

Abstract
Dynamic modification of heptad-repeats with the consensus sequence Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7 of RNA polymerase II (RNAPII) C-terminal domain (CTD) regulates transcription-coupled processes. Mass spectrometry analysis revealed that K7-residues in non-consensus repeats of human RNAPII are modified by acetylation, or mono-, di-, and tri-methylation. K7ac, K7me2, and K7me3 were found exclusively associated with phosphorylated CTD peptides, while K7me1 occurred also in non-phosphorylated CTD. The monoclonal antibody 1F5 recognizes K7me1/2 residues in CTD and reacts with RNAPIIA. Treatment of cellular extracts with phosphatase or of cells with the kinase inhibitor flavopiridol unmasked the K7me1/2 epitope in RNAPII0, consistent with the association of K7me1/2 marks with phosphorylated CTD peptides. Genome-wide profiling revealed high levels of K7me1/2 marks at the transcriptional start site of genes for sense and antisense transcribing RNAPII. The new K7 modifications further expand the mammalian CTD code to allow regulation of differential gene expression.
Keywords: CTD code, C-terminal domain (CTD), lysine methylation, RNA polymerase II
Introduction
The C-terminal domain (CTD) of eukaryotic RNA polymerase II (RNAPII) consists of an array of heptad-repeats with the consensus sequence Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7.1,2 The CTD undergoes dynamic changes of its phosphorylation pattern during the transcription cycle.3-5 RNAPII with an unphosphorylated CTD is recruited to the pre-initiation complex6,7 and subsequently phosphorylated by Cdk7 kinase at serine 5 and serine 7 residues in a not yet exactly determined number of heptad-repeats.8-10 The release of RNAPII in the transcription mode is associated with phosphorylation of CTD serine 2 residues by Cdk9, and other factors, as DSIF and NELF.11-13 Phosphorylation marks in CTD further coordinate the recruitment of RNA maturation factors, as capping enzyme, splicing factors, and the 3′ RNA processing machinery.14,15 Together, the phosphorylation and dephosphorylation of CTD heptad-repeats facilitate the coordinated recruitment and dissociation of cellular factors during the transcription cycle.16-18
The CTD of yeast S. cerevisiae and S. pombe is composed of 26 and 29 heptad-repeats, which almost all comply with the consensus sequence. In contrast, vertebrates have evolved a 52 repeats long CTD with 21 consensus repeats in the proximal and 31 non-consensus repeats in the distal part of CTD.4,19 Serine at position 7 is replaced by lysine (K) in 8 non-consensus repeats (repeat 35, 38, 39, 40, 42, 45, 47, and 49). The number and positions of K7 residues is conserved throughout in vertebrates suggesting a specific function in gene regulation.19,20 K7 residues are targets of posttranslational modifications including acetylation, mono-, di-, and tri-methylation, but also ubiquitination, SUMOylation, and neddylation. Acetylation of K7 residues in CTD has recently been reported to be required for the transcriptional activation of the immediately response genes c-Fos and Egr.21 Acetylation of CTD of transcribing RNAPII in c-Fos and Egr genes occurs downstream of the transcriptional start and is probably mediated by the cellular acetyltransferase p300. Knockdown of p300 or replacement of all 8 lysine residues in CTD strongly impaired the induction of c-Fos and Egr genes upon EGF receptor signaling, but did not affect expression of other house keeping genes.21
Here we show that K7 residues in CTD are also target of mono-, di-, and tri-methylation. We further show that K7 residues in the same CTD-heptad can be alternatively acetylated or methylated. While acetylated and di- and tri-methylated K7 residues are present in the hyperphosphorylated form of RNAPII, monomethylation of K7 residues occurs also in the CTD of the hypophosphorylated form of RNAPII. Finally, we show combined methylation and acetylation of K7 residues in adjacent CTD heptads. We conclude that K7 residues in CTD, similar to K residues in histone tails, are targets of complex posttranscriptional modification.
Results
Mass spectrometric analysis of lysine modifications in CTD
The mammalian CTD of RNAPII contains 8 lysine (K) and 2 arginine (R) residues (Fig. 1, Figure S1A). To study modification of lysine residues in CTD we performed mass spectrometric analysis of the large subunit Rpb1. Rpb1 was enriched from extracts of 3 × 108 Raji cells by immunoprecipitation (IP) with anti-CTD specific antibodies, separated by polyacrylamide (PAA) gel electrophoresis, and digested with trypsin (Fig. 1, Figure S1B). The IP enriched the hyperphosphorylated II0 form of RNAPII but also the IIA form. Enrichment of the IIA form occurs probably due to few or a single serine 5 phosphorylation in RNAPIIA being sufficient for the precipitation of the IIA form. Trypsin digestion fragmented the CTD in 10 peptides of various lengths (Figure S1A). The fragment consisting of repeats 2 – 31 contains an arginine at the C-terminus but was too large for a modification specific analysis. Similarly, the coverage rate for peptides containing heptad-repeats 32 – 35 was consistently very low in our analysis and possible modification states of K7 residues in heptad-repeat 35 remain therefore uncertain. All other peptides with K7 residues in heptad-repeats 38, 39, 40, 42, 45, 47, and 49 were covered and therefore included in our analysis (Fig. 1). Trypsin cleaves peptide chains mainly at the carboxyl side of the amino acids lysine and arginine. The proteolytic digest of proteins by trypsin is sensitive to methylation of K residues. Cleavage is observed after unmethylated and monomethylated K residues but not after di- or tri-methylated K residues. An example for the inhibition of trypsin digestion by trimethylation is seen for the K7 residue of heptad-repeats 40, which remains connected with its N-terminal repeat, if the K residue is trimethylated (Fig. 1B). The mass spectrometric analysis revealed modification of K7 residues in CTD by acetylation in repeats 39 and 42. Acetylation has also been assigned to K7 in heptad-repeats 46/47 and 48/49, which cannot be discriminated using mass spectrometry. However, the MS/MS spectra could potentially also be explained by the acetylation residing at Ser5 within this repeat. However, Ser5 acetylation of CTD has never been described before. Monomethylation of K7 residues was detected in repeats 38, 39, 40, 42, and 47/49, dimethylation in repeat 38, and trimethylation in repeat 40 (Fig. 1B, C; Figures S2 – S5). Acetylation or mono-, di-, and tri-methylation were not detectable for all K7 residues in CTD. This lack of modification could have technical, cell type specific, or other reasons. Therefore, the list in Figure 1B, C has to be considered as a positive list and does not exclude the occurrence of acetylation and methylation in other K residues in heptad-repeats of CTD.
Figure 1.

Mass spectrometry analysis of RNAPII CTD. (A) Scheme of RNAPII and distribution of lysine (K) and arginine (R) residues in CTD. (B) Modified peptides identified for CTD after tryptic digestion of purified Rpb1. Phosphorylated amino acid residues are marked by blue, acetylated by yellow, and mono-, di-, and tri-methylated residues by green boxes. Peptides containing heptad-repeats 46 – 47 and 48 – 49 are identical and cannot be discriminated by mass spectrometry. All peptides were also detected mono- and di-phosphorylated without K modification (data not shown). (C) Overview of modifications determined for various K residues in CTD.
Interestingly, acetylation and di- or tri-methylation of K7 residues was detectable exclusively in phosphorylated CTD peptides. This stringent association with phosphorylated peptides cannot be explained by the prior enrichment of phospho-peptides on a TiO2 column for our analysis, because a large portion (>50 %) of detected CTD peptides still turned out to be non-phosphorylated even after the phospho-enrichment. The peptides shown in Figure 1B can also be detected phosphorylated without modification of K7 residues (data not shown). Therefore, acetylation as well as di- and tri-methylation of K7 residues either occur after phosphorylation of CTD or CTD phosphorylation prevents extensive demethylation of the K7 residues. We conclude that the degree of K7 modification may be coupled to phosphorylation of CTD and occur mainly in transcriptionally engaged polymerases.
We next asked whether acetylation and methylation of K7 residues correlate with specific phosphorylation patterns in CTD? For heptad-repeats 47 and 49, acetylation occurred in combination with phosphorylation of Ser2 and Ser5, while monomethylation of K7 residues occurred combined with Tyr1 and Thr4 phosphorylation. This could be a first example for the interdependence of CTD phosphorylation and methylation/acetylation.
The K7 residue in repeats 39 and 42 can alternatively be acetylated or methylated. Acetylation and monomethylation of K7 residues does occur in 2 consecutive K7 residues of the heptads 39 and 40 in the same peptide. We conclude that acetylation and mono-, di-, and tri-methylation of K7 residues are common features of mammalian CTD that are likely to occur in the transcriptionally engaged II0 form of RNAP, whereas monomethylation also appears in the RNAPIIA form. This in depth mass spectrometry analysis of K7 residue modifications provides new insights in the huge complexity of CTD regulated cellular processes.
Generation of CTD Kme1/Kme2 specific monoclonal antibody 1F5
To analyze the modifications of lysine residues in CTD in more detail, we attempted to raise monoclonal antibodies (mAbs) against methylated K7 residues in CTD. We immunized rats with synthetic CTD peptides modified by acetylation, or mono-, di-, and tri-methylation at lysine residues (Fig. 2A). The K residues in these peptides were flanked by CTD consensus sequences (see Methods for details). The mAb 1F5 was isolated from rats immunized with a CTD peptide containing a dimethylated K residue. Titration of mAb 1F5 in ELISA experiments revealed a 10-fold higher affinity toward dimethylated compared to monomethylated K residues (Fig. S6), while trimethylated K residues or other CTD peptides were not recognized (Fig. S7). The mAb 1F5 recognized wild-type RNAPII containing K residues in CTD, whereas recombinant versions of RNAPII having replaced all 8 K residues by alanine or containing only consensus repeats were not recognized (Fig. S8). Further analysis of mAb 1F5 in ELISA experiments revealed that phosphorylation of the N-terminal serine 5 residue can block the binding, while phosphorylation of the C-terminal serine 2 residue was not inhibitory (Fig. S7).
Figure 2.
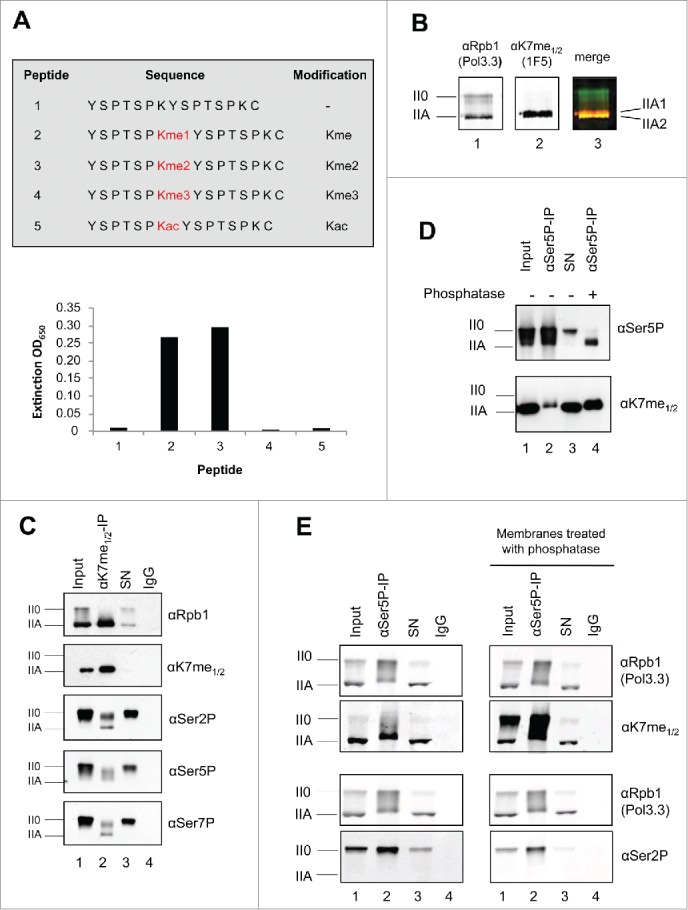
Kme1/2 specific mAb production for CTD. (A) Survey of synthetic CTD peptides used for immunization and characterization of CTD specific mAbs. mAb 1F5 was raised against a dimethylated K residue. mAb 1F5 showed reactivity toward mono- and di-methylated K, but not toward unmethylated, trimethylated, or acetylated K residues of CTD in ELSIA experiments. (B) mAb 1F5 strongly reacts with the IIA form of RNAPII in Western blot analysis, but not with the II0 form. RNAPIIA is composed of a slowly migrating, strongly methylated IIA1 and a faster migrating, unmethylated or weakly methylated IIA2 form. mAb Pol3.3 recognizes Rpb1 outside the CTD. (C) mAb 1F5 strongly precipitates the IIA form of RNAPII, and partly the II0 form phosphorylated at Ser2, Ser5, and Ser7 residues. (D) Phosphatase treatment unmasks the 1F5 epitope in RNAPII0. Rpb1 was precipitated with a Ser5P specific mAb from whole cell extract (Input), treated with alkaline phosphatase, and subsequently analyzed for K methylation with mAb 1F5. SN, supernatant of IP. (E) RNAPII was precipitated with a Ser5P specific mAb. CTD K7 methylation of RNAPII0 was analyzed with and without treatment of membranes with alkaline phosphatase.
Methylation of the IIA and II0 form of RNAP
We next studied the reactivity of mAb 1F5 for the various phosphorylated forms of RNAPII in cellular extracts. The antibody strongly recognized the IIA but hardly the II0 form of RNAPII (Fig. 2B). Interestingly, the IIA form separated in a slowly migrating IIA1 form with high reactivity (red) toward the mAb 1F5 and a faster migrating less reactive IIA2 form (yellow). This suggests that already the CTD of the IIA form is methylated and that the methylation can be correlated to the slower migrating IIA2 form in PAA gel electrophoresis. Immunoprecipitation experiments revealed that the mAb 1F5 precipitates almost the entire fraction of RNAPIIA, whereas only minor fractions of the Ser2, Ser5, and Ser7 phosphorylated RNAPII0 were precipitated (Fig. 2C). The weak immunoprecipitation efficiency of mAb 1F5 for RNAPII0 was unexpected, because many of the K7 methylated CTD peptides were also phosphorylated in our mass spectrometry analysis and should be derived from the RNAPII0 form. We wondered therefore, if the methylated K7 residues in RNAP II0 might be masked by phosphorylation of adjacent residues. To investigate this possibility we precipitated RNAPII with a Ser5P specific mAb, incubated the precipitate with alkaline phosphatase, and tested subsequently the reactivity of the 1F5 mAb. Phosphatase treatment shifted the Ser5P positive RNAPII0 form to the RNAPIIA form and thereby strongly increased the reactivity of the mAb 1F5 (Fig. 2D, compare lane 2 and 4). The masking of K7 methylation in the RNAPII0 form was further confirmed when western blot membranes were treated with alkaline phosphatase (Fig. 2E). A strong increase for the reactivity of mAb 1F5 for the RNAPII0 form was seen after phosphatase treatment. This strong increase was detectable although the removal of phospho marks in CTD was incomplete (see Ser2-P marks). Treatment of membranes with phosphatase did not affect the reactivity of mAb Pol3.3 toward Rpb1. The result suggests that the accessibility of the mAb 1F5 to methylated K7 residues is blocked by CTD phosphorylation and that the mass spectrometry analysis results in regard to CTD K7 methylation are reliable.
We next asked whether methylation of CTD occurs in the cytoplasm of cells? For this purpose we fractionated cytoplasmic and nuclear extracts of cells. We found that a large portion of the RNAPIIA was present in the cytoplasm, while the entire RNAPII0 and a small sub-fraction of RNAPIIA were present in the nucleus. Concordant with the distribution of the RNAPIIA form, methylation of CTD K7 residues was highest in the cytoplasmic RNAPII (Fig. 3A). The proper fractionation of cytoplasmic and nuclear proteins was controlled by mAbs for CTD Ser2P, H3K4me3, and tubulin. The reactivity of mAb 1F5 with cytoplasmic RNAPIIA was further confirmed in immunofluorescence (IF) experiments after fixation of cells with formaldehyde. While CTD Ser2P, Ser5P, and Ser7P specific mAbs recognized only nuclear RNAPII, mAb 1F5 stained in addition also RNAPII in the cytoplasm of cells (Fig. 3B, white arrow).
Figure 3.
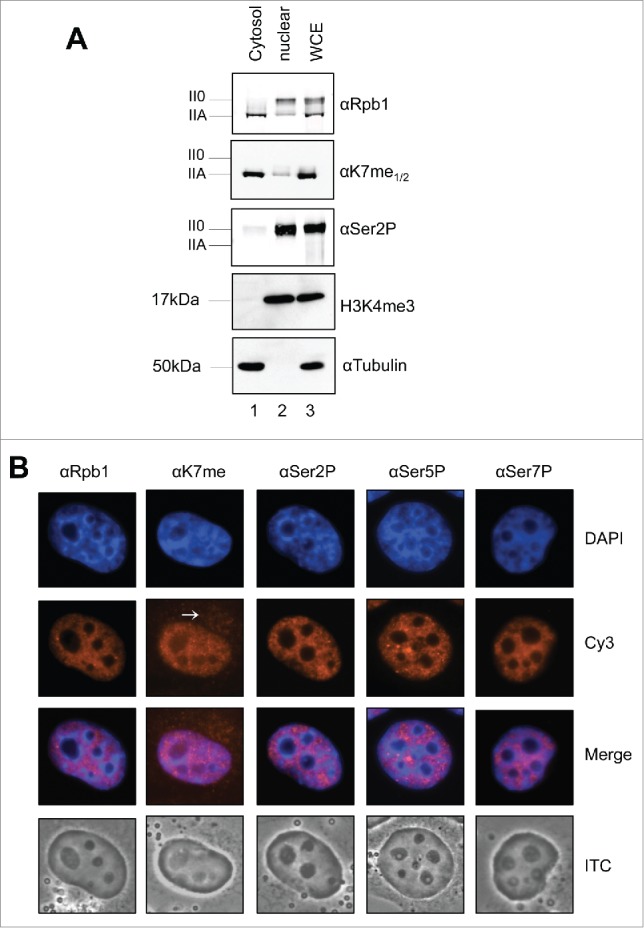
Cytoplasmic - nuclear distribution of Kme1/2 methylated RNAPIIA. (A) Cytoplasmic and nuclear extracts of H1299 cells were prepared and analyzed in Western blots. The ratio of RNAPIIA to RNAPII0 in the cytoplasm and nucleus was visualized with mAbs Pol3.3 and Ser2P. H3K4me3 served as nuclear markers and tubulin as cytoplasmic marker. (B) Immunofluorescence staining of formaldehyde fixed H1299 cells. Ser2P, Ser5P, and Ser7P modified CTD was observed exclusively in the nucleus, while significant CTD Kme1/2 staining was observed also for the cytoplasm. Merge: overlay of DAPI and Cy3; PhC: phase contrast.
Stability of CTD K7 methylation
We next studied the stability of K7 methylation in cells, if phosphorylation of CTD was inhibited by a kinase inhibitor or upon salt stress. We asked, if the removal of phospho-marks in CTD and the shift from the II0 to the IIA form can uncover methylated K residues in CTD in vivo and increase the reactivity for mAb 1F5. Treatment of cells with the kinase inhibitor flavopiridol induced a dramatic change in the phosphorylation stage of RNAPII and a rapid shift from the II0 to the IIA form (Fig. 4A). The increase of RNAPIIA goes in hand with the reactivity of mAb 1F5 toward the IIA form and is in concordance with the observation above that CTD phosphorylation can mask the recognition of methylated K7 residues by mAb 1F5. A shift from the II0 to the IIA of RNAPII is also induced after stressing cells with high salt concentration (2-fold PBS) (Fig. 4B). Again, the increase of levels of the IIA form after 15 to 45 min salt stress was accompanied with an enhanced reactivity of the mAb 1F5 for the IIA form. Even though less likely, for the flavopiridol and salt stress experiments we cannot exclude de novo methylation of Rpb1 after shift of RNAP from the II0 to the IIA form. All together, from the results we conclude that the II0 form of RNAPII is substantially modified by K7 methylation in vivo, but that the K7me1/2 mark is masked in RNAPII0. Our results further suggest that the K7me1/2 marks can remain in the CTD, if the II0 shifts to the IIA form. Finally, the results are in agreement with our mass spectrometry analysis, which revealed extensive phosphorylation of CTD peptides in combination with the K7me1/2 mark. We should note, however, that all the K7 containing peptides were also found phosphorylated in our mass spectrometric analysis without being modified at K7 residues.
Figure 4.
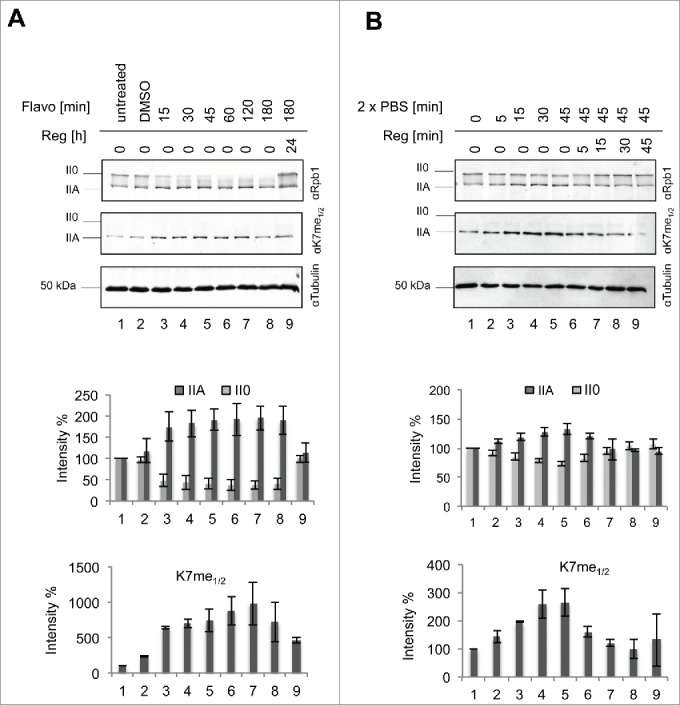
Dephosphorylation of RNAPII0 by kinase inhibitor or salt stress increase RNAPIIA levels and the reactivity of mAb 1F5 toward the CTD specific epitope Kme1/2. H1299 cells were treated (A) with the CTD specific kinase inhibitor flavopiridol (800 nM) or (B) double concentration of PBS for indicated time periods. For regeneration cells were washed after 180 min (flavopiridol) or 45 min (2 x PBS) and analyzed at indicated time points. Levels of the IIA and II0 of RNAP and of K7me1/2 modification were determined by Western blot analysis and blotted as intensity.
Genome-wide distribution of CTD K7 methylated RNAPII
Despite the failure of mAb 1F5 to recognize and immunoprecipitate the II0 form of RNAPII we asked, if this antibody is able to recognize RNAPII associated with chromatin in chromatin immunoprecipitation (ChIP) experiments? The occupancy profiles of total RNAPII and of the CTD K7me1/2 marks were examined by ChIP-seq in the human B cell line Raji. The genome-wide occupancy analysis revealed high levels of RNAPII and CTD Kme1/2 marks at transcription start sites (TSS) (Fig. 5A). While substantial amounts of RNAPII were detectable also in the body of genes and 3′ of the polyadenylation signal, the signals for K7me1/2 were low in the body of genes and increased only slightly 3′ of the polyA site. This result is consistent with the limitations of mAb 1F5 to recognize methylated K7 residues in II0 form of RNAPII. We previously showed increased CTD Tyr1 phosphorylation for antisense transcribing RNAPII at promoters,22,23 while the rates of Ser5 and Ser7 phosphorylation for sense and anti-sense (AS) RNAPII were similar. For K7me1/2 marks we also noticed increased levels for antisense RNAPII (Fig. 5B). Examples are shown for the highly expressed genes RPL22L1, IER5 and EIF1B in Figure 5C. However, although this result seemed pronounced for several model genes, we could not confirm this status genome-wide as we did for Tyr1P.22,23 Because mAb 1F5 recognizes only the IIA form of RNAPII in IP experiments we propose that RNAPII detected at the transcription start sites (TSS) corresponds largely to the IIA form and has not yet been shifted by CTD hyperphosphorylation to the II0 form. Unfortunately, the methylation state of the RNAPII0 form could not be analyzed in ChIP experiments with mAb 1F5. The accessibility of modification specific epitopes by antibodies is a general issue of IP experiments. Epitopes can be impaired by other adjacent modifications or simply masked by binding to other cellular factors.
Figure 5.

Genome-wide RNAPII and CTD K7me1/2 methylation profiles. (A) Diagram summarizing the average transcription unit with representative clusters for RNAPII and CTD K7me1/2 occupancy profiles of top 5%, 20%, 20–80% of signals across the genome using average transcription unit analysis (see Methods). (B) Distribution of RNAPII upstream and downstream of TSS for the top 20% of genes >4kb. (C) Examples of gene RPL22L1 (Ribosomal Protein L22-like-1), IER5 (immediate early response 5) and EIF1B (eukaryotic translation initiation factor 1B) showing RNAPII distribution and signals for Tyr1P, Ser7P, Ser5P, Ser2P, and CTD Kme1/2. Strand (+) shows genes in 5' – 3′ and strand ( − ) in 3′ − 5' orientation.
Discussion
Modifications of K7 residues in non-consensus heptad-repeats of CTD represent a new layer of gene regulatory mechanisms of the RNAPII transcription machinery in mammals. Mass spectrometry analyses revealed modification of K7 residues in CTD by mono-, di-, and tri-methylation, and confirmed the previously reported acetylation.21 Six of 8 K7 residues revealed the potential of multiple modifications, while the K7 residues in heptad-repeats 35 and 45 were devoid of modifications. However, we cannot rule out that the lack of modifications in these 2 K7 residues might have technical reasons.
Interestingly, the large majority of K7 modifications were found only in phosphorylated CTD peptides. The bias for phosphorylated peptides cannot result from the TiO2-enrichment protocol that we applied for enrichment of CTD phospho-peptides. Even with this protocol the detection rate for unphosphorylated CTD peptides was still higher than for phosphorylated CTD peptides. K7 modification in non-phosphorylated CTD peptides was restricted to monomethylation, which was detectable only in heptad-repeats 47 and 49. Thus, the strong reactivity of mAb 1F5 toward the IIA form of RNAP is caused probably by monomethylation of these 2 K7 residues. All other modifications for K7 were detectable only in phosphorylated RNAPII0 derived CTD peptides. Di-, and tri-methylation as well as K7 acetylation occurred in different mixtures with phosphorylation of CTD peptides. For example, K7 monomethylation and acetylation occurred in various combinations with phosphorylation in heptad-repeats 46/47 and 48/49, and a bias of K7 acetylation and monomethylation with specific phosphorylation signature of heptad-repeats was observed. An interesting future question will be, how the transition from monomethylation of K7 residues in heptad-repeats 47 and 49 to acetylation is regulated, and where in the transcription cycle this transition occurs.
The presence of monomethylated but not acetylated K7 residues in the IIA form of RNAPII suggests that K7 acetylation may occur only after phosphorylation of CTD. This assumption would further imply that K7 acetylation occurs only in transcriptionally engaged RNAPII and may be involved in gene regulatory mechanisms (Fig. 6).
Figure 6.
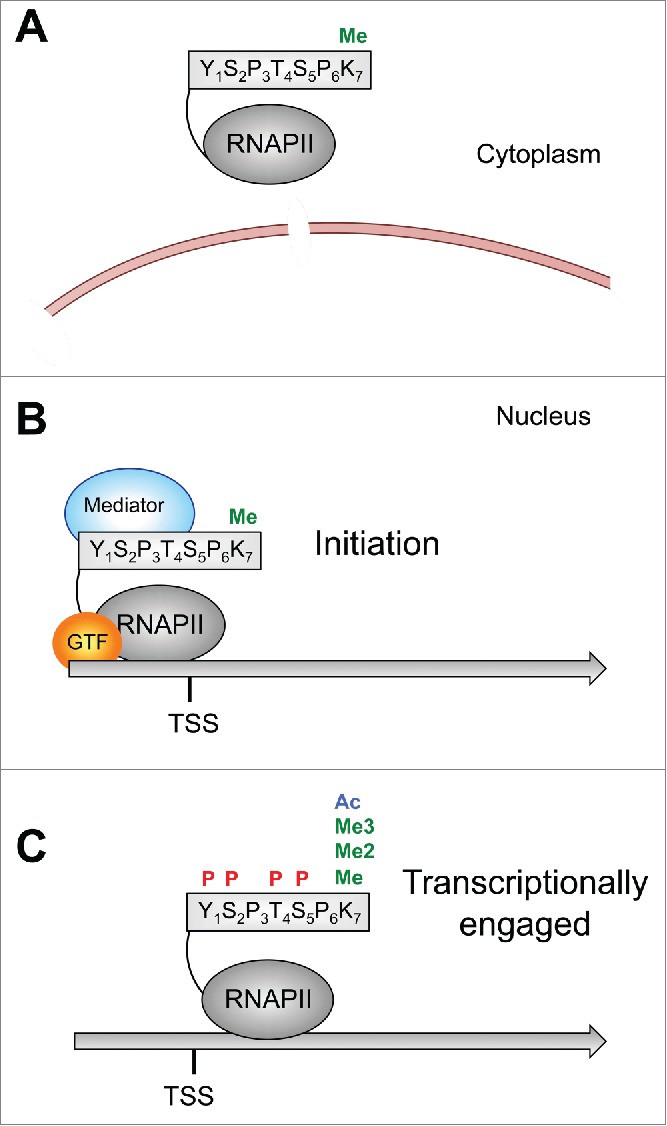
Model for CTD K7 modifications. (A) Monomethylation of CTD K7 occurs already in the cytoplasm. (B) K7 monomethylated RNAPIIA binds to TSS sequences. (C) After initiation RNAPIIA becomes increasingly phosphorylated and shifts to the RNAPII0 form. The II0 form can be further modified at K7 residues by di- and tri-methylation or acetylation.
This model is supported by a recent study describing a function of CTD K7 acetylation for growth factor induced gene expression.21 CTD acetylation specifically enriched downstream of the TSS of RNAPII occupied genes genome-wide. Mutation of K7 residues in CTD or treatment of cells with an inhibitor of the acetyltransferase p300 caused a loss of epidermal growth factor induced expression of c-Fos and Egr2, 2 immediate-early genes with promoter-proximal paused RNAPII, while expression of house keeping genes was not affected.21 Thus, CTD K7 methylation and acetylation might be involved in the regulation of specific cellular genes, which are not essential for cell viability. This is in line with our previous observation that K7 residues in mammalian CTD can be replaced by serine residues without affecting cell viability in cell culture.24
We also observed di- and tri-methylation of K7 residues in several phosphorylated CTD peptides, or that monomethylation and acetylation can occur in K7 residues in adjacent heptads of the same peptide. The different K7 residue modifications observed in mammalian CTD have many facets in common with modification of K residues in histone tails.25 K residues in histone tails display a large and complex repertoire of modifications, which can be associated with activation and repression of gene activity.26 While many of the enzymes involved in K modification of histone tails are well studied, only little is known about the enzymes involved in modifying K7 residues in CTD. So far, only p300 has been identified as a K7 specific acetyltransferase.21 The identification of other K7 acetyltransferase, and the enzymes involved in deacetylation, methylation, and demethylation of K7 residues in CTD is still open. However, it would be no surprise, if in addition to p300, other histone modifying enzymes would be involved also in CTD K7 modification.
What could be the function of the various K7 modifications in CTD, if K7 residues are not essential for cell viability? The heptad-repeat structure is a hallmark of all RNAPII molecules of metazoa and the CTD probably contributed significantly to the evolution of complex multicellular organisms. All vertebrates have a CTD with 52 heptad-repeats and this number of repeats remained stable for several hundred millions of years. In addition to the number of heptad-repeats also the number and the position of heptad-repeats with K7 residues remained constant. This raises the questions of whether non-consensus heptad-repeats in CTD, including those with K7 residues, could be important for the developmental program of vertebrates and if the spacing and number of K7 residues, as well as the different K7 modifications, are critical for cell type specific differentiation programs.
The notion that non-consensus heptad-repeats in CTD can regulate specific gene classes is supported by the recent observation that the asymmetrical methylation of a single arginine residue in heptad-repeat 31 of mammalian CTD by methyltransferase CARM1 regulates the expression of a variety of small snRNAs and snoRNAs.27 In analogy, it appears likely that the various modifications of K7 residues identified in this analysis may similarly contribute to fine tuning of gene expressing by RNAPII, and that these modifications are placed in K7 residues of CTD dependent on cell signaling and epigenetic programming of cells.
Materials and Methods
Cell culture
H1299 cells were grown in Dulbecco's modified Eagle's medium (DMEM) and Raji B-cells in RPMI medium supplemented with 10% FCS, 100 µg/ml streptomycin, 100 U/ml penicillin and 2 mM L-glutamine at 37°C with 8% or 5% CO2. Stable transfected Raji cell lines were selected and maintained by G418 (Invitrogen).28 The expression of the recombinant Rpb1 was induced by the removal of tetracycline. The endogenous RNAPII was inhibited by α-amanitin (2 µg/ml).
Antibodies
Antibodies α-tubulin (T6557) (Sigma), HA (3F10) (Roche), H3K4me3 (Ab8580) (Abcam) were commercially available. Monoclonal antibodies against CTD Tyr1P (3D12),29 Ser2P (3E10), Ser5P (3E8), Ser7P (4E12) and Rpb1 (Pol 3.3),30 Pes1 (8E9), Bop1 (6H11)31 were described previously. Pol 3.3 recognizes an epitope in Rpb1 outside of the CTD. The rat monoclonal antibody 1F5 (IgG2a) against CTD K7me1/2 was produced toward the CTD-peptide (YSPTSPKme2YpSPTSPSC) coupled to ovalbumin for immunization (Peptide Specialty Laboratories GmbH, Heidelberg, Germany).
Cloning of the CTD mutants
The CTD sequences of (rec WT, rec WT K/S, Con48) with an optimized codon usage were synthesized by Gene Art (Regensburg) and cloned into the LS*mock vector.32 The constructs were transfected into Raji cells by electroporation using 1 × 107 cells (10 µg plasmid, 960 µF, 250 V) and selected with G418 (1 mg/ml).
Immunofluorescence microscopy
H1299 cells were seeded on coverslips and grown over night with complete DMEM medium. Cells were washed once with 1 x PBS, fixed with 2% paraformaldehyde for 5 min and permeabilized with 0.15% Triton X-100 for 15 min. Samples were blocked in PBS+ (1% BSA, 0.15% glycine in PBS) for 30 min. All steps were performed at room temperature. The coverslips were incubated with the primary antibody over night at 4°C. Afterwards the samples were washed once with PBS (5 min), 0.15% Triton X-100 (10 min), blocked in PBS+ (7 min) and incubated with the secondary antibody (Cy3-conjugated goat anti-rat, Dianova) for 45 min at room temperature in the dark. The coverslips were washed once with 0.15% Triton X-100 (10 min), PBS (15 min) and stained with DAPI (90 sec). Samples were washed with PBS and mounted on slides using fluorescent mounting medium (Dako). Fluorescence microscopy was performed on Axiovert 200M fluorescence microscope (objective 63x). Images were taken and processed with OpenLab.
Immunoprecipitation (IP)
H1299 cells {1 × 106 per 100 µl lysis buffer [50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40 (Roche), 1 × PhosStop (Roche), 1 × protease cocktail (Roche)]} were lysed for 30 min on ice. Afterwards the samples were sonicated on ice with the BRANSON Sonifier 250 (15 sec on, 15 sec off, 50% duty) and centrifuged (14000 rpm, 10 min, 4°C). The supernatants were incubated with antibody coupled sepharose A/G beads (1:1). Antibodies (2.5 µg) were coupled for 4 h at 4°C followed by 2 washing steps with 750 µl lysis puffer. Supernatant of the lysate and beads were incubated over night at 4°C on a shaker. Finally the beads were washed 5 times with 750 µl lysis buffer and the proteins were boiled off the beads with laemmli buffer for SDS-PAGE.
Phosphatase treatment of nitrocellulose membranes
Proteins were transferred to a nitrocellulose membrane. Membranes were washed with water and treated either with alkaline phosphatase buffer (NEB) and or with 10 µl alkaline phosphatase in phosphatase buffer (60 min, room temperature). Membranes were washed again with water (5 min), blocked with 5% milk-TBS-T (60 min) and incubated with the primary antibodies over night.
Western blots
Laemmli buffer (2 x) was added to protein samples, which were separated by SDS-PAGE (6.5% gel for Rpb1, 15% gel for histones, 10% gel for α-tubulin) and transferred to a nitrocellulose membrane (GE Healthcare). The membrane was blocked with 5% milk/TBS-T for 1 h and incubated with the primary antibody (over night, 4°C). Afterwards the membranes were stained either with IR-labeled secondary rat-specific (680 nm, Invitrogen), or mouse-specific antibodies (800 nm, Rockford, Biomol) and analyzed by using an Odyssey Imaging system (Licor), or stained with HRP-conjugated secondary antibodies against rat (Dianova), mouse (Promega), or rabbit (Dianova) and detected by chemiluminescence.
Chromatin preparation and ChIP-seq
ChIP-seq was essentially performed as previously described.22,24 For K7me1/2 Chip-seq, 5 × 107 Raji cells were crosslinked at room temperature by adding 1/10th volume of crosslinking solution (11% formaldehyde, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 50 mM Hepes pH 7.8) to the cell culture medium for 10 min. The reaction was stopped by the addition of 250 mM glycine for 5 min. Afterwards, the cells were washed twice with PBS and lysed in 2.5 ml LB1 [50 mM Hepes, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8.0, 10% Glycerol, 1% NP-40, 0.5% Triton X-100, 1 × phosphatase inhibitors, 1 × EDTA‐free protease inhibitors (Roche)] for 20 min at 4°C on a shaker. Lysates were spun down (5 min, 4°C, 1350 g) and washed with LB2 (200 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 10 mM Tris pH 8.0, 1 × phosphatase inhibitors, 1 × EDTA‐free protease inhibitors) for 20 min at 4°C. The samples were spun down again and suspended in 1.5 ml LB3 (1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 10 mM Tris pH 8.0, 100 mM NaCl, 0.1 % Na-Deoxycholate, 0.5% N-lauroylsarcosine, 1 × phosphatase inhibitors, 1 × EDTA‐free protease inhibitors). The samples were sonicated with a Misonix 4000 (Misonix Inc.) sonicator for 14 cycles (30 sec on, 30 sec off, amplitude 40). Triton X-100 was added to a final concentration of 1% and the chromatin was collected by centrifugation (20 000 g, 20 min, 4°C). Chromatin IP were performed as described22 with 1F5 monoclonal antibody. An aliquot of 50 µl was taken as input control. The input was mixed with an equal volume of 2 × elution buffer (100 mM Tris pH 8.0, 20 mM EDTA pH 8.0, 2 % SDS) and incubated at 65°C for 13–15 h. The sample was diluted with same volume of TE (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0) and digested by RNase A (0.2 µg/ml, 2 h, 37°C) and Proteinase K (0.2 µg/ml, 2 h, 55°C). DNA was purified by 2 phenol:chloroform:isoamylalcohol (25:24:1, pH 8.0) extraction steps and a subsequent PCR column purification (Qiagen). DNA fragmentation of input and ChIP samples was controlled on a bioanalyser DNA chip to shear between 200–300 bp. Processing of the RNAPII ChIP-seq data was performed as described previously.22 Data reported for RNAPII, Ser2P, Ser5P, Ser7P, and Tyr1P can be found at Gene Expression Omnibus database under the accession number GSE52914 and under GSEXXX for K7me1/2.
Cell fractionation
For fractionating the cell in cytosol and nucleus, 1 × 108 cells were washed with PBS and suspended in 5 ml buffer A [10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 × EDTA‐free protease inhibitors (Roche)] and kept on ice for 5 min. Cells were opened by using a Dounce homogenizer. Dounced cells were centrifuged (228 g, 5 min, 4°C) and the supernatant was collected as cytoplasmic fraction. The cytoplasmic fraction was mixed with 5 × RIPA buffer (250 mM Tris pH 7.5, 750 mM NaCl, 5% NP-40, 2.5% deoxycholate, 1 × EDTA‐free protease inhibitors) and again centrifuged (2800 g, 10 min, 4°C) to remove solids. The pellet containing the nuclei was suspended in 3 ml buffer S1 (0.25 mM Sucrose, 10 mM MgCl2, 1 × EDTA‐free protease inhibitors) and overlaid with 3 ml S3 (0.88 mM Sucrose, 0.5 mM MgCl2, 1 × EDTA‐free protease inhibitors). Finally the sample was resuspended in 1 × RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate), sonicated, centrifuged (2800 g, 10 min, 4°C), and the supernatant was processed further as nuclear fraction.
Phosphatase treatment
RNAPII was immunoprecipitated with the Ser5P specific antibody (3E8). The IP was washed 5 × with lysis buffer [50 mM Tris-HCL, pH 8.0, 150 mM NaCl, 1% NP-40 (Roche), 1 × PhosStop (Roche), 1 × protease cocktail (Roche)] and subsequently treated with 10 µl alkaline phosphatase (NEB) for 30 min at 30°C. The sample was resuspended and boiled in laemmli buffer (3 min, 95°C).
Purification of Rpb1 for mass spectrometric analysis
Cells (3 × 108) were lysed in lysis buffer [50 mM Tris-HCL, pH 8.0, 150 mM NaCl, 1% NP-40 (Roche), 1 × PhosStop (Roche), 1 × protease cocktail (Roche)] for 30 min on ice. Samples were sonified (Sonifier 250 BRANSON, 3 × 20 cycles, output 7, duty cycle 70) and incubated on a shaker for 1 h at 4°C. Subsequently, samples were centrifuged (10500 g, 15 min) and the supernatants were incubated with antibody-coupled sepharose G beads (Ser2P, Ser5P in a 1:1 ratio) over night on a shaker. The beads were washed 3 times with lysis buffer and boiled with 2 × laemmli buffer (95°C, 8 min). Proteins were loaded onto SDS-PAGE, separated and stained with EZBlue Coomassie brilliant blue G-250 (Sigma).
In-gel cleavage and phospho-peptide enrichment
The different steps of the in-gel digest including destaining, cysteine reduction, alkylation, trypsin digestion, and extraction of the peptides were performed as previously described.33,34 The extracted peptides were subjected to phospho-peptide enrichment. Therefore the sample volume was reduced to 5 µl by evaporation and subsequently suspended in 100 µl loading buffer (80% acetonitrile, 5%TFA and 1M glycolic acid). Samples were mixed with 10 µl TiO2 beads (0.03 mg/µl TiO2 beads in 100% ACN) and incubated for 10 min at room temperature on a shaker. The beads were spun down and washed with washing buffer 1 (80% acetonitrile, 1% TFA), washing buffer 2 (10% acetonitrile, 0.2% TFA), respectively, and dried for 10 min. The phospho-peptides were eluted with 50 µl elution buffer [40 µl ammonia solution (28%) in 960 µl H2O] on a shaker at room temperature and reduced to a final volume of 5 µl by evaporation. Finally, samples were suspended in 30 µl (0.1% formic acid) and stored at −20°C until measuring.
Liquid chromatography coupled to tandem TiO2 mass spectrometry (LC-MS/MS)
Phospho-enriched peptides were automatically injected into in Ultimate 3000 HPLC system (Dionex), desalted on-line in a C18 micro column (300 µm i.d. × 5mm, PepMap100 C18 5 µm, 100 Å, Dionex) and separated on an analytical column C18 micro column (75 µm i.d. × 10 cm, packed in-house with Reprosil Pur C18 AQ 2.4 µm, Doctor Maisch) using a 60 min gradient from 5 to 30 % acetonitrile in 0.1% formic acid. The effluent from the HPLC was subsequently electrosprayed into a LTQ Orbitrap XL mass spectrometer (Thermo). The MS instrument was operated in a data dependent mode to automatically switch between full scan MS and MS/MS acquisition. Survey full scan MS spectra (from m/z 300 – 2000) were acquired in the Orbitrap with a resolution of R = 60,000 at m/z 400 (after accumulation to a ‘target value’ of 500,000 in the linear ion trap). The three most intense peptide ions with charge state between 1 and 4 were sequentially isolated to a target value of 10,000 and fragmented in the linear ion trap by collision induced dissociation (CID). The fragmentation method was multistage activation (MSA) to automatically select and fragment the ions originated from the loss of one or 2 phosphate groups from the parent ion. For all measurements with the Orbitrap mass analyzer, 3 lock-mass ions from ambient air (m/z = 371.10123, 445.12002, 519.13882) were used for internal calibration as described before.35 Usual MS conditions were: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 200°C; normalized collision energy 35% for CID in LTQ. The threshold for ion selection was 10,000 counts for MS2. The used activation was 0.25 and activation time 30 ms. The following MS data were analyzed with Proteome Discoverer 1.4/PhosphoRS 2.0 [MS tol, 10ppm; MS/MS tol, 0.8Da, Variable modifications, Oxidation (M) and Phosphorylation (S, T, Y); Fixed modifications, Carbamidomethyl (C); FDR Peptide, 0.01].36
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Ana Pombo for discussion of unpublished results.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
The laboratories of D. Eick and A. Imhof were supported by grants from the Deutsche Forschungsgemeinschaft DFG (SFB1064). The laboratory of J.-C. Andrau was supported by a grant of the Fondation pour la Recherche Médicale AJE20130728183, supporting the salary costs of M.A. Maqbool. N. Descostes was supported by a grant from the ministère de la recherche and by the Ligue nationale contre le cancer (LNCC). Cost of high-throughput sequencing experiments were partially covered by a European ESGI translational access grant.
References
- 1.Corden JL. RNA polymerase II C-terminal domain: Tethering transcription to transcript and template. Chem Rev 2013; 113:8423-55; PMID:24040939; http://dx.doi.org/ 10.1021/cr400158h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eick D, Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 2013; 113:8456-90; PMID:23952966; http://dx.doi.org/ 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- 3.Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell 2009; 36:541-6; PMID:19941815; http://dx.doi.org/ 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egloff S, Dienstbier M, Murphy S. Updating the RNA polymerase CTD code: adding gene-specific layers. Trends Genet 2012; 28:333-41; PMID:22622228; http://dx.doi.org/ 10.1016/j.tig.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Heidemann M, Hintermair C, Voss K, Eick D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta 2013; 1829:55-62; PMID:22982363; http://dx.doi.org/ 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 6.Conaway RC, Bradsher JN, Conaway JW. Mechanism of assembly of the RNA polymerase II preinitiation complex. Evidence for a functional interaction between the carboxyl-terminal domain of the largest subunit of RNA polymerase II and a high molecular mass form of the TATA factor. J Biol Chem 1992; 267:8464-7; PMID:1569096. [PubMed] [Google Scholar]
- 7.Lu H, Flores O, Weinmann R, Reinberg D. The nonphosphorylated form of RNA polymerase II preferentially associates with the preinitiation complex. Proc Natl Acad Sci U S A 1991; 88:10004-8; PMID:1946417; http://dx.doi.org/ 10.1073/pnas.88.22.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, Ansari AZ. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell 2009; 34:387-93; PMID:19450536; http://dx.doi.org/ 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boeing S, Rigault C, Heidemann M, Eick D, Meisterernst M. RNA polymerase II C-terminal heptarepeat domain Ser-7 phosphorylation is established in a mediator-dependent fashion. J Biol Chem 2010; 285:188-96; PMID:19901026; http://dx.doi.org/ 10.1074/jbc.M109.046565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature 1995; 374:283-7; PMID:7533895; http://dx.doi.org/ 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 11.Kwak H, Lis JT. Control of transcriptional elongation. Annu Rev Genet 2013; 47:483-508; PMID:24050178; http://dx.doi.org/ 10.1146/annurev-genet-110711-155440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell 2006; 23:297-305; PMID:16885020; http://dx.doi.org/ 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi Y, Shibata H, Handa H. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim Biophys Acta 2013; 1829:98-104; PMID:23202475; http://dx.doi.org/ 10.1016/j.bbagrm.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 2014; 15:163-75; PMID:24514444; http://dx.doi.org/ 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 2012; 26:2119-37; PMID:23028141; http://dx.doi.org/ 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jasnovidova O, Stefl R. The CTD code of RNA polymerase II: a structural view. Wiley Interdiscip Rev RNA; 2013; 4:1-16. [DOI] [PubMed] [Google Scholar]
- 17.Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. A structural perspective of CTD function. Genes Dev 2005; 19:1401-15; PMID:15964991; http://dx.doi.org/ 10.1101/gad.1318105. [DOI] [PubMed] [Google Scholar]
- 18.Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 2006; 20:2922-36; PMID:17079683; http://dx.doi.org/ 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 19.Chapman RD, Heidemann M, Hintermair C, Eick D. Molecular evolution of the RNA polymerase II CTD. Trends Genet 2008; 24:289-96; PMID:18472177; http://dx.doi.org/ 10.1016/j.tig.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Simonti CN, Pollard KS, Schroder S, He D, Bruneau BG, Ott M, Capra JA. Evolution of lysine acetylation in the RNA polymerase II C-terminal domain. BMC Evol Biol 2015; 15:35; PMID:25887984; http://dx.doi.org/ 10.1186/s12862-015-0327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder S, Herker E, Itzen F, He D, Thomas S, Gilchrist DA, Kaehlcke K, Cho S, Pollard KS, Capra JA, et al.. Acetylation of RNA polymerase II regulates growth-factor-induced gene transcription in mammalian cells. Mol Cell 2013; 52:314-24; PMID:24207025; http://dx.doi.org/ 10.1016/j.molcel.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Descostes N, Heidemann M, Spinelli L, Schuller R, Maqbool MA, Fenouil R, Koch F, Innocenti C, Gut M, Gut I, et al. Tyrosine phosphorylation of RNA polymerase II CTD is associated with antisense promoter transcription and active enhancers in mammalian cells. Elife 2014; 3:e02105; PMID:24842994; http://dx.doi.org/ 10.7554/eLife.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsin JP, Li W, Hoque M, Tian B, Manley JL. RNAP II CTD tyrosine 1 performs diverse functions in vertebrate cells. Elife 2014; 3:e02112; PMID:24842995; http://dx.doi.org/ 10.7554/eLife.02112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hintermair C, Heidemann M, Koch F, Descostes N, Gut M, Gut I, Fenouil R, Ferrier P, Flatley A, Kremmer E, et al.. Threonine-4 of mammalian RNA polymerase II CTD is targeted by Polo-like kinase 3 and required for transcriptional elongation. EMBO J 2012; 31:2784-97; PMID:22549466; http://dx.doi.org/ 10.1038/emboj.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Sabari BR, Garcia BA, Allis CD, Zhao Y. SnapShot: histone modifications. Cell 2014; 159(2):458-458; http://dx.doi.org/ 10.1016/j.cell.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta 2013; 1829:84-97; PMID:22982198; http://dx.doi.org/ 10.1016/j.bbagrm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sims RJ 3rd, Rojas LA, Beck D, Bonasio R, Schuller R, Drury WJ 3rd, Eick D, Reinberg D. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 2011; 332:99-103; PMID:21454787; http://dx.doi.org/ 10.1126/science.1202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman RD, Conrad M, Eick D. Role of the mammalian RNA polymerase II C-terminal domain (CTD) nonconsensus repeats in CTD stability and cell proliferation. Mol Cell Biol 2005; 25:7665-74; PMID:16107713; http://dx.doi.org/ 10.1128/MCB.25.17.7665-7674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 2012; 336:1723-5; PMID:22745433; http://dx.doi.org/ 10.1126/science.1219651. [DOI] [PubMed] [Google Scholar]
- 30.Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 2007; 318:1780-2; PMID:18079404; http://dx.doi.org/ 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 31.Holzel M, Rohrmoser M, Schlee M, Grimm T, Harasim T, Malamoussi A, Gruber-Eber A, Kremmer E, Hiddemann W, Bornkamm GW, et al.. Mammalian WDR12 is a novel member of the Pes1-Bop1 complex and is required for ribosome biogenesis and cell proliferation. J Cell Biol 2005; 170:367-78; PMID:16043514; http://dx.doi.org/ 10.1083/jcb.200501141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meininghaus M, Chapman RD, Horndasch M, Eick D. Conditional expression of RNA polymerase II in mammalian cells. Deletion of the carboxyl-terminal domain of the large subunit affects early steps in transcription. J Biol Chem 2000; 275:24375-82; PMID:10825165; http://dx.doi.org/ 10.1074/jbc.M001883200. [DOI] [PubMed] [Google Scholar]
- 33.Shevchenko A, Chernushevich I, Wilm M, Mann M. De Novo peptide sequencing by nanoelectrospray tandem mass spectrometry using triple quadrupole and quadrupole/time-of-flight instruments. Methods Mol Biol 2000; 146:1-16; PMID:10948493. [DOI] [PubMed] [Google Scholar]
- 34.Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 1996; 379:466-9; PMID:8559255; http://dx.doi.org/ 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- 35.Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 2005; 4:2010-21; PMID:16249172; http://dx.doi.org/ 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 2011; 10:5354-62; PMID:22073976; http://dx.doi.org/ 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.