu S. ROS‐mediated differential effects of the human atrial natriuretic peptide T2238C genetic variant on endothelial cells in vitro. J Hypertens. 2009;27:1804–1813. [DOI] [PubMed] [Google Scholar]
- 20. Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ. Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blu native PAGE. Electrophoresis. 2007;28:1036–1045. [DOI] [PubMed] [Google Scholar]
- 21. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. [DOI] [PubMed] [Google Scholar]
- 22. Drew B, Leeuwenburgh C. Method for measuring ATP production in isolated mitochondria: ATP production in brain and liver mitochondria of Fischer‐344 rats with age and calorie restriction. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1259–R1267. [DOI] [PubMed] [Google Scholar]
- 23. Geurts AM, Cost GJ, Rémy S, Cui X, Tesson L, Usal C, Ménoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene‐specific mutated rats using zinc‐finger nucleases. Methods Mol Biol. 2010;597:211–225. [DOI] [PubMed] [Google Scholar]
- 24. Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Ménoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc‐finger nucleases. Science. 2009;325:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rubattu S, Ferrari M, Evangelista A, Beccia M, Stanzione R, Egidy Assenza G, Volpe M, Rasura M. A role of TNF‐α gene variant on juvenile ischemic stroke: a case‐control study. Eur J Neurol. 2005;12:989–993. [DOI] [PubMed] [Google Scholar]
- 26. Giusti B, Saracini C, Bolli P, Magi A, Martinelli I, Peyvandi F, Rasura M, Volpe M, Lotta LA, Rubattu S, Mannucci PM, Abbate R. Early‐onset ischaemic stroke: analysis of 58 polymorphisms in 17 genes involved in methioninemetabolism. Thromb Haemost. 2010;104:231–242. [DOI] [PubMed] [Google Scholar]
- 27. Lotta LA, Giusti B, Saracini C, Vestrini A, Volpe M, Rubattu S, Peyvandi F. No association between chromosome 12p13 single nucleotide polymorphisms and early‐onset ischemic stroke. J Thromb Haemost. 2010;8:1858–1860. [DOI] [PubMed] [Google Scholar]
- 28. Rubattu S, Giusti B, Lotta LA, Peyvandi F, Cotugno M, Stanzione R, Marchitti S, Palombella AM, Di Castro S, Rasura M, Mannucci PM, Volpe M. Association of a single nucleotide polymorphism of the NPR3 gene promoter with early onset ischemic stroke in an Italian cohort. Eur J Intern Med. 2013;24:80–82. [DOI] [PubMed] [Google Scholar]
- 29. Frezzato M, Tosetto A, Rodeghiero F. Validated questionnaire for the identification of previous personal or familial venous thromboembolism. Am J Epidemiol. 1996;143:1257–1265. [DOI] [PubMed] [Google Scholar]
- 30. Fuss IJ, Kanof ME, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol. 2009;Chapter 7:Unit7.1. [DOI] [PubMed] [Google Scholar]
- 31. Schreiber S, Bueche CZ, Garz C, Kropf S, Kuester D, Amann K, Heinze H‐J, Goertler M, Reymann KG, Braun H. Kidney pathology precedes and predicts the pathological cascade of cerebrovascular lesions in stroke prone rats. PLoS One. 2011;6:e26287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Franco AA, Odom RS, Rando TA. Regulation of antioxidant enzyme gene expression in response to oxidative stress and during differentiation of mouse skeletal muscle. Free Radic Biol Med. 1999;27:1122–1132. [DOI] [PubMed] [Google Scholar]
- 33. Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta. 2012;1817:851–862. [DOI] [PubMed] [Google Scholar]
- 35. Shapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. [DOI] [PubMed] [Google Scholar]
- 36. Gershoni M, Levin L, Ovadia O, Toiw Y, Shani N, Dadon S, Barzilai N, Bergman A, Atzmon G, Wainstein J, Tsur A, Nijtmans L, Glaser B, Mishmar D. Disrupting mitochondrial‐nuclear coevolution affects OXPHOS complex I integrity and impacts human health. Genome Biol Evol. 2014;6:2665–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dekker Nitert M, Dayeh T, Volkov P, Elgzyri T, Hall E, Nilsson E, Yang BT, Lang S, Parikh H, Wessman Y, Weishaupt H, Attema J, Abels M, Wierup N, Almgren P, Jansson PA, Rönn T, Hansson O, Eriksson KF, Groop L, Ling C. Impact of an exercise intervention on DNA methylation in skeletal muscle from first‐degree relatives of patients with type 2 diabetes. Diabetes. 2012;61:3322–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Olsson AH, Ronn T, Ladenvall C, Parikh H, Isomaa B, Groop L, Ling C. Two common genetic variants near nuclear‐encoded OXPHOS genes are associated with insulin secretion in vivo. Eur J Endocrinol. 2011;164:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chin SF, Teschendorff AE, Marioni JC, Wang Y, Barbosa‐Morais NL, Thorne NP, Costa JL, Pinder SE, van de Wiel MA, Green AR, Ellis IO, Porter PL, Tavaré S, Brenton JD, Ylstra B, Caldas C. High‐resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007;8:R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Igci YZ, Bozgeyik E, Borazan E, Pala E, Suner A, Ulasli M, Gurses SA, Yumrutas O, Balik AA, Igci M. Expression profiling of SCN8A and NDUFC2 genes in colorectal carcinoma. Exp Oncol. 2015;37:77–80. [PubMed] [Google Scholar]
- 41. Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, Zhao JH, Ramasamy A, Zhai G, Vitart V, Huffman JE, Igl W, Albrecht E, Deloukas P, Henderson J, Granell R, McArdle WL, Rudnicka AR; Wellcome Trust Case Control Consortium , Barroso I, Loos RJ, Wareham NJ, Mustelin L, Rantanen T, Surakka I, Imboden M, Wichmann HE, Grkovic I, Jankovic S, Zgaga L, Hartikainen AL, Peltonen L, Gyllensten U, Johansson A, Zaboli G, Campbell H, Wild SH, Wilson JF, Gläser S, Homuth G, Völzke H, Mangino M, Soranzo N, Spector TD, Polasek O, Rudan I, Wright AF, Heliövaara M, Ripatti S, Pouta A, Naluai AT, Olin AC, Torén K, Cooper MN, James AL, Palmer LJ, Hingorani AD, Wannamethee SG, Whincup PH, Smith GD, Ebrahim S, McKeever TM, Pavord ID, MacLeod AK, Morris AD, Porteous DJ, Cooper C, Dennison E, Shaheen S, Karrasch S, Schnabel E, Schulz H, Grallert H, Bouatia‐Naji N, Delplanque J, Froguel P, Blakey JD; NSHD RespiratoryStudy Team , Britton JR, Morris RW, Holloway JW, Lawlor DA, Hui J, Nyberg F, Jarvelin MR, Jackson C, Kähönen M, Kaprio J, Probst‐Hensch NM, Koch B, Hayward C, Evans DM, Elliott P, Strachan DP, Hall IP, Tobin MD. Genome‐wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soler Artigas M, Loth DW, Wain LV, Gharib SA, Obeidat M, Tang W, Zhai G, Zhao JH, Smith AV, Huffman JE, Albrecht E, Jackson CM, Evans DM, Cadby G, Fornage M, Manichaikul A, Lopez LM, Johnson T, Aldrich MC, Aspelund T, Barroso I, Campbell H, Cassano PA, Couper DJ, Eiriksdottir G, Franceschini N, Garcia M, Gieger C, Gislason GK, Grkovic I, Hammond CJ, Hancock DB, Harris TB, Ramasamy A, Heckbert SR, Heliövaara M, Homuth G, Hysi PG, James AL, Jankovic S, Joubert BR, Karrasch S, Klopp N, Koch B, Kritchevsky SB, Launer LJ, Liu Y, Loehr LR, Lohman K, Loos RJ, Lumley T, Al Balushi KA, Ang WQ, Barr RG, Beilby J, Blakey JD, Boban M, Boraska V, Brisman J, Britton JR, Brusselle GG, Cooper C, Curjuric I, Dahgam S, Deary IJ, Ebrahim S, Eijgelsheim M, Francks C, Gaysina D, Granell R, Gu X, Hankinson JL, Hardy R, Harris SE, Henderson J, Henry A, Hingorani AD, Hofman A, Holt PG, Hui J, Hunter ML, Imboden M, Jameson KA, Kerr SM, Kolcic I, Kronenberg F, Liu JZ, Marchini J, McKeever T, Morris AD, Olin AC, Porteous DJ, Postma DS, Rich SS, Ring SM, Rivadeneira F, Rochat T, Sayer AA, Sayers I, Sly PD, Smith GD, Sood A, Starr JM, Uitterlinden AG, Vonk JM, Wannamethee SG, Whincup PH, Wijmenga C, Williams OD, Wong A, Mangino M, Marciante KD, McArdle WL, Meibohm B, Morrison AC, North KE, Omenaas E, Palmer LJ, Pietiläinen KH, Pin I, Pola Sbreve Ek O, Pouta A, Psaty BM, Hartikainen AL, Rantanen T, Ripatti S, Rotter JI, Rudan I, Rudnicka AR, Schulz H, Shin SY, Spector TD, Surakka I, Vitart V, Völzke H, Wareham NJ, Warrington NM, Wichmann HE, Wild SH, Wilk JB, Wjst M, Wright AF, Zgaga L, Zemunik T, Pennell CE, Nyberg F, Kuh D, Holloway JW, Boezen HM, Lawlor DA, Morris RW, Probst‐Hensch N; International Lung Cancer Consortium; GIANT consortium , Kaprio J, Wilson JF, Hayward C, Kähönen M, Heinrich J, Musk AW, Jarvis DL, Gläser S, Järvelin MR, Ch Stricker BH, Elliott P, O'Connor GT, Strachan DP, London SJ, Hall IP, Gudnason V, Tobin MD. Genome‐wide association and large‐scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011;43:1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]