

Abstract
The structures of the cytosolic portion of voltage activated sodium channels (CTNav) in complexes with calmodulin and other effectors in the presence and the absence of calcium provide information about the mechanisms by which these effectors regulate channel activity. The most studied of these complexes, those of Nav1.2 and Nav1.5, show details of the conformations and the specific contacts that are involved in channel regulation. Another voltage activated sodium channel, Nav1.4, shows significant calcium dependent inactivation, while its homolog Nav1.5 does not. The available structures shed light on the possible localization of the elements responsible for this effect. Mutations in the genes of these 3 Nav channels are associated with several disease conditions: Nav1.2, neurological conditions; Nav1.4, syndromes involving skeletal muscle; and Nav1.5, cardiac arrhythmias. Many of these disease-specific mutations are located at the interfaces involving CTNav and its effectors.
Keywords: calmodulin, calcium inactivation, Nav, SCN5A, SCN4A, SCN2A, voltage gated sodium channels
Abbreviations
- Nav
Voltage activated sodium channels
- CIP
channel interacting proteins
- CaM
calmodulin
- Cav
voltage activated calcium channels
- CTNav
C-terminal Nav
- CDI
calcium dependant inactivation
- CaM C-lobe
C-terminal lobe of calmodulin
- CaM N-lobe
N-terminal lobe of calmodulin
- EFL
EF hand like domain
- FHF
fibroblast growth factor homologous factor
- rmsd
root mean square deviation.
Voltage activated sodium channels (Nav) are the major participants in the depolarization and electrical conductance of neurons, muscle cells and heart cells. They consist of a major subunit (α) whose activity is modulated by several channel interacting proteins (CIP). The α subunit contains 4 pseudo-repeats, each containing 6 transmembrane helices which form the Na+ conducting pore and the voltage sensor, plus a C-terminal region that includes an EF-hand-like motif (EFL) and a long helix (helix αVI) followed by a region of variable length depending on the isoform. The different isoforms (Nav1.1 to Nav1.9 and Nax) also differ in their tissue distribution, cellular localization and the specific effectors that control of their function. Recent publications have provided structural insight into the regulation of several sodium channels (Nav1.2, Nav1.4, and Nav1.5) by 2 of the most important effectors: calmodulin—a CIP— and Ca2+.1-4 Another CIP, the fibroblast growth factor homologous factor (FHF, such as FGF11, FGF12, FGF13 or FGF14), which has an inhibitory effect on channel activity, is present in several relevant sructures.3 The cytoplasmic C-terminal is the region of the channels that interacts with these effectors. This region is also the locus of many of disease causing mutations.
Mutations in the genes coding for these 3 Nav channels have been associated with neurological conditions (Nav1.2; autism,5 infantile spasms and bitemporal glucose hypermetabolism,6 febrile seizures7,8), with skeletal muscle disorders (Nav1.4; myotonia9-11 and paralysis12,13), and cardiac arrhythmias (Nav1.5; Long QT syndromes,14 Brugada syndrome,15,16 cardiac conduction defect, atrial fibrillation) and dilated cardiomyopathy.14
Here we review and discuss the regulation of these channels by calmodulin (CaM) and Ca2+ with emphasis on the specific structural characteristics of the C-terminal region that may be responsible for their differences in functional behavior of the isoforms.
Comparison of the Nav1.5 and Nav1.2 structures in their CaM complexes
The structure of the Nav1.5 C-terminal fragment in complex with CaM (CTNav1.5-CaM) was determined in the presence of Mg2+ (PDB ID 4OVN) 4 and CTNav1.2 was crystallized in complex with CaM and FGF-13 in the presence of Ca2+ (PDB ID 4JPZ).3 These 2 structures show a high conservation of secondary structure elements, as expected from their high sequence similarity (91% sequence similarity, 80% identity) (Fig. 1A). However, despite the high sequence homology, there are differences between the 2 structures as shown by the main chain trace of Nav1.5 (PDB ID 4OVN) with the tube radius representing the mean r.m.s. deviation of the α-carbons of the equivalent residues of the 2 chains 3,4 (Fig. 1B). Most of the differences in Cα positions result from a change in the orientation of helix αVI with respect to the EFL between the Nav1.5 and the Nav1.2 structures (distance between the ends of the helices of ∼14 Å; see Figure 2). A similar distance is observed when the CTNav in the structure of the CTNav1.5-CaM complex is aligned with that in the Nav1.5-CaM-FHF (Fig. 2).
Figure 1.

Sequence and structural similarity of Nav1.5 and Nav1.2 in their cytoplasmic C-terminal region. (A) Red background displays sequence identity, yellow background sequence conservation of charge and white background, non-homologous residues. The relative accessibility (acc), calculated by Endspript/DSSP, is shown in white for buried residues and in blue for accessible residues. 39 Residues involved in the intermolecular contact between 2 Nav1.5 molecules are labeled “H” (contacts with distances less 3.2 Å, black H; those with distances between 3.2 and 5.0 Å, red H). Helix αIII interacts with the CaM N-lobe and helix αVI with the CaM C-lobe (“D”; colored as for the “H”). Residues at the end of helix αVI involved in intermolecular contact between two Nav1.5 molecules (in CTNav1.5-CaM) or with CaM N-lobe in CTNav1.5-CaMCa-FHF are labeled “G.” The end of helix αVI interacts with another molecule of Nav1.5. The light gray boxes show the residues of Nav1.2 that interact with FHF in the ternary complex (PDB ID 4JPZ). (B) Cα trace of CTNav1.5 in which the thickness of the tube is proportional to the rmsd of the Cα residues in the structural overlap of CTNav1.5 (PDB ID 4OVN) with the CTNav1.2 (PDB ID 4JPZ); (non-conserved residues in gray). (C) As (B) with CaM bound as in 4OVN. (D) Surface representation of the Nav1.5 showing complete sequence conservation at the surface that binds FHF (green); Nav1.5 is 180 degrees from the orientation in (B and C).
Figure 2.

Snapshots of Nav1.5 structures aligned by their EFL domain. (A) Heterodimer of the structure of Nav1.5-CaM. (B) Nav1.5 bound to FHF and with CaM in extended conformation. (C) Nav1.5 bound to FHF with CaCaM as in 4JQ0. In (A–C), the structures are aligned by the EFL domain. (D) Close-up of helix αVI displaying the position of K1922, R1910 and K1899. (E) Similar to panel (B) but showing the same side chains as (D) to display the rotation of 90° around the helix αVI axis relative to the position of the other Nav1.5 molecule (light gray). (F) Displacement of helix aVI of the CTNav1.5-CaM-Ca-FHF (yellow) with respect to the CTNav1.5-CaM-FHF (green).
Residues involved in the interface EFL-FHF, in particular the loops between αII and αIII, αIII and β2, and loop L4 and strand β4, are conserved between Nav1.5 and Nav1.2 suggesting that FHF may play a similar role in the regulation of the long-term inactivation of both isoforms by interacting with the EFL of CTNav (Fig. 1C, D).2,3
Interaction of Nav with calmodulin
In complexes involving Nav, CaM lobes have been observed in all 3 conformations described previously: open, semi-open, and closed.17 CaM has 2 lobes (N- and C-lobe) each containing 2 EF-hands both of which can bind Ca2+. Each lobe can, in principle, adopt any of the 3 conformations. The open conformation is observed when Ca2+ is bound to the EF-hands, while the other 2 are observed in the structurally equivalent apoCaM and Mg2+ bound CaM (Table 1).
Table 1.
Available CTNav structures. The table includes the residues of CTNav used in the study, the residues and lobe of CaM, whether or not Ca2+ was present in the experimental conditions, the orientation of CaM with respect to CTNav and the PDB ID of the structure. All the structures listes have the CaM C-lobe bound to the Nav IQ motif in a semi-open conformation
| Identifier | Amino Acids | FGF | CaM* | Ca2+ | N-lobe Config. | N-lobe Target | CaM Orient. | Method | PDB ID |
|---|---|---|---|---|---|---|---|---|---|
| IQNav1.5-CaM | 1901–1927 | no | 1–148 | apo | closed | none | NA§ | NMR | 2L5321 |
| IQNav1.2-CaM | 1901–1927 | no | 76–148# | apo | NA | NA | NA† | NMR | 2KXW18 |
| CTNav1.5-CaM-FGF | 1773–1940 | yes | 1–148 | apo | closed‡ | none | NA§ | X-ray | 4DCK2 |
| CTNav1.5-CaM-Ca-FGF | 1773–1940 | yes | 1–148 | Ca2+ | open | post-IQ | anti-parallel | X-ray | 4JQ03 |
| CTNav1.2-CaM-Ca-FGF | 1777–1937 | yes | 1–148 | Ca2+ | open‡ | post-IQ | anti-parallel | X-ray | 4JPZ3 |
| CTNav1.5-CaM | 1773–1929 | no | 1–148 | apo | closed | EFL | parallel | X-ray | 4OVN4 |
Hs: Homo sapiens, unless indicated.
Pt: Paramecium tetraurelia
CaM N-lobe is not bound, resulting in no determination of CaM lobes' orientation relative to Nav.
Only the CaM C-lobe was used, resulting in no determination of CaM lobes' orientation relative to Nav.
The semi-open conformation of the CaM C-lobe in the CTNav1.5-CaM structure (PDB ID 4OVN) is similar to that of apo CaM bound to Nav peptides such as the apo CaM C-lobe complex with the Nav1.2 IQ motif peptide, (PDB ID 2KXW; rmsd 1.0 Å; Fig. 3).18 Although the CTNav1.2-CaM-Ca-FHF and the CTNav1.5-CaM-Ca-FHF are reported as Ca2+ bound, the arrangement of the CaM helices in the C-lobe does not resemble the open conformation typically described in bound CaM-Ca2+ structures such as those observed in Cav-CaM-Ca structures.19,20 It is noteworthy that the authors indicate that the anomalous signal of Ca2+ in the C-lobe was weak. Given the weakness of the anomalous signal and the fact that the data extend only to 3.8 Å, it is possible that a majority of the CaM C-lobe molecules do not contain Ca2+.3 Alternatively, the strong interaction of the C-lobe with helix αVI may prevent the lobe from adopting the open conformation even in the presence of Ca2+.
Figure 3.

CTNav1.2 and CTNav1.5 aligned by their CaM C-lobe. This alignment displays the different contacts of the CaM N-lobe: parallel in CTNav1.5-CaM (olive) and antiparallel in CTNav1.2-CaM-Ca-FHF (marine). The alignment shows that the EFL domains do not align due to the 90° degree rotation of helix αVI. The FHF domains have been omitted for clarity. (A) Alignment of CTNav1.5-CaM with CTNav1.5-CaM-Ca-FHF. (B) CTNav1.5-CaM (CTNav1.5, purple; CaM, marine). (C) CTNav1.5-CaM-Ca-FHF (CTNav1.5, yellow; CaM, olive). (D) CTNav1.2-CaM-Ca-FHF (CTNav1.2, orange; CaM, teal).
The N-terminal lobe of CaM (CaM N-lobe) is observed in different conformations in the complexes with CTNav: closed, and open (Fig. 4).17 The structural similarity of the CaM N-lobe in the structure of the CTNav1.5-CaM-FHF (PDB ID 4DCK) and the structure of the isolated CaM N-lobe without Ca2+ (rmsd ˜1.0 Å with PDB ID 3UCW; Fig. 4A, B) is in agreement with, that as reported, this structure does not contain Ca2+ (rmsd ∼2.7Å with 1CDM, a complex with Ca2+; Fig. 4C, D). Comparison of the alignment of CTNav1.5-CaM N-lobe with CaM N-lobe-Mg (rmsd 0.4 Å, Fig. 4 E, K) vs with CaM-Ca (rmsd 4.1 Å, Fig. 4 H,N) emphasizes the lack of Ca2+, and the presence of Mg2+ in the CTNav1.5-CaM structure. On the other hand, alignment of the CaM N-lobe with Ca2+ to either CTNav1.5-Ca-CaM-FHF (rmsd 1.4 Å, Fig. 4I, O) or CTNav1.2-Ca-CaM-FHF (rmsd 0.8 Å, Fig. 4J, P) compared to the alignment of the same 2 structures with the CaM N-lobe-Mg (rmsd 3.4 Å Fig. 4 F,L; and rmsd 3.2 Å Fig. 4G, M) agrees with the presence of Ca2+ in the N-lobe of both structures (Fig. 4F–P) .
Figure 4.

CaM N-lobe conformations in Nav-CaM complexes. The structures of the CaM N-lobe in the different complexes are compared with the isolated CaM N-lobe with Mg2+ (green, 3UCW 40,41) or CaM-Ca (magenta, 1CDM 40). Helix αVI is shown in the structures in which there is an interaction between the CaM N-lobe and the helix. In the figure, CTNav1.5-CaM-FHF (navy blue, PDB ID 4DCK), CTNav1.5-CaM (marine, PDB ID 40VN), CTNav1.5-CaM-Ca-FHF (olive, PDB ID 4JQ0), CTNav1.2-CaM-Ca-FHF (teal, PDB ID 4JPZ), and CaM-Ca (magenta, PDB ID 1CDM) are shown. (A) Overlap of the CaM N-lobe as seen in CaM N-lobe with Mg2+ (PDB ID 3UCW) with CTNav1.5-CaM-FHF (navy, PDB ID 4DCK). (B) 90° rotation of A. (C) Overlap of the CaM N-lobe as seen in CaM-Ca (PDB ID 1CDM) with CTNav1.5-CaM.FHF (navy). (D) 90° rotation from C. (E, K) Alignment of the CTNav1.5-CaM N-lobe with CaM N-lobe-Mg (rmsd 0.4 Å). (H, N) Alignment of the CTNav1.5-CaM with CaM-Ca (rmsd 4.1 Å). (F, L) Alignment of the CTNav1.5-CaM-Ca-FHF N-lobe with CaM N-lobe-Mg (rmsd 3.4 Å). (I, O) Same as (F, L)with CaM-Ca (rmsd 1.4 Å). G, M. Alignment of the CTNav1.2-CaM-Ca-FHF N-lobe with CaM N-lobe-Mg (rmsd 3.2 Å). (J, P) Same as (G, M)with CaM-Ca (rmsd 1.45).
The C-terminal lobe of apoCaM (CaM C-lobe) interacts with the Ile-Gln of the IQ motif of helix α-VI of the C-terminus of all Nav channels. This interaction seems to serve as an anchor for the control of activation of the channels by CaM (Fig. 3). The apoCaM C-lobe recognition of the CTNav helix αVI shows structural characteristics typical of the complexes of CaM with the helices of the large number of other CaM regulated proteins (Fig. 5).18,20-25 The residues that participate in this interaction in the case of Nav have been identified in several structures. 2,4,18,21,26 These residues are mostly conserved in Nav1.2, Nav1.4, and Nav1.5 (Fig. 1A and Fig. 6). In addition to the isoleucine and the glutamine of the IQ motif, several residues in the middle of helix αVI are also conserved (Fig. 1 and Fig. 6). Of the 14 residues of helix αVI in contact with the CaM C-lobe, 10 are conserved and 4 are similar (Fig. 1A and Fig. 6), underscoring the equivalence of the interactions between the helix αVI of the 3 Nav channels and the CaM C-lobe.
Figure 5.

CaM C-lobe conformations bound to Nav C-termini. The structures display a semi-open, not the open conformation typical of Ca-CaM. In the figure the colors are CaM C-lobe (yellow, PDB ID 2KXW), CTNav1.5-CaM (marine, PDB ID 40VN), CTNav1.5-CaM-FHF (navy blue, PDB ID 4DCK), CTNav1.2-CaM-Ca-FHF (teal, PDBID 4JPZ), IQNav1.2-apoCaM-(yellow, PDBID 2KXW), and CaM-Ca (Magenta, PDB ID 1CDM). (A) Overlap of the semi-open CaM C-lobe (PDB ID 2KXW) as seen IQNav1.2-CaM with CTNav1.5-CaM-FHF (PDB ID 4DCK). (B) 90° rotation from A. (C) Overlap of CaM C-lobe as seen in CaM-Ca (PDB ID 1CDM) with CTNav1.5-CaM-FHF (PDB ID 4DCK). (D) 90° rotation from (C). (E-G) and (K–M). CaM C-lobe displaying the lack of change in conformation of the helices upon Ca2+ binding with respect to semi open CaM C-lobe (rmsd for all pairwise aligments is ˜1.0 Å). (E, K)Alignment of the CTNav1.5-CaM C-lobe (4OVN) with CaM C-lobe (2KXW, rmsd 1.3 Å). (H, N) Alignment of the CTNav1.5-CaM C-lobe with CaM-Ca C-lobe (1CDM, rmsd 3.1 Å). (F, L) Alignment of the CTNav1.5-CaM-Ca-FHF C-lobe with CaM C-lobe (2KXW, rmsd 1.3 Å). (I, O) Alignment of the the CTNav1.5-CaCaM-FHF C-lobe with CaM-Ca C-lobe (rmsd 3.1 Å). (G, M) Alignment of the CTNav1.2-CaM-Ca-FHF C-lobe with CaM C-lobe (2KWC, rmsd 1.0 Å). (J, P) CTNav1.2-CaM-Ca-FHF C-lobe with CaMCa C-lobe (1CDM, rmsd 3.1 Å).
Figure 6.

Comparison of Nav1.5, Nav1.2 and Nav1.4. (A) Alignment of the sequences of Nav1.5, Nav1.2, and Nav1.4. The orange background indicates sequence identity, red letters sequence conservation of charge and white background, non-homologous residues. (B) Structure of CTNav1.5-CaM showing the residues of αIII at the interface with CaM-N-lobe (marine) that are not conserved in Nav1.4. Differences in these contacts may be responsible for the CDI differences between Nav1.4 and Nav1.5.
In the structure of the CTNav1.5-CaM complex, the N-terminal lobe of CaM (CaM N-lobe) interacts with the CTNav1.5 EFL (Fig. 1C, 2A).4 This interaction involves contacts of helices α1 and α2 of the CaM N-lobe with helix αIII of the EFL (residues 1832–1838) and the beginning of helix αVI (residues 1895–1899) of the CTNav1.5 (Fig. 1A, Fig. 2A). Binding of FHF is not compatible with this interaction (see below). The sequences of both Nav1.5 regions involved in contacting the CaM N-lobe are conserved in the 3 Nav channels considered in this review. The helix αIII residues that contact the CaM N-lobe in the Nav1.5-CaM complex (QISLI) have sequences in Nav1.2 (KVQLI) and Nav1.4 (KIKLI) that appear to be compatible with the same EFL/CaM N-lobe interaction. The sequences of the other contact region involving the N-terminal of helix αVI (TL(R/K)RK) is highly conserved among the 3 proteins. These observations suggest that the interaction of the CaM N-lobe with the CTNav is a common feature of the 3 Nav channels (Fig. 6).
The structure of the CTNav1.5-CaM complex can be described as a “parallel” arrangement to reflect the fact that the N-lobe of CaM is located toward the N-terminal EFL domain of CTNav1.5 (Fig. 2A, B and 3A, B). On the other hand the CTNav1.5-CaM-Ca-FHF is an “antiparallel” arrangement with the N-lobe of CaM located toward the C-terminus of CTNav1.5 (Fig. 3C, D). This change in orientation of the CaM N lobe has been explained by its binding of Ca2+ that appears to favor post IQ binding.3 In the structure of CTNav1.5-CaM-FHF, determined in the absence of Ca2+ by the same group, the N-lobe of CaM does not contact Nav1.5 (Fig. 2B).
In summary, residues observed interacting with CaM in the structure of the CTNav1.5-CaM are conserved in the 3 Nav channels with a small number of exceptions, some participating in contacts of the parallel arrangement with the CaM N-lobe [1832-1834, 1837, 1900 (Fig. 3 and Fig. 6)], and some of the antiparallel arrangement [1915, 1918–1920 (Fig. 1, Fig. 3C and D)].
Interaction of Nav with another molecule of Nav: Nav-Nav dimerization
An intermolecular Nav1.5-Nav1.5 interaction was observed in the structure of the CTNav1.5-CaM complex between the EFL domain of one Nav1.5 molecule (helices αI and αV) and the end of helix αVI of another CTNav1.5. Residues involved in this interaction are conserved between Nav1.5 and Nav1.2 (marked with H in Fig. 1A) suggesting that this CTNav-CTNav interaction may be have a similar functional role in both channels. Noteworthy is the fact that the only EFL residues that differ between Nav1.5 and Nav1.2 at this CTNav-CTNav interface are in contact with each other in the CTNav1.5-CaM crystal (S1920 and A1882 in Nav1.5, are K1147 and N1109 in Nav1.2; Fig. 2). Although the other component of this interaction, the end of helix αVI, displays less sequence identity between the 2 channels, the conservation of the EFL residues suggests that the intermolecular CTNav-CTNav interaction may be functional in both Nav channels, even though this interaction has not been observed in the published Nav1.2 structures.3,18
Conformations of CTNav in the structures of CTNav with CaM and FHF
The binary complex of CT Nav1.5 with CaM (CTNav1.5-CaM) and the ternary with FHF (CTNav1.5-CaM-FHF) provide snapshots of the states of the cytoplasmic C-terminal domain during the cycle of opening and closing the channel. Structural alignment of the EFL domain of the 3 known structures shows that helix αVI is not in the same orientation with respect to the CTNav EFL in all the structures: in the structures containing FHF helix αVI is rotated 90° about its long axis (Fig. 2). A change in the conformation of residues 1892–1895 (Loop L4), which link the EFL to helix αVI allows the 90° rotation of the helix. As a result, the positively charged residues of helix αVI that interact with the EFL (helix αV) of the other Nav1.5 molecule (Arg1910, Lys1899 and Arg1922) are rotated, most likely preventing or disrupting this interaction (Fig. 2D–F). The change of conformation in loop L4 which results in the 90° rotation of helix VI has been proposed to reflect a change from the inactivated state to the state that is poised for activation.4
In the structure of the CTNav1.5-CaM-Ca-FHF complex, helix αVI forms a different angle with the CTNav EFL than in the ternary CTNav1.5-CaM-FHF complex (Fig. 2F). This difference has not been suggested to be associated with a unique physiological state of the channel.
Residues involved in the EFL-FHF interface observed in the CTNav1.2(or 1.5)-CaM-FHF structures, in particular strand β4 and the preceding loop L4, are conserved between Nav1.5 and Nav1.2 suggesting a similar role for FHF in the regulation of both channels (Fig. 1C).2,3
Ca2+ dependent inactivation
Calcium dependent inactivation (CDI) in the presence of CaM is an essential property of voltage activated Ca2+ channels (Cav). Recently, it was shown that the same phenomenon is exhibited by Nav1.4.1,27 Surprisingly, Nav1.5 does not show this inactivation. Figure 6 highlights the location of the residues that are different between the Nav1.4 and Nav1.5 channels. In the portion of the Nav cytoplasmic region that is present in the CTNav1.5-CaM structure there are a small number of sequence differences between Nav1.4 and Nav1.5: the major differences are beyond the end of helix αVI. If the structural elements that contribute to CDI are located in the C-terminal of the channels, they must include residues that differ between Nav1.5 and Nav1.4 to account for their differences in CDI. Since CDI is likely to involve CaM, at least part of the effect must reside in the residues that differ between the 2 channels in the portion of the channels that interact with CaM—i.e., the residues showed in Figure 6.
Nav mutations and disease
Mutations in SCN5A, the gene that encodes the Nav1.5 channel, have been found in patients with LQT3 or Brugada Syndrome.14,15,28,29 The mutations in the CT include changes at positions 1774, 1779, 1784, 1787, 1795, 1825, 1839, 1895, 1901,1909, 1924 (LQT3; Fig. 7) and positions 1774, 1795, 1812, 1861, 1837, 1872, 1901, 1904, 1919, 1924 (Brugada Syndrome).30-37 Some of these mutations occur at the CTNav1.5-CaM-C-lobe interface (1901, 1909), the CTNav1.5-FHF (1839 LQT3), or close to this interface (1839, LQT3; 1872, Brugada Syndrome). Other mutations affect the interaction of one CTNav1.5 with another CTNav1.5 (1790, 1795 and 1913 in LQT3; 1787, 1919 and 1924 in Brugada Syndrome).
Figure 7.
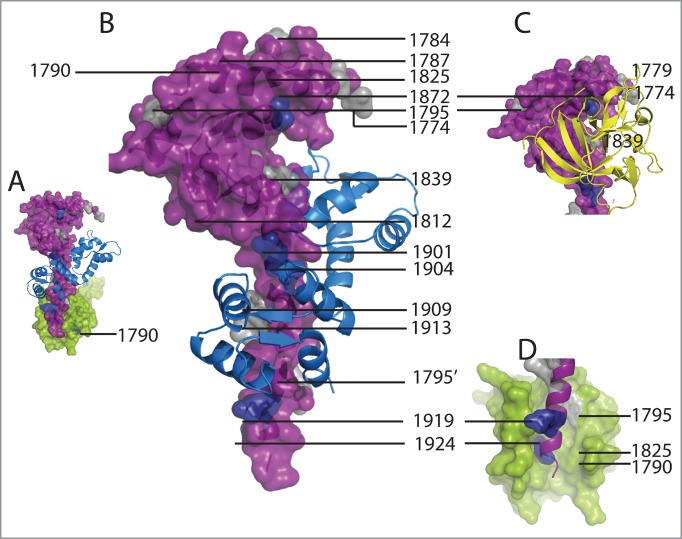
Location of mutations of Nav1.5 observed in Brugada Syndrome and LQT3. (A) Surface representation of one CTNav1.5 monomer (purple) interacting with another CTNav1.5 (green). CaM is indicated only in the foreground CTNav1.5. This state was postulated to be poised for activation. (B) Brugada Syndrome mutations are at the CTNav1.5-CaM-C-lobe interface (colored as is A). CTNav1.5 mutations present in LQT3 (1774, 1779, 1784, 1787, 1795, 1825, 1839, 1872, 1901, 1909, 1903, 1924) are shown in gray and Brugada Syndrome mutations (1774, 1812, 1826, 1837, 1861, 1872, 1795, 1919, 1924) in navy blue. (C) Inset showing the FHF to display the mutations that are at the interface of the EFL domain of CTNav1.5 and the FHF (1839 LQT3) or close to it (1840, LQT3; 1872, Brugada Syndrome). (D) Close-up of the interaction of CTNav1.5-CTNav1.5 (1795 and 1913, gray, LQT3; 1919 and 1924, blue, Brugada Syndrome).
Mutations in the gene coding for Nav1.4 (SCN4A) result in myotonias and other syndromes involving skeletal muscles. Residue Gln1633 of Nav1.4 is equivalent to residue 1807 of Nav1.5, also a Gln (Fig. 6). Assuming similar roles for this residue in both proteins, the mutation Gln1633 of Nav1.4 would introduce a charge at the interface between 2 Nav channels that may be incompatible with maintaining this contact. The same interface is affected by the F1705I mutation (F1705 is equivalent to F1879 of Nav1.5; Figures 1, 6). Similarly, if FHF does interact with Nav1.4, the E1702K mutation of Nav1.4 (Glu1876 in Nav1.5, Fig. 6) would interfere with this interaction. Interestingly, these mutations occur in residues that are conserved among the 3 isoforms.
Mutations in the SCN2A gene, which encodes for the neuronal channel Nav1.2, underlie seizure and autism spectrum disorders. 6,7,38 Mutation D1793N, observed in epilepsy patients, is located in helix αI of the EFL domain that interacts with the CaM N-lobe (Fig. 8).8 Arg1918, which interacts with the linker of the N-lobe the C-lobe when Ca2+ is bound to CaM, was observed mutated to His in a patient with febrile seizures.7 In the CTNav1.2-CaM-Ca-FHF complex, Arg1918 interacts with CaM residues Asp79 and Asp81.3 This interaction may act as a sensor of the presence of Ca2+ in CaM: compared with the structure of CTNav1.5-CaM in the absence of Ca2+ this portion of the linker is the pivot for positioning the CaM N-lobe. In particular, CaM Asp79 is in the same position and CaM Asp81 is 7 Å away (Fig. 8D). The mutation R1902C seems to impair Ca2+ binding 38 to the CaM in the complex, but its position in the Ca2+ bound structure does not support this proposal (Fig. 8).
Figure 8.

Location of mutations of Nav1.2 observed in autism and febrile seizures. (A) Surface representation of CTNav1.2 (top view, orange) interacting with CaM (teal) and FHF (lime green). Mutation of residue 1793 of the EFL domain is shown in gray. (B) CTNav1.2 mutations in gray (1902, 1918). (C) Close-up of residue R1902 close to the interface of CTNav1.2 with FHF. (D) Close-up of the position of residues Asp79 and Asp81 of CaM in the CTNav1.2-CaM-Ca-FHF (antiparallel; teal) vs CTNav1.5-CaM (parallel; marine).
Concluding Remarks
Atomic resolution structural information is accumulating on the conformations of the cytoplamic C-terminal fragments of Nav channels and CaM lobes in Nav-CaM and Nav-CaM-FHF complexes in the presence and absence of Ca2+. Some of these structures have been proposed to be associated with specific functional states of the channels. The structures contain direct information about the specific interactions of the C-terminal domain of the channels that govern the control of channel activity by CIPs and by Ca2+. As a consequence they are providing insights about the mechanisms of altered channel function by mutations associated with the phenotypically variant sodium channelopathies. Further experimentation, including electrophysiological measurements, binding experiments, and determination of additional structures will be essential for the identification of all the states of the cytoplasmic components of the Nav channels needed to provide a full understanding of their regulation of channel activity.
Funding
This work was funded in part by NIH HL050411.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This review is dedicated to the memory of David Yue who introduced us to the differences between Nav1.4 and Nav1.5 in Ca2+-dependent inactivation.
References
- 1.Ben-Johny M, Yang PS, Niu J, Yang W, Joshi-Mukherjee R, Yue DT. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels, Cell 2014; 157:1657-1670; PMID:24949975; http://dx.doi.org/ 10.1016/j.cell.2014.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang C, Chung BC, Yan H, Lee SY, Pitt GS. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin, Structure 2012; 20:1167-1176; PMID:22705208; http://dx.doi.org/ 10.1016/j.str.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C, Chung BC, Yan H, Wang HG, Lee SY, Pitt GS. Structural analyses of Ca(2)(+)/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation, Nat Commun 2014; 5:4896; PMID:25232683; http://dx.doi.org/ 10.1038/ncomms5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gabelli SB, Boto A, Kuhns VH, Bianchet MA, Farinelli F, Aripirala S, Yoder J, Jakoncic J, Tomaselli GF, Amzel LM. Regulation of the NaV1.5 cytoplasmic domain by calmodulin, Nat Commun 2014; 5:5126; PMID:25370050; http://dx.doi.org/ 10.1038/ncomms6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al.. De novo mutations revealed by whole-exome sequencing are strongly associated with autism, Nature 2012; 485:237-241; PMID:22495306; http://dx.doi.org/ 10.1038/nature10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundaram SK, Chugani HT, Tiwari VN, Huq AH. SCN2A mutation is associated with infantile spasms and bitemporal glucose hypometabolism, Pediat Neurol 2013; 49:46-49; PMID:23827426; http://dx.doi.org/ 10.1016/j.pediatrneurol.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haug K, Hallmann K, Rebstock J, Dullinger J, Muth S, Haverkamp F, Pfeiffer H, Rau B, Elger CE, Propping P, Heils A. The voltage-gated sodium channel gene SCN2A and idiopathic generalized epilepsy, Epilepsy Res 2001; 47:243-246; PMID:11738931; http://dx.doi.org/ 10.1016/S0920-1211(01)00312-6 [DOI] [PubMed] [Google Scholar]
- 8.Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, et al.. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy, Cell 2011; 145:1036-1048; PMID:21703448; http://dx.doi.org/ 10.1016/j.cell.2011.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groome JR, Larsen MF, Coonts A. Differential effects of paramyotonia congenita mutations F1473S and F1705I on sodium channel gating, Channels (Austin) 2008; 2:39-50; PMID:18690054; http://dx.doi.org/ 10.4161/chan.2.1.6051 [DOI] [PubMed] [Google Scholar]
- 10.Kubota T, Kinoshita M, Sasaki R, Aoike F, Takahashi MP, Sakoda S, Hirose K. New mutation of the Na channel in the severe form of potassium-aggravated myotonia, Muscle & nerve 2009; 39:666-673; PMID:19347921; http://dx.doi.org/ 10.1002/mus.21155 [DOI] [PubMed] [Google Scholar]
- 11.Biswas S, DiSilvestre DA, Dong P, Tomaselli GF. Mechanisms of a human skeletal myotonia produced by mutation in the C-terminus of NaV1.4: is Ca2+ regulation defective?, PLoS One 2013; 8:e81063; PMID:24324661; http://dx.doi.org/ 10.1371/journal.pone.0081063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu FF, Gordon E, Hoffman EP, Cannon SC. A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation, J Physiol 2005; 565:371-380; PMID:15774523; http://dx.doi.org/ 10.1113/jphysiol.2005.082909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Y, Wang H, Luo XG, Ren Y. Permanent myopathy caused by mutation of SCN4A Metl592Val: observation on myogenesis in vitro and on effect of basic fibroblast growth factor on the muscle, Neurosci Bull 2009; 25:61-66; PMID:19290024; http://dx.doi.org/ 10.1007/s12264-009-0926-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmer T, Surber R. SCN5A channelopathies-an update on mutations and mechanisms, Prog Biophys Mol Biol 2008; 98:120-136; PMID:19027780; http://dx.doi.org/ 10.1016/j.pbiomolbio.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 15.Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel, Circ Res 2011; 108:884-897; PMID:21454796; http://dx.doi.org/ 10.1161/CIRCRESAHA.110.238469 [DOI] [PubMed] [Google Scholar]
- 16.Hoshi M, Du XX, Shinlapawittayatorn K, Liu H, Chai S, Wan X, Ficker E, Deschenes I. Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations, Circul Cardiov Genet 2014; 7:123-131; PMID:24573164; http://dx.doi.org/ 10.1161/CIRCGENETICS.113.000292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houdusse A, Cohen C. Target sequence recognition by the calmodulin superfamily: implications from light chain binding to the regulatory domain of scallop myosin, Proc Natl Acad Sci U S A 1995; 92:10644-10647; PMID:7479857; http://dx.doi.org/ 10.1073/pnas.92.23.10644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldkamp MD, Yu L, Shea MA. Structural and energetic determinants of apo calmodulin binding to the IQ motif of the Na(V)1.2 voltage-dependent sodium channel, Structure 2011; 19:733-747; PMID:21439835; http://dx.doi.org/ 10.1016/j.str.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F, Minor DL Jr. Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation, Structure 2008; 16:1455-1467; PMID:18940602; http://dx.doi.org/ 10.1016/j.str.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Petegem F, Chatelain FC, Minor DL Jr. Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex, Nat Struct Mol Biol 2005; 12:1108-1115; PMID:16299511; http://dx.doi.org/ 10.1038/nsmb1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chagot B, Chazin WJ. Solution NMR structure of Apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J Mol Biol 2011; 406:106-119; PMID:21167176; http://dx.doi.org/ 10.1016/j.jmb.2010.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fallon JL, Halling DB, Hamilton SL, Quiocho FA. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Ca(v)1.2 calcium channel, Structure 2005; 13:1881-1886; PMID:16338416; http://dx.doi.org/ 10.1016/j.str.2005.09.021 [DOI] [PubMed] [Google Scholar]
- 23.Halling DB, Georgiou DK, Black DJ, Yang G, Fallon JL, Quiocho FA, Pedersen SE, Hamilton SL. Determinants in CaV1 channels that regulate the Ca2+ sensitivity of bound calmodulin, J Biol Chem 2009; 284:20041-20051; PMID:19473981; http://dx.doi.org/ 10.1074/jbc.M109.013326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houdusse A, Gaucher JF, Krementsova E, Mui S, Trybus KM, Cohen C. Crystal structure of apo-calmodulin bound to the first two IQ motifs of myosin V reveals essential recognition features, Proc Natl Acad Sci U S A 2006; 103:19326-19331; PMID:17151196; http://dx.doi.org/ 10.1073/pnas.0609436103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori MX, Vander Kooi CW, Leahy DJ, Yue DT. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+, Structure 2008; 16:607-620; PMID:18400181; http://dx.doi.org/ 10.1016/j.str.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chichili VP, Xiao Y, Seetharaman J, Cummins TR, Sivaraman J. Structural basis for the modulation of the neuronal voltage-gated sodium channel NaV1.6 by calmodulin, Sci Rep 2013; 3:2435; PMID:23942337; http://dx.doi.org/ 10.1038/srep02435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin, Circ Res 2002; 90:E49-57; PMID:11884381; http://dx.doi.org/ 10.1161/01.RES.0000012502.92751.E6 [DOI] [PubMed] [Google Scholar]
- 28.Bankston JR, Sampson KJ, Kateriya S, Glaaser IW, Malito DL, Chung WK, Kass RS. A novel LQT-3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences, Channels (Austin) 2007; 1:273-280; PMID:18708744; http://dx.doi.org/ 10.4161/chan.4956 [DOI] [PubMed] [Google Scholar]
- 29.Tomaselli GF, Aiba T. LQTS: better with age?, J Cardiov Electrophysiol 2008; 19:800-801; PMID:18399970; http://dx.doi.org/ 10.1111/j.1540-8167.2008.01158.x [DOI] [PubMed] [Google Scholar]
- 30.Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes, J Biol Chem 2001; 276:30623-30630; PMID:11410597; http://dx.doi.org/ 10.1074/jbc.M104471200 [DOI] [PubMed] [Google Scholar]
- 31.Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease, Hum Mutat 2003; 21:651-652; PMID:14961552; http://dx.doi.org/ 10.1002/humu.9144 [DOI] [PubMed] [Google Scholar]
- 32.Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, Towbin JA. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome, Jama 2001; 286:2264-2269; PMID:11710892; http://dx.doi.org/ 10.1001/jama.286.18.2264 [DOI] [PubMed] [Google Scholar]
- 33.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, Kamakura S, Kyndt F, Koopmann TT, Miyamoto Y, Pfeiffer R, Pollevick GD, Probst V, Zumhagen S, Vatta M, Towbin JA, Shimizu W, Schulze-Bahr E, Antzelevitch C, Salisbury BA, Guicheney P, Wilde AA, Brugada R, Schott JJ, Ackerman MJ. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing, Heart Rhythm: Off J Heart Rhythm Soc 2010; 7:33-46; PMID:20129283; http://dx.doi.org/ 10.1016/j.hrthm.2009.09.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test, Heart Rhythm: Off J Heart Rhythm Soc 2009; 6:1297-1303; PMID:19716085; http://dx.doi.org/ 10.1016/j.hrthm.2009.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tester DJ, Ackerman MJ. Genetic testing for cardiac channelopathies: ten questions regarding clinical considerations for heart rhythm allied professionals, Heart Rhythm: Off J Heart Rhythm Soc 2005; 2:675-677; PMID:15922282; http://dx.doi.org/ 10.1016/j.hrthm.2004.09.024 [DOI] [PubMed] [Google Scholar]
- 36.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing, Heart Rhythm: Off J Heart Rhythm Soc 2005; 2:507-517; PMID:15840476; http://dx.doi.org/ 10.1016/j.hrthm.2005.01.020 [DOI] [PubMed] [Google Scholar]
- 37.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice, Jama 2005; 294:2975-2980; PMID:16414944; http://dx.doi.org/ 10.1001/jama.294.23.2975 [DOI] [PubMed] [Google Scholar]
- 38.Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH. Sodium channels SCN1A, SCN2A and SCN3A in familial autism, Mol Psychiat 2003; 8:186-194; PMID:12610651; http://dx.doi.org/ 10.1038/sj.mp.4001241 [DOI] [PubMed] [Google Scholar]
- 39.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server, Nucleic Acids Res 2014; 42:W320-324; PMID:24753421; http://dx.doi.org/ 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures, Science 1993; 262:1718-1721; PMID:8259515; http://dx.doi.org/ 10.1126/science.8259515 [DOI] [PubMed] [Google Scholar]
- 41.Senguen FT, Grabarek Z. X-ray structures of magnesium and manganese complexes with the N-terminal domain of calmodulin: insights into the mechanism and specificity of metal ion binding to an EF-hand, Biochemistry 2012; 51:6182-94; PMID:22803592 [DOI] [PMC free article] [PubMed] [Google Scholar]