

Abstract
Dysplastic cerebellar gangliocytoma or Lhermitte-Duclos disease (LDD) especially in children are extremely rare. In this report we add one further case to this rare entity. A three year old boy with a history of cerebellar and brain stem compression signs was presented in unconscious condition. Computerized tomography (CT) scan revealed a well defined lesion mixed with area of calcification in the right cerebellum and severe obstructive hydrocephalus. Ventricular shunting was performed followed by gross total tumor removal three days after shunting. In the follow up period, the patient showed almost total resolution of all neurological deficits. MRI has been an imaging tool to preoperatively diagnose this disease; otherwise it would be uncertain preoperative diagnosis. However, in this report we would like to emphasize that not all uncertain diagnosis of LDD leads to palliative treatment. Well demarcated lesion may account for the safe surgical resection of this disease.
Keywords: Cerebellar tumor, dysplastic gangliocytoma, Lhermitte-Duclos disease, surgical resection
Introduction
The unique anatomical features of LDD, which include preservation and widening of the gyral pattern can be diagnosed preoperatively with MRI.[1,2,3,4,5,6,7,8] Other imaging studies such as CT scan are not considered sufficiently sensitive to make the diagnosis.[2,3,9] However, there are reports of successful surgical resection of this particular disease even the preoperative diagnosis is uncertain.[9,10,11]
We reported a case of LDD in a child who was preoperatively diagnosed as cerebellar tumor based on CT scan.
Case Report
A three year old boy with two weeks history of right facial nerve paralysis, gait disturbance and difficulty swallowing, presented in an unconscious condition. CT scan revealed a well defined lesion with area of calcification in the right cerebellum [Figure 1]. There was a right to left shift of the fourth ventricle and severe obstructive hydrocephalus. After a shunting procedure the patient regained full consciousness. Further neurological examination revealed paralysis of right facial nerve, right sided dysmetria and dysdiadokokinesia neck stiffness and nystagmus. Deep reflexes were increased bilaterally. There was no papilledema. There were no cutaneous lesions or significant family history suggesting the diagnosis. A diagnosis of right cerebellar tumor was made and surgical removal was performed three days following shunting procedure.
Figure 1.
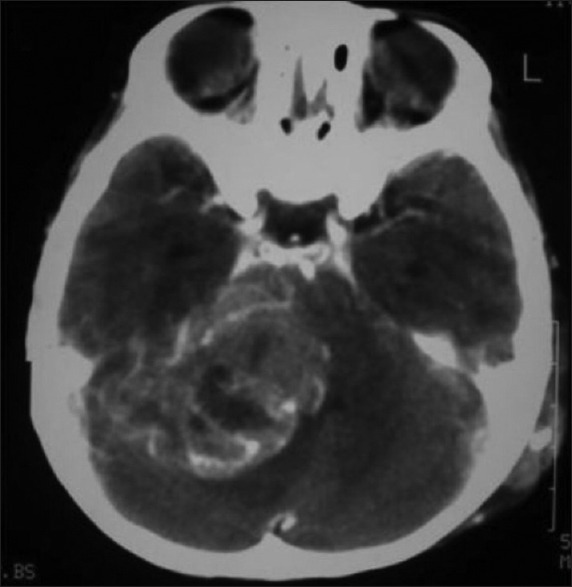
Preoperative CT scan of the patient with LDD shows a well defined lesion mixed with area of calcification in the right cerebellar hemisphere. There is a right to left shift of the fourth ventricle
Right sub occipital craniotomy was performed. After retracting normal cerebellar tissue, we observed widened and well preserved cerebellar folia with a consistency comparable to a glioma. The tumor tissue was pale gray and poorly vascularized. As we went deeper, the border between tumor and normal cerebellar tissue was clearly distinguished. Further surgical exploration revealed that the lesion involved only the cerebellar folia; there was no infiltration of the medulla, pons and cranial nerves. Gross total tumor removal was achieved in this case.
The patient made uneventful recovery. Facial nerve paralysis remained but swallowing difficulties were reduced. On discharge, the patient was able to walk although a little unstable and had no difficulty swallowing. Over the following six month period there was complete resolution of all neurological deficits except for very mild paralysis of the right facial nerve. Control MRI three months after surgery at a private hospital revealed total tumor removal and the brain stem has resumed almost normal size and position [Figure 2a and b].
Figure 2.
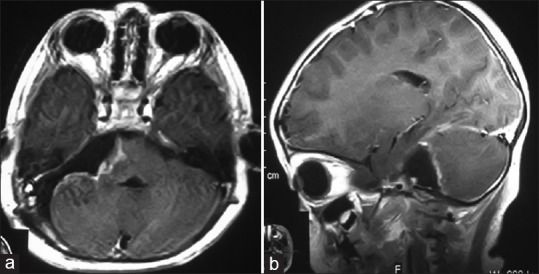
Axial (a) and sagittal (b) three months postoperative T1-weighted MRI of the patient with LDD reveals total tumor resection. The brain stem resumes almost normal size and position. The fourth ventricle is open
Section of the tumor mass revealed relatively well preservation of the cerebellar architecture with widened and distorted folia [Figure 3a]. There was diffuse enlargement of the molecular and internal granular layers, which were filled with dispersed of ganglionic cells of varying sizes [Figure 3b]. Purkinje cells were absent throughout the entire specimen. On the basis of this pathological report, the patient was diagnosed as LDD. We did not do immunohistochemistry study in this case.
Figure 3.
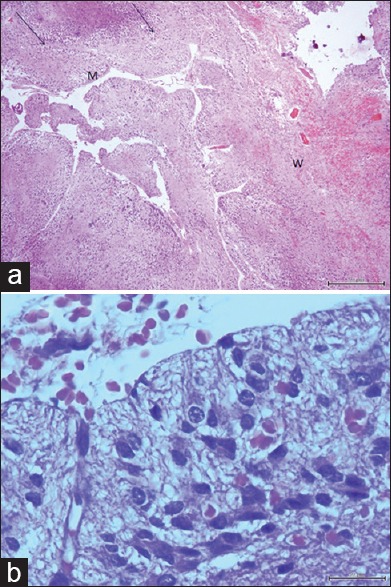
(a) Microphotograph of tumor tissue reveals widened of cerebellar folia (black arrows) and thickened of the molecular layer (m) and thinned of central white matter (w) (H and E, × 40). (b) Microphotograph of tumor tissue shows numerous of dysplastic ganglion cells (H and E, × 400)
Discussion
Patients with LDD usually present with a long standing history of ill defined neurological signs related to increased intracranial pressure with brainstem compression and cerebellar signs, such as cranial nerve palsies and unsteadiness of gait.[2,3,10,12] Acute-onset of cerebellar symptoms, characterized by a rapid neurological deficit is rarely reported.[4,5] Our patient showed this acute-onset symptom.
Microscopic pathology of LDD is unique and well described,[1,3,13] therefore we did not do immunohistochemistry study in our present case. The histopathological findings consist of widening of folia and molecular layer, which is occupied by abnormal ganglion cell, absence of Purkinje cell layer and hypertrophy of granular cell layer.[1,2,4,12]
Familial occurrence of LDD or associated hereditary syndromes such as Cowden disease has been reported.[12,14] LDD manifested in the second or third decade of life is most likely associated with Cowden disease.[14] Our patient was a child and had no history of familial LDD or hereditary diseases.
On MRI, the widening of cerebellar folia appears as parallel linear striation on the surface of the lesion. This appearance is called “tiger striping” or “striated cerebellum”, which is a characteristic appearance of LDD.[6,7,15] Establishing preoperative diagnosis of LDD with MRI obviates the need for biopsy and this allows neurosurgeons to plan for appropriate treatment.[4,5,7] The CT scan appearance of LDD, consists of hypo-dense and calcified areas is nonspecific for this disease.[2,3,9] In our case, preoperative diagnosis was uncertain because MRI was not available yet at our hospital. However, clinical and CT scan findings in our case had shown an emergency situation. This situation had led us to perform posterior fossa decompression by gross total tumor removal. Our literature review suggests that surgical resection is the treatment of choice for this particular disease.[1,2,3,5,6,7] Many reports have confirmed that decompressive craniotomy for LDD has been successful not only in relieving symptoms[9] but also improving long-term survival.[3,10]
The relative merits of MRI and CT in delineating the margin and the location of this lesion have been discussed previously.[1,2,3,10,13] However, the precise margin of abnormal tissue of this tumor should be assessed under surgical microscope since well border lesion seen on MRI is not always apparent during surgical exploration.[13] During surgical exploration in some cases of LDD, the lesion is clearly distinguishable from the surrounding normal cerebellar tissue.[2,3] A well-circumscribed lesion allows neurosurgeons to safely and totally excise this lesion.[3] Reeder et al. observed one of the three cases with LDD was well-demarcated lesion noted on both MRI and during operation.[3] Nowak et al. reported one of the two cases of LDD was well-circumscribed lesion observed on both MRI and during surgical exploration.[2] The well- demarcated lesion in this particular case may account for the successful surgical resection. In our present case the well-defined lesion was fortuitously found during surgical exploration.
Since MRI is sensitive in imaging this disease,[1,2,3,4,7,15] we sent the patient for a postoperative MRI at a private hospital. This was done to assess the success of surgical resection and the possibility of tumor recurrence.
Conclusion
Encountering patient with acute-onset symptoms of cerebellar tumor and significant cerebellar mass effect on CT scan, surgical tumor removal should be performed even with diagnosis uncertainty. There are reports of successful posterior fossa decompression of this particular disease even the preoperative diagnosis is uncertain.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Carter JE, Merren MD, Swann KW. Preoperative diagnosis of Lhermitte-Duclos disease by magnetic resonance imaging: Case report. J Neurosurg. 1989;70:135–7. doi: 10.3171/jns.1989.70.1.0135. [DOI] [PubMed] [Google Scholar]
- 2.Nowak DA, Trost HA, Porr A, Stölzle A, Lumenta CB. Lhermitte-Duclos disease (Dysplastic gangliocytoma of the cerebellum) Clin Neurol Neurosurg. 2001;103:105–10. doi: 10.1016/s0303-8467(01)00124-x. [DOI] [PubMed] [Google Scholar]
- 3.Reeder RF, Saunders RL, Roberts DW, Fratkin JD, Cromwell LD. Magnetic resonance imaging in the diagnosis and treatment of Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum) Neurosurgery. 1988;23:240–5. doi: 10.1227/00006123-198808000-00022. [DOI] [PubMed] [Google Scholar]
- 4.Hariri OR, Khachekian A, Muilli D, Amin J, Minassian T, Berman B, et al. Acute-onset cerebellar symptoms in Lhermitte-Duclos disease: Case report. Cerebellum. 2012;11:1–4. doi: 10.1007/s12311-012-0394-2. [DOI] [PubMed] [Google Scholar]
- 5.Yağci-Küpeli B, Oguz KK, Bilen MA, Yalçin B, Akalan N, Buyukpamukcu M. An unusual cause of posterior fossa mass: Lhermitte-Duclos disease. J Neurol Sci. 2010;290:138–41. doi: 10.1016/j.jns.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 6.Nair P, Pal L, Jaiswal AK, Behari S. Lhermitte-Duclos disease associated with dysembryoplastic neuroepithelial tumor differentiation with characteristic magnetic resonance appearance of “Tiger Striping”. World Neurosurg. 2011;75:699–703. doi: 10.1016/j.wneu.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 7.Moenninghoff C, Kraff O, Schlamann M, Ladd ME, Katsarava Z, Gizewski ER. Assessing a dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) with 7T MR imaging. Korean J Radiol. 2010;11:244–8. doi: 10.3348/kjr.2010.11.2.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takei H, Dauser R, Su J, Chintagumpala M, Bhattacharjee MB, Jones J, et al. Anaplastic ganglioglioma arising from a Lhermitte-Duclos-like lesion. J Neurosurg. 2007;107(2 Suppl):137–42. doi: 10.3171/PED-07/08/137. [DOI] [PubMed] [Google Scholar]
- 9.Roski RA, Roessmann U, Spetzler RF, Kaufman B, Nulsen FE. Clinical and pathological study of dysplastic gangliocytoma: Case report. J Neurosurg. 1981;55:318–32. doi: 10.3171/jns.1981.55.2.0318. [DOI] [PubMed] [Google Scholar]
- 10.Di Lorenzo N, Lunardi P, Fortuna A. Granulomolecular hypertrophy of the cerebellum (Lhermitte-Duclos disease): Case report. J Neurosurg. 1984;60:644–6. doi: 10.3171/jns.1984.60.3.0644. [DOI] [PubMed] [Google Scholar]
- 11.Leech RW, Christoferson LA, Gilbertson RL. Dysplastic gangliocytoma (Lhermitte-Duclos disease) of the cerebellum: Case report. J Neurosurg. 1977;47:609–12. doi: 10.3171/jns.1977.47.4.0609. [DOI] [PubMed] [Google Scholar]
- 12.Rainov NG, Holzhausen HJ, Burkert W. Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease): Case report. Clin Neurol Neurosurg. 1995;97:175–80. doi: 10.1016/0303-8467(95)00017-e. [DOI] [PubMed] [Google Scholar]
- 13.Prestor B. Dysplastic gangliocytoma of the cerebellum (Lhermitte- Duclos disease) J Clin Neurosci. 2006;13:877–81. doi: 10.1016/j.jocn.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 14.Robinson S, Cohen AR. Cowden disease and Lhermitte-Duclos disease (an update): Case report and review of literature. Neurosurg Focus. 2006;20:E6. doi: 10.3171/foc.2006.20.1.7. [DOI] [PubMed] [Google Scholar]
- 15.Shinagare AB, Patil NK, Sorte SZ. Case 144: Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) Radiology. 2009;25:298–303. doi: 10.1148/radiol.2511071390. [DOI] [PubMed] [Google Scholar]