

Abstract
The modulation and regulation of voltage-gated Ca2+ channels is affected by the pore-forming segments, the cytosolic parts of the channel, and interacting intracellular proteins. In this study we demonstrate a direct physical interaction between the N terminus (NT) and C terminus (CT) of the main subunit of the L-type Ca2+ channel CaV1.2, α1C, and explore the importance of this interaction for the regulation of the channel. We used biochemistry to measure the strength of the interaction and to map the location of the interaction sites, and electrophysiology to investigate the functional impact of the interaction. We show that the full-length NT (amino acids 1-154) and the proximal (close to the plasma membrane) part of the CT, pCT (amino acids 1508-1669) interact with sub-micromolar to low-micromolar affinity. Calmodulin (CaM) is not essential for the binding. The results further suggest that the NT-CT interaction regulates the channel's inactivation, and that Ca2+, presumably through binding to calmodulin (CaM), reduces the strength of NT-CT interaction. We propose a molecular mechanism in which NT and CT of the channel serve as levers whose movements regulate inactivation by promoting changes in the transmembrane core of the channel via S1 (NT) or S6 (pCT) segments of domains I and IV, accordingly, and not as a kind of pore blocker. We hypothesize that Ca2+-CaM-induced changes in NT-CT interaction may, in part, underlie the acceleration of CaV1.2 inactivation induced by Ca2+ entry into the cell.
Keywords: binding, calcium channel, calmodulin, C-terminus, CaV1.2, inactivation, L-type, N-terminus
Introduction
Intracellular Ca2+ concentration is maintained at very low levels under resting conditions, below 100 nM. 1 However, it rises sharply (to tens or hundreds of µM within channel’s nanodomain) 2 upon stimulation. The principal Ca2+ entryways of nerve, muscle, and some endocrine cells are VGCCs (voltage-gated Ca2+ channels). Because Ca2+ ions are chemical messengers, 1 influx through VGCCs can directly link membrane potential changes to stimulation of intracellular signaling cascades.3 Since excess Ca2+ influx is toxic, Ca2+ entry into the cell is tightly regulated. Hence, VGCC activity is controlled by self-regulatory and extrinsic mechanisms that tune their action. Inactivation, i.e. loss of conductance during stimulation, is an important negative feedback mechanism in which VGCCs are regulated by entering Ca2+ and internal Ca2+ levels (Ca2+-dependent inactivation) and by conformational changes induced by the membrane potential (voltage-dependent inactivation).4 Channel inactivation generates short and accurate Ca2+ signals, which prevent high toxic levels of Ca2+. Visionary studies of David Yue and collaborators, and of other research groups, revealed the crucial role of VGCC cytosolic domains and calmodulin (CaM) in the process of inactivation, and provided formidable quantitative and qualitative information about this complex machinery. Nonetheless, many details of the molecular mechanisms of inactivation remain incompletely understood.
The main subunit of the ubiquitous L-type VGCC CaV1.2, α1C, has cytosolic amino and carboxyl termini (NT and CT) (Fig. 1). NT and CT play an important role in the inactivation of the channel, and in gating in general. The pCT contains EF hand, preIQ and IQ motifs, and is necessary for Ca2+-dependent inactivation, Ca2+-dependent facilitation and voltage-dependent inactivation.5-8 Deletion of the NT results in changes of the inactivation properties of the channel.9 Truncation of a major part of both termini from the pore forming α1C subunit causes changes in the properties of the current of the channel,4,10-12 which are more than additive compared to changes caused by each truncation.10
Figure 1.
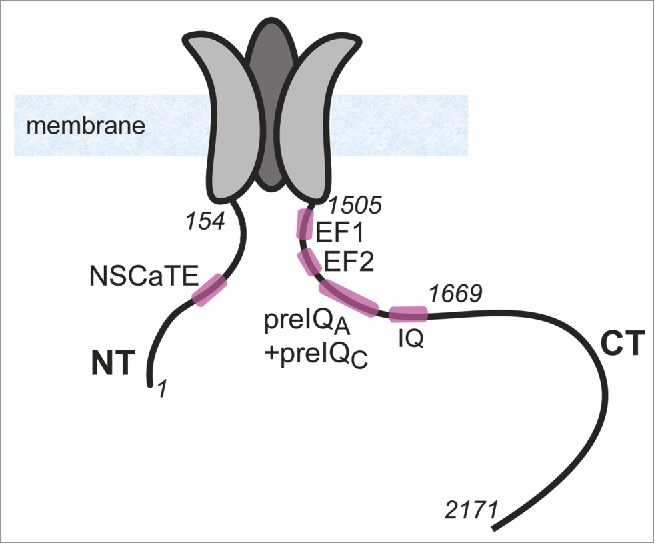
Illustration of the structure of α1C subunit of CaV1.2. NT and CT and 3 out of the 4 transmembrane domains of α1C are shown schematically. Cytosolic loops I, II and III are not shown. In italics are numbers of selected a.a. residues in NT and CT, shown for orientation.
CaM is a small (148 amino acids (a.a.), 17 kDa), soluble, Ca2+-binding protein with N-terminal and C-terminal lobes (CaMN-lobe and CaMC-lobe, respectively). Its flexible structure enables CaM to serve as a Ca2+ sensor for a wide range of target proteins of various structures involved in a wide array of functions in the cell.8,13-15 Ca2+ binding to CaM induces a conformational change in CaM, in turn altering either the association with target proteins or the tertiary structure of the CaM:target complex, thus transforming changes of intracellular Ca2+ levels into modulation of function.16-18 A number of regulatory mechanisms of CaV1.2 by CaM have been proposed, all of which utilize its plasticity in interacting with its target proteins. CaM has several interaction sites on CaV1.2 (Fig. 1). In the CT, they comprise the IQ motif, essential for CaM regulation of CaV1.2, 19-22 and the PreIQ, which contains 2 potential Ca2+-CaM binding sites known as preIQA and preIQC.23-25 The NT contains the NSCaTE motif,4,10 a short linear segment in the NT found only in the CaV1.2 and CaV1.3 L-type Ca2+ channels (a.a. ˜82-90, numbering by rabbit cardiac α1C). CaM binds NSCaTE in a Ca2+-dependent manner10 and undergoes a significant structural change, from unstructured to α-helix upon binding CaM.26,27
The CaV1.2 NT and CT play an important role in gating. They have been the subject of many studies which demonstrated how various NT and pCT deletions and mutations dramatically change inactivation.6,9-11,28-34 It has been proposed that, following channel opening and Ca2+ entry, the previously CT-anchored and now Ca2+-bound CaM binds the NSCaTE, bridging the NT and CT, tightening an “NT-CT scaffold”10 and promoting the inactivation process.4 However, this attractive hypothesis lacks direct proof, and has been challenged by the finding that Ca2+-bound CaM does not foster a direct binding between the NSCaTE and the CT.35 We, therefore, sought evidence for a CaM-independent interaction between the NT and CT. Our data indicate that the 2 cytosolic segments indeed interact directly, possibly affecting inactivation. Furthermore, Ca2+-CaM alters the NT-CT interaction, providing a new insight into the underlying mechanism and leading us to suggest revisions to previously proposed models.4,10
Results
NT-pCT interaction
The inactivation of the CaV1.2 channel involves different parts of the channel. We focused on the role of the cytosolic parts of the channel, and the NT (NT1-154) and pCT (pCT1505-1671) of the α1C subunit in particular (Fig. 1). We first performed pull-down assays in order to test for potential interactions. We synthesized 35S-labeled pCT1505-1671 proteins (target) in vitro in reticulocyte lysate and pulled them down with GST-NT1-154 protein (bait). We note that the reticulocyte lysate undoubtedly contains intrinsic CaM, but its amount relative to the newly synthesized protein could not be estimated.
The results revealed a Ca2+-regulated interaction between the NT and pCT (Fig. 2). The robust NT-pCT interaction observed in a Ca2+-free buffer was reduced in the presence of a high level of Ca2+ (1 mM, Fig. 2, right) but not in the presence of a lower Ca2+ concentration (0.2 mM, Fig. 2, left). However, in the presence of purified CaM, the interaction was reduced by both 0.2 and 1 mM Ca2+. These results indicate the possibility of a Ca2+/CaM-dependent weakening of the NT-pCT interaction upon physiologically relevant Ca2+ increase within the channel nanodomain. Without the added CaM, the endogenous CaM derived from the reticulocyte lysate was probably insufficient to saturate all the overexpressed pCT, and this probably explains the inability of 200 µM Ca2+ to significantly reduce the observed NT-pCT binding.
Figure 2.
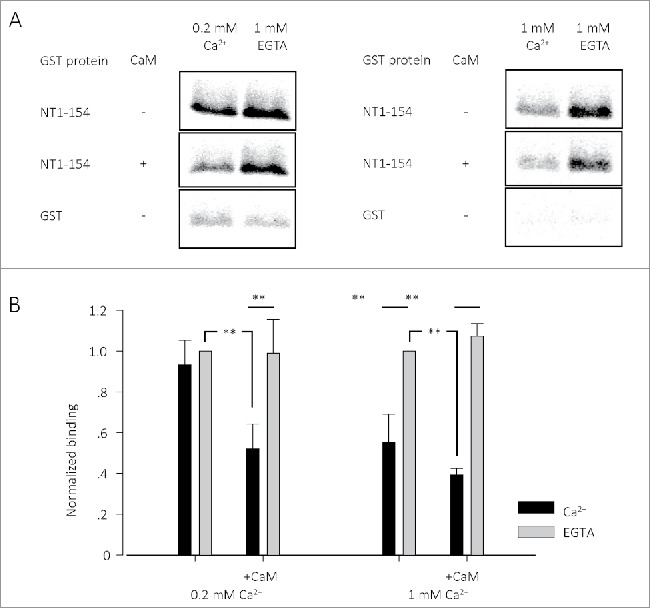
CT1505-1671 (IVT) interaction with GST-NT1-154 in the presence of Ca2+ or EGTA, with or without CaM. (A) Pull down experiment of radioactively labeled pCTs, pulled by GST-NT1-154. Radiograms of the labeled proteins are shown. (B) Average of 4-6 pull down experiments as in A. Each group of experiments is normalized to the conditions of EGTA with no CaM of the same group. 0.2 mM was calculated with the winmaxc32 software. ** p < 0.01. All the experiments were done with 3 µg GST-NT1-154 and CaM.
To confirm and quantitate the NT-pCT interaction, we used recombinant purified proteins. pCT expressed in E. coli separately or in the presence of CaM was unstable at low Ca2+ concentrations (data not shown, and see below). We engineered a construct in which CaM was fused to the C-terminal end of pCT separated by a 14 a.a. residue linker (Fig. 3A and Methods). A mutant C1564S pCT1508-1669 was used in order to generate a protein containing a single Cys for the labeling (CaM sequence does not contain Cys residues). This protein, termed pCT1508-1669(C1564S)-lnk-CaM, was stable at all Ca2+ concentrations. As the interaction partner, we used the full NT (MBP-NT1-154) and NT segments as a fusion to maltose binding protein (MBP). This fusion facilitates protein expression and stability. All MBP-fused proteins also contained a His-tag (Fig. 3A).
Figure 3.

Interaction between pCT1508-1669(C1564S)-lnk-CaM with MBP-NT1-154 studied by MST. MST monitoring of the interaction was done by titrating labeled pCT1508-1669(C1564S)-lnk-CaM (50 nM) with increasing doses of MBP-NT1-154. Each individual data point shows mean±SEM from 2-3 independent determinations. The Ca2+ concentration was adjusted using EDTA and CaCl2 (for 100 µM, 12 µM, 100 nM and 20 nM) at 25° C. The binding curves in B, C and E were fitted to data using the Monolith software. (A) Schematic presentation of the proteins used. (B) Titrating MBP-NT1-154 in virtual absence of Ca2+. The binding buffer contained 1 mM EDTA and no added Ca2+. (C) Titrating MBP-NT1-154 at 100 nM Ca2+. (D) Titrating MBP-NT1-154 at 1 mM Ca2+. The raw data shown indicate a biphasic event. (E) For 1 mM Ca2+, the KD of the low-affinity phase (4340 ± 309 nM) was determined from the fitting of data for MBP-NT1-154 concentrations above 0.1 µM. The high-affinity phase yields a KD of 41.2 ± 4.4 nM (fit not shown). (F) Ca2+-dependent changes in KD of pCT1508-1669(C1564S)-lnk-CaM with MBP-NT1-154 interaction. For 1 mM Ca2+, only the low-affinity KD point is shown.
Interaction between MBP-NT1-154 and pCT1508-1669(C1564S)-lnk-CaM was studied using microscale thermophoresis (MST), at Ca2+ concentrations between 0 to 1 mM. In the virtual absence of free Ca2+ (1 mM EDTA), the MST signal showed a monophasic binding curve with a dissociation constant (KD) of 0.72 µM (Fig. 3B). For all Ca2+ concentrations tested except 1 mM, monophasic binding curves were obtained. The KD mildly increased (pointing at a decrease in the affinity of interaction) until levelling off at 3-4 µM between 10 and 100 µM Ca2+ (Fig. 3C, 3F). At 1 mM Ca2+ a biphasic MST signal curve was obtained (Fig. 3D), with a descending high-affinity component (apparent KD of ˜0.2 µM) and an ascending component, like at all other Ca2+ concentrations, of lower affinity (KD = 4.34 µM; Fig. 2E). Whereas the interpretation of the high-affinity component of binding seen at 1 mM Ca2+ is unclear, it can be safely stated that in the physiologically relevant Ca2+ range, NT and CaM-fused CT associate with sub-micromolar to low micromolar affinity, which weakens as Ca2+ concentration rises (Fig. 3F).
We also pursued characterization of NT-CT interactions using pCT constructs bacterially coexpressed with CaM rather than as a fusion protein. We prepared His-tagged pCT1508-1669 (CaV1.2 CT a.a. 1508-1669) containing the C1564S mutation and coexpressed it with CaM in E. coli. pCT1508-1669 co-purifies with CaM on both Ni2+ chelation and gel-filtration columns (see Methods), as a pCT1508-1669/CaM complex. Nevertheless, this protein complex tends to precipitate at low Ca2+ concentrations, therefore, all experiments were performed at 1 mM Ca2+.
To further quantitate the NT-CT interaction and map the important sites using the pCT1508-1669/CaM complex, we employed a fluorescence-based binding assay (Fig. 4A). A fluorescent pCT1508-1669 was produced by chemically conjugating a fluorescein to the C1570 thiol group of pCT1508-1669. Both C1564 and C1570 are located in pCT1508-1669's predicted EF-hand helix bundle, but are not part of the putative Ca2+- or Mg2+- binding positions.36 Binding of the NT to the pCT1508-1669/CaM complex was assessed using fluorescence polarization (FP) measurements at the fluorescein emission peak. The dependence of FP on molecular mobility enables accurate measurement of the binding of a fluorescent group to another molecular entity which, when associated, form a larger fluorescent complex.
Figure 4 (See previous page).

The NT-pCT interaction site in the NT. (A) Binding isotherms of CaV1.2 MBP-NT proteins with a fluorescein-labeled pCT1508-1669/CaM complex measured by fluorescence polarization (FP). a. Schematic illustration of the fluorescein labeled ‘bait’ and MBP fused ‘target’ molecules used in the assay (label is on pCT1508-1669 C1570). b. Full length MBP-NT1-154 binds pCT1508-1669 with KD = 0.29 ± 0.05 μM. c. MBP-NT1-154 binding is specific to pCT1508-1669/CaM. FP signal is abolished with the addition of excess unlabeled (‘cold’) pCT1508-1669/CaM (5 μM, 100 fold higher than labeled protein). d. No binding was observed for MBP-NT60-120. e. Binding is also observed for a converse FP experiment where a fluorescein labeled NT47-154(C136S) is titrated with the pCT1508-1669/CaM complex. f. MBP-NT1-134 binds pCT1508-1669/CaM with KD = 0.21 ± 0.06 μM. g. MBP-NT135-154 only weakly binds pCT1508-1669/CaM with KD =16.67 ± 28.58 μM. All measurements were performed at 10° C. Solutions contained 1 mM CaCl2 and 25% glycerol. Fitting model assumed a single binding site + non-specific binding. mP denotes the change from baseline (ΔP) polarization in thousandth FP unit. (B) Pull-down experiment of pCT1508-1669 and CaM by MBP-fused NT fragments. a. Schematic illustration of the proteins used: His-MBP-NTs, CaM and His-pCT. b. Final elution from amylose resin used for immobilization is presented. (C) a. NT 129-154 mutations arranged in triples or quadruples which were mutated to alanines. b-c. Top, pull down experiments of radioactive labeled NTs, pulled by GST-pCT in 1 mM Ca2+ (b) or 1 mM EGTA (c). Bottom, average of 3 of pull down experiments normalized to WT NT of each experiment. * P < 0.05, ** P < 0.01. d. Non-normalized average of 3 pull down experiments with EGTA (blue bars) or 1 mM Ca2+ (dark red bars). ** P < 0.01.
The FP assay also demonstrates the interaction between NT and pCT. The binding curve of the fluorescein-labeled pCT1508-1669/CaM and MBP-NT1-154 was monophasic with KD = 0.29 ± 0.05 µM (Fig. 4Ab). When excess unlabeled pCT1508-1669/CaM was added, the FP returned to baseline, proving the interaction to be specific (Fig. 4Ac). The apparently higher interaction affinity and the absence of 2 binding phases, as compared to the MST data in 1 mM Ca2+, may stem from the use of different methods and protein constructs (fused vs. non-fused CaM). Nonetheless, both methods provide clear evidence for a sub-micromolar to low-micromolar affinity interaction between NT and pCT/CaM, even in high Ca2+.
Mapping the sites of NT-pCT-CaM interaction
We mapped the location of the NT-CT binding determinants using the FP assay with pCT/CaM and MBP-fused segments of the NT. As observed in Figure 4Ad, MBP-NT60-120, which includes the NT CaM-binding site, NSCaTE, did not bind the pCT1508-1669/CaM complex. This finding is not consistent with a previous proposal that Ca2+-CaM may be the bridging element between the NT and CT of the channel, binding with its C-lobe to the CT IQ domain and with its N-lobe to the NT CaM binding site. 4 In addition, this result rules out a possible interaction between pCT and the MBP moiety of the tested NT fragments.
Figure 4Ae shows a FP assay in an inverse configuration, where NT segment lacking the first 46 a.a., NT47-154(C136S), was titrated by pCT/CaM (Cys136 in the NT was mutated to Ser leaving only one thiol group in NT, in C147). This truncated NT bound unlabeled pCT1508-1669/CaM with a KD of 0.85±0.37 µM, indicating that the first 46 a.a. of the NT are not essential for the NT-CT interaction. Further FP assays with fluorescein-labeled pCT1508-1669/CaM explored the role of the last 20 a.a. of NT (residues 135-154) which appeared functionally important for the inactivation process (see below). Deletion of this region has little effect on NT-pCT binding (KD = 0.21 ± 0.06 µM for MBP-NT1-134, Fig 4Af). Nevertheless, the MBP-fused NT135-154 alone bound pCT1508-1669/CaM albeit with a much lower apparent affinity (KD = 16.67 ± 28.58 µM, Fig. 4Ag). Thus, the last 20 amino acids of the NT135-154, are sufficient for binding the pCT and may participate in NT-CT binding, but do not constitute the only or the major binding determinant, at least under high-Ca2+ conditions. The low affinity of interaction may also indicate that the construct used lacked additional amino acid residues, located N-terminally to the last 20 a.a., which may constitute a part of the binding site (see below).
In order to further map the CaV1.2 NT-pCT interaction sites, a pull down analysis was performed (Fig. 4B). pCT1508-1669 and CaM were co-expressed together with a particular MBP-fused NT truncation in E.coli (Fig. 4Ba). The interaction with the NT was detected using pulldown with amylose resin in the presence of 1 mM Ca2+. The experiments were performed in 2 steps. For the first step, both the pCT and MBP-NT proteins were pulled down using a Ni2+-chelate resin, as both proteins contained His-tags. Subsequently, the eluted proteins were incubated with amylose beads which pull-down only the MBP-NT moiety. Since CaM is tag-less, it was pulled down via the NSCaTE-containing segments of NT or via its interactions with the pCT. Pull-down results (Fig. 4Bb) showed ternary NT-pCT-CaM complex formation for MBP-NT1-154, whereas no binding of pCT/CaM was observed for the MBP control. The results also demonstrate that the shortest segment that successfully pulled down pCT is NT135-154. MBP-NT60-120, MBP-NT30-99 and MBP-NT60-99, the NSCaTE-containing segments,4 also did not bind to pCT but, as expected, pulled-down CaM by direct binding. In addition, binding was observed for NT with a triple ‘WIR’ mutation (W82A, I86A and R90A), previously shown to abrogate CaM binding to NSCaTE.4,35 These results are in full agreement with the FP mapping. First, they again prove that NSCaTE does not take part in the NT-pCT interaction. Second, they confirm that the 135-154 a.a. segment is sufficient to bind pCT/CaM. Moreover, they underscore the importance of this segment (despite the apparently low affinity seen in direct FP assay), and hint that there are multiple structural determinants on the NT, skirting NSCaTE, that participate in the interaction with the CT. Binding of pCT1508-1669 was observed for MBP-NT1-120 and MBP-NT60-154 but not for MBP-NT60-120. These results imply a complex arrangement of NT. Certain NT-pCT binding determinants are located both within NT1-47 and NT135-154, and possibly in additional parts of the NT.
Studying the role of NT segment proximal to the plasma membrane by mutagenesis
Next, we mutated a.a. triplets or quadruplets to alanine in NT1-154, covering residues 129-154, in order to locate critical a.a. within this segment pivotal for interaction with pCT (Fig. 4Ca ). We used pull-down assays to pull mutated [S35]-Met labeled NT1-154 with GST-pCT/CaM in the presence of 1 mM Ca2+ (high Ca2+) or 1 mM EGTA (Ca2+-free). For the GST-pCT/CaM production, we co-expressed GST-pCT in E. coli with CaM; CaM remains bound to the pCT in the production process. The changes in interaction caused by the alanine mutations are different in high Ca2+ and Ca2+-free buffers. Under high Ca2+ conditions, mutated NT1-154RPPR (a.a. 129-132), NT1-154PIRR (a.a. 142-145) and NT1-154ACI (a.a 146-148) significantly increased the NT-pCT interaction (Fig. 4Cb). In contrast, under Ca2+-free conditions, all mutations except NT1-154RPPR and NT1-154CLT reduced the NT-pCT interaction (Fig. 4Cc). These results suggest that the part of NT close to the plasma membrane contributes to NT-CT binding under low-Ca2+ conditions. This segment, which we provisionally term “the proximal NT,” or pNT, comprises the last 20 a.a. of the NT explored in the FP and pulldown assays, probably together with a few preceding residues, in particular A133 and L134. Since most pNT mutations reduce NT-pCT/CaM interaction in low Ca2+ but not in high-Ca2+ (where some mutations even improve the binding), it is plausible that the affinity of pNT to pCT/CaM is higher in low-Ca2+ than in high-Ca2+. This may in part underlie the apparent low affinity of pCT interaction with the last 20 a.a. of NT observed in the FP assay in the presence of high-Ca2+.
Full analysis of pull-down results for all mutations is shown in Fig. 4Cd. For each radioactively labeled NT1-154 protein, binding is shown as % of input (not normalized to any standard group). These results confirm that high Ca2+ reduces the interaction between NT1-154 and GST-pCT/CaM compared to Ca2+-free conditions (Fig. 4Cd), as seen previously (Figures 2 and 3). Strikingly, all pNT mutations except NT1154RPPR eliminate the Ca2+-sensitivity of NT-pCT binding; the mutated NT1-154 binds pCT/CaM similarly both in the absence of Ca2+ and in high Ca2+ (compare blue and dark red bars in Fig. 4Cd). Importantly, these are the same mutations that, by themselves, reduce the NT-pCT binding in the absence of Ca2+ (NT1-154ALL, NT1-154PIRR, NT1-154ACI and NT1-154SIV). In other words, many mutations along the pNT appear to mimic the effect of high-Ca2+ on NT-pCT/CaM binding. A parsimonious interpretation for all the data of Fig. 4C is that the pNT not only participates in the pCT binding, but also takes part in Ca2+-induced reduction in NT-pCT/CaM interaction.
Functional effects of mutations that alter NT-CT interaction
Given the importance of the pNT for the Ca2+-induced changes in NT-pCT interaction, as revealed in the experiments of Figure 4C, we tested the effects of the pNT mutations on channel function. We introduced the same NT mutations into the intact channel and tested activation (current-voltage relation, I-V, and conductance-voltage relation, G-V) and inactivation (kinetics) properties of the Ca2+ and Ba2+ currents (ICa and IBa, respectively) of mutated α1C in Xenopus oocytes (Fig. 5). None of the mutations significantly affected the I-V and G-V relations (Fig. 5B, second column, and data not shown).
Figure 5.

Effect of NT mutations on ICa and IBa. (A) 129-154 NT a.a. arranged in triples or quadruples which were mutated to alanines. (B) Inactivation of the channel with Ba2+ and Ca2+. From left to right: 1st column, normalized averaged IBa (black) and ICa (red) currents (3-15 oocytes in each group) measured at +20 mV. 2nd column, voltage-conductance (G-V) curve. 3rd column, fraction of peak current after 400 ms depolarization (r400) of IBa and ICa at different voltages. 4th column, fraction of peak current after 400 ms depolarization (r400) of IBa and ICa at 20 mV, *P < 0.05. 5th column, the difference between r400 of IBa and ICa (f400), *P < 0.05.
Both IBa and ICa inactivate during depolarization 37 (see Figure 5B, 1st column from the left). Kinetics of inactivation were routinely characterized using the r400 parameter, calculated as the ratio [current 400 ms after the beginning of the depolarizing pulse]/[ peak current amplitude]. 29,30 Low r400 reports strong inactivation; r400=1 when there is no inactivation. The dependence of r400 on Vm of all the mutated channels has the typical U-shaped form in ICa which disappears in IBa (Fig. 5B, 3rd column). r400 of the mutated channels at 20 mV (Fig. 5B, 4th column) indicates that the mutated channels α1CCLT and α1CPIR/R have significantly stronger IBa and ICa inactivation (changes in IBa were more robust) compared to the control channel (WT channel). The stronger inactivation suggests that replacing these a.a. with alanine removes some kind of obstacle that normally slows the inactivation. Overall, all the pNT mutations either accelerated the inactivation or did not change it, implying that the NT plays a general role in suppression of inactivation. f400, the difference between r400 of IBa and ICa which indicates the Ca2+ component of inactivation, 10,37 was reduced in CLT and PIR/R and also in ALL and SIV mutants (Fig. 5B, note the f400 values on the right). Thus, mutations CLT, PIR/R, ALL and SIV reduce the Ca2+-dependent component of total inactivation.
In summary, the binding experiments showed that several mutations weaken the NT-pCT interaction under Ca2+-free conditions, and some mutations strengthen the interaction in the presence of Ca2+. We compared the changes in binding to altered inactivation of ICa and IBa. This comparison indicates 3 trends caused by the majority of mutations: (i) reduction in NT-pCT binding under Ca2+-free conditions, (ii) an acceleration of IBa inactivation, and (iii) a reduction in f400, which corresponds to a reduction in Ca2+-induced acceleration of inactivation.
Further comparison of IBa inactivation of the mutated channels to the control channel at different time points reveals that the pNT mutations generally have only a small effect on the fast component of IBa inactivation (after 400 ms, Fig. 5 and Fig. 6A), and a stronger effect on the slow component (after 2000 ms or 10000 ms, Fig. 6B and C, respectively) of IBa inactivation. Generally, all mutations in pNT (especially CLT and PIR/PIRR) accelerated the inactivation of IBa, suggesting that this region is important for a slow component of voltage-dependent inactivation (whose nature is not known).
Figure 6.
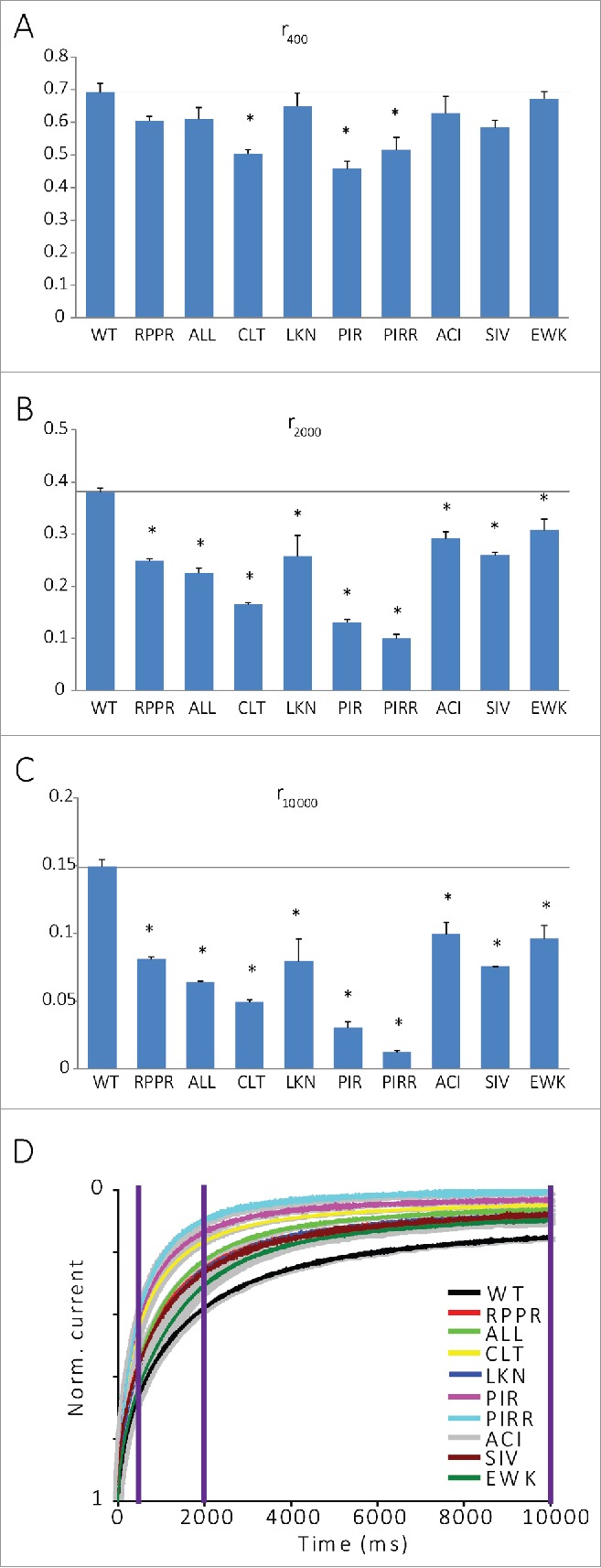
NT mutations have little effect on the fast component of IBa inactivation and a stronger effect on the slow component. (A) Fraction of peak current after 400 ms depolarization (r400) of IBa. *P < 0.05. (B) Fraction of peak current after 2000 ms depolarization (r2000) of IBa. *P < 0.05. (C) Fraction of peak current after 10000 ms depolarization (r10000) of IBa., N is between 3 and 10 cells. *P < 0.05. (D) Normalized averaged IBa currents over 10000 ms.
Discussion
NT-CT interaction and its dependence on Ca2+
Interaction between NT and CT of a channel is neither a new idea nor exclusive for Ca2+ channels. For example, the 4 subunits of the K+ channels KCNQ2 and KCNQ3 have an NT-CT interaction38 and in the rat olfactory CNG channel, the interaction of NT with CT promotes channel activation.39 The involvement of CaM in the NT-CT interaction also appears in other channels, such as CNG, where CaM binding to the NT disrupts the NT-CT interaction, leading to channel inactivation.39,40 However, Ca2+ channels, unlike CNG or K+ channels composed of 4 separate subunits (each with an NT and a CT), have only one NT and CT. The idea of an interaction between NT and CT in α1C, and for a role of this interaction (within a hypothetical “NT-CT scaffold”)10 in channel gating and inactivation, has been already raised in the past.4,10 However, our current work shows that the details of this interaction and its function are different from the previously envisioned ones.
In this study we demonstrate, for the first time, that there is a direct physical interaction between the NT and pCT of CaV1.2 α1C subunit. Furthermore, we show the formation of a ternary complex of NT-pCT-CaM in vitro. Importantly, the NT-pCT/CaM interaction is reduced in the presence of high Ca2+. Quantitative experiments using microscale thermophoresis (MST) indicate an approximately 4-fold reduction in interaction affinity when going from nominally Ca2+-free conditions to 12-100 µM (Fig. 3), which corresponds to physiological Ca2+ levels attained in the Ca2+-CT interaction nanodomain upon Ca2+ entry that follows channel’s opening.2 Ca2+-induced reduction in NT-pCT binding was confirmed in pull-down experiments using different protocols and with purified as well as in vitro synthesized NT and pCT segments.
We were able to assess the affinity of NT-pCT-CaM interaction in the presence of 0.1-1 mM Ca2+ using 2 different purified recombinant pCT constructs, where CaM was either present as a separate but co-purified protein, or it was fused to the pCT via a peptide linker. The apparently higher affinity observed with the non-fused pCT/CaM (˜0.3 µM) vs. the fused pCT-lnk-CaM (˜3-4 µM) could be due to the difference in the protein construct itself or in the method used (FP vs. MST, respectively). Whatever the reason for this difference, it is evident that the binding affinity of NT-CT/CaM interaction in solution is in the sub-micromolar or low-micromolar range. The affinity of interaction in Ca2+-free conditions, as estimated by MST, is sub-micromolar (Fig. 3). Since NT and CT are parts of the same protein in the native channel, the relative effective concentration of these channel termini is very high, thus a robust interaction is predicted between these regions in the intact CaV1.2.
Mapping of the NT-pCT interaction determinants within the NT by deletion and single a.a. mutants points to a complex binding surface comprising the initial (distal) and proximal (close to plasma membrane, pNT) parts of the NT, and possibly additional segments. Furthermore, mutagenesis of the last 25 a.a. of the NT suggests that pNT contributes to the NT-pCT interaction particularly in low Ca2+. Structural elucidation of the NT and pCT interface and the Ca2+-induced changes are an important challenge for the future.
NT-CT interaction may be involved in Ca2+-induced inactivation process
In this study, we have begun the exploration of the importance of the NT-CT interaction for the regulation of the channel. At this stage, we explored the functional consequences of mutations within the pNT, a segment which appears to play an important role in Ca2+-induced changes in NT-pCT interaction. Our results suggest that the NT-CT interaction does not alter the voltage-dependent activation of CaV1.2 but it regulates the channel's inactivation. We compared IBa inactivation (generally considered to represent the voltage-dependent inactivation) and the faster ICa inactivation. The latter occurs when both depolarization and Ca2+ entry take place and is usually viewed as an acceleration, by Ca2+, of the voltage-dependent process, although it is not known whether and how the 2 processes are coupled.41,42 Several pNT mutations, in particular LL134-135, CLT136-138 and PIRR142-145 to alanines, accelerated IBa inactivation, whereas the inactivation of ICa was only slightly affected (Figs. 5, 6). Notably, the same mutations both reduced the NT-pCT interaction in the absence of Ca2+, and eliminated the Ca2+-induced reduction in NT-pCT binding (Fig. 4). All mutations that weakened NT-CT binding in the absence of Ca2+ reduced the net effect of Ca2+ on inactivation kinetics, as indicated by the reduction in f400 parameter (Fig. 5). Thus, mutations that mimicked the effect of Ca2+ on NT-pCT binding, also partially recapitulated the accelerating effect of Ca2+ on inactivation. These findings indicate coupling between Ca2+-induced changes in NT-CT interaction and acceleration of inactivation by Ca2+ entry in the CaV1.2 channel.
We propose a model in which Ca2+ entry may induce a weakening of the NT-CT interaction, and the ensuing rearrangement of the cytosolic domain is allosterically transmitted to the channel's gate(s) to accelerate inactivation. Accordingly, perturbations (such as mutations) that weaken the NT-CT interaction accelerate inactivation already in the absence of Ca2+ and, consequently, diminish the relative accelerating effect of Ca2+. This mechanism may account for all of the Ca2+-induced acceleration of inactivation, or be a part of a more complex mechanism where additional Ca2+/CaM-induced processes take place.
Ca2+ entry may cause a rearrangement of the cytosolic area of the channel
The domain-swapped architecture of the transmembrane domain of voltage-dependent channels places the pore (S5-S6) from domain IV, which is connected to pCT, next to the voltage sensor (S1-S4) from domain I (connected to NT) and ensures cooperativity between these 2 domains and the 2 termini.43 Binding experiments in vitro indicate a weaker NT-pCT interaction under high-Ca2+ vs. Ca2+-free (EGTA) conditions. The interaction between the NT and the pCT in Ca2+ buffer still persists, albeit with a reduced affinity, perhaps in a different configuration. Most of the pNT mutations that we introduced reduced NT-CT binding under Ca2+-free conditions, and weakened the Ca2+-induced reduction of NT-CT binding. This finding implies that it will be easier to produce the hypothesized NT-CT rearrangement in the mutated channels, making it easier for the mutants to reach the inactivated state, resulting in the observed acceleration in IBa inactivation seen in most mutants. Thus, according to our model, the more avid NT-pCT interaction under Ca2+-free conditions may also explain the difference between ICa inactivation vs. IBa inactivation. When Ba2+ is the carrier ion and [Ca2+] remains at its resting low level, the NT-pCT domains are bound tightly and consequently the IBa inactivation is slower than the ICa inactivation.
The role of CaM
Our results argue against the formation of an NT-CT bridge by CaM, previously proposed as a part of the mechanism of Ca2+-CaM-dependent inactivation.4,10 Pull-down experiments show that NT60-99 (which includes the NSCaTE area) binds CaM but does not bind the pCT. We also saw no interaction between NT60-120 and pCT under Ca2+-free conditions or in the presence of Ca2+-CaM 35 (Fig. 4). In contrast, the NT1-154 WIR mutant in which the mutated NSCaTE domain cannot bind CaM, does form such a ternary NT-pCT-CaM complex. The lack of evidence for the formation of a ternary NT60-120-pCT-CaM complex, even in pull down experiments where all 3 proteins are present at over-expression levels, suggests that the NT-pCT interaction occurs directly through a region outside the NSCaTE.
Nevertheless, since CaM is the uncontested Ca2+ sensor for Ca2+-induced inactivation,8 the first event in this process must be the binding of Ca2+ to CaM, already docked at pCT as apo-CaM, followed by structural rearrangements at the pCT.44 We propose that this change is followed by a rearrangement of the NT-pCT-CaM scaffold. Our in vitro experiments do support a major role for CaM in mediating the Ca2+-induced rearrangement of the NT-CT scaffold, because CaM was essential for the Ca2+-induced reduction in NT-CT binding at physiologically relevant Ca2+ levels (Fig. 2).
It is not clear whether and how the NT’s CaM-binding segment NSCaTE is involved in the mechanism described here. Binding of CaM (the same one anchored at the CT, or a separate additional CaM molecule) to the NT, in the presence of elevated Ca2+ concentration, could contribute to the weakening of CaM-independent NT-CT interaction. The possibility that an additional CaM binds to the ternary NT-CT-CaM complex in the presence of Ca2+ is theoretically plausible, because the VGCC acts as a ‘magnet’ for CaM molecules and is locally enriched with CaM in the channel’s nanodomain, up to an estimated 2.5 mM.45 This second CaM can interact with NSCaTE. The affinity of the NScaTE to Ca2+-CaM is below 1 µM. 35 Although this is ˜1000 times lower compared to the IQ domain,46 after channel opening there is enough Ca2+-CaM to saturate the NSCaTE.
Proposed molecular mechanism of CaV1.2 modulation through NT-pCT interaction
We propose a molecular mechanism based on findings from this work and other published studies (Fig. 7). Under conditions of low Ca2+ levels, apoCaM interacts with the IQ and preIQ domains of the closed channel. While CaMC-lobe interacts with IQ, CaMN-lobe interacts with preIQ.47 We cannot rule out the possibility of CaMN-lobe not interacting with the pCT at all, as in the NaV channels.48 We demonstrated an interaction between NT and pCT/CaM; Ca2+ reduces the affinity of this interaction. Depolarization opens the channel, Ca2+ enters through the pore raising the Ca2+ levels in the nano-domain of the channel. The CaM bound to the pCT, now with 4 Ca2+ (Ca2+-CaM), changes its conformation and embraces the IQ with both lobes,49 driving rearrangements within NT-CT scaffold (presented in Figure 7, for illustration purposes, as distancing of NT from the CT). This rearrangement promotes inactivation. How this happens is not yet known, but we further propose that the NT and CT channel domains serve as levers whose movements promote changes in the transmembrane core of the channel (narrowing or broadening it) via S1 (NT) or S6 (pCT), akin to our observations regarding the I-II linker 28 and not as a kind of physical gate blocker. CaM can promote or restrain these movements and thus influence the inactivation of the channel. Mutations in these cytoplasmic areas can change the inactivation either by weakening/strengthening the interaction between distinct cytoplasmic areas, or by disrupting the structural elements involved in the transfer of the movements of the cytoplasmic parts to the pore/gate.
Figure 7.
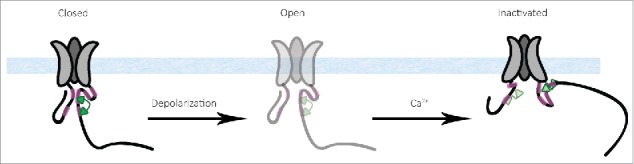
Proposed molecular mechanism of CaV1.2 modulation. CaM is bound to the CT at preIQ and IQ sites, the NT and CT of the channel are interacting, the channel is closed (left). The channel opens (middle), Ca2+ enters, binds to CaM, the NT and CT rearrange and the channel is inactivated (right). In the inactivated conformation, the possibility that a second CaM (light green) becomes bound to the NSCaTE is indicated.
Experimental Procedures
Molecular biology
DNA constructs
cDNA constructs of rabbit heart α1C (X15539), β2b (L06110), α2δ1 (P13806), CaM, CaBP1 and the segments (NT and pCT) are in the pGEM vector which contains 5′ and 3′ untranslated regions (UTRs) from Xenopus β-globin. GST protein constructs are in the pGEX-4 vector. New cDNA constructs were obtained using standard PCR procedures and fully sequenced.
RNA preparation
RNA was prepared by in vitro transcription from linearized DNA (6.5μg), at 37°C. T7 polymerase was used for the preparation of pGEM constructs RNA. The RNAs were kept separately at −80°C and mixed before the injection for the expression of many proteins.
Heterologous expression system in Xenopus oocytes
Maintenance of frogs (Xenopus laevis) and preparation of oocytes was as described.50 Experiments were approved by Tel Aviv University Institutional Animal Care and Use Committee (permits M-08-081 and M-13-002). mRNA (0.5-5 ng/oocyte) of each subunit was injected into the oocyte which was incubated at 20-22°C in NDE solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM HEPES, PH=7.5) for 3-5 days before measuring currents. In case of Ca2+ currents or massive Cl- currents, the oocytes were injected with 25-50 nl of the Ca2+ chelator BAPTA about 15 min prior to the measurement. The BAPTA effect lasts about 2 hours after the injection.
Electrophysiology
Two-electrode voltage clamp (TEVC) techniques were used to measure Ba2+ or Ca2+ currents in Xenopus oocytes, using inactivation and IV protocols. Ba2+ currents were measured in a high Ba2+ solution (40mM Ba(OH)2, 5mM HEPES, 50mM NaOH, 2mM KOH, pH=7.5) and Ca2+ currents were measured with a high Ca2+ solution (40mM Ca(OH)2, 5mM HEPES, 50mM NaOH, 2mM KOH, pH=7.5), which were titrated to pH 7.5 with methanesulfonic acid. Each protocol was measured with a high Ba2+/Ca2+ solution with or without addition of 200 μM Cd2+ (Ca2+ channel blocker, which allows measurement of non-Ca2+ channels currents in the oocyte). The net current was obtained by subtracting the Cd2+ current from the one without Cd2+. Data analysis was performed using the pCLAMP software (Axon Instruments). Graphs and statistical analysis were performed with SigmaPlot (SPSS, Inc.., Chicago, IL).
The current–voltage (I-V) curve was fitted to the Boltzmann equation:
where Ka is the slope factor, V1/2 is the voltage that causes half maximal activation, Gmax is the maximal macroscopic conductance, Vm is membrane voltage, IBa is the current measured at the same voltage, and Vrev is the reversal potential of IBa. The obtained parameters of Gmax and Vrev were then used to calculate fractional conductance at each Vm, G/Gmax, using the equation:
Where G is the total macroscopic conductance at Vm. The conductance–voltage (G-V) curves were plotted with the values of V1/2 and Ka obtained from the fit of the I-V curves, using the following form of the Boltzmann equation:
Protein presparation
GST-fused proteins
The GST-fused proteins were expressed in Escherichia coli BL21 cells. Cultures were grown in a shaker device, in a 2xYT medium (16g/L Trypton, 10g/L Yeast extract, 5g/L NaCl), induced at OD 0.4-0.6 with 0.1 mM IPTG, and followed by further 37°C incubation for 3 h. The cells were separated from the medium by centrifugation at 7000 RPM for 10 minutes at 4°C and preserved for at least one night at −80°C. After thawing, the cells were suspended in Phosphate Buffered Saline (PBS), 1 mM PMSF and 1 mM DNAse, and disrupted using a homogenizer, followed by a micro-fluidizer (Microfluidics, Newton, MA). Next, we separated the proteins from the non-soluble precipitates of the cell by centrifugation at 12000 RPM for 40 minutes at 4°C. All the proteins were produced from the cell’s soup. The proteins were purified on ÄKTAprime (GE Healthcare, USA) with GST-affinity chromatography, followed by gel-filtration chromatography in the experiment buffer. The protein concentrations were determined by Bradford at λ = 595 nm or by absorbance at λ = 280 nm.
pCT1508-1669/CaM and pCT1508-1669 (C1564S)/CaM
E. coli pellet was resuspended in 50 mM sodium phosphate pH 8.0, 0.35 M NaCl, 20% glycerol, DNase (15 units/ml), lysozyme and 1 mM PMSF. Clarified lysate was loaded onto a Ni2+ chelate column (Ni2+CaM, Sigma), pre-equilibrated with buffer A. The column was extensively washed to remove non-specifically bound contaminating proteins prior to elution by a linear gradient of imidazole (5-150 mM). Eluted fractions were immediately collected, concentrated and loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare), pre-equilibrated with GF1 buffer (20 mM Tris pH 7.5, 0.5 M NaCl, 5 mM Imidazole, 25 % glycerol, 10 μM CaCl2). Collected fractions were subjected to overnight TEV proteolysis at 4°C. Proteins were then run again through a Ni2+ chelate column pre-equilibrated with GF1 buffer. Concentrated column flow through was then loaded onto a Superdex-200 gel filtration column pre-equilibrated with GF2 buffer (20 mM Tris-HCl pH 7.5, 125 mM NaCl, 5 mM 2-mercaptoethanol). Selected fractions were then collected, concentrated and stored at −80°C. For the pCT1508-1669 (C1564S)/CaM complex, the protein was subjected to fluorescein labeling prior to loading onto the gel filtration column. For FP measurements, proteins were labeled via thiol modifications at cysteine residues using the reactive dye fluorescein-5-maleimide (Anaspec). Fluorescent dye was freshly dissolved in DMSO and mixed at 1:100 dye:protein ratio with the protein solution (˜100 μM). Following a 2 hour incubation at dark in room temperature, the reaction was stopped by the addition of 1 mM glutathione. Separation of protein from excess dye was then attained by gel filtration/desalting as described for each protein.
pCT1508-1669(C1564S)-lnk-CaM
Rabbit α1C 1508-1669 was fused with calmodulin by a peptide linker (AAADLEVLFQGPLH). The upstream of proximal C terminus has MGSHHHHHHHHGSDYDIPTTENLYFQGS. E. coli pellet was suspended at a ratio of 10 ml buffer per 1 gm pellet in lysis buffer (20 mM Tris-HCl buffer pH-7.5, 0.5 M NaCl, 20% glycerol, 10 mM 2-mercaptoethanol, DNase (15 units/ml), lysozyme, 0.1 % TritonX and 1 mM PMSF). The lysate was homogenized and cells were lysed by ultrasonication. Cell debris were removed by centrifugation for 50 min at 15000 rpm at 4° C. Subsequently, supernatant was loaded on a Ni2+-NTA (Qiagen) column, pre-equilibrated with buffer A (20 mM Tris-HCl buffer pH-7.5, 0.5 M NaCl, 20% glycerol, 10 mM 2-mercaptoethanol) at a flow rate of 1ml/min. The column was extensively washed by buffer A, containing 9 mM imidazole to remove non-specifically bound contaminating proteins prior to elution by buffer A containing 150 mM imidazole. Elution profiles were monitored at 280 nm. Eluted fractions were immediately collected, concentrated and loaded onto a Superdex 200 gel filtration column (GE Healthcare), pre-equilibrated with GF1 buffer. Selected fractions were collected, concentrated and stored at −80° C. These fractions were further used for dye labeling via thiol modification at cysteine residues using the reactive NT-647-Maleimide fluorescent dye (Nano Temper Technologies). The buffer exchange of the protein was done using the provided buffer and spin column following the procedures mentioned by the manufacturer. Fluorescent dye was freshly dissolved in DMSO and mixed at 10:1 dye:protein ratio with the protein solution (˜10 μM) following one hour incubation at dark in room temperature. The unreacted dyes were separated from the protein using the gravity flow column using the buffer 20 mM Tris-HCl pH-7.5, 200 mM NaCl, 25% glycerol, 1 mM DTT. The labeled proteins were flash-frozen in liquid nitrogen in several aliquots and stored at −80° C and subsequently used for microscale thermophoresis experiments.
MBP-NT fusion proteins
MBP-NT60-120, MBP-NT1-134 and MBP-NT1-154 were co-expressed with CaM while MBP-NT135-154 and MBP-NT100-154 were expressed alone. Cells were resuspended in Ni2+ column buffer (50 mM Tris-HCl pH 7.5, 0.3 M NaCl, 20% glycerol, 5 mM CaCl2 (only for MBP-NT/CaM)). Ni2+ chelate column eluted protein was then diluted by the addition an equivalent volume of: 20 mM Tris-HCl pH 7.5, 0.3 M NaCl, 20% glycerol, 20 mM 2-mercaptoethanol, 2 mM EDTA. For the MBP-NTx/CaM proteins, dilution solution also contained 10 mM EGTA in order to assure the separation of the complex. Diluted protein was loaded onto an amylose resin (NEB) column pre-equilibrated with: 20 mM Tris-HCl pH 7.5, 0.3 M NaCl, 20% glycerol, 10 mM 2-mercaptoethanol, 1 mM EDTA. Isolated MBP-NTx proteins were eluted by the addition of 10 mM maltose. Further purification was performed using Superdex 200 gel filtration in: 20 mM Tris-HCl pH 7.5, 200 mM NaCl, 1 mM DTT.
MST analysis
Binding experiments were carried out with Monolith NT.115 (Nano Temper Technologies GMBH, München, Germany). Ten microliters of 100 nM NT-647 labeled pCT1508-1669(C1564S)-linker-CaM were mixed with 10 μl of various concentrations of MBP-NT1-154 (2 nM to 20 μM) and mixed in PCR tubes. Subsequently, the samples were loaded on standard treated capillaries. All the binding experiments were carried out at 25° C, using 60% LED power with 80% MST power. Experiments were carried out in 20 mM Tris-HCl buffer pH-7.5, 200 mM NaCl, 1 mM DTT, 25% glycerol, 0.05% Tween 20. The concentration of Ca2+ was varied from 1 to 0 mM and the concentrations of Ca2+ were maintained using EDTA with CaCl2 solutions. Data analysis was performed with the Monolith software using the thermophoresis and T-jump process.
Fluorescence polarization measurements
Increasing concentrations of unlabeled target protein were mixed with nM concentrations of fluorescein tagged “bait” proteins and incubated on ice in the dark for ˜5 minutes. Steady-state fluorescence polarization measurements were then taken using either an ISS K2 or a Horiba Jobin-Yvon FluoroLog-3 spectrofluorometer at 10° C using excitation and emission wavelengths of 492 and 522 nm, respectively. Five to 10 repeated polarization measurements were taken for each sample with integration times and instrumental parameters yielding a maximum standard deviation of 5% between repeats. Triplicates were taken for each data point in most cases although in some cases, a duplicate was taken due to lack of material. Polarization data was analyzed using Sigmaplot (Systat Software, Inc.). Polarization change from baseline (ΔP) to target protein concentration relations were fitted by nonlinear regression (ligand binding macro) to a one site saturation model by the following equation: ΔP = NX +Bmax x X / (KD + X), where X is the concentration of free target protein (ligand); Bmax, the maximum change in polarization upon saturation; KD, the concentration of ligand required to reach half-maximal binding; N, non-specific binding between the bait and target proteins.
Pull down with S35-Met labeled proteins experiments
Different parts of the CaV1.2 channel were translated in vitro from the appropriate RNA using rabbit reticulocyte lysate, according to standard protocols. The S35-Met labeled lysate and GST-fused proteins were incubated in high Ca2+/Ca2+-free (EGTA) binding buffer (150 mM KCl, 50 mM Tris, 5 mM MgCl2, 1 mM CaCl2/EGTA, pH=7.0) with 0.5% CHAPS (3-[(3-cholamidopropyl) dimethylammonio] - 1-propanesulfonic acid). After incubation for 30 minutes at 30°C, the total volume was completed to 300 µl with the same buffer and incubated for another 30 minutes. Next, a sample of the input (5 µl of the 300 µl inserted into the reaction, 1/60) was removed, to be loaded later on the gel as “input,” and 30 µl glutathione-Sepharose beads (Amersham Pharmacia Biotech) were added. The reaction was incubated again for 30 minutes at 4°C, with rotation, and then washed 3 times from non-specific binding with the same buffer. The GST-fused proteins and any associated molecules were eluted with excess free glutathione in the elution buffer (120 mM NaCl, 100 mM Tris-HCl, pH 8, 30 μl). The proteins were resolved on a 12% SDS polyacrylamide gel, stained with 12.5% Coomassie brilliant blue R-250 (Bio-rad) and processed for further analysis by autoradiography for at least 3 days exposure before the identification of the labeled products with Phosphor-imager (Molecular dynamics).
Pull-down with MBP proteins experiments
In order to co-express the HisTagged pCT1508-1669, CaM and an MBP (and His) tagged fragment of the CaV1.2 NT (MBP-NTx), E.coli cells were transformed with a mixture of either the pET-Duet pCT1508-1669/CaM vector and pET28 MBP-NTx or pET-Duet pCT1508-1669 with CDF MBP-NTx/CaM expression vectors. Cell growth and protein expression induction were similar to the described above. Harvested E. coli pellet was resuspended in binding buffer (50 mM Tris-HCl pH 7.5, 0.2 M NaCl, 5 mM CaCl2, 20% glycerol). Cell lysis was then performed by sonication and was followed by a 30 min centrifugation at 14000 rpm, 4°C. The soluble fraction (˜1 ml) was then removed and incubated with 40 µl of buffer equilibrated Ni2+ chelate beads at 4°C for 1-2 h. Subsequently, beads were pelleted and washed with binding buffer prior to protein elution by 500 µl of binding buffer added with 250 mM imidazole. Eluted protein was then incubated again with 40 µl of buffer equilibrated amylose resin at 4°C for 1-2 h. Subsequent to incubation, beads were washed for 3 times with binding buffer to remove unbound proteins. A sample from the last wash was collected to analyze for traces of the unbound proteins as a control. Proteins were then eluted with 30 µl of binding buffer plus 10 mM maltose and analyzed by Tricine–SDS–PAGE.
Statistics and Data Presentation
Comparison between several groups was performed using one-way analysis of variance (ANOVA) followed by Dunnet's test, using the SigmaPlot software (SPSS Corp.). Data presentation was carried out using Excel (Microsoft) and SigmaPlot (SPSS Corp.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the German-Israeli Foundation for Scientific Research and Development, grant # I-1210-286.13/2012, the Fields Funds for Cardiovascular Research to ND, and by DIP (DFG) and Israel Science Foundation (1519/12) grants to JAH.
References
- 1.Clapham DE. Calcium signaling. Cell 2007; 131:1047-58; PMID:18083096; http://dx.doi.org/ 10.1016/j.cell.2007.11.028 [DOI] [PubMed] [Google Scholar]
- 2.Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 2008; 133:1228-40; PMID:18585356; http://dx.doi.org/ 10.1016/j.cell.2008.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 2000; 16:521-55; PMID:11031246; http://dx.doi.org/ 10.1146/annurev.cellbio.16.1.521 [DOI] [PubMed] [Google Scholar]
- 4.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of Cav channels. Nature 2008; 451:830-4; PMID:18235447; http://dx.doi.org/ 10.1038/nature06529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science 1995; 270:1502-6; PMID:7491499; http://dx.doi.org/ 10.1126/science.270.5241.1502 [DOI] [PubMed] [Google Scholar]
- 6.Zhou J, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Feedback inhibition of Ca2+ channels by Ca2+ depends on a short sequence of the C terminus that does not include the Ca2+-binding function of a motif with similarity to Ca2+-binding domains. Proc Natl Acad Sci U S A 1997; 94:2301-5; PMID:9122189; http://dx.doi.org/ 10.1073/pnas.94.6.2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim EY, Rumpf CH, Van Petegem F, Arant RJ, Findeisen F, Cooley ES, Isacoff EY, Minor DL Jr. Multiple C-terminal tail Ca2+/CaMs regulate CaV1.2 function but do not mediate channel dimerization. EMBO J 2010; 29:3924-38; PMID:20953164; http://dx.doi.org/ 10.1038/emboj.2010.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol 2014; 143:679-92; PMID:24863929; http://dx.doi.org/ 10.1085/jgp.201311153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanevsky N, Dascal N. Regulation of maximal open probability is a separable function of CaVb subunit in L-type Ca2+ channel, dependent on NH2 terminus of a1C (CaV1.2a). J Gen Physiol 2006; 128:15-36; PMID:16801381; http://dx.doi.org/ 10.1085/jgp.200609485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by Gbg and calmodulin via interactions with N- and C-termini of a1C. J Biol Chem 2000; 275:39846-54; PMID:10995757; http://dx.doi.org/ 10.1074/jbc.M005881200 [DOI] [PubMed] [Google Scholar]
- 11.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of CaV1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem 2005; 280:29612-9; PMID:15980432; http://dx.doi.org/ 10.1074/jbc.M504167200 [DOI] [PubMed] [Google Scholar]
- 12.Kobrinsky E, Schwartz E, Abernethy DR, Soldatov NM. Voltage-gated mobility of the Ca2+ channel cytoplasmic tails and its regulatory role. J Biol Chem 2003; 278:5021-8; PMID:12473653; http://dx.doi.org/ 10.1074/jbc.M211254200 [DOI] [PubMed] [Google Scholar]
- 13.Chou JJ, Li S, Klee CB, Bax A. Solution structure of Ca2+-calmodulin reveals flexible hand-like properties of its domains. Nat Struct Biol 2001; 8:990-7; PMID:11685248; http://dx.doi.org/ 10.1038/nsb1101-990 [DOI] [PubMed] [Google Scholar]
- 14.Halling DB, Aracena-Parks P, Hamilton SL. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE 2006; 2006:er1; PMID:16685765 [DOI] [PubMed] [Google Scholar]
- 15.Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends Cell Biol 2000; 10:322-8; PMID:10884684; http://dx.doi.org/ 10.1016/S0962-8924(00)01800-6 [DOI] [PubMed] [Google Scholar]
- 16.Mruk K, Shandilya SM, Blaustein RO, Schiffer CA, Kobertz WR. Structural insights into neuronal K+ channel-calmodulin complexes. Proc Natl Acad Sci U S A 2012; 109:13579-83; PMID:22869708; http://dx.doi.org/ 10.1073/pnas.1207606109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biswas S, DiSilvestre DA, Dong P, Tomaselli GF. Mechanisms of a human skeletal myotonia produced by mutation in the C-terminus of NaV1.4: is Ca2+ regulation defective? PLoS One 2013; 8:e81063; PMID:24324661; http://dx.doi.org/ 10.1371/journal.pone.0081063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Johny M, Yang PS, Niu J, Yang W, Joshi-Mukherjee R, Yue DT. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell 2014; 157:1657-70; PMID:24949975; http://dx.doi.org/ 10.1016/j.cell.2014.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 2001; 411:484-9; PMID:11373682; http://dx.doi.org/ 10.1038/35078091 [DOI] [PubMed] [Google Scholar]
- 20.Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of CaV2.1 channels. Proc Natl Acad Sci U S A 2003; 100:16059-64; PMID:14673106; http://dx.doi.org/ 10.1073/pnas.2237000100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F, Minor DL Jr. Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure 2008; 16:1455-67; PMID:18940602; http://dx.doi.org/ 10.1016/j.str.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mori MX, Vander Kooi CW, Leahy DJ, Yue DT. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+. Structure 2008; 16:607-20; PMID:18400181; http://dx.doi.org/ 10.1016/j.str.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton SL. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem 2000; 275:39786-92; PMID:11005820; http://dx.doi.org/ 10.1074/jbc.M007158200 [DOI] [PubMed] [Google Scholar]
- 24.Mouton J, Feltz A, Maulet Y. Interactions of calmodulin with two peptides derived from the c-terminal cytoplasmic domain of the CaV1.2 Ca2+ channel provide evidence for a molecular switch involved in Ca2+-induced inactivation. J Biol Chem 2001; 276:22359-67; PMID:11294864; http://dx.doi.org/ 10.1074/jbc.M100755200 [DOI] [PubMed] [Google Scholar]
- 25.Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem 2001; 276:30794-802; PMID:11408490; http://dx.doi.org/ 10.1074/jbc.M104959200 [DOI] [PubMed] [Google Scholar]
- 26.Taiakina V, Boone AN, Fux J, Senatore A, Weber-Adrian D, Guillemette JG, Spafford JD. The calmodulin-binding, short linear motif, NSCaTE is conserved in L-type channel ancestors of vertebrate Cav1.2 and Cav1.3 channels. PLoS One 2013; 8:e61765; PMID:23626724; http://dx.doi.org/ 10.1371/journal.pone.0061765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Vogel HJ. Structural basis for the regulation of L-type voltage-gated calcium channels: interactions between the N-terminal cytoplasmic domain and Ca2+-calmodulin. Front Mol Neurosci 2012; 5:38; PMID:22518098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Almagor L, Chomsky-Hecht O, Ben-Mocha A, Hendin-Barak D, Dascal N, Hirsch JA. The role of a voltage-dependent Ca2+ channel intracellular linker: a structure-function analysis. J Neurosci 2012; 32:7602-13; PMID:22649239; http://dx.doi.org/ 10.1523/JNEUROSCI.5727-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 1999; 399:159-62; PMID:10335846; http://dx.doi.org/ 10.1038/20200 [DOI] [PubMed] [Google Scholar]
- 30.Zuhlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the a1C subunit. J Biol Chem 2000; 275:21121-9; PMID:10779517; http://dx.doi.org/ 10.1074/jbc.M002986200 [DOI] [PubMed] [Google Scholar]
- 31.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci U S A 1999; 96:2435-8; PMID:10051660; http://dx.doi.org/ 10.1073/pnas.96.5.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron 2004; 41:745-54; PMID:15003174; http://dx.doi.org/ 10.1016/S0896-6273(04)00081-9 [DOI] [PubMed] [Google Scholar]
- 33.Bernatchez G, Talwar D, Parent L. Mutations in the EF-hand motif impair the inactivation of Ba2+ currents of the cardiac a1C channel. Biophys J 1998; 75:1727-39; PMID:9746514; http://dx.doi.org/ 10.1016/S0006-3495(98)77614-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stephens GJ, Page KM, Bogdanov Y, Dolphin AC. The a1B Ca2+ channel amino terminus contributes determinants for b subunit-mediated voltage-dependent inactivation properties. J Physiol 2000; 525 Pt 2:377-90; PMID:10835041; http://dx.doi.org/ 10.1111/j.1469-7793.2000.t01-1-00377.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benmocha A, Almagor L, Oz S, Hirsch JA, Dascal N. Characterization of the calmodulin-binding site in the N terminus of CaV1.2. Channels (Austin) 2009; 3:337-42; PMID:19713738; http://dx.doi.org/ 10.4161/chan.3.5.9686 [DOI] [PubMed] [Google Scholar]
- 36.Brunet S, Scheuer T, Klevit R, Catterall WA. Modulation of CaV1.2 channels by Mg2+ acting at an EF-hand motif in the COOH-terminal domain. J Gen Physiol 2005; 126:311-23; PMID:16157690; http://dx.doi.org/ 10.1085/jgp.200509333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 1999; 22:549-58; PMID:10197534; http://dx.doi.org/ 10.1016/S0896-6273(00)80709-6 [DOI] [PubMed] [Google Scholar]
- 38.Etzioni A, Siloni S, Chikvashvilli D, Strulovich R, Sachyani D, Regev N, Greitzer-Antes D, Hirsch JA, Lotan I. Regulation of neuronal M-channel gating in an isoform-specific manner: functional interplay between calmodulin and syntaxin 1A. J Neurosci 2011; 31:14158-71; PMID:21976501; http://dx.doi.org/ 10.1523/JNEUROSCI.2666-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varnum MD, Zagotta WN. Interdomain interactions underlying activation of cyclic nucleotide-gated channels. Science 1997; 278:110-3; PMID:9311913; http://dx.doi.org/ 10.1126/science.278.5335.110 [DOI] [PubMed] [Google Scholar]
- 40.Trudeau MC, Zagotta WN. Mechanism of calcium/calmodulin inhibition of rod cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A 2002; 99:8424-9; PMID:12048242; http://dx.doi.org/ 10.1073/pnas.122015999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cens T, Rousset M, Leyris JP, Fesquet P, Charnet P. Voltage- and calcium-dependent inactivation in high voltage-gated Ca2+ channels. Prog Biophys Mol Biol 2006; 90:104-17; PMID:16038964; http://dx.doi.org/ 10.1016/j.pbiomolbio.2005.05.013 [DOI] [PubMed] [Google Scholar]
- 42.Grandi E, Morotti S, Ginsburg KS, Severi S, Bers DM. Interplay of voltage and Ca-dependent inactivation of L-type Ca current. Prog Biophys Mol Biol 2010; 103:44-50; PMID:20184915; http://dx.doi.org/ 10.1016/j.pbiomolbio.2010.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J. Domain-domain interactions in ion channels. J Gen Physiol 2013; 142:347-50; PMID:24043858; http://dx.doi.org/ 10.1085/jgp.201311090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minor DL Jr., Findeisen F. Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 2010; 4:459-74; PMID:21139419; http://dx.doi.org/ 10.4161/chan.4.6.12867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science 2004; 304:432-5; PMID:15087548; http://dx.doi.org/ 10.1126/science.1093490 [DOI] [PubMed] [Google Scholar]
- 46.Findeisen F, Rumpf CH, Minor DL Jr. Apo states of calmodulin and CaBP1 control CaV1 voltage-gated calcium channel function through direct competition for the IQ domain. J Mol Biol 2013; 425:3217-34; PMID:23811053; http://dx.doi.org/ 10.1016/j.jmb.2013.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asmara H, Minobe E, Saud ZA, Kameyama M. Interactions of calmodulin with the multiple binding sites of CaV1.2 Ca2+ channels. J Pharmacol Sci 2010; 112:397-404; PMID:20308803; http://dx.doi.org/ 10.1254/jphs.09342FP [DOI] [PubMed] [Google Scholar]
- 48.Wang C, Chung BC, Yan H, Wang HG, Lee SY, Pitt GS. Structural analyses of Ca2+/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat Commun 2014; 5:4896; PMID:25232683; http://dx.doi.org/ 10.1038/ncomms5896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Petegem F, Chatelain FC, Minor DL Jr. Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol 2005; 12:1108-15; PMID:16299511; http://dx.doi.org/ 10.1038/nsmb1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kahanovitch U, Tsemakhovich V, Berlin S, Rubinstein M, Styr B, Castel R, Peleg S, Tabak G, Dessauer CW, Ivanina T, et al.. Recruitment of Gβγ controls the basal activity of GIRK channels: crucial role of distal C-terminus of GIRK1. J Physiol London 2014; 592:5373-90; PMID:25384780; http://dx.doi.org/ 10.1113/jphysiol.2014.283218 [DOI] [PMC free article] [PubMed] [Google Scholar]