

Abstract
Background
Resolving the short phylogenetic branches that result from rapid evolutionary diversification often requires large numbers of loci. We collected targeted sequence capture data from 585 nuclear loci (541 ultraconserved elements and 44 protein-coding genes) to estimate the phylogenetic relationships among iguanian lizards in the North American genus Sceloporus. We tested for diversification rate shifts to determine if rapid radiation in the genus is correlated with chromosomal evolution.
Results
The phylogenomic trees that we obtained for Sceloporus using concatenation and coalescent-based species tree inference provide strong support for the monophyly and interrelationships among nearly all major groups. The diversification analysis supported one rate shift on the Sceloporus phylogeny approximately 20–25 million years ago that is associated with the doubling of the speciation rate from 0.06 species/million years (Ma) to 0.15 species/Ma. The posterior probability for this rate shift occurring on the branch leading to the Sceloporus species groups exhibiting increased chromosomal diversity is high (posterior probability = 0.997).
Conclusions
Despite high levels of gene tree discordance, we were able to estimate a phylogenomic tree for Sceloporus that solves some of the taxonomic problems caused by previous analyses of fewer loci. The taxonomic changes that we propose using this new phylogenomic tree help clarify the number and composition of the major species groups in the genus. Our study provides new evidence for a putative link between chromosomal evolution and the rapid divergence and radiation of Sceloporus across North America.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-016-0628-x) contains supplementary material, which is available to authorized users.
Keywords: Gene tree, Lizards, Phrynosomatidae, Phylogenomics, Species tree, Systematics
Background
Rapid radiations represent some of the most intriguing and well-studied biological systems. They also present some of the most difficult phylogenetic problems. The short time intervals separating the speciation events that occur during a rapid radiation leave few opportunities for molecular evolutionary changes to become established in the genome. This lack of phylogenetic information typically leads to large-scale gene tree discordance and a lack of resolution for the phylogenetic relationships [1]. Species involved in rapid radiations are typically partitioned into major clades with clear support from multiple sources of data, yet the interrelationships among the major clades are often ambiguous. This basic conundrum repeats itself across the Tree of Life (e.g., the root of life [2, 3], major bird orders [4, 5], Mammals [6, 7], and Neobatrachian frogs [8]). Attempting to resolve rapid radiations using a combination of large numbers of loci together with coalescent-based species tree inference methods [9–14] represents an important new direction in systematic biology this is expected to help resolve difficult phylogenetic problems.
There are at least three fundamental challenges confronting the resolution of rapid radiations using molecular genetic data: 1) quick bursts of speciation limit the opportunities for character changes to accumulate across the genome [1], 2) long-branch attraction artifacts during phylogeny estimation [15], and 3) incomplete lineage sorting [16]. Increasing the number of loci used to estimate the phylogeny can sometimes help alleviate the first problem [17–19]. However, depending on the method and the model, increasing the amount of data can be positively misleading when faced with long branch attraction and/or incomplete lineage sorting [15, 20, 21]. Overcoming these collective challenges, which are not mutually exclusive and are difficult to distinguish, requires the acquisition of large datasets composed of many independent loci together with the implementation of coalescent models of phylogenetic inference; however, analyzing large datasets is computationally demanding, and this problem is amplified when utilizing complex coalescent-based models. Our ability to generate sequence data is quickly outpacing our capacity to analyze genetic data under complex models such as the multispecies coalescent [22]. Coalescent methods that utilize gene trees instead of sequence data can dramatically decrease computation times [23], but this comes at the cost of information loss as uncertainty in the sequence data is not taken into account.
The phrynosomatid lizard genus Sceloporus is a diverse clade containing 90 + species with a broad distribution across North America [24]. Developing a robust phylogenetic framework for comparative studies of Sceloporus has been of interest for decades (reviewed by [24–30]). Previous phylogenetic studies of Sceloporus based on a few nuclear genes suggest that the group has experienced a period of rapid evolutionary diversification [27]. These successive and rapid speciation events have resulted in bursts of speciation that have impeded the inference of a fully-resolved and strongly supported phylogeny [25, 28, 29]. Differentiation in the fundamental number of chromosomes among species and species groups is hypothesized to be a primary factor responsible for driving the rapid radiation of Sceloporus [27, 31]. The genus is comprised of 19 species groups containing anywhere from one species (two of the species groups are monotypic) to 15 species (Table 1). Most of the polytypic species groups have been the focus of detailed phylogeographic and phylogenetic study, including the formosus group [32], grammicus group [33], torquatus and poinsettii groups [34, 35], magister group [36], scalaris group [37], spinosus group [38], undulatus group [39, 40], and the variabilis group [41]. These systematic studies have advanced our knowledge of the interrelationships within many species groups; however, resolving the phylogenetic relationships among the species groups has remained difficult [28, 29].
Table 1.
Specimens included in the study
| Species | Voucher | RAW reads | Clean pairs | idba contig | idba loci |
|---|---|---|---|---|---|
| angustus group (2/2) | |||||
| Sceloporus angustus | LACM 134741 | 3,788,618 | 3,457,044 | 1205 | 579 |
| Sceloporus grandaevus | ROM 26215 | 1,342,322 | 1,134,076 | 700 | 521 |
| clarkii group (2/2) | |||||
| Sceloporus clarkii | MVZ 245876 | 2,844,162 | 2,543,574 | 1223 | 579 |
| Sceloporus melanorhinus | MZFC 7454 | 3,384,030 | 2,971,666 | 1298 | 580 |
| formosus group (11/15) | |||||
| Sceloporus acanthinus | ANMO1932 | 3,734,696 | 3,350,382 | 1971 | 575 |
| Sceloporus adleri | UWBM 6608 | 8,137,738 | 7,364,486 | 4460 | 576 |
| Sceloporus cryptus | MZFC 7438 | 2,350,112 | 2,133,386 | 1036 | 577 |
| Sceloporus druckercolini | JAC 25172 | 2,760,330 | 2,463,818 | 1357 | 559 |
| Sceloporus formosus | RVT 76 | 3,354,056 | 2,960,696 | 1550 | 569 |
| Sceloporus formosus | ANMO 1248 | 2,192,170 | 1,693,102 | 948 | 541 |
| Sceloporus formosus scitulus | UWBM 6623 | 5,476,146 | 4,950,836 | 2243 | 577 |
| Sceloporus internasalis | JAC 22552 | 4,393,164 | 3,792,716 | 2119 | 573 |
| Sceloporus lunaei | not sampled | – | – | – | – |
| Sceloporus lundelli | not sampled | – | – | – | – |
| Sceloporus malachiticus | MVZ 263420 | 5,373,316 | 4,945,178 | 2673 | 578 |
| Sceloporus salvini | not sampled | – | – | – | – |
| Sceloporus smaragdinus | unknown | 1,891,310 | 1,461,336 | 938 | 546 |
| Sceloporus stejnegeri | MZFC 7452 | 3,981,586 | 3,470,518 | 3492 | 383 |
| Sceloporus subpictus | MZFC 8028 | 6,456,814 | 5,737,156 | 5349 | 572 |
| Sceloporus taeniocnemis | MVZ 264322 | 1,892,698 | 1,423,322 | 771 | 247 |
| Sceloporus tanneri | not sampled | – | – | – | – |
| gadoviae group (2/2) | |||||
| Sceloporus gadoviae | UWBM 7309 | 6,519,836 | 5,991,130 | 1835 | 577 |
| Sceloporus maculosus | JAM 650 | 3,140,176 | 2,691,774 | 2339 | 339 |
| graciosus group (3/3) | |||||
| Sceloporus arenicolus | ADL 047 | 2,637,380 | 2,266,994 | 946 | 579 |
| Sceloporus graciosus | MVZ 240898 | 6,241,172 | 5,631,350 | 3413 | 576 |
| Sceloporus vandenburgianus | TWR 430 | 4,961,440 | 4,386,238 | 1435 | 581 |
| grammicus group (6/6) | |||||
| Sceloporus anahuacus | AMH684 | 3,986,550 | 3,219,532 | 1512 | 573 |
| Sceloporus asper | JAC23686 | 7,225,354 | 4,490,778 | 2723 | 573 |
| Sceloporus grammicus | UWBM 6585 | 4,406,008 | 3,910,096 | 2054 | 576 |
| Sceloporus grammicus microlepidotus | UOGV2525 | 1,315,156 | 1,051,322 | 702 | 518 |
| Sceloporus heterolepis | MZFC 8017 | 4,824,734 | 3,952,154 | 4777 | 344 |
| Sceloporus palaciosi | UWBM 7313 | 10,419,276 | 9,501,996 | 2546 | 578 |
| Sceloporus shannonorum | JADE 220 | 5,377,848 | 5,003,792 | 4205 | 563 |
| jalapae group (2/2) | |||||
| Sceloporus jalapae | UWBM 7318 | 4,557,808 | 4,128,130 | 1228 | 575 |
| Sceloporus ochoterenae | UWBM 6641 | 3,716,194 | 3,242,038 | 1449 | 579 |
| magister group (6/6) | |||||
| Sceloporus hunsakeri | ADG 98 | 2,336,356 | 1,724,688 | 1168 | 580 |
| Sceloporus licki | ADG 73 | 5,951,168 | 5,355,664 | 4806 | 554 |
| Sceloporus lineatulus | unknown | 8,289,082 | 7,293,688 | 7954 | 567 |
| Sceloporus magister cephaloflavus | UWBM 7395 | 4,391,974 | 3,903,318 | 1457 | 578 |
| Sceloporus magister bimaculosus | DGM 924 | 1,818,256 | 1,472,176 | 1015 | 549 |
| Sceloporus magister magister | ROM 14488 | 8,938,692 | 8,446,102 | 5308 | 561 |
| Sceloporus magister uniformis | DGM 474 | 11,528,354 | 10,328,510 | 8009 | 574 |
| Sceloporus magister uniformis | MVZ 162077 | 4,856,264 | 3,818,418 | 2286 | 562 |
| Sceloporus orcutti | UWBM 7654 | 5,061,334 | 4,346,088 | 1695 | 579 |
| Sceloporus orcutti | RWM 798 | 3,769,420 | 2,886,214 | 1486 | 564 |
| Sceloporus zosteromus | ADG 49 | 3,256,502 | 2,843,448 | 1274 | 577 |
| Sceloporus zosteromus | ADG 74 | 14,267,908 | 12,650,332 | 5117 | 579 |
| megalepidurus group (2/3) | |||||
| Sceloporus halli | not sampled | – | – | – | – |
| Sceloporus megalepidurus | MZFC 8026 | 4,443,820 | 3,916,852 | 3922 | 546 |
| Sceloporus pictus | LCM 1149 | 3,581,154 | 3,310,488 | 2674 | 576 |
| merriami group (1/1) | |||||
| Sceloporus merriami | LSUMZ 48844 | 3,786,064 | 3,181,240 | 1134 | 578 |
| poinsettii group (10/12) | |||||
| Sceloporus aureolus | RVT 54 | 5,861,080 | 5,350,974 | 2627 | 566 |
| Sceloporus aureolus | JAC 22409 | 5,310,894 | 4,798,000 | 2361 | 568 |
| Sceloporus cyanogenys | FMQ 3250 | 3,588,644 | 3,028,816 | 3126 | 378 |
| Sceloporus cyanostictus | unknown | 2,276,708 | 1,830,554 | 1122 | 549 |
| Sceloporus dugesii | UTAR 23955 | 3,147,022 | 2,445,678 | 1887 | 236 |
| Sceloporus macdougalli | MZFC 7017 | 5,546,916 | 4,884,484 | 3456 | 559 |
| Sceloporus minor | UOGV 1369 | 6,196,168 | 5,486,782 | 3883 | 564 |
| Sceloporus mucronatus | UWBM 6636 | 4,831,958 | 4,464,672 | 2783 | 578 |
| Sceloporus oberon | not sampled | – | – | – | – |
| Sceloporus ornatus | JAM 652 | 3,298,578 | 2,602,280 | 2533 | 311 |
| Sceloporus poinsettii | LSUMZ 48847 | 3,518,050 | 3,088,022 | 3029 | 309 |
| Sceloporus serrifer | UTAR 39870 | 3,255,154 | 2,896,292 | 2649 | 579 |
| Sceloporus sugillatus | not sampled | – | – | – | – |
| pyrocephalus group (2/2) | |||||
| Sceloporus nelsoni | ANMO 3749 | 1,786,424 | 1,489,126 | 1380 | 578 |
| Sceloporus pyrocephalus | unknown | 2,960,302 | 1,701,102 | 723 | 567 |
| Sceloporus pyrocephalus | UTAR 53473 | 2,510,072 | 1,987,732 | 1711 | 576 |
| scalaris group (10/11) | |||||
| Sceloporus aeneus | RWB 769 | 3,950,636 | 3,201,546 | 1868 | 541 |
| Sceloporus aurantius | RWB 1024 | 5,101,362 | 4,469,276 | 2943 | 544 |
| Sceloporus bicanthalis | UWBM 7307 | [45] | [45] | [45] | 583 |
| Sceloporus brownorum | RWB 6136 | 4,763,078 | 3,880,564 | 2676 | 559 |
| Sceloporus chaneyi | RWB 6199 | 5,669,414 | 4,989,592 | 3018 | 543 |
| Sceloporus goldmani | not sampled | – | – | – | – |
| Sceloporus samcolemani | JJW 698 | 7,011,786 | 6,414,192 | 6966 | 511 |
| Sceloporus samcolemani | RWB 6263 | 1,252,600 | 1,174,070 | 865 | 509 |
| Sceloporus scalaris | UWBM 6589 | 6,473,054 | 5,873,872 | 3253 | 575 |
| Sceloporus scalaris | RWB 6247 | 3,654,246 | 3,176,608 | 2603 | 536 |
| Sceloporus slevini | RWB 741 | 4,044,246 | 3,096,622 | 1329 | 526 |
| Sceloporus subniger | RWB 686 | 2,348,238 | 2,073,796 | 3028 | 517 |
| Sceloporus subniger "West" | RWB 645 | 7,806,116 | 7,173,556 | 1532 | 577 |
| Sceloporus unicanthalis | JJ UANL 2/11 | 3,823,644 | 3,613,842 | 1651 | 558 |
| siniferus group (3/4) | |||||
| Sceloporus carinatus | UWBM 6614 | 5,530,392 | 5,075,056 | 3202 | 577 |
| Sceloporus cupreus | not sampled | – | – | – | – |
| Sceloporus siniferus | UWBM 6653 | 2,520,248 | 2,276,060 | 971 | 560 |
| Sceloporus siniferus | MVZ 236299 | 4,928,056 | 4,507,972 | 1750 | 569 |
| Sceloporus squamosus | UTAR 39846 | 2,220,578 | 1,990,330 | 1855 | 579 |
| spinosus group (3/3) | |||||
| Sceloporus edwardtaylori | UWBM 6588 | 3,662,374 | 3,288,800 | 1769 | 578 |
| Sceloporus horridus | MZFC 7458 | 2,687,744 | 2,205,246 | 2100 | 241 |
| Sceloporus spinosus | UWBM 6672 | 3,349,020 | 3,069,336 | 1682 | 571 |
| torquatus group (5/5) | |||||
| Sceloporus bulleri | FMQ 2815 | 4,561,554 | 4,287,202 | 2990 | 554 |
| Sceloporus insignis | ANMO 1130 | 2,778,082 | 2,445,878 | 1275 | 554 |
| Sceloporus insignis | unknown | 8,364,794 | 7,792,166 | 6936 | 571 |
| Sceloporus jarrovii | LSUMZ 48786 | 3,875,608 | 3,536,668 | 3359 | 535 |
| Sceloporus lineolateralis | MZFC 6650 | 4,062,766 | 3,406,354 | 3365 | 264 |
| Sceloporus torquatus | UWMB 6600 | 8,116,338 | 7,474,646 | 4694 | 576 |
| Sceloporus torquatus | UOGV 2526 | 3,381,124 | 3,072,114 | 2917 | 578 |
| undulatus group (10/10) | |||||
| Sceloporus cautus | MZFC 7414 | 8,275,890 | 7,070,754 | 1828 | 581 |
| Sceloporus consobrinus | ADL105 | 1,417,172 | 1,185,840 | 773 | 534 |
| Sceloporus cowlesi | AMNH 154059 | 4,275,658 | 3,889,018 | 2001 | 575 |
| Sceloporus exsul | UWBM 6590 | 6,472,236 | 5,934,742 | 3189 | 574 |
| Sceloporus occidentalis | UWBM 6281 | [13] | [13] | [13] | 540 |
| Sceloporus occidentalis | MVZ 245697 | 2,319,900 | 2,137,418 | 1469 | 564 |
| Sceloporus olivaceus | UWBM 7968 | 2,263,854 | 1,994,096 | 1088 | 580 |
| Sceloporus tristichus | ADL189 | 5,483,728 | 5,010,016 | 2721 | 572 |
| Sceloporus undulatus | ADL182 | 742,440 | 661,418 | 743 | 577 |
| Sceloporus virgatus | MVZ 150112 | 2,818,968 | 2,451,942 | 905 | 575 |
| Sceloporus woodi | MVZ 10643 | 1,430,400 | 1,277,004 | 870 | 574 |
| utiformis group (0/1) | |||||
| Sceloporus utiformis | not sampled | – | – | – | – |
| variabilis group (6/7) | |||||
| Sceloporus chrysostictus | UTAR 53535 | 3,682,608 | 3,321,192 | 1536 | 572 |
| Sceloporus cozumelae | not sampled | – | – | – | – |
| Sceloporus couchii | MZFC 6676 | 4,611,012 | 3,855,118 | 2613 | 394 |
| Sceloporus parvus | MZFC 6664 | 4,973,960 | 4,489,574 | 5075 | 561 |
| Sceloporus smithi | UWBM 6662 | 2,046,916 | 1,901,554 | 998 | 575 |
| Sceloporus teapensis | UTAR 52778 | 4,352,968 | 3,926,160 | 3545 | 565 |
| Sceloporus variabilis | UWBM 6678 | 6,819,682 | 6,201,808 | 2537 | 577 |
| Sceloporinae outgroups | |||||
| Petrosaurus thalassinus | MVZ 161183 | [13] | [13] | [13] | 523 |
| Urosaurus ornatus | UWBM 7587 | [13] | [13] | [13] | 577 |
| Uta stansburiana | UWBM 7605 | [13] | [13] | [13] | 538 |
| Callisaurini outgroups | |||||
| Callisaurus draconoides | MVZ 265543 | [13] | [13] | [13] | 575 |
| Callisaurus draconoides | MVZ unknown | 10,414,104 | 9,871,780 | 44,419 | 574 |
| Cophosaurus texanus | UWBM 7347 | [13] | [13] | [13] | 573 |
| Holbrookia maculata | UWBM 7362 | [13] | [13] | [13] | 573 |
| Uma notata | SDSNH 76166 | [13] | [13] | [13] | 577 |
| Phrynosomatini outgroups | |||||
| Phrynosoma asio | UWBM 7281 | [45] | [45] | [45] | 565 |
| Phrynosoma blainvillii | CAS 200652 | [45] | [45] | [45] | 565 |
| Phrynosoma braconnieri | UWBM7282 | [45] | [45] | [45] | 561 |
| Phrynosoma cerroense | MVZ 161206 | [45] | [45] | [45] | 579 |
| Phrynosoma cornutum | MVZ 238582 | [45] | [45] | [45] | 575 |
| Phrynosoma coronatum | UABC 1007 | [45] | [45] | [45] | 364 |
| Phrynosoma ditmarsi | RRM 2459 | [45] | [45] | [45] | 576 |
| Phrynosoma douglasii | UWBM 7227 | [45] | [45] | [45] | 515 |
| Phrynosoma goodei | CAS 229922 | [45] | [45] | [45] | 574 |
| Phrynosoma hernandesi | MVZ 245875 | [45] | [45] | [45] | 573 |
| Phrynosoma mcallii | CAS 229923 | [45] | [45] | [45] | 538 |
| Phrynosoma modestum | MVZ 238583 | [45] | [45] | [45] | 545 |
| Phrynosoma orbiculare | UWBM 7285 | [45] | [45] | [45] | 508 |
| Phrynosoma platyrhinos | MVZ 161495 | [45] | [45] | [45] | 563 |
| Phrynosoma sherbrookei | MZFC 28101 | [13] | [13] | [13] | 579 |
| Phrynosoma solare | MVZ 241510 | [45] | [45] | [45] | 410 |
| Phrynosoma taurus | UWBM 7296 | [45] | [45] | [45] | 559 |
| Iguanidae outgroups | |||||
| Gambelia wislizenii | UWBM 7353 | [13] | [13] | [13] | 549 |
| Liolaemus darwinii | LJAMM-CNP 14634 | [13] | [13] | [13] | 581 |
In order to try to resolve the Sceloporus phylogeny and understand the relationship between chromosome evolution and diversification we sought near complete taxon sampling and a broad sampling of loci from throughout the genome. We estimated a phylogenomic tree for Sceloporus using targeted sequence capture data that includes a combination of ultraconserved elements [42] and protein-coding genes used in previous studies of squamate phylogeny [43]. These new data are analyzed using concatenation and coalescent-based species tree inference methods. We conduct a diversification analysis to estimate the number of rate shifts and their locations on the phylogeny. These patterns of diversification are then discussed in relation to chromosomal diversity. The results suggest that differentiation in the fundamental number of chromosomes among species groups may be linked to Sceloporus diversification.
Results
Targeted sequence capture data
We obtained targeted sequence capture (TSC) data from 44 Squamate Tree of Life (ToL) loci and 541 ultraconserved elements (UCE’s; Table 2). Summaries of the sequence capture loci were generated using scripts available from https://github.com/dportik/Alignment_Assessment [44]. and frequency distributions summarizing the properties of the phylogenomic data on a per locus basis are shown in Fig. 1. Although we included 131 samples in our analysis (129 phrynosomatids and two outgroup species), the final sequence alignments for the Squamate ToL loci contained 118 individuals on average (46 min. – 129 max.), and the UCE alignments contained 121 individuals on average (15 min. – 131 max.). Some of the phylogenomic data were taken from previous studies, including 11 samples from a study of phrynosomatid lizards [13] and 17 samples from a study of the genus Phrynosoma [45]. Sequence capture inefficiency during the probe hybridization step and low sequencing effort are two likely reasons for the lack of data for some individuals across loci. A summary of the variation in the TSC data is provided in Table 2. On average, the Squamate ToL loci are longer compared to the UCEs (538 base pairs [bp] vs. 482 bp, respectively), contain more variation (31 % vs. 19 %), and contain more parsimony informative characters (104 vs. 47).
Table 2.
Summary of the variation in the targeted sequence capture data
| Loci | Number of loci | Number of samples | Length (bp) | Variation (%) | PIC |
|---|---|---|---|---|---|
| Ultraconserved elements | 541 | 121 (15–131) | 482 (284–713) | 19 (2–45) | 47 (0–146) |
| Squamate ToL loci | 44 | 118 (46–129) | 538 (355–664) | 31 (16–57) | 104 (48–181) |
Values are reported as the mean and minimum-maximum values. PIC refers to parsimony informative characters
Fig. 1.
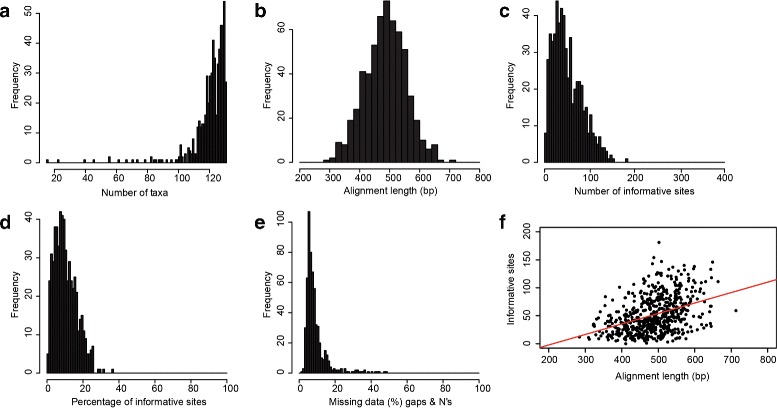
Properties of the targeted sequence capture data collected for phrynosomatid lizards. Frequency distributions summarize the properties of the phylogenomic data on a per locus basis, including number of taxa (a), alignment length (b), number of informative sites (c), percentage of informative sites (d), and missing data including both gaps and N characters (e). There is a positive correlation between alignment length and informative sites, adjusted R2 = 0.1523, p<0.000 (f)
Phylogenetic analysis
The phylogenetic trees that we estimated for Sceloporus using the 585 loci using concatenation (RAxML; [46]) and a coalescent-based species tree approach (SVDquartets; [47]) are shown in Fig. 2. The phylogenetic relationships inferred at the base of Sceloporus differ between the two approaches. Using concatenation, a clade containing the angustus and siniferus species groups is sister to the remaining members of Sceloporus, whereas in the coalescent tree the variabilis group is sister to the rest of the genus. This discrepancy has weak support in the concatenation and coalescent trees (68 and 26 % bootstraps, respectively). The phylogenetic relationships for the remaining species groups are consistent starting at the point in the phylogeny where S. merriami diverges. The major relationships include a clade containing the pyrocephalus, gadoviae, and jalapae groups, a clade containing the graciosus and magister groups, a 22-chromosome clade containing the undulatus, formosus, and spinosus groups (sister to the scalaris group), and a 32-chromosome clade containing the megalepidurus, torquatus, and poinsettii groups (sister to the grammicus group and the clarkii group). The support for these clades varies between the concatenation tree (these relationships all have high support) and the coalescent tree (only the 22 and 32 chromosome clades have significant support). One notable difference is that the concatenation tree fails to support the monophyly of the spinosus group, whereas the coalescent tree provides weak support (62 % bootstrap) for this group.
Fig. 2.
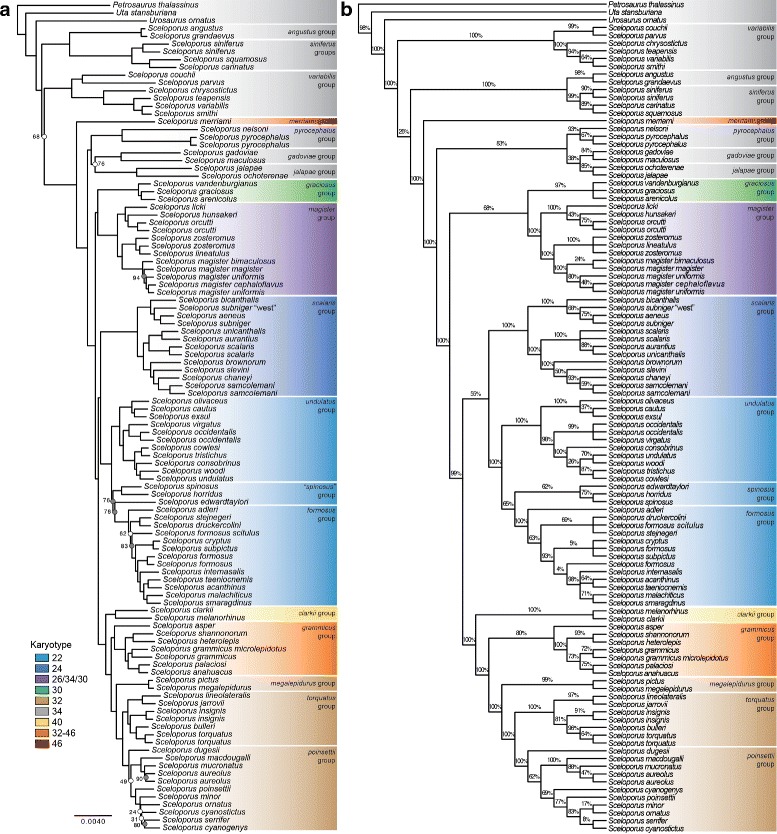
Sceloporus phylogeny estimated using targeted sequence capture data. Phylogenetic relationships among Sceloporus estimated with concatenation (a) and with a coalescent approach (b)
Our time-calibrated phylogeny estimated using the Squamate ToL loci in BEAST [48] (Fig. 3) indicates that the crown age for the family Phrynosomatidae is approximately 54 million years (mean = 54.12, highest posterior density [HPD] = 46.13–61.65 Ma). The age estimate for the genus Sceloporus is 37 million years (mean = 37.02, HPD = 30.71–43.71). Both estimates are consistent with previous estimates [30], but this might not be unexpected given that we used a similar prior. In addition, it is likely that the use of a concatenated data matrix in BEAST is causing divergence time overestimation, and that a species tree approach would provide more accurate estimates. The topology of the BEAST tree is largely similar to the concatenation and coalescent trees shown in Fig. 2, but there are several key differences. First, the BEAST tree places the scalaris group sister to the magister and graciosus groups instead of sister to the 22 chromosome clade. Second, the spinosus group is paraphyletic and S. edwardtaylori is and placed at the base of the 22-chromosome clade. Third, the grammicus group is paraphyletic as a result of moving S. asper to the base of a group containing the 32-chromosome clade and the grammicus group. These differences in topology are likely the result of excluding the ultraconserved elements from the phylogenetic analysis instead of modeling differences between the phylogenetic methods.
Fig. 3.
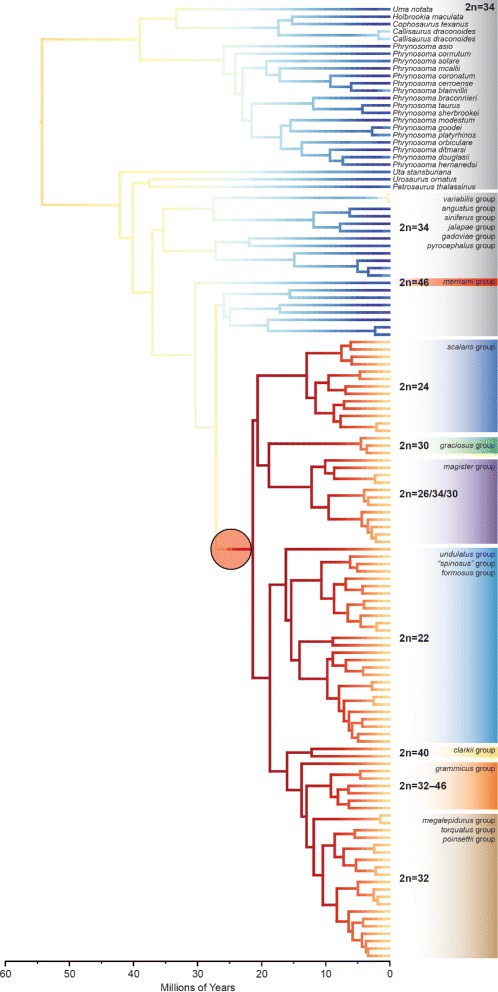
The BAMM analysis supports a single rate shift in Sceloporus that coincides with the rapid radiation of species groups containing different numbers of chromosomes
Gene tree congruence and rapid radiation
Rapid radiations are expected to produce increased gene tree discordance. We investigated congruence between the 585 gene trees (estimated using RAxML) and the estimated species tree by quantifying the number of gene trees that supported the major relationships obtained in the species tree analysis. This approach for measuring congruence does not distinguish between gene tree discordance resulting from a lack of genetic variability versus incomplete lineage sorting. Three Sceloporus species groups have gene tree congruence that exceeds 50 % (i.e., at least 50 % of the 585 gene trees support their monophyly): the angustus, siniferus, and graciosus groups (Fig. 4). The remaining species groups have higher levels of gene tree discordance, and some are supported by <10 % of the loci, including the undulatus group (40 loci), poinsettii group (36 loci), torquatus group (30 loci), grammicus group (24 loci), and spinosus group (9 loci). The 22-chromosome and 32-chromosome clades are supported by 28 and 18 loci, respectively.
Fig. 4.

Gene tree congruence in the Sceloporus rapid radiation. The majority of species groups in Sceloporus are supported by fewer than 50 % of the gene trees (a), and there is a positive correlation between gene tree congruence and the duration of a branch (b)
There is a strong correlation between the amount of gene tree congruence for a taxon bipartition (e.g., a species group) and the branch length for a taxon bipartition (Fig. 4). We explored this relationship using the branch duration estimates (measured in millions of years) obtained from the BEAST analysis (Fig. 3). As expected, the branches with the shortest time intervals had low gene tree congruence, and branches with longer time intervals had high gene tree congruence.
Diversification analysis
Diversification analyses conducted using BAMM [49] recovered an average speciation rate (λ) of 0.09 species/Ma across the phrynosomatid tree. The analysis also found a positive extinction rate (μ) of 0.02 species/Ma that has been relatively consistent throughout the history of phrynosomatids (Fig. 5). We found strong evidence for heterogeneous diversification dynamics with a single acceleration in speciation rate at 20–25 million years ago (Fig. 5). The posterior probability for this rate shift occurring on the branch leading to Sceloporus species groups exhibiting increased diversity in the fundamental chromosome number is 0.997 (Fig. 3). The following species groups are included in this rapid radiation: graciosus and magister groups, a 22-chromosome clade containing the undulatus, formosus, and spinosus groups, the scalaris group, a 32-chromosome clade containing the megalepidurus, torquatus, and poinsettii groups, and the grammicus and clarkii groups. Furthermore, we calculated the Bayes factor (BF) for a shift on this branch by incorporating the probability of a rate shift at that branch under the prior alone, and found overwhelming evidence for a shift (BF >139,000). When examined separately, the increased speciation rate for the rapid radiation clade is 0.15 species/Ma, which is double that of the background rate (0.06 species/Ma).
Fig. 5.

Speciation and extinction rate changes in Sceloporus. The speciation rate shift in Sceloporus occurred approximately 20–25 million years ago (a), but the magnitude of the shift is low (0.05). Extinction rates appear to be constant through time (b)
Discussion
Chromosome evolution and diversification
The link between chromosomal evolution and diversification in Sceloporus has been recognized for decades (reviewed by [24, 31]. A previous study of Sceloporus diversification and chromosomal evolution using a Bayesian cross-validation predictive density approach found that species diversity was significantly higher in some parts of the phylogeny than predicted in comparison to background diversification rates [27]. Instead of using a local approach to test hypotheses about diversification rate shifts on pre-specified sections of the phylogeny where chromosomal changes occurred, the BAMM analyses presented here take a global approach with the goal of detecting significant speciation rate shifts anywhere on the phylogeny (Fig. 3; Table 2). The single significant rate shift is estimated to have occurred during the rapid radiation leading to a clade of Sceloporus species groups with high diversity in fundamental chromosome number (Fig. 3). The estimated background rates of diversification are similar between the two methods (approximately 0.06 species/Ma), and this rate doubles in the clade containing increased chromosomal diversity (Fig. 5).
Common methods for testing for trait-dependent diversification are the “state speciation and extinction” models (e.g., BiSSE, MuSSE, QuaSSE, etc.) [50]. This family of methods attempts to identify significant speciation or diversification rate differences between species in relation to a trait of interest. This approach sounds appealing for testing the link between chromosome evolution and diversification in Sceloporus. However it is important to note that detecting trait-dependent speciation is prone to errors from model violations and model inadequacies, and that these problems have led to an excess of trait-dependent speciation associations in the literature [51, 52]. New statistical tests aimed at distinguishing false associations are available, but these tests are currently limited to binary and continuous characters [53]. In Sceloporus, attempting to coerce the multistate karyotype data into a binary model results in few independent associations between the character state and diversification, and this type of problematic character state distribution is expected to return a false positive association [53, 54]. As expected, BiSSE provides strong support for karyotype-dependent diversification in Scelporus (results not shown).
Vertebrate radiations, including Sceloporus, tend to diversify following a semi-predictable trajectory of divergence [55] along axes of habitat [56], trophic morphology [57], and communication [34, 58–60]. Chromosomal variation is a prominent feature of Sceloporus diversity that is putatively linked to their rapid diversification. Disentangling these factors (i.e., ecology, morphology, diet, chromosomes, etc.) to determine their separate and joint contributions to diversification will be an interesting route to take in future studies (see [61] for an example).
Based on a cursory examination of the current geographic distributions of species in relation to their karyotypes, closely-related species of Sceloporus with the same karyotype formula are not typically found in sympatry [24]. Instead, communities with multiple species of Sceloporus tend to contain species with different karyotypes. The relationship between community assembly and chromosome number has not been formally tested, but we predict that communities of Sceloporus will be over-dispersed on the phylogeny and support the observation that species with similar karyotypes are typically not sympatric.
The ancestral karyotype for phrynosomatid lizards is 2n = 34 (12 macrochromosomes, 20 microchromosomes, and an XY sex chromosome pair), and only Sceloporus shows variation around this karyotype formula, which ranges from 2n = 22 to 2n = 46 [31]. The speciation rate shift that we detected on the phylogeny (Fig. 3) is located at the base of a clade containing high chromosome number diversity. There are changes in the karyotypes of Sceloporus that are not associated with this particular clade, including minor modifications such as inversions and/or secondary constrictions near the centromeres of the macrochromosomes [27]. The most dramatic example of a chromosomal change in a species that is outside of the rapid radiation is Sceloporus merriami, which has a karyotype formula of 2n = 46 resulting from the fission of 6 macrochromosomes. The chromosomal changes observed in the species/species groups falling outside of the rapid radiation do not appear to be correlated with any significant shift in speciation rate.
The evolutionary changes in autosomes and sex chromosomes that have produced karyotypic diversity that is distinctive from the ancestral 2n = 34 formula require a reevaluation on our new phylogeny (Figs. 2 and 3). Previous studies suggesting that the magister and graciosus groups were not sister taxa assumed that they must have independently evolved several unique karyotype features. These groups each have missing or indistinct sex chromosomes, and they each contain 2n = 30 chromosomes (although the magister group also contains species with other arrangements). The new phylogenetic trees presented here support these species groups as sister taxa, and therefore the presence of indistinct or missing sex chromosomes presumably evolved once in the common ancestor, and the ancestral karyotype is most likely 2n = 30. The sister group relationship between the 22-chromosome clade and the 2n = 24 scalaris group is unchanged (this clade received 100 % support from concatenation, but only 55 % support from coalescent analysis), and this further supports the notion that multiple fusion events are responsible for progressively reducing the number of chromosomes in these groups. The new phylogeny also supports a 32-chromosome clade composed of the torquatus group, poinsettii group, and megalepidurus group. The 32-chromosome clade is sister to the grammicus group (2n = 32 – 46), and this clade is sister to the clarkii group (2n = 40).
Resolving rapid radiations
Resolving rapid radiations using molecular phylogenetic techniques requires sequencing a very large number of nucleotides. However, there is an important distinction between obtaining enough nucleotides to resolve a gene tree versus sequencing enough loci to resolve a species tree. Resolving a gene tree should be feasible if enough nucleotides are available at the locus and as long as the rate of evolution is adequate for the scope of the investigation. The extreme of this approach is taken when full sequences are obtained for non-recombining animal mitochondrial DNA (mtDNA) genomes (e.g., amphibians [62], birds [63], mammals [64]) or plant chloroplast genomes [65]. The gene trees estimated from these studies typically provide strong support for phylogenetic relationships, even for species involved in rapid radiations. Despite the strong appeal of obtaining a robust tree from just a single locus, there are many reasons to be suspicious of the relationships in gene trees, including problems associated with incomplete lineage sorting, gene duplication and extinction, and horizontal gene transfer [66], as well as issues related to inaccurate phylogenetic model assumptions (reviewed by [67]). The advantage of sampling independent loci from across the genome, rather than focusing effort on obtaining long sequences from one or a few loci, is that some of these problems can be circumvented in an attempt to obtain a more accurate phylogeny.
In Sceloporus lizards, previous studies using mtDNA obtained a fairly well-resolved and strongly supported phylogeny [29], but large discrepancies in relationships were apparent in comparison to a species tree estimated from a few nuclear loci, presumably as a consequence of mtDNA introgression [28]. Instead of sequencing more mtDNA aimed at obtaining an even more robust mtDNA gene tree, we leveraged our resources towards obtaining a large number of independent loci from across the genome using a targeted sequence capture approach. Not all of these loci that we selected were particularly useful for resolving the rapid radiation in Sceloporus. Only 3 % of the 585 loci that we obtained supported the rapid radiation that corresponds to the period of increased chromosomal diversification in Sceloporus (Fig. 4). The lizard-specific probe set that we designed for this project appears to have been barely capable of resolving this rapid radiation, and it is likely that this same set of markers will be incapable of resolving more difficult phylogenetic problems. Aside from developing a new probe set that targets more loci, two ways to increase the percentage of loci that contribute useful phylogenetic information in a targeted sequence capture experiment are to invest in longer sequence reads and/or optimize the lab protocol to obtain longer loci. Overall, the new paradigm of sequencing 100s or 1000s of loci in order to obtain a few loci that resolve a rapid radiation seems highly inefficient. Developing more refined locus selection methods that can identify loci with optimal evolutionary rates for a specific question, and thereby increase the probability that a loci will contribute useful phylogenetic signal, is an important direction for the future of phylogenomic studies.
Systematics of Sceloporus
The phylogenomic estimates for Sceloporus obtained using 585 loci (Fig. 2) provide strong support for relationships that have been difficult to elucidate using smaller amounts of data. At a higher taxonomic level, we find strong support for relationships among genera in the Sceloporine (i.e., [[[Urosaurus + Sceloporus],Uta,],Petrosaurus]) that are consistent with a recent study using the same data [13], and restriction site associated DNA sequencing (RADseq) data [68], but conflict with previous estimates that combine mtDNA and nuclear genes [29]. Within Sceloporus, the relationships at the base of the phylogeny are weak and differ depending on the analysis type (e.g., concatenation vs. coalescent analysis). More data may be necessary to obtain a definitive placement for the initial divergences within the genus. The composition of the early diverging groups is clear [69], including the variabilits group and the close relationships between the siniferus, angustus, and utiformis groups (this group was not sampled in our study), and determining which was the first to diverge requires further study.
The addition of loci has helped provide strong support for some species groups relationships that were unresolved with smaller nuclear datasets. For example, the clade containing the pyrocephalus, gadoviae, and jalapae groups that we obtained with the TSC data is also supported by analyses of mtDNA [28]. However, previous analyses of smaller nuclear gene datasets did not support the monophyly of this group [28, 29]. Several species group relationships have been difficult to determine because of the influence of gene tree conflict between nuclear and mtDNA [28]. One example of this problem pertains to the relationship between S. clarkii and S. melanorhinus, which differs between mtDNA and nuclear genes [28]. The mtDNA gene tree separates these species across the phylogeny, whereas analyses of nuclear data support them as a clade. We find strong support for a clade containing S. clarkii and S. melanorhinus, and since species groups are intended to provide names for monophyletic groups, we recommend naming this clade the clarkii species group.
We find strong support for a clade containing the poinsettii and torquatus groups, and we recommend referring to all species included in these groups as members of the torquatus species group. The poinsettii group was erected to deal with non-monophyly of the torquatus group in relation to the megalepidurus group [29]. Given that there is no longer any evidence of paraphyly in the torquatus group it does not seem necessary to retain the poinsettii group.
Monophyly of the spinosus group is weak or missing depending on the type of analysis and source of molecular data. A recent phylogeographic study of this species group revealed mtDNA introgression and gene flow between species [38]. These processes are likely responsible for the discordant phylogenetic relationships that have been described for these species [28, 29]. Gene flow and introgression play a prominent role in the evolution of Sceloporus [28, 70–72], and future phylogenetic studies of the group will benefit from new analytical approaches that can identify gene flow during species tree estimation.
Methods
Specimen collection
The family Phrynosomatidae is a diverse group of lizards with a broad North American distribution from Canada to Panama. Much of their diversity is centered in the arid regions of the southwestern United States and Mexico. The group has approximately 148 species arranged into nine genera. We sampled 129 phrynosomatid individuals, including one sample of Cophosaurus, Holbrookia, Petrosaurus, Uma, and Uta, two specimens of Callisaurus, all 17 species of Phrynosoma, and 86 species of Sceloporus (see Table 1 for voucher details). We sampled all species groups within Sceloporus with the exception of S. utiformis. We used Gambelia wislizenii and Liolaemus darwinii to root the tree. Specimens collected for this project from Mexico and the United States are deposited at the Burke Museum of Natural History and Culture at the University of Washington and the Museo de Zoologia “Alfonso L. Herrera” at the Universidad Nacional Autónoma de México. Specimens were collected with approval from the University of Washington Institutional Animal Care and Use Committee (IACUC #4209-01). Scientific specimens were collected in México with permission from the Secretariat of Environment and Natural Resources (SEMARNAT Permit No. 05034/11 to ADL, and Permit No. FAUT 0093 to ANMO). We also obtained tissue and/or DNA loans from the following genetic resource collections and herpetology collections: Museum of Vertebrate Zoology (University of California, Berkeley), Burke Museum of Natural History and Culture (University of Washington), California Academy of Sciences, Ambrose Monell Cryo Collection (American Museum of Natural History), Los Angeles County Museum, Royal Ontario Museum, University of Texas at Arlington, and the Museo de Zoologia “Alfonso L. Herrera” (Universidad Nacional Autónoma de México).
Targeted sequence capture data
We collected targeted sequence capture data using a set of RNA probes specific for iguanian lizards (Leaché et al., 2015). We synthesized custom probes that target 585 loci (2X tiling; two 120 bp probes per locus) using the MYbaits target enrichment kit (MYcroarray Inc., Ann Arbor, MI, USA). The probes target 541 ultraconserved elements (UCEs) used in the Tetrapods-UCE-5Kv1 probes (ultraconserved.org; [42]) and 44 nuclear loci used for the Squamate ToL project [43].
Whole genomic DNA was extracted from tissues using a NaCl extraction method [73]. Genomic DNA (400 ng) was sonicated to a target peak of 400 bp using a Bioruptor Pico (Diagenode Inc.). Genomic libraries were prepared using the Illumina TruSeq Nano library preparation kit. The samples were hybridized to the RNA-probes in the presence of a blocking mixture composed of forward and reverse compliments of the Illumina TruSeq Nano Adapters, with inosines in place of the indices, as well as chicken blocking mix (Chicken Hybloc, Applied Genetics Lab Inc.) and salmon blocking mix to reduce repetitive DNA binding to beads. Libraries were incubated with the RNA probes for 24 hours at 65 °C. Post-hybridized libraries were enriched using TruSeq adapter primers with Phusion®; High-Fidelity DNA Polymerase (New England Biolabs Inc.) for 20 cycles. Enriched libraries were cleaned with AMPure XP beads. We quantified enriched libraries using qPCR (Applied Biosystems Inc.) with primers targeting five loci mapping to different chromosomes in the Anolis genome. Library quality was verified using an Agilent Tape-station 2200 (Agilent Technologies). These samples were pooled in equimolar ratios and sequenced using an Illumina HiSeq2000 at the QB3 facility at UC Berkeley.
Bioinformatics
The raw DNA sequence reads were demultiplexed based on unique sequence tags using Casava (Illumina). We removed low-quality reads, trimmed low-quality ends, and removed adapter sequences using Trimmomatic [74]. The clean reads were assembled for each species using the de novo assembler IDBA [75]. We ran IDBA iteratively over k-mer values from 50 to 90 with a step length of 10. We used phyluce [42] to assemble loci across species. We performed multiple sequence alignments for each locus using MAFFT [76], and we trimmed long ragged-ends to reduce missing or incomplete data. The final data assemblies are often fragmentary as a result of poor sequence quality and/or lack of sequencing coverage across a locus. As a result, sequence alignments tend to contain large regions of gaps separating relatively few nucleotides. Checking large numbers of loci by eye for these artifacts is difficult, and there is a need for the development of new bioinformatic tools that can help increase the accuracy of sequence alignments obtained from large phylogenomic datasets.
Phylogenetic analysis
We estimated phylogenetic trees using concatenation and coalescent-based species tree inference. For the concatenation analyses, we conducted unpartitioned maximum likelihood (ML) analyses with RAxML v8.0.2 [46]. We used the GTRGAMMA model, and branch support was estimated using 1000 bootstrap replicates. The RAxML tree was rooted with Gambelia wislizenii and Liolaemus darwinii. We estimated a species tree using SVDquartets [47], a coalescent-based species tree inference method that uses the full sequence data. This method infers the topology among randomly sampled quartets of species using a coalescent model, and then a quartet method is used to assemble the randomly sampled quartets into a species tree. Reducing the species tree inference problem into quartets makes the analysis of large numbers of loci feasible. We randomly sampled 100,000 quartets from the data matrix, and used the program Quartet MaxCut v.2.1.0 [77] to infer a species tree from the sampled quartets. We measured uncertainty in relationships using nonparametric bootstrapping with 100 replicates. The bootstrap values were mapped to the species tree estimated from the original data matrix using SumTrees v.3.3.1 [78].
Divergence times were estimated using BEAST v1.8 [48] using the Squamate ToL loci. The ultraconserved elements were removed from the analysis to help decrease the computation time. We assigned an uncorrelated lognormal relaxed clock site model and use a single calibration with a normal distribution of 55 ± 4 mya over the family Phrynosomatidae [30]. We ran two analyses, one with a Yule (birth only) tree prior and one with a birth-death prior, using random starting trees [79, 80]. The concatenated dataset was run for 10 million generations under the GTR+I+ Γ model with four gamma categories. We sampled every 1000 generations and discarded the first 25 % as burnin. Convergence statistics were examined using Tracer v1.6 [81], and assumed to have been met when effective sample sizes (ESS) were greater than 200 for all statistics. We used TreeAnnotator v1.8 to produce the maximum clade credibility (MCC) tree from all post-burnin trees and the 95 % highest posterior density (HPD) for each node.
A prediction from rapid radiations is that gene tree discordance will be high. To investigate the level of congruence between the 585 gene trees and the estimated species tree, we quantified the number of gene trees that supported the major relationships obtained in the species tree analysis. First, we estimated phylogenetic trees for each locus separately using RAxML with the HKY model [82]. Next, we used PAUP v4.0b10 [83] to quantify the number of loci that supported taxon bipartitions that were present in the MCC tree. The taxon bipartitions of interest included the Sceloporus species groups and all of the relationships along the backbone of the Sceloporus phylogeny. Each taxon bipartition was loaded into PAUP as a monophyly constraint prior to loading the gene tree. Species that were absent from a gene tree were removed from the monophyly constraint. We tallied the total number of gene trees (out of 585 total) that supported each taxon bipartition of interest. Finally, we used the branch duration estimates (in millions of years) from the MCC tree to test for a relationship between the number of gene trees supporting a taxon bipartition and the duration of a branch.
Diversification analysis
We tested for shifts in diversification rate through time using Bayesian Analysis of Macroevolutionary Models (BAMM v.2.1.0 [49]). BAMM models speciation and extinction rates by simulating rate shift configurations using reversible-jump Markov chain Monte Carlo (rjMCMC). This approach relaxes the assumption of time-homogeneous diversification and allows a vast space of candidate models to be explored [49]. We ran BAMM for 10 million generations on the MCC tree, sampling parameters every 1000 generations. BAMM incorporates incomplete taxon sampling into the likelihood equation; we assume to be missing 20 % of species diversity for the famliy. We used default prior settings, though results are generally robust to the choice of prior under a compound Poisson process [49]. We assessed convergence by computing the effective sample sizes of the log-likelihoods and number of evolutionary rate shifts in R using the package coda [84].
Availability of supporting data
The data set supporting the results of this article are available in Additional file 1. Sequence reads can be accessed through GenBank under the Accession Numbers KU765209–KU820629.
Acknowledgements
We thank the following institutions for tissue loans: Museum of Vertebrate Zoology (University of California, Berkeley), Burke Museum of Natural History and Culture (University of Washington), California Academy of Sciences, Ambrose Monell Cryo Collection (American Museum of Natural History), Los Angeles County Museum, Royal Ontario Museum, University of Texas at Arlington, and the Museo de Zoologia “Alfonso L. Herrera” (Universidad Nacional Autónoma de México). The University of Washington eSciences Institute provided computing infrastructure. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. For assistance with data collection we thank N. Bouzid, R. Bryson, A. Chavez, J. Grummer, L. Jones, and N. Porcino. The manuscript benefitted from feedback received from the UW Biology Phylogenetics Seminar group, F. Burbrink, J. Oaks, P. Wood, J. Wiens, P. Wood, and three anonymous reviewer. Support for this project was provided by grants from the National Science Foundation (DBI-1144630) and the University of Washington Royalty Research Fund (A61649) awarded to ADL.
Abbreviations
- BF
bayes factor
- bp
base pairs
- HPD
highest posterior density
- Ma
million years
- MCC
maximum clade credibility
- ML
maximum likelihood
- mtDNA
mitochondrial DNA
- RADseq
restriction site associated DNA sequencing
- rjMCMC
reversible-jump Markov chain Monte Carlo
- ToL
Tree of Life
- TSC
targeted sequence capture
- UCE
ultraconserved elements
Additional file
Sequence alignment and phylogenetic trees. Full alignment of the targeted sequence capture data (541 ultraconserved elements and 44 Squamate ToL loci) with data partitions in NEXUS format. The file is compatible with multiple programs; it can be imported into BEAUti and exported in XML format for BEAST [48], executed in PAUP [83], or viewed with a text editor. Phylogenetic trees are embedded at the top of the file; these can be copied to a new file and viewed in FigTree. (ZIP 9751 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Data collection and bioinformatics were performed by CWL. Diversification analyses were conducted by BLB. Phylogenetic analyses were performed by all authors. Fieldwork was organized and led by ADL and ANMO. All authors contributed to writing the manuscript. All authors have read and approved the final version of the manuscript.
References
- 1.Whitfield JB, Lockhart PJ. Deciphering ancient rapid radiations. Trends Ecol Evol. 2007;22(5):258–65. doi: 10.1016/j.tree.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 2.De Duve C. The origin of eukaryotes: a reappraisal. Nature Reviews Gene. 2007;8(5):395–403. doi: 10.1038/nrg2071. [DOI] [PubMed] [Google Scholar]
- 3.Poole AM, Penny D. Evaluating hypotheses for the origin of eukaryotes. Bioessays. 2007;29(1):74–84. doi: 10.1002/bies.20516. [DOI] [PubMed] [Google Scholar]
- 4.Poe S, Chubb AL. Birds in a bush: five genes indicate explosive evolution of avian orders. Evol. 2004;58(2):404–15. doi: 10.1111/j.0014-3820.2004.tb01655.x. [DOI] [PubMed] [Google Scholar]
- 5.Hackett SJ, Kimball RT, Reddy S, Bowie RC, Braun EL, Braun MJ, Chojnowski JL, Cox WA, Han KL, Harshman J, et al. A phylogenomic study of birds reveals their evolutionary history. Sci. 2008;320(5884):1763–8. doi: 10.1126/science.1157704. [DOI] [PubMed] [Google Scholar]
- 6.Springer MS, Burk-Herrick A, Meredith R, Eizirik E, Teeling E, O’Brien SJ, Murphy WJ. The adequacy of morphology for reconstructing the early history of placental mammals. Syst Biol. 2007;56(4):673–84. doi: 10.1080/10635150701491149. [DOI] [PubMed] [Google Scholar]
- 7.Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007;17(4):413–21. doi: 10.1101/gr.5918807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roelants K, Gower DJ, Wilkinson M, Loader SP, Biju S, Guillaume K, Moriau L, Bossuyt F. Global patterns of diversification in the history of modern amphibians. Proc Natl Acad Sci. 2007;104(3):887–92. doi: 10.1073/pnas.0608378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawford NG, Faircloth BC, McCormack JE, Brumfield RT, Winker K, Glenn TC. More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol Lett. 2012;8(5):783–6. doi: 10.1098/rsbl.2012.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCormack JE, Faircloth BC, Crawford NG, Gowaty PA, Brumfield RT, Glenn TC. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res. 2012;22(4):746–54. doi: 10.1101/gr.125864.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, Ho SY, Faircloth BC, Nabholz B, Howard JT, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346(6215):1320–31. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leaché AD, Wagner P, Linkem CW, Böhme W, Papenfuss TJ, Chong RA, Lavin BR, Bauer AM, Nielsen SV, Greenbaum E, et al. A hybrid phylogenetic–phylogenomic approach for species tree estimation in African Agama lizards with applications to biogeography, character evolution, and diversification. Mole Phylogenet Evol. 2014;79:215–30. doi: 10.1016/j.ympev.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Leaché AD, Chavez AS, Jones LN, Grummer JA, Gottscho AD, Linkem CW. Phylogenomics of phrynosomatid lizards: conflicting signals from sequence capture versus restriction site associated DNA sequencing. Genome Biol Evol. 2015;7(3):706–19. doi: 10.1093/gbe/evv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giarla TC, Esselstyn JA. The challenges of resolving a rapid, recent radiation: empirical and simulated phylogenomics of Philippine shrews. Syst Biol. 2015;64:727–40. doi: 10.1093/sysbio/syv029. [DOI] [PubMed] [Google Scholar]
- 15.Swofford DL, Waddell PJ, Huelsenbeck JP, Foster PG, Lewis PO, Rogers JS. Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst Biol. 2001;50:525–539. doi: 10.1080/106351501750435086. [DOI] [PubMed] [Google Scholar]
- 16.Pollard DA, Iyer VN, Moses AM, Eisen MB. Widespread discordance of gene trees with species tree in drosophila: evidence for incomplete lineage sorting. PLoS Gene. 2006;2(10):173. doi: 10.1371/journal.pgen.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, Seaver E, Rouse GW, Obst M, Edgecombe GD, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452(7188):745–9. doi: 10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- 18.Thomson RC, Shaffer HB. Sparse supermatrices for phylogenetic inference: taxonomy, alignment, rogue taxa, and the phylogeny of living turtles. Syst Biol. 2009;59:075. doi: 10.1093/sysbio/syp075. [DOI] [PubMed] [Google Scholar]
- 19.Wiens JJ, Kuczynski CA, Smith SA, Mulcahy DG, Sites JW, Townsend TM, Reeder TW. Branch lengths, support, and congruence: testing the phylogenomic approach with 20 nuclear loci in snakes. Syst Biol. 2008;57(3):420–31. doi: 10.1080/10635150802166053. [DOI] [PubMed] [Google Scholar]
- 20.Degnan JH, Rosenberg NA. Discordance of species trees with their most likely gene trees. PLoS Gene. 2006;2(5):68. doi: 10.1371/journal.pgen.0020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linkem CW, Minin VN, Leaché A. D.Detecting the anomaly zone in species trees and evidence for a misleading signal in higher-level skink phylogeny (Squamata: Scincidae). Syst Biol. 2016;:001. doi:10.1093/sysbio/syw001. [DOI] [PMC free article] [PubMed]
- 22.Ogilvie HA, Heled J, Xie D, Drummond AJ. Computational performance and statistical accuracy of *BEAST and comparisons with other methods. Syst Biol. 2016. doi:10.1093/sysbio/syv118. [DOI] [PMC free article] [PubMed]
- 23.Liu L, Yu L, Edwards SV. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol Biol. 2010;10(1):302. doi: 10.1186/1471-2148-10-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sites JW, Archie JW, Cole CJ, Flores-Villela O. A review of phylogenetic hypotheses for lizards of the genus Sceloporus (Phrynosomatidae): implications for ecological and evolutionary studies. Bulletin of the AMNH; no. 213. 1992.
- 25.Wiens JJ, Reeder TW. Phylogeny of the spiny lizards (Sceloporus) based on molecular and morphological evidence. Herpetol Monogr. 1997;11:1–101. doi: 10.2307/1467007. [DOI] [Google Scholar]
- 26.Harmon LJ, Schulte JA, Larson A, Losos JB. Tempo and mode of evolutionary radiation in iguanian lizards. Science. 2003;301(5635):961–4. doi: 10.1126/science.1084786. [DOI] [PubMed] [Google Scholar]
- 27.Leaché AD, Sites Jr JW. Chromosome evolution and diversification in North American spiny lizards (genus Sceloporus) Cytogenet Genome Res. 2009;127(2–4):166–81. doi: 10.1159/000293285. [DOI] [PubMed] [Google Scholar]
- 28.Leaché AD. Species trees for spiny lizards (Genus Sceloporus): Identifying points of concordance and conflict between nuclear and mitochondrial data. Mole Phylogenet Evol. 2010;54(1):162–71. doi: 10.1016/j.ympev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Wiens JJ, Kuczynski CA, Arif S, Reeder TW. Phylogenetic relationships of phrynosomatid lizards based on nuclear and mitochondrial data, and a revised phylogeny for Sceloporus. Mole Phylogenet Evol. 2010;54(1):150–61. doi: 10.1016/j.ympev.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Wiens JJ, Kozak KH, Silva N. Diversity and niche evolution along aridity gradients in North American lizards (Phrynosomatidae) Evol. 2013;67(6):1715–28. doi: 10.1111/evo.12053. [DOI] [PubMed] [Google Scholar]
- 31.Hall WP. Chromosome variation, genomics, speciation and evolution in Sceloporus lizards. Cytogenet Genome Res. 2009;127(2–4):143–65. doi: 10.1159/000304050. [DOI] [PubMed] [Google Scholar]
- 32.Smith EN. Species boundaries and evolutionary patterns of speciation among the malachite lizards (Formosus group) of the genus Sceloporus (Squamata: Phrynosomatidae). PhD thesis, University of Texas at Arlington. 2001.
- 33.Arèvalo E, Davis SK, Sites JW. Mitochondrial DNA sequence divergence and phylogenetic relationships among eight chromosome races of the Sceloporusgrammicus complex (Phrynosomatidae) in central Mexico. Systematic Biology. 1994;43(3):387–418. doi: 10.1093/sysbio/43.3.387. [DOI] [Google Scholar]
- 34.Wiens JJ, Reeder TW, Nieto-Montes de Oca A. Molecular phylogenetics and evolution of sexual dichromatism among populations of the Yarrow’s spiny lizard (Sceloporus jarrovii) Evol. 1999;53:1884–97. doi: 10.2307/2640448. [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Mendez N, Mendez-De la Cruz F. R. Molecular phylogeny of the Sceloporus torquatus species-group (Squamata: Phrynosomatidae) Zootaxa. 2007;1609:53–68. [Google Scholar]
- 36.Leache AD, Mulcahy DG. Phylogeny, divergence times and species limits of spiny lizards (Sceloporus magister species group) in western North American deserts and Baja California. Mole Ecol. 2007;16(24):5216–33. doi: 10.1111/j.1365-294X.2007.03556.x. [DOI] [PubMed] [Google Scholar]
- 37.Grummer JA, Bryson RW, Reeder TW. Species delimitation using Bayes factors: simulations and application to the Sceloporus scalaris species group (Squamata: Phrynosomatidae) Syst Biol. 2014;63(2):119–33. doi: 10.1093/sysbio/syt069. [DOI] [PubMed] [Google Scholar]
- 38.Grummer JA, Calderón-Espinosa ML, Nieto-Montes de Oca A, Smith EN, Méndez-de la Cruz FR, Leaché AD. Estimating the temporal and spatial extent of gene flow among sympatric lizard populations (genus Sceloporus) in the southern Mexican highlands. Mole Ecol. 2015;24(7):1523–42. doi: 10.1111/mec.13122. [DOI] [PubMed] [Google Scholar]
- 39.Leaché AD, Reeder TW. Molecular systematics of the eastern fence lizard (Sceloporus undulatus): a comparison of parsimony, likelihood, and Bayesian approaches. Syst Biol. 2002;51(1):44–68. doi: 10.1080/106351502753475871. [DOI] [PubMed] [Google Scholar]
- 40.Leaché AD. Species tree discordance traces to phylogeographic clade boundaries in North American fence lizards (Sceloporus) Syst Biol. 2009;58(6):547–59. doi: 10.1093/sysbio/syp057. [DOI] [PubMed] [Google Scholar]
- 41.Mendoza-Quijano F, Flores-Villela O, Sites Jr JW. Genetic variation, species status, and phylogenetic relationships in rose-bellied lizards (Variabilis group) of the genus Sceloporus (Squamata: Phrynosomatidae). Copeia. 1998; 1998:354–66.
- 42.Faircloth BC, McCormack JE, Crawford NG, Harvey MG, Brumfield RT, Glenn TC. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst Biol. 2012;61(5):717–26. doi: 10.1093/sysbio/sys004. [DOI] [PubMed] [Google Scholar]
- 43.Wiens JJ, Hutter CR, Mulcahy DG, Noonan BP, Townsend TM, Sites JW, Reeder TW. Resolving the phylogeny of lizards and snakes (Squamata) with extensive sampling of genes and species. Biol Lett. 2012;8(6):1043–6. doi: 10.1098/rsbl.2012.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Portik DM, Smith LL, Bi K. An evaluation of transcriptome-based exon capture for frog phylogenomics across multiple scales of divergence (Class: Amphibia, Order: Anura). Mole Ecol Res. 2015. in press. [DOI] [PubMed]
- 45.Leaché AD, Linkem CW. Phylogenomics of horned lizards (Genus: Phrynosoma) using targeted sequence capture data. Copeia. 2015;103(3):586–94. doi: 10.1643/CH-15-248. [DOI] [Google Scholar]
- 46.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma. 2014;30(9):1312–13. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chifman J, Kubatko L. Quartet inference from SNP data under the coalescent model. Bioinforma. 2014;30:3317–24. doi: 10.1093/bioinformatics/btu530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mole Biol Evol. 2012;29(8):1969–73. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabosky DL. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE. 2014;9(2):89543. doi: 10.1371/journal.pone.0089543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.FitzJohn RG. Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol Evol. 2012;3(6):1084–92. doi: 10.1111/j.2041-210X.2012.00234.x. [DOI] [Google Scholar]
- 51.Davis MP, Midford PE, Maddison W. Exploring power and parameter estimation of the BiSSE method for analyzing species diversification. BMC Evol Biol. 2013;13(1):38. doi: 10.1186/1471-2148-13-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabosky DL, Goldberg EE. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst Biol. 2015;64(2):340–55. doi: 10.1093/sysbio/syu131. [DOI] [PubMed] [Google Scholar]
- 53.Rabosky DL, Huang H. A robust semi-parametric test for detecting trait-dependent diversification. Syst Biol. 2015; 066. [DOI] [PubMed]
- 54.Maddison WP, FitzJohn RG. The unsolved challenge to phylogenetic correlation tests for categorical characters. Syst Biol. 2015;64(1):127–36. doi: 10.1093/sysbio/syu070. [DOI] [PubMed] [Google Scholar]
- 55.Streelman JT, Danley PD. The stages of vertebrate evolutionary radiation. Trends Ecology & Evolution. 2003;18(3):126–31. doi: 10.1016/S0169-5347(02)00036-8. [DOI] [Google Scholar]
- 56.Wiens JJ, Kozak KH, Silva N. Diversity and niche evolution along aridity gradients in North American lizards (Phrynosomatidae) Evol. 2013;67(6):1715–28. doi: 10.1111/evo.12053. [DOI] [PubMed] [Google Scholar]
- 57.Galindo-Gil S, de Jesus Rodriguez-Romero F, Velazquez-Rodriguez AS, Moreno-Barajas R. Morphological correlations between head shape, diet and habitat use of some Sceloporus of Mexico: A quantitative analysis. Int J Morphology. 2015;33(1):295–300. doi: 10.4067/S0717-95022015000100047. [DOI] [Google Scholar]
- 58.Martins EP. A comparative study of the evolution of Sceloporus push-up displays. 994–1018. 1993. [DOI] [PubMed]
- 59.Ossip-Klein AG, Fuentes JA, Hews DK, Martins EP. Information content is more important than sensory system or physical distance in guiding the long-term evolutionary relationships between signaling modalities in Sceloporus lizards. Behav Ecol Sociobiol. 2013;67(9):1513–22. doi: 10.1007/s00265-013-1535-4. [DOI] [Google Scholar]
- 60.Martins EP, Ossip-Klein AG, Zúñiga-Vega JJ, García CV, Campos SM, Hews DK. Evolving from static to dynamic signals: evolutionary compensation between two communicative signals. Animal Behaviour. 2015;102:223–9. doi: 10.1016/j.anbehav.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagner CE, Harmon LJ, Seehausen O. Ecological opportunity and sexual selection together predict adaptive radiation. Nature. 2012;487(7407):366–9. doi: 10.1038/nature11144. [DOI] [PubMed] [Google Scholar]
- 62.Wielstra B, Arntzen JW. Unraveling the rapid radiation of crested newts (Triturus cristatus superspecies) using complete mitogenomic sequences. BMC Evol Biology. 2011;11(1):162. doi: 10.1186/1471-2148-11-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paton T, Haddrath O, Baker AJ. Complete mitochondrial DNA genome sequences show that modern birds are not descended from transitional shorebirds. Proc Royal Soc London B: Biol Sci. 2002;269(1493):839–46. doi: 10.1098/rspb.2002.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiong Y, Brandley MC, Xu S, Zhou K, Yang G. Seven new dolphin mitochondrial genomes and a time-calibrated phylogeny of whales. BMC Evol Biol. 2009;9(1):20. doi: 10.1186/1471-2148-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Straub SC, Moore MJ, Soltis PS, Soltis DE, Liston A, Livshultz T. Phylogenetic signal detection from an ancient rapid radiation: Effects of noise reduction, long-branch attraction, and model selection in crown clade Apocynaceae. Mole Phylogenet Evol. 2014;80:169–85. doi: 10.1016/j.ympev.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 66.Maddison WP. Gene trees in species trees. Syst Biol. 1997;46(3):523–36. doi: 10.1093/sysbio/46.3.523. [DOI] [Google Scholar]
- 67.Philippe H, Brinkmann H, Lavrov DV, Timothy J Littlewood D, Manuel M, Wörheide G, Baurain D. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 2011;9(3):402. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leaché A. D, Banbury BL, Felsenstein J, Nieto-Montes de Oca A, Stamatakis A. Short tree, long tree, right tree, wrong tree: exploring biases in phylogenetic analyses of RAD loci. Syst Biol. 2015. doi:10.1093/sysbio/syv053. [DOI] [PMC free article] [PubMed]
- 69.Flores-Villela O, Kjer KM, Benabib M, Sites JW. Multiple data sets, congruence, and hypothesis testing for the phylogeny of basal groups of the lizard genus Sceloporus (Squamata, Phrynosomatidae) Syst Biol. 2000;49(4):713–39. doi: 10.1080/106351500750049798. [DOI] [PubMed] [Google Scholar]
- 70.Rosenblum EB, Hickerson MJ, Moritz C. A multilocus perspective on colonization accompanied by selection and gene flow. Evol. 2007;61(12):2971–85. doi: 10.1111/j.1558-5646.2007.00251.x. [DOI] [PubMed] [Google Scholar]
- 71.Leaché AD. Multi-locus estimates of population structure and migration in a fence lizard hybrid zone. PloS ONE. 2011;6(9):25827–5827. doi: 10.1371/journal.pone.0025827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leaché AD, Harris RB, Maliska ME, Linkem CW. Comparative species divergence across eight triplets of spiny lizards (Sceloporus) using genomic sequence data. Genome Biol Evol. 2013;5(12):2410–19. doi: 10.1093/gbe/evt186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacManes M. Macmanes salt extraction protocol. 2013. https://dx.doi.org/10.6084/m9.figshare.658946.v1. Accessed 3 March 2016.
- 74.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinforma. 2014;30(15):2114–0120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peng Y, Leung HC, Yiu SM, Chin FY. Research in Computational Molecular Biology. Berlin Heidelberg: Springer; 2010. IDBA–a practical iterative de Bruijn graph de novo assembler. [Google Scholar]
- 76.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mole Biol Evol. 2013;30(4):772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Snir S, Rao S. Quartet MaxCut: a fast algorithm for amalgamating quartet trees. Mole Phylogenet Evol. 2012;62(1):1–8. doi: 10.1016/j.ympev.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 78.Sukumaran J, Holder MT. Dendropy: a Python library for phylogenetic computing. Bioinforma. 2010;26(12):1569–71. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 79.Gernhard T. The conditioned reconstructed process. J Theor Biol. 2008;253(4):769–78. doi: 10.1016/j.jtbi.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 80.Yule GU. A mathematical theory of evolution, based on the conclusions of Dr. J.C. Willis, F.R.S. Philos Trans R Soc Lond B: Biol Sci. 1925;213(402–410):21–87. doi: 10.1098/rstb.1925.0002. [DOI] [Google Scholar]
- 81.Rambaut A, Drummond A. Tracer 1.6. Edinburgh, UK: University of Edinburgh; 2013. [Google Scholar]
- 82.Hasegawa M, Kishino H, Yano TA. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mole Evol. 1985;22(2):160–74. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 83.Swofford DL. PAUP*. phylogenetic analysis using parsimony (* and other methods). Version 4.0. 2003.
- 84.Plummer M, Best N, Cowles K, Vines K. Coda: Convergence diagnosis and output analysis for mcmc. R News. 2006;6(1):7–11. [Google Scholar]
- 85.Uetz P, Hallermann J. The Reptile Database: Peter Uetz; 1995. http://www.reptile-database.org. Accessed 3 March 2016.