

ABSTRACT
The third marsupial genome was sequenced from the Tasmanian devil (Sarcophilus harrisii), a species that currently is driven to extinction by a rare transmissible cancer. The transposable element (TE) landscape of the Tasmanian devil genome revealed that the main driver of retrotransposition the Long INterspersed Element 1 (LINE1) seem to have become inactivated during the past 12 million years. Strangely, the Short INterspersed Elements (SINE), that normally hijacks the LINE1 retrotransposition system, became inactive prior to LINE1 at around 30 million years ago. The SINE inactivation was in vitro verified in several species. Here I discuss that the apparent LINE1 inactivation might be caused by a genome assembly artifact. The repetitive fraction of any genome is highly complex to assemble and the observed problems are not unique to the Tasmanian devil genome.
Keywords: genome assembly, inactivation, LINE1, marsupials, retrotransposition, Tasmanian devil
Transposable elements and evolution
Genomic analyses of TEs offer an enormous source of information in the genome about their biology, evolution and for phylogenetic reconstruction. The large abundance of TEs, their continuous propagation and diversity in genomes make them a treasure trove for evolutionary studies.1-2 The analyses of TE insertions and their sequences are a valuable aid in the understanding of relationships among species, populations, and morphological adaption and give insights into the dynamics of structural changes of a genome.3-8 The biggest obstacle in the analysis of TE sequences has been the difficulty to extract information about TE insertions from a genome without a high quality reference genome sequence. The human genome is currently the best-assembled genome with the highest sequence coverage from different sequencing technologies, and therefore TE research focused on this resource.9-10 However, despite the high quality of the human genome, there are still some contigs and genes that have not been anchored in the assembly yet. These are mostly highly repetitive regions, such as large segmental duplications found in the pericentromeres but also protein-coding genes.11
Currently mammalian genomes are becoming available at an increasing speed, but the sequences are nearly exclusively produced from placental mammals.12 It is essential to understand the great diversity of TEs in as many different mammalian orders and infraclasses as possible and not only human or placental mammals. Mammals consist of about 5,000 living species divided into 3 very different infraclasses, the placental, marsupial and monotreme mammals.13 Marsupials diverged 160 million years (Ma) ago from placental mammals14 and the 320 living Australian and South American marsupial species are so far only represented by 3 published genomes: that of the Australian tammar wallaby (Macropus eugenii),15 the South American opossum (Monodelphis domestica),16 and the Australian Tasmanian devil (Sarcophilus harrisii).17 In marsupial and placental mammals, the autonomous LINE1 is the main driver of retrotransposition.18-19 A full-length coding mammalian LINE1 is about 6,000-7,000 nucleotides (nt) long and has 2 open-reading frames (ORF). LINE1 encodes for reverse transcriptase (RT) and an endonuclease that are essential for propagation of LINE1 and non-autonomous SINEs.19 As a result of incomplete reverse transcription most LINE1 copies are 5′-truncated. Of the ∼500,000 LINE1 copies in the human genome only 80-100 copies are retrotranspositionally competent.19
The third marsupial genome was sequenced from the Tasmanian devil
After the extinction of the Tasmanian tiger (Thylacinus cynocephalus) in the 1930s, the Tasmanian devil remained as the largest living marsupial carnivore.13 Taxonomically, the Tasmanian devil belongs to Dasyuromorphia, which is one of the 7 marsupial orders. Dasyuromorphia consists of 69 species found in Australia and New Guinea.13 Fossil evidence shows that Tasmanian devils were once widespread across the Australian continent, but for unknown reasons survived only on the Tasmanian island.20 The shrinkage of their geographical range resulted in a severe genetic bottleneck that drastically reduced the genetic diversity of the Tasmanian population.21 More alarmingly, however, is a recent outbreak of a rare transmissible cancer, the devil facial tumor disease (DFTD). The DFTD cancer threatens the survival of the species and was one of the reasons to sequence the Tasmanian devil genome.17,21-22 The first Australian marsupial to have its genome sequenced was that of the tammar wallaby, but the assembly is currently highly fragmented15 and has not allowed detailed TE analyses. Thus, the genome data of the Tasmanian devil offer a chance to analyze the TE composition and dynamics in an Australian marsupial genome and compare it to other mammalian species.
Tasmanian devil genome may be void of active LINE1
One of the more surprising findings from the TE screen of the Tasmanian devil genome strongly suggested that LINE1 activity might have gone extinct.23 The in silico screening of the Tasmanian devil genome did not identify any LINE1 copies with intact ORFs and only very few full-length elements.23 As a functional RT sequence is essential for retrotransposition to occur, alternative methods were used to identify RT motifs in the Tasmanian devil genome. Hidden Markov Models (HMM) were constructed from RT sequences from a spectrum of organisms and autonomous TEs to identify such coding regions. The results indicated that only very few short remnants of RT-coding ORFs were left in the Tasmanian devil genome. The HMM approach was also not able to identify intact RT genes from other autonomous TEs, such as LINE2, LINE3 or RTE. However, as a positive control, the same approach identified, as expected, a large amount of RT-derived ORFs the South American opossum genome.
Given the presence of active LINE1 elements in most marsupial and placental mammalian genomes, the finding of a LINE1 inactivation event in the Tasmanian devil genome was unexpected. The absence of LINE1 activity in mammals has so far only been found in species from 2 mammalian orders: rodents (Rodentia) and bats (Chiroptera). In rodents, LINE1 inactivation apparently occurred independently, in the South American sigmodontine rodents (∼300 species) and in the 13-lined squirrel (Ictidomys tridecemlineatus) genome.24-26 In bats, the inactivation happened in the ancestor to the megabat group that consists of ∼800 living species.27 Further, the LINE1 length landscape of 8 mammals was compared to investigate the overall distribution of 5’-truncated versus full-length LINE1 copies in the entire genome.23 The results show a distinct difference between species with documented LINE1 activity (human, mouse, dog, opossum) and species that have suspected LINE1 inactivation (megabats, 13-lined squirrel and Tasmanian devil). The LINE1 active mammals have in addition to 5’-truncated LINE1 copies, a sizable fraction of full-length elements. The suspected LINE1 inactive mammals lack or have very few full-length elements in what resembles depletion.23 Based on these results, one could conclude that the Tasmanian devil is the fourth species/group to be added to the list of mammals without LINE1 activity.
Retrotransposition of SINEs was inactivated prior to LINE1 in Dasyuromorphia
Besides the in silico screen for LINE1 activity in the Tasmanian devil genome, TEs were used to reconstruct the phylogenetic relationship among the species of the order Dasyuromorphia. A phylogenetic screen is also useful for gathering independent information about the past retrotransposition activity. When examining an orthologous locus in 2 closely related species, with one species having a TE insertion that is missing from the other species, it is obvious that the TE must have integrated after their divergence.2 Thus, the respective TE family was propagating in the genome at that point and if this event was recent may still do so. The same principle can be applied for older nodes in the evolutionary tree. Together with divergence time estimates from fossil data and the molecular clock, a specific age can be placed on the past activity of different TEs. For the phylogenetic screen in the Tasmanian devil genome, TEs were pre-selected for presence in short introns, and each locus was experimentally PCR amplified in-vitro for additional dasyuromorphian species that are not yet covered by genome sequences. This allowed investigating TE insertions in an evolutionary perspective. Apart from the novel phylogenetic information, it became obvious that (a) the SINEs in the genome seem to have stopped mobilizing around 30 Ma and (b) that LINE1 continued to propagate up until relative recent times (12-0 Ma). The reason for this is that unique LINE1 insertions were found in the Tasmanian devil. All Tasmanian devil SINE and LINE1 insertions that were analyzed had a 100% sequence similarity to the Tasmanian devil reference genome sequence and the experimentally sequenced and analyzed Tasmanian devil individual.23 This suggests that for most introns, SINEs and truncated LINE1s are sequenced and assembled correctly in the Tasmanian devil reference genome. Thus, 30 Ma something disabled LINE1 to recognize the SINEs in the ancestral dasyuromorphian genome. SINE inactivation prior to LINE1 inactivation is not unique to Dasyuromorphia but has also been described for the sigmodontine rodents.28 Known factors that contribute to the death of SINE propagation is the length of its poly(A)-tail as well as the nucleotide composition of the poly(A)-tail.29
Transposable elements and genome assembly issues
Unexpected findings from in silico genome studies, such as the Tasmanian devil LINE1 inactivation23 may be suspicious.30 To date, reference genome sequences from most species may not represent ‘true’ genome sequences, because sequencing and assembly quality might not be optimal. Even if the quality of a genome assembly was sufficient to analyze the initial question, it may not be suitable for detailed TE analyses. The repetitive nature of TE sequences in addition to short sequencing reads ranging between 125–150 nt from the currently dominating Illumina technique, makes it tremendously difficult to correctly assemble TEs. Similar sequence reads might end up being mapped to the same location, even though they have vastly different genomic origins. In particular, the evolutionary young TEs from recent transposition events with more than 97% sequence similarity cause the most problems for assembly programs, because they are nearly identical.31 The task to assemble long TEs, such as LINEs or ERVs (Endogenous RetroVirus) from short sequence reads is even more complicated, because the sequences of short Illumina reads cover only a small fraction of the element. In addition to that, assembly artifacts can lead to a complete absence of full-length elements, if the element is placed between scaffolds (Fig. 1A). The internal regions of LINE1 elements are problematic to assemble, because these sequences occur at multiple genomic locations. Sequence reads that cover the genomic flanks, and the beginning and end of the TE, provide a set of anchoring sequence reads. The internal region of the TE is then left out from the assembly, and an artificial break point is created in the scaffold (Fig. 1A). This leads to a set of fragmented ‘ghost LINE1s’ that are found at the ends of scaffolds. As a consequence, the overall copy number increases as each copy will be counted as 2 individual copies. The problem of ‘ghost LINE1s’ was observed when comparing the 2 available Tasmanian devil assemblies.17,21,23 With help of the LINE1 flanking genomic sequence in the reference genome assembly17 the location and a small stretch of the orthologous LINE1 copy could be identified in a second genome assembly from the individual ‘Cedric’.21 The majority of the scarce full-length LINE1 copies found in the reference assembly was found divided across 2 scaffolds in the Cedric assembly. Another assembly challenge is the unintentional incorporation of substitutions in the ORFs of LINE1 sequences (Fig. 1B) when trying to merge the reads into a consensus sequence of the elements at each unique location in the assembly. As TE reads map to multiple genomic locations, at each location there will be a mixture of reads originating from different copies. This can lead to the incorporation of substitutions or ambiguous nucleotides (Ns) in the LINE1 copies in genome assemblies. Furthermore, LINE1 copies are flanked by identical target site duplications (TSD) of variable length.19 At such loci an assembly program can collapse the LINE1 sequence completely by mistakenly assembling the 2 TSDs into a single sequence, leaving the entire remaining LINE1 sequence out of the assembly (Fig. 1C). Finally, artifacts can occur from problems sequencing through long homopolymeric stretches. Young LINE1 copies are characterized by long poly(A)-tails which are up to 100 nt long. These result in sequencing stops at the poly(A)-tail so that short reads are filtered out even before the assembly step. Additional TE related assembly artifacts have been described previously31, and this represents a selection that might have influenced the LINE1 content in the Tasmanian devil assembly as well. These problems have been observed in the initial release of the cat (Felis catus) genome with 1.9X coverage32 and the tammar wallaby genome with 2X coverage15. In the initial genome releases in neither the cat nor the wallaby had assembly full-length LINE1 copies with intact ORFs. Similarly, only 4 copies of LINE1 sequences, with intact ORFs, could be retrieved from the initial version of the dog genome (Canis familiaris).33
Figure 1.
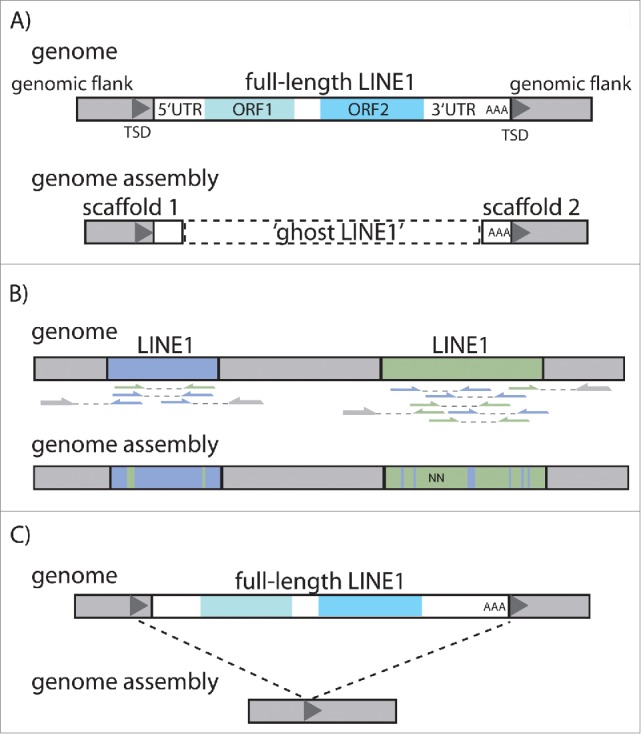
Selected problems that can affect TE sequences and copy numbers in a final genome assembly. (A) Due to different assembly algorithms implemented in the programs, problematic regions might be left out of assemblies all together due to the lack of supporting or conflicting reads leading to ghost LINE1s. In such cases, full-length LINE1s are split across 2 different scaffolds. (B) Paired-end reads (opposing arrows connected by dashed lines) originating from multiple LINE1 copies can falsely be incorporated into one single consensus sequence, leading to unintentional substitutions as well as ambiguous nucleotides (N) in the TE sequence in the assembly. (C) The identical flanking target site duplications (TSDs) can be falsely assembled into a single sequence, collapsing the entire LINE1 (or ERV). TSD: target site duplication; AAA: poly(A)-tail; ORF: open reading frame, UTR: untranslated region.
Future work and conclusions
Genomes are rarely sequenced with the purpose to investigate the TE content. Therefore, the current resolution of most genome assemblies may be insufficient to give a detailed picture of the active long autonomous TEs in the genomes such as LINE1 and ERVs. Often genome assemblies are sufficient to analyze most protein-coding sequences, but improving the assembly still leads to identifying additional genes and improving annotations.34-36 The problem is not exclusive to non-primate genome data. Even the genomes of primates are often inadequately assembled, despite being closely related to humans.37 Thus, reliance on in silico data for investigation of active LINEs, ERVs or even SINE activity in a genome is problematic and makes in-vitro experimental methods necessary to verify new and unexpected findings.23
Understanding and evaluating unexpected findings such as the possible LINE1 inactivation in the Tasmanian devil genome require verification by high-throughput experimental methods that need to be developed. The experimental method that was used to uncover the LINE1 inactivation38 in sigmodontine rodents as well as in megabats27 marks a beginning, but needs to be streamlined. The method is based on screening a ∼600 nt part of the LINE1 reverse transcriptase coding region of ORF2 for stop codons by PCR amplification with conserved primers, targeting, young LINE1 copies. Cloning follows the PCR screen, which is a tedious procedure. In addition, this method only analyzes a small region (∼15%) of the ORF2 locus. Despite its shortcomings, we have applied the PCR-screen method to the Tasmanian devil and its close relatives to gain a first experimental understanding of the properties of young LINE1 sequences in the genome of the Tasmanian devil (Gallus et al. in preparation).
We are currently experiencing the dawn of TE analyses in diverse vertebrates. Despite shortcomings, genome data are already a valuable source to study the properties and dynamics of TEs. In silico analyses of TE insertions are revolutionizing phylogenetic analyses39-40 from painstakingly in-vitro analysis of TE insertions, to being able to extract hundreds such events on a genomic scale. Biological relevant data will become available with new sequencing techniques such as PacBio that sequence longer genome fragments. Then the crucial role of TEs in disease, genome structure and evolutionary studies will become more prominent on a comparative scale.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Axel Janke, Susanne Gallus and Fritjof Lammers provided thoughtful comments on the manuscript.
References
- [1]. Ray DA. SINEs of progress: mobile element applications to molecular ecology. Mol Ecol 2007; 16:19-33; PMID:17181718; http://dx.doi.org/ 10.1111/j.1365-294X.2006.03104.x [DOI] [PubMed] [Google Scholar]
- [2]. Nishihara H, Okada N. Retroposons: genetic footprints on the evolutionary paths of life. Methods Mol Biol 2008; 422:201-25; PMID:18629669; http://dx.doi.org/ 10.1007/978-1-59745-581-7_13 [DOI] [PubMed] [Google Scholar]
- [3]. Churakov G, Kriegs JO, Baertsch R, Zemann A, Brosius J, Schmitz J. Mosaic retroposon insertion patterns in placental mammals. Genome Res 2009; 19:868-75; PMID:19261842; http://dx.doi.org/ 10.1101/gr.090647.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Chalopin D, Naville M, Plard F, Galiana D, Volff JN. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol Evol 2015; 7:567-80; PMID:25577199; http://dx.doi.org/ 10.1093/gbe/evv005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Dodsworth S, Chase MW, Kelly LJ, Leitch IJ, Macas J, Novák P, Piednoël M, Weiss-Schneeweiss H, Leitch AR. Genomic repeat abundances contain phylogenetic signal. Syst Biol 2015; 64:112-26; PMID:25261464; http://dx.doi.org/ 10.1093/sysbio/syu080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol 2013; 22:1503-17; PMID:23293987; http://dx.doi.org/ 10.1111/mec.12170 [DOI] [PubMed] [Google Scholar]
- [7]. Barrón MG, Fiston-Lavier AS, Petrov DA, González J. Population genomics of transposable elements in Drosophila. Annu Rev Genet 2014; 48:561-81; PMID:25292358; http://dx.doi.org/ 10.1146/annurev-genet-120213-092359 [DOI] [PubMed] [Google Scholar]
- [8]. Lee SI, Kim NS. Transposable elements and genome size variations in plants. Genomics Inform 2014; 12:87-97; PMID:25317107; http://dx.doi.org/ 10.5808/GI.2014.12.3.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009; 10:691-703; PMID:19763152; http://dx.doi.org/ 10.1038/nrg2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Hancks DC, Kazazian HH, Jr. Active human retrotransposons: variation and disease. Curr Opin Genet Dev 2012; 22:191-203; PMID:22406018; http://dx.doi.org/ 10.1016/j.gde.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]. Genovese G, Handsaker RE, Li H, Altemose N, Lindgren AM, Chambert K, Pasaniuc B, Price AL, Reich D, Morton CC, et al. Using population admixture to help complete maps of the human genome. Nat Genet 2013; 45:406-14; PMID:23435088; http://dx.doi.org/ 10.1038/ng.2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. Koepfli KP, Paten B. Genome 10K Community of Scientists, O'Brien SJ. The Genome 10K Project: a way forward. Annu Rev Anim Biosci 2015; 3:57-111; PMID:25689317; http://dx.doi.org/ 10.1146/annurev-animal-090414-014900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Nowak RM. 2005. Walker's mammals of the world. Baltimore: The Johns Hopkins University Press. [Google Scholar]
- [14]. Luo Z-X, Yuan C-X, Meng Q-J, Ji Q. A Jurassic eutherian mammal and divergence of marsupials and placentals. Nature 2011; 476:442-45; PMID:21866158; http://dx.doi.org/ 10.1038/nature10291 [DOI] [PubMed] [Google Scholar]
- [15].Renfree MB, Papenfuss AT, Deakin JE, Lindsay J, Heider T, Belov K, Rens W, Waters PD, Pharo EA, Shaw G, et al. Genome sequence of an Australian kangaroo, Macropus eugenii, provides insight into the evolution of mammalian reproduction and development. Genome Biol 2011; 12:R81; PMID:21854559; http://dx.doi.org/ 10.1186/gb-2011-12-8-r81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 2007; 447:167-77; PMID:17495919; http://dx.doi.org/ 10.1038/nature05805 [DOI] [PubMed] [Google Scholar]
- [17]. Murchison EP, Schulz-Trieglaff OB, Ning Z, Alexandrov LB, Bauer MJ, Fu B, Hims M, Ding Z, Ivakhno S, Stewart C, et al. 2012. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell 2012; 148:780-91; PMID:22341448; http://dx.doi.org/ 10.1016/j.cell.2011.11.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Gentles AJ, Wakefield MJ, Kohany O, Gu W, Batzer MA, Pollock DD, Jurka J. Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica. Genome Res 2007; 17:992-1004; PMID:17495012; http://dx.doi.org/ 10.1101/gr.6070707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr 2015; 3: MDNA3-0061-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Brown OJF. Tasmanian devil (Sarcophilus harrisii) extinction on the Australian mainland in the mid-Holocene: multicausality and ENSO intensification. Alcheringa 2006; 31:49-57; http://dx.doi.org/ 10.1080/03115510608619574 [DOI] [Google Scholar]
- [21]. Miller W, Hayes VM, Ratan A, Petersen DC, Wittekindt NE, Miller J, Walenz B, Knight J, Qi J, Zhao F, et al. Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc Natl Acad Sci U S A 2011; 108:12348-53; PMID:21709235; http://dx.doi.org/ 10.1073/pnas.1102838108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Grueber CE, Peel E, Gooley R, Belov K. Genomic insights into a contagious cancer in Tasmanian devils. Trends Genet 2015; 31:528-35; PMID:26027792; http://dx.doi.org/ 10.1016/j.tig.2015.05.001 [DOI] [PubMed] [Google Scholar]
- [23]. Gallus S, Hallström BM, Kumar V, Dodt WG, Janke A, Schumann GG, Nilsson MA. Evolutionary histories of transposable elements in the genome of the largest living marsupial carnivore, the Tasmanian devil. Mol Biol Evol 2015; 32:1268-83; PMID:25633377; http://dx.doi.org/ 10.1093/molbev/msv017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Casavant NC, Scott L, Cantrell MA, Wiggins LE, Baker RJ, Wichman HA. The end of the LINE?: lack of recent L1 activity in a group of South American rodents. Genetics 2000; 154:1809-17; PMID:10747071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Grahn RA, Rinehart TA, Cantrell MA, Wichman HA. Extinction of LINE-1 activity coincident with a major mammalian radiation in rodents. Cytogenet Genome Res 2005; 110:407-415; PMID:16093693; http://dx.doi.org/ 10.1159/000084973 [DOI] [PubMed] [Google Scholar]
- [26]. Platt RN, 2nd, Ray DA. A non-LTR retroelement extinction in Spermophilus tridecemlineatus. Gene 2012; 500:47-53; PMID:22465530; http://dx.doi.org/ 10.1016/j.gene.2012.03.051 [DOI] [PubMed] [Google Scholar]
- [27]. Cantrell MA, Scott L, Brown CJ, Martinez AR, Wichman HA. Loss of LINE-1 activity in the megabats. Genetics 2008; 178:393-404; PMID:18202382; http://dx.doi.org/ 10.1534/genetics.107.080275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Rinehart TA, Grahn RA, Wichman HA. SINE extinction preceded LINE extinction in sigmodontine rodents: implications for retrotranspositional dynamics and mechanisms. Cytogenet Genome Res 2015; 110:416-25; PMID:16093694; http://dx.doi.org/ 10.1159/000084974 [DOI] [PubMed] [Google Scholar]
- [29]. Comeaux MS, Roy-Engel AM, Hedges DJ, Deininger PL. Diverse cis factors controlling Alu retrotransposition: what causes Alu elements to die?. Genome Res 2009; 19:545-55; PMID:19273617; http://dx.doi.org/ 10.1101/gr.089789.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Birney E. Assemblies: the good, the bad, the ugly. Nat Methods 2011; 8:59-60; PMID:21191376; http://dx.doi.org/ 10.1038/nmeth0111-59 [DOI] [PubMed] [Google Scholar]
- [31]. Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nature Rev Genet 2011; 13:36-46; PMID:22124482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Pontius JU, Mullikin JC, Smith DR, Agencourt Sequencing Team , Lindblad-Toh K, Gnerre S, Clamp M, Chang J, Stephens R, Neelam B, et al. Initial sequence and comparative analysis of the cat genome. Genome Res 2007; 17:1675-89; PMID:17975172; http://dx.doi.org/ 10.1101/gr.6380007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Wang W, Kirkness EF. Short interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res 2005; 15:1798-08; PMID:16339378; http://dx.doi.org/ 10.1101/gr.3765505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Elsik CG, Worley KC, Bennett AK, Beye M, Camara F, Childers CP, de Graaf DC, Debyser G, Deng J, Devreese B, et al. Finding the missing honey bee genes: lessons learned from a genome upgrade. BMC Genomics 2014; 15:86; PMID:24479613; http://dx.doi.org/ 10.1186/1471-2164-15-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Zhang X, Goodsell J, Norgren RB, Jr. Limitations of the rhesus macaque draft genome assembly and annotation. BMC Genomics 2012; 13:206; PMID:22646658; http://dx.doi.org/ 10.1186/1471-2164-13-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Florea L, Souvorov A, Kalbfleisch TS, Salzberg SL. Genome assembly has a major impact on gene content: a comparison of annotation in two Bos taurus assemblies. PLoS One 2011; 6:e21400; PMID:21731731; http://dx.doi.org/ 10.1371/journal.pone.0021400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Norgren RB, Jr. Improving genome assemblies and annotations for nonhuman primates. ILAR J 2013; 54:144-53; PMID:24174438; http://dx.doi.org/ 10.1093/ilar/ilt037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Cantrell MA, Grahn RA, Scott L, Wichman HA. Isolation of markers from recently transposed LINE-1 retrotransposons. Biotechniques 2000; 29:1310-16; PMID:11126134 [DOI] [PubMed] [Google Scholar]
- [39]. Suh A, Smeds L, Ellegren H. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol 2015; 13:e1002224; PMID:26284513; http://dx.doi.org/ 10.1371/journal.pbio.1002224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Doronina L, Churakov G, Shi J, Brosius J, Baertsch R, Clawson H, Schmitz J. Exploring massive incomplete lineage sorting in arctoids (Laurasiatheria, Carnivora). Mol Biol Evol 2015; 32:3194-204; PMID:26337548 [DOI] [PubMed] [Google Scholar]