

Abstract
Calcium ions are Nature's most widely used signaling mechanism, mediating communication between pathways at virtually every physiological level. Ion channels are no exception, as the activities of a wide range of ion channels are intricately shaped by fluctuations in intracellular Ca2+ levels. Mirroring the importance and the breadth of Ca2+ signaling, free Ca2+ levels are tightly controlled, and a myriad of Ca2+ binding proteins transduce Ca2+ signals, each with its own nuance, comprising a constantly changing symphony of metabolic activity. The founding member of Ca2+ binding proteins is calmodulin (CaM), a small, acidic, modular protein endowed with gymnastic-like flexibility and E-F hand motifs that chelate Ca2+ ions. In this review, I will trace the history that led to the realization that CaM serves as the Ca2+-gating cue for SK channels, the experiments that revealed that CaM is an intrinsic subunit of SK channels, and itself a target of regulation.
Keywords: curing, calmodulin, Ca2+-gating, E-F hands, intrinsic subunit, Paramecium, SK channel
Abbreviations
- CaM
calmodulin
- EDTA
ethylenediaminetetraacertic acid
- EGTA
ethylene glycol tetraacetic acid
- SK channels
small conductance calcium-activated potassium channels
- BK channels
big conductance calcium-activated potassium channels
- GST
glutathione S-transferase
- CaMBD
calmodulin binding domain
- PP2A
protein phosphatase 2A
The Ca2+-K+ Connection
In 1958, Gardos1 presented the first evidence that Ca2+ ions modulate K+ permeability by showing that when glycolysis was inhibited K+ efflux from red blood cells increased 10–30-fold. Additional Ca2+ increased the rate of efflux and this dramatic increase in K+ permeability was inhibited by addition of EDTA. Today, we know that Gardos was measuring the activity of the intermediate conductance calcium-activated K+ channel, IK1 (SK4, KCNN4).2
In the 1970s, studies using mulluscan neurons such as from Aplysia and Helix revealed that injection of Ca2+ results in a rapid hyperpolarization of the membrane that was dependent upon the concentration of external potassium,3 and was abolished by injection of EGTA. Further experiments showed that there is a long lasting increase in K+ conductance following a train of action potentials that decays as cytoplasmic Ca2+ levels, increased during the train, slowly decline. The increased K+ conductance was abolished by EGTA.4-6 Similar results were found in frog motor neurons7 and cat spinal neurons.8,9
Apamin – the magic bullet
It has been known for more than 70 y that bee venom has neurotoxic effects. The principle neurotoxin was isolated from honey bee venom (Apis mellifera) and appropriately named ‘apamin’.10 It is a pear-shaped, 18 amino acid peptide with 2 disulfide bridges. Apamin readily crosses the blood-brain barrier and iodo-apamin studies reveal a binding sites map similar to the distribution of SK2 and SK3 in rodent brain.11,12 Sub-lethal intravenous injections into rodents cause motor coordination deficits and hyperactivity, symptoms similar to those seen in SK2 null mice (unpublished). Early studies using guinea pig liver and visceral smooth muscle preparations showed that apamin blocked the hyperpolarizing effects of ATP or adrenergic agonists, due to block of a Ca2+-mediated increase in K+ permeability.13 While early studies using preparations of apamin isolated from bee venom suggested possible other targets, only SK channels are the targets of highly purified apamin. In a landmark paper in 1986, Blatz and Magleby presented single channel recordings of apamin-blocked SK channels from cultured rat skeletal muscle.14 The remarkable selectivity and specificity of apamin have enabled many insights into the physiological roles of SK channels.
The calmodulin connection
The first indication that CaM regulates K+ channels was presented in 1983 by Orlov and Kravtsov.15 Like Gardos before them, Orlov and Kravtsov worked with erythrocyte membranes. They showed that the Ca2+ ionophore, A23187, caused a hyperpolarization that was due to increased K+ permeability, and this effect required calmodulin. Pape and Kristensen presented a similar conclusion in 1984.16 In 1987, Klaerke and colleagues used a calmodulin affinity column to partially purify a Ca2+-activated K+ channel from luminal membranes of outer renal medulla.17
But the thread that most directly led to the SK channel story came from the laboratory of Ching Kung, who later would say, “Serendipity played a role in the rediscovery of CaM in channels from a line of hypothesis-free research in vivo.”18 Kung and Saimi were performing behavioral experiments with ciliated Paramecium. These unicellular machines swim freely in search of food and sense their environment. However, upon encountering a noxious stimulant they display a stereotypical ‘avoiding reaction’. A Ca2+-based action potential causes the cell to reverse its ciliary motion, stop, turn, and then resume swimming in a new direction. The action potential is the result of sequential opening of a Ca2+-activated Na+ channel and then a Ca2+-activated K+ channel. The avoiding reaction is complete in just a few seconds.18 Saimi and Kung used this phenotype as the basis for mutant selection and found at least 2 classes of mutants. One class they called Pantophobiacs (pnt) (originally paranoiacs) that over-react to the noxious stimulant and swim backward for a long time.19 The other class was called Fast-2, and these animals under-react and swim backward only briefly, if at all.20 Using electrophysiology they found that Pantophobiacs lacked the Ca2+-activated K+ channel, while Fast-2 lacked the Ca2+-activated Na+ channel.
This was exciting and Kung thought they might be able to isolate the 2 channels based on these mutants. But when they mapped the loci, they were shocked and initially confused to find that Pantophobiacs and Fast-2 are allelic - the mutations underlying both sets of mutants are in the same gene. To solve this mystery, Kung and colleagues performed an elegant, remarkable experiment (Fig. 1). They made cytoplasmic extracts from wild type Paramecium and injected it into the cytoplasm of Pantophobiac animals. What they found was a transient restoration of the wild type avoiding reaction. Using this assay, they fractionated the wild type extracts and ultimately found that the molecule responsible for the ‘curing’ was CaM. When they injected wild type CaM into Pantophobiacs the animals were transiently cured and their avoiding reaction was like wild type, but when they injected Pantophobiac CaM into Pantophobiacs the mutant phenotype was unaffected. The pièce de résistance (Fig. 2) was that they could record the transiently restored Ca2+-activated K+ current from Pantophobiacs injected with wild type CaM.21
Figure 1.
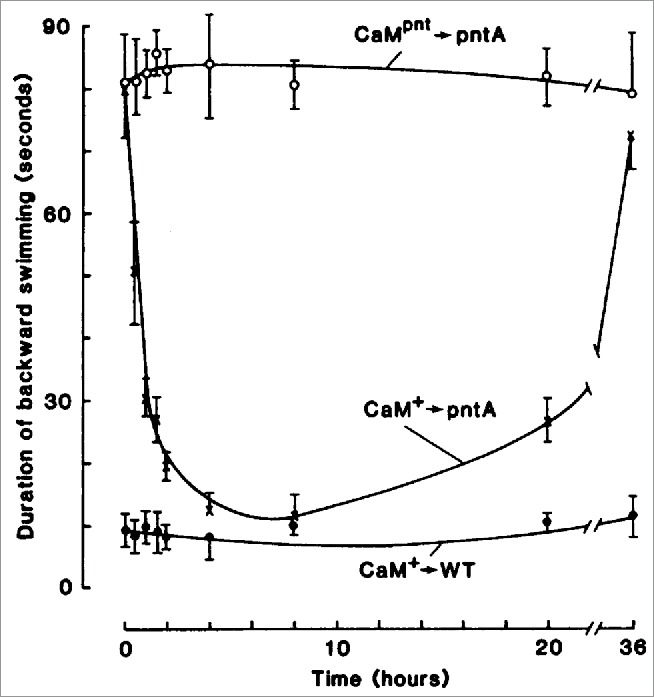
The time course for the restoration of pntA after injection of calmodulin from wild-type Paramecium (CaM+). At each time point, the cells were tested for the duration of their backward swimming. The control pntA cells were injected with calmodulin prepared from pntA cells (CaMpnt); the control wild-type cells (WT) were injected with buffer solution. Each point represents 4 cells (± SD). © [AAAS]. Reproduced by permission of Dr. Ching Kung. Permission to reuse must be obtained from the rightsholder.
Figure 2.
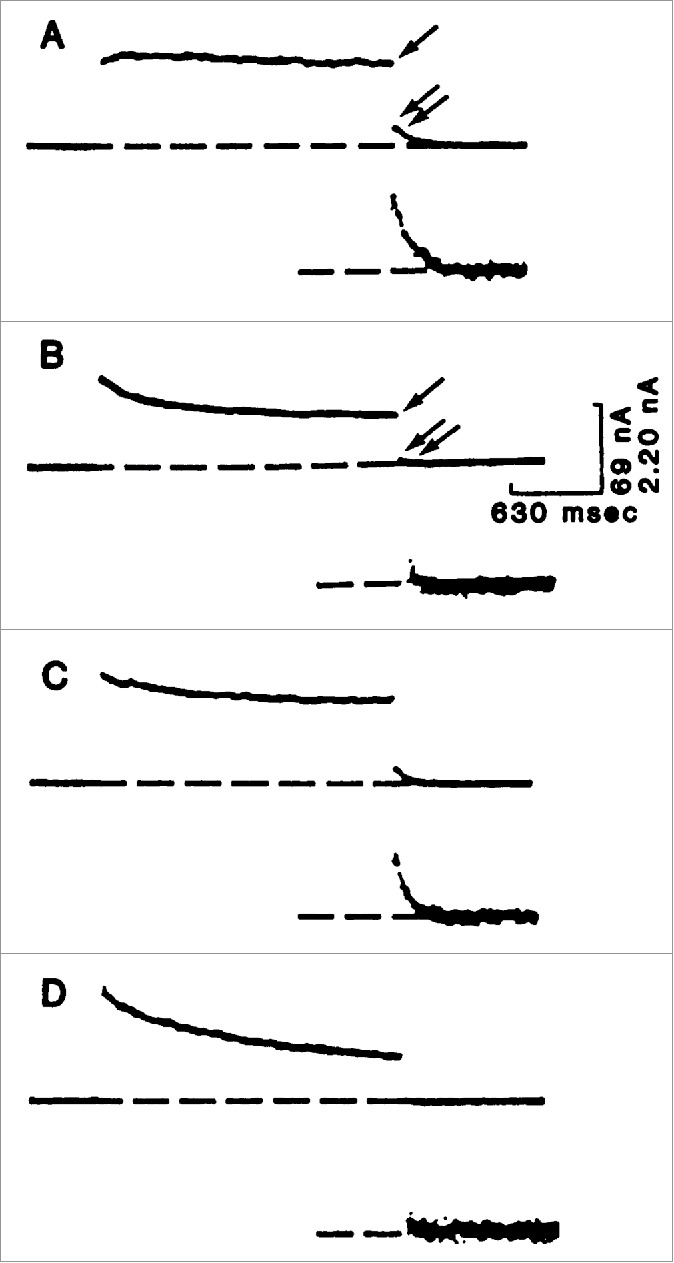
Voltage clamp analysis of the Ca2+-dependent K+ current in Paramecium cells injected with calmodulin or buffer. A depolarization step was given from a resting level of −40 mV to −5 mV and held for 2 seconds before returning to −40 mV. The cells were microinjected with either buffer solution or calmodulin. In (A–D), the inset shows the tail current amplified 4 times. (A) Wild-type cells injected with buffer solution showed the Ca2+-dependent K+ current. The single arrow indicates the late outward current upon depolarization, primarily through the Ca2+-dependent K+ channel. Upon repolarization, there was an obvious tail current (double arrows and inset). (B) The pntA mutant, injected with buffer solution, virtually devoid of the Ca2+-dependent K+ current. There is little or no tail current upon repolarization (double arrows and inset). (C) The pntA mutant after injection of wild-type calmodulin, showed a return of the tail current. (D) The pntA mutant after injection with pntA calmodulin showed no restoration. © [AAAS]. Reproduced by permission of Dr. Ching Kung. Permission to reuse must be obtained from the rightsholder.
Kung's initial reaction reflected his feeling that this enterprise that had seemed so promising turned out to be, apparently, pedestrian. “By fractionating the wild-type cytoplasm, the curing element turned out to be CaM, much to the disappointment of one of us (C. Kung) who was hoping for a new find.”18
But the story does not end there. Kung and colleagues went on to map the individual mutations underlying each of the Pantohobiac and Fast-2 mutants. What they found was prescient. All of the Pantophobiac mutations resided in the C-lobe domain of CaM, while all of the Fast-2 mutations mapped to the N-lobe domain of CaM. These data clearly indicated that the N- and C-terminal Ca2+-binding lobes of CaM were functionally distinct.22 Thus CaM is essential for Ca-activated K+ channel activity in Paramecium, but the exact mechanism was not clear. While these findings were presented in high visibility journals, Kung wondered if they would be appreciated. “Anthropocentric physiologists might question whether this Paramecium finding, like the earlier one by Brehm and Eckert, can also be applied to mammals.”18
Almost 20 y later, in landmark work, David Yue would bring together these 2 discoveries from Paramecium, Ca2+-dependent inactivation of Ca2+ channels23 and the distinct roles of the N- and C-terminal lobes of CaM, to show how local and global Ca2+ signals, important for neuronal and cardiac physiology, are transduced.24,25
SK channels and calmodulin
Prior to the cloning of Ca2+-activated K+ channels, BK channels had been extensively studied. Their large unitary conductance and the ease with which they could be reconstituted into planar lipid bilayers facilitated biophysical studies that revealed their Ca2+ and voltage dependent gating.26 We now appreciate that BK channels are voltage-gated channels with voltage dependent Po modulated by internal Ca2+.27,28 Nevertheless, because of the intimate interactions between voltage and Ca2+, very complex gating models could be constructed, and hampered the interpretation of structure-function studies designed to understand gating.26,29 However, once the SK channels were cloned30 and showed to be strictly Ca2+ gated, much simpler models could account for SK channel gating31 and suggested that the underlying mechanism of Ca2+ gating might be more easily approached than was the case for BK channels.
Heterologously expressed homomeric rat SK2, SK3, and human SK1 channels demonstrated similar Ca2+ dose response relationships with apparent Kd values of ∼0.5 μM and Hill slopes of ∼4.30,32,33 These results suggested that the mechanisms of Ca2+gating for the 3 channels was the same and that this would be reflected by conserved structural domains. Rapid application of saturating Ca2+ to inside-out membrane patches revealed that Ca2+ gating was rapid with τon ∼5–10 msec.32 Importantly, this suggested that Ca2+ gating was mediated by a direct interaction of Ca2+ ions with the SK channel proteins, and not secondary to a Ca2+ dependent signaling cascade that would be expected to take tens or hundreds of msec. In light of the high apparent Ca2+ sensitivity, we scoured the coding sequences for E-F hand motifs, but could not identify them. If Ca2+ ions were directly interacting with the SK channel proteins, perhaps through a novel structural motif, negatively charged residues were likely to be involved. Therefore, we systematically mutated each of the 21 conserved intracellular glutamate (E-to-Q) or aspartate (D-to-N) residues, and measured their effects on Ca2+ sensitivity. To our surprise, none of these mutations dramatically altered Ca2+ gating.32
Faced with the daunting possibility that we would need to construct and analyze thousands of combinatorial mutations, we first performed more major molecular surgery and analyzed truncations of SK2. This revealed that for Ca2+ gating the distal cytoplasmic domain of the channel was dispensable for Ca2+ gating, but the membrane proximal half of this region was essential. Focusing our attention on these ∼100 amino acids showed us that this was the most highly conserved domain on the channel protein, not only among the rodent and human channels but across many species as well.32
Then we remembered Kung's work. This suspicious domain carries a net positive charge and the concept emerged that perhaps this domain bound the acidic Ca2+ binding protein, CaM, and Ca2+ binding to CaM triggered SK channel gating. To begin to test this hypothesis, we constructed a set of yeast 2 hybrid probes that interrogated the ability of different domains of the SK channel proteins to interact with CaM. The results clearly showed that CaM could indeed interact with the proximal C-terminal domain, which we then called the CaM binding domain, CaMBD.32
But one piece of the puzzle remained. If Ca2+ binds to CaM and this results in the canonical mechanism of CaM-mediated Ca2+ signaling, a change in CaM conformation and subsequent interaction with the SK channel, then how could Ca2+ gating take place on the order of a few milliseconds? The only way to envision this was to hypothesize that CaM was pre-associated with the SK channel, an intrinsic subunit that rapidly triggers gating. To test this, we constructed a series of GST fusion proteins, representing different domains of the SK channels and tested them for the ability to bind purified CaM or to pull down CaM from cytoplasmic extracts; binding reactions were performed either in the presence or absence of Ca2+. The results showed that CaM efficiently bound to the CaMBD whether Ca2+ was present or not. Moreover, SK2 antibody co-immunoprecipitated CaM.32 Kung had been right all along, although he had not envisioned the constitutive association between the channels and CaM. The constitutive interaction was also reflected functionally in that washing Ca2+ on and off inside-out patches to open and close the channels can routinely be performed as long as the patch integrity is maintained, even when leaving patches in Ca2+ free - and CaM free - solution for long periods of time. But how could we directly demonstrate that CaM was mediating Ca2+ gating for SK channels?
Then we remembered work by Trish Davis who had been studying yeast CaM. Davis and colleagues had made the provocative finding that yeast deleted for the CaM gene cannot survive, but yeast harboring CaM that cannot bind Ca2+ ions are viable.34 This showed that CaM is required for function beyond Ca2+ binding. To demonstrate this, Davis had introduced point mutations into each of the E-F hand motifs, rendering them essentially unable to chelate Ca2+ ions. Therefore, we debilitated either all 4 (1,2,3,4), or 3 of the 4, E-F hands (2,3,4), and expressed them in Xenopus oocytes (Fig. 3). When wild type CaM binds Ca2+, there is a dramatic conformational change that results in faster migration through SDS gels. Running protein extracts either with or without Ca2+ showed that the mutations were effective, essentially eliminating the migration shift. Finally, we co-expressed SK channels with the mutant CaMs. We found that for E-F(2,3,4), harboring only a single intact E-F hand in the N-lobe of CaM, the Ca2+ sensitivity of SK channel gating was dramatically reduced and the Hill slope was also reduced. Co-expression of SK channels with CaM(1,2,3,4), with no intact E-F hand motifs, resulted in very small currents with essentially normal Ca2+ sensitivity, reflecting the small population of channels that had acquired endogenous Xenopus CaM. Collectively these data convincingly demonstrated that CaM is an intrinsic subunit of SK channels and mediates Ca2+ gating.32
Figure 3.
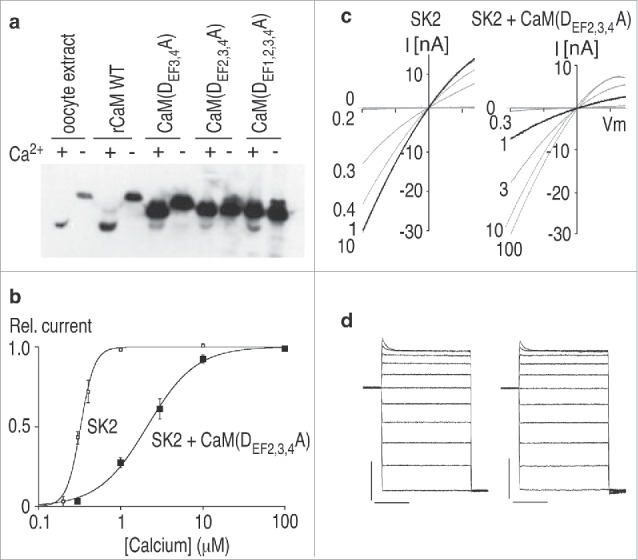
Calmodulin mediates Ca2+ gating of SK channels. (A) Western blot prepared rom oocytes expressing the indicated CaM mutants. Samples were electrophoresed in loading buffer containing either Ca2+ (+) or EGTA (−), and the blot was probed with anti-calmodulin antibody. r, rat; WT, wild-type. (B) Ca2+ dose-response relation for SK2 channels expressed alone or with CaM(DEF2,3,4A). Relative current amplitudes measured at -100 mV are plotted as a function of Ca2+ concentration. Lines represent a fit to the Hill equation to the data, yielding K0.5 and Hill coefficient of 32 μM and 5.1 for SK2, and 2.1 μM and 1.5 for SK2 co-expressed with CaM(DEF2,3,4A). (C) Current responses to voltage ramps from −100 mV to 70 mV in the presence of the indicated intracellular Ca2+ concentrations. Traces recorded in the presence of 1 μM Ca2+ are shown by dark lines. (D) Currents evoked by voltage steps in an inside-out patch expressing SK2 (left) or with CaM(DEF2,3,4A) (right) in the presence of 10 μM Ca2+. Voltage steps were from −100 to 100 mV in 20 mV increments from a holding potential of 0 mV. The current and time calibrations are 10 nA and 20 ms, respectively. Adapted from ref.31 © [Nature]. Permission to reuse must be obtained from the rightsholder.
Then once again we remembered Kung. Could the different E-F hands on the 2 lobes of CaM serve distinct functions for SK channels? To test this, we made several combinations of mutant E-F hands and co-expressed them with SK channels. This revealed that mutating either E-F(1) or E-F(2), in the N-lobe of CaM reduced the apparent Ca2+ sensitivity of the SK channels, shifting the dose-response curve to the right, while the double mutant, E-F(1,2) completely abolished channel activity. In striking contrast, mutating either of the C-lobe E-F hand motifs, E-F(3) or E-F(4), or both E-F(3,4), had no effect on Ca2+ gating.35 The crystal structure of the CaMBD in complex with Ca2+-CaM later showed that the C-lobe E-F hands are rendered dysfunctional due to anchoring interactions with the CaMBD that disrupt the geometry of the E-F hand motifs.36
The intimate relationship between SK channels and CaM held yet another surprise. A proteomics approach that used the C-terminal domain of SK2 as bait to identify protein binding partners revealed that in addition to CaM, protein kinase CK2 and protein phosphatase 2A (PP2A) also bound to the SK channels. Further studies showed that these interactions were, like CaM, constitutive. Unexpectedly, the target of the kinase/phosphatase regulation was not the SK channel α subunit, but threonine 80 (T80) within the linker domain of CaM. Phosphoryation of T80 shifts the Ca2+ dose-response to the right while dephosphorylation shifts the Ca2+ dose-response to the left. Remarkably, the actions of CK2 and PP2A are strictly state-dependent; CK2 only phosphorylates T80 when the channels are closed, and PP2A only dephosphorylates T80 when the channels are open. This state-dependence mirrors the relative position of a single lysine residue, K121, in the N-terminal domain of the SK2 channels. Thus, in neurons, the Ca2+ sensitivity of the SK channels is itself activity dependent.37,38
During the past 15 years, the field of calmodulation of ion channels has expanded dramatically. The number of channels involved and the variety of regulatory mechanisms mediated by CaM is far more than we or others initially expected. Indeed, ‘calmodulation’ is no longer truly accurate as many distinct members of the superfamily of E-F hand Ca2+ binding proteins have been shown to mediate their own brands of ion channel modulation. This area of research stems from seminal findings from seemingly unrelated sources - K+ flux assays with erythrocyte ghosts, behavioral studies of Paramecium, viability studies in yeast – that colluded to open the concepts and provide the initial tools to dissect the molecular details of the relationships between Ca2+ binding proteins and a wide range of ion channels. Even with all of these discoveries I am certain that there are more surprises yet to come. We all should stay tuned.
A personal note David, perhaps more than any other individual, took the field of calmodulation of ion channels to another level, not once but several times with his series of papers on CaM and Ca2+ channels. Indeed, his final installment in Cell late last year accomplished this yet again, showing that apoCaM upregulates Ca2+ channel opening and that Ca2+ dependent inactivation, mediated by Ca2+-CaM, may simply reverse this effect.39 Everyone who knew David appreciated his passion, his eloquence and above all the beauty and creativity of his science. When reading a paper of David's for the first time, I was always certain of 3 things. The tapestry of techniques would be awesome, the results would be extremely provocative, and it would take at least 4 readings to come close to understanding the paper. I will miss David.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I am forever indebted to my close colleague, Jim Maylie, and to the students and postdoctoral fellows who worked with dedication on the SK-CaM story. Thanks also to Ms. Lori Vaskalis for superb graphics.
Funding
I am grateful for the continued support from NIH over many years that made this work possible.
References
- 1. Gárdos G. The function of calcium in the potassium permeability of human erythrocytes. Biochim Biophys Acta 1958; 30:653-4; PMID:13618284; http://dx.doi.org/ 10.1016/0006-3002(58)90124-0 [DOI] [PubMed] [Google Scholar]
- 2. Maher AD, Kuchel PW. The Gardos channel: a review of the Ca2+-activated K+ channel in human erythrocytes. Int J Biochem Cell Biol 2003; 35:1182-97; PMID:12757756; http://dx.doi.org/ 10.1016/S1357-2725(02)00310-2 [DOI] [PubMed] [Google Scholar]
- 3. Meech RW. Intracellular calcium injection causes increased potassium conductance in Aplysia nerve cells. Comp Biochem Physiol A Comp Physiol 1972; 42:493-9; PMID:4404379; http://dx.doi.org/ 10.1016/0300-9629(72)90128-4 [DOI] [PubMed] [Google Scholar]
- 4. Meech RW. The sensitivity of Helix aspersa neurones to injected calcium ions. J Physiol 1974; 237:259-77; PMID:4825448; http://dx.doi.org/ 10.1113/jphysiol.1974.sp010481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meech RW, Standen NB. Calcium-mediated potassium activation in Helix neurones. J Physiol 1974; 237:43P-4P; PMID:4825461; http://dx.doi.org/ 10.1113/jphysiol.1974.sp010481 [DOI] [PubMed] [Google Scholar]
- 6. Thomas MV, Gorman AL. Internal calcium changes in a bursting pacemaker neuron measured with arsenazo III. Science 1977; 196:531-3; PMID:850795; http://dx.doi.org/ 10.1126/science.850795 [DOI] [PubMed] [Google Scholar]
- 7. Barrett EF, Barret JN. Separation of two voltage-sensitive potassium currents, and demonstration of a tetrodotoxin-resistant calcium current in frog motoneurones. J Physiol 1976; 255:737-74; PMID:1083431; http://dx.doi.org/ 10.1113/jphysiol.1976.sp011306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ito M, Kostyuk PG, Oshima T. Further study on anion permeability of inhibitory post-synaptic membrane of cat motoneurones. J Physiol 1962; 164:150-6; PMID:13956993; http://dx.doi.org/ 10.1113/jphysiol.1962.sp007009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krnjevic K, Puil E, Werman R. Evidence for Ca2+-activated K+ conductance in cat spinal motoneurons from intracellular EGTA injections. Can J Physiol Pharmacol 1975; 53:1214-8; PMID:816435; http://dx.doi.org/ 10.1139/y75-171 [DOI] [PubMed] [Google Scholar]
- 10. Habermann E. Apamin. Pharmacol Ther 1984; 25:255-70; PMID:6095335; http://dx.doi.org/ 10.1016/0163-7258(84)90046-9 [DOI] [PubMed] [Google Scholar]
- 11. Janicki PK, Horvath E, Seibold G, Habermann E. Quantitative autoradiography of [125I] apamin binding sites in the central nervous system. Biomed Biochim Acta 1984; 43:1371-5; PMID:6335967 [PubMed] [Google Scholar]
- 12. Stocker M, Pedarzani P. Differential distribution of three Ca(2+)-activated K(+) channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci 2000; 15:476-93; PMID:10833304; http://dx.doi.org/ 10.1006/mcne.2000.0842 [DOI] [PubMed] [Google Scholar]
- 13. Vladimirova IA, Shuba MF. [Effect of strychnine, hydrastine and apamin on synaptic transmission in smooth muscle cells]. Neirofiziologiia 1978; 10:295-9; PMID:673075 [PubMed] [Google Scholar]
- 14. Blatz AL, Magleby KL. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat skeletal muscle. Nature 1986; 323:718-20; PMID:2430185; http://dx.doi.org/ 10.1038/323718a0 [DOI] [PubMed] [Google Scholar]
- 15. Orlov SM KG. Participation of calmodulin in the regulation of plasma membrane electric potential by intracellular calcium. Biokhimiia 1983; 48:1447-55; PMID:6414535 [PubMed] [Google Scholar]
- 16. Pape L, Kristensen BI. A calmodulin activated Ca2+-dependent K+ channel in human erythrocyte membrane inside-out vesicles. Biochim Biophys Acta 1984; 770:1-6; PMID:6320879; http://dx.doi.org/ 10.1016/0005-2736(84)90065-8 [DOI] [PubMed] [Google Scholar]
- 17. Klaerke DA, Petersen J, Jorgensen PL. Purification of Ca2+-activated K+ channel protein on calmodulin affinity columns after detergent solubilization of luminal membranes from outer renal medulla. FEBS Lett 1987; 216:211-6; PMID:2438163; http://dx.doi.org/ 10.1016/0014-5793(87)80691-9 [DOI] [PubMed] [Google Scholar]
- 18. Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annu Rev Physiol 2002; 64:289-311; PMID:11826271; http://dx.doi.org/ 10.1146/annurev.physiol.64.100301.111649 [DOI] [PubMed] [Google Scholar]
- 19. van Houten J, Chang SY, Kung C. Genetic analyses of "paranoiac" mutants of Paramecium tetraurelia. Genetics 1977; 86:113-20; PMID:885338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Satow Y, Kung C. A mutant of Paramecium with increased relative resting potassium permeability. J Neurobiol 1976; 7:325-38; PMID:956817; http://dx.doi.org/ 10.1002/neu.480070405 [DOI] [PubMed] [Google Scholar]
- 21. Hinrichsen RD, Burgess-Cassler A, Soltvedt BC, Hennessey T, Kung C. Restoration by calmodulin of a Ca2+-dependent K+ current missing in a mutant of paramecium. Science 1986; 232:503-6; PMID:2421410; http://dx.doi.org/ 10.1126/science.2421410 [DOI] [PubMed] [Google Scholar]
- 22. Kink JA, Maley ME, Preston RR, Ling K-Y, Wallen-Friedman MA, Saimi Y, Kung C. Mutations in paramecium calmodulin indicate functional differences between the C-terminal and N-terminal lobes in vivo. Cell 1990; 62:165-74; PMID:2163766; http://dx.doi.org/ 10.1016/0092-8674(90)90250-I [DOI] [PubMed] [Google Scholar]
- 23. Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science 1978; 202:1203-6; PMID:103199; http://dx.doi.org/ 10.1126/science.103199 [DOI] [PubMed] [Google Scholar]
- 24. DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 2001; 411:484-9; PMID:11373682; http://dx.doi.org/ 10.1038/35078091 [DOI] [PubMed] [Google Scholar]
- 25. Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron 1999; 22:549-58; PMID:10197534; http://dx.doi.org/ 10.1016/S0896-6273(00)80709-6 [DOI] [PubMed] [Google Scholar]
- 26. Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, Milesi V, Neely A, Alvarez O, Ferreira G, Gonzalez C, et al. . A BK (Slo1) channel journey from molecule to physiology. Channels (Austin) 2013; 7:442-58; PMID:24025517; http://dx.doi.org/ 10.4161/chan.26242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol 1997; 110:257-81; PMID:9276753; http://dx.doi.org/ 10.1085/jgp.110.3.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J Gen Physiol 1997; 109:647-73; PMID:9154910; http://dx.doi.org/ 10.1085/jgp.109.5.647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol 2002; 120:267-305; PMID:12198087; http://dx.doi.org/ 10.1085/jgp.20028605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science 1996; 273:1709-14; PMID:8781233; http://dx.doi.org/ 10.1126/science.273.5282.1709 [DOI] [PubMed] [Google Scholar]
- 31. Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J Gen Physiol 1998; 111:565-81; PMID:9524139; http://dx.doi.org/ 10.1085/jgp.111.4.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia X-M, fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, et al. . Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 1998; 395:503-7; PMID:9774106; http://dx.doi.org/ 10.1038/26758 [DOI] [PubMed] [Google Scholar]
- 33. Adelman JP, Maylie J, Sah P. Small-Conductance Ca(2+)-Activated K(+) Channels: Form and Function. Annu Rev Physiol 2011; PMID:21942705 [DOI] [PubMed] [Google Scholar]
- 34. Geiser JR, Tuinen DV, Brockerhoff SE, Neff MM, Davis TN. Can calmodulin function without binding calcium?. Cell 1991; 65:949-59; PMID:2044154; http://dx.doi.org/ 10.1016/0092-8674(91)90547-C [DOI] [PubMed] [Google Scholar]
- 35. Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, Janowsky A, Fakler B, Adelman JP, Maylie J. Domains responsible for constitutive and Ca(2+)-dependent interactions between calmodulin and small conductance Ca(2+)-activated potassium channels. J Neurosci 1999; 19:8830-8; PMID:10516302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 2001; 410:1120-4; PMID:11323678; http://dx.doi.org/ 10.1038/35074145 [DOI] [PubMed] [Google Scholar]
- 37. Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, et al. . Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron 2004; 43:847-58; PMID:15363395; http://dx.doi.org/ 10.1016/j.neuron.2004.08.033 [DOI] [PubMed] [Google Scholar]
- 38. Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci 2007; 27:2369-76; PMID:17329434; http://dx.doi.org/ 10.1523/JNEUROSCI.3565-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams PJ, Ben-Johny M, Dick IE, Inoue T, Yue DT. Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell 2014; 159:608-22; PMID:25417111; http://dx.doi.org/ 10.1016/j.cell.2014.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]