

Abstract
Background
Gram-positive bacteria in the phylum Firmicutes synthesize the low molecular weight thiol bacillithiol rather than glutathione or mycothiol. The bacillithiol transferase YfiT from Bacillus subtilis was identified as a new member of the recently discovered DinB/YfiT-like Superfamily. Based on structural similarity using the Superfamily program, we have determined 30 of 31 Staphylococcus aureus strains encode a single bacillithiol transferase from the DinB/YfiT-like Superfamily, while the remaining strain encodes two proteins.
Methods
We have cloned, purified, and confirmed the activity of a recombinant bacillithiol transferase (henceforth called BstA) encoded by the S. aureus Newman ORF NWMN_2591. Moreover, we have studied the saturation kinetics and substrate specificity of this enzyme using in vitro biochemical assays.
Results
BstA was found to be active with the co-substrate bacillithiol, but not with other low molecular weight thiols tested. BstA catalyzed bacillithiol conjugation to the model substrates monochlorobimane, 1-chloro-2,4-dinitrobenzene, and the antibiotic cerulenin. Several other molecules, including the antibiotic rifamycin S, were found to react directly with bacillithiol, but the addition of BstA did not enhance the rate of reaction. Furthermore, cells growing in nutrient rich medium exhibited low BstA activity.
Conclusions
BstA is a bacillithiol transferase from Staphylococcus aureus that catalyzes the detoxification of cerulenin. Additionally, we have determined that bacillithiol itself might be capable of directly detoxifying electrophilic molecules.
General Significance
BstA is an active bacillithiol transferase from Staphylococcus aureus Newman and is the first DinB/YfiT-like Superfamily member identified from this organism. Interestingly, BstA is highly divergent from Bacillus subtilis YfiT.
Keywords: Bacillithiol, bacillithiol transferase, detoxification, Staphylococcus aureus
1. Introduction
Aerobic organisms synthesize low molecular weight thiols (LMWTs) to mediate redox reactions and neutralize electrophiles produced intracellularly and extracellularly. The most well studied detoxification system is in mammals, which utilize multiple organs including the liver, which contains many glutathione transferases (GSTs)[1]. Although less studied, GSTs are also found in aerobic Gram-negative bacteria, which produce glutathione (GSH) as well. These bacterial GSTs catalyze many types of detoxification reactions including reactions with epoxides, halogens, quinones, enals and a range of xenobiotics[2, 3].
Detoxification of xenobiotics is less well understood in the Gram-positive bacteria because the thiol co-substrate GSH for GSTs is absent in most of these bacteria[4]. In the Actinobacteria, mycothiol (MSH) appears to be the dominant LMWT, with functions similar to GSH in Gram-negative bacteria[5, 6]. Until recently, the Firmicutes were known to only produce coenzyme A and cysteine as cellular LMWTs. In 2009 bacillithiol (BSH; 1, Equation 1), the α-anomeric glycoside of l-cysteinyl-d-glucosamine with l-malic acid, was described and found to have properties similar to GSH[7, 8]. BSH has been found to be required for resistance to the antibiotic fosfomycin, acid stress, and salt stress in Bacillus subtilis[8]. Furthermore, during treatment with oxidants or sodium hypochlorite, cysteine residues of proteins are bacillithiolated[9, 10]. Bacilliredoxins that reverse protein bacillithiolation in the redox sensitive proteins such as organic hydroperoxide reductase regulator (OhrR) and methionine synthase (MetE) have been recently described in B. subtilis[11].
Equation 1.

Bacillithiol transferases (BSTs) are enzymes that catalyze the transfer of bacillithiol to electrophilic substrates. The most recently identified BSTs are members of the DinB/YfiT-like Superfamily of thiol transferases, here referred to as the YfiT-like Superfamily [12]. Enzymes in this Superfamily are related by structure rather than sequence based on predictions using the Superfamily database (http://supfam.org/SUPERFAMILY/). FosB, the first bacillithiol transferase described, is unrelated by structure or sequence to the YfiT-like bacillithiol transferases and is involved in detoxification of the antibiotic fosfomycin[13–15]. The YfiT-like Superfamily includes enzymes that utilize thiol cofactors glutathione, mycothiol, and bacillithiol, and the Superfamily members from the Firmicutes are predicted to be bacillithiol transferases. The B. subtilis YfiT was the first the BST in the YfiT-like Superfamily to be identified and is the only BST from this family that has been characterized to date[12].
The natural substrates and the physiological roles of the YfiT-like thiol transferases are currently unknown. Interestingly, there is a correlation between the number of predicted YfiT-like thiol transferases (using the Superfamily database) and the number of predicted secondary metabolite operons (using the NCBI database). This observation has led to the hypothesis that the YfiT-like Superfamily BSTs play a role in the detoxification of endogenously produced toxins. This was observed in a Streptomyces coelicolor A3(2) mutant in the actinorhodin biosynthesis pathway. During the production of this major polyketide secondary metabolite, a mycothiol conjugate of a toxic intermediate was observed[16]. This mycothiol-dependent detoxification reaction may have been catalyzed by a mycothiol transferase, possibly one of the 26 YfiT-like Superfamily members present in the S. coelicolor genome[12].
A search of the S. aureus genome using the antibiotics and Secondary Metabolite Analysis SHell (antiSMASH, http://www.secondarymetabolites.org/) platform has revealed that S. aureus has only 5 predicted secondary metabolite operons, encoding 2 siderophores, 1 non-ribosomal peptide synthase, 1 terpene, and 1 lantipeptide. Thus, it is possible that the S. aureus BST detoxifies intermediates or final products from one or more of the predicted antibiotics or secondary metabolites. In addition to detoxification reactions, LMWTs have also been found to act as the thiol cofactor in halide displacement reactions, detoxification of reactive oxygen and nitrogen species, and isomerization reactions[4], so other roles are possible for this putative enzyme.
A Superfamily database search of YfiT related proteins in S. aureus revealed that 30 of 31 strains in the database encode one BST, while the remaining strain encodes two YfiT related BSTs. In this study, we describe the purification and characterization of the single predicted YfiT-like bacillithiol transferase from Staphylococcus aureus Newman, ORF NWMN_2591, which is identical in sequence to that of ORF SAUSA300_2626 from S. aureus USA 300 LAC, a community-associated methicillin-resistant strain of S. aureus. This ORF was predicted to encode a thiol transferase because of its structural similarity to the YfiT protein from B. subtilis, although there is little sequence similarity. We show that NWMN_2591 is a bacillithiol transferase, henceforth called BstA. Furthermore, phylogenetic analysis of a subset of predicted YfiT-like Superfamily members revealed that BstA and B. subtilis YfiT do not cluster into the same family. We have here used the recombinant purified enzyme and in vitro biochemical assays to characterize the substrate specificity and to identify inhibitors of BstA.
2. Materials and Methods
2.1. Chemicals
Bacillithiol and mycothiol were produced as previously described[13, 17]. The following chemicals were from Sigma Aldrich: α-cyano-4-hydroxycinnamic acid, azithromycin, cerulenin, cephalexin, coumaric acid, caffic acid, 1-chloro 2,4-dinitrobenzene, dithiothreitol, ethidium bromide, ferulic acid, fosfomycin, glutathione, gramicidin D, lincomycin, 2-mercaptoethanol (2-ME), mitomycin C, mupirocin, p-nitrophenyl acetate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), plumbagin, N-α-p-tosyl-L-phenylalanylchloromethyl ketone (TPCK), N-α-p-tosyl-L-lysinechloromethyl ketone (TLCK), rhodamine 6G, tetracycline, tiamulin, triclosan, and vancomycin. Ampicillin, antimycin A, daunomycin, 4-hydroxynonenal, and streptozotocin were from Calbiochem. Spectinomycin and methanesulfonic acid were from Fluka. Monochlorobimane and Hoechst 33342 were from Invitrogen. Etacrynic acid was from Enzo Life Sciences. Clindamycin and 1,10-phenanthroline were from MP Biomedicals. Nitrofurazone was from TCI and 2-methoxy-4-vinylphenol was from Alfa Aesar. All other buffers and reagents were from Fisher except as noted. The following kinase inhibitors were purchased from Life Chemicals and used without further purification: 3- F1374-0140, 5-bromo-N-[5-(4-fluorophenyl)-1,3,4-oxadiazol-2-yl]thiophene-2-carboxamide; 4-F1374-0808, N-(5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)-2-methylbenzamide, N-[5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl]-2-methylbenzamide; 5-F1374-0081, N-(5-(2-chlorophenyl)-1,3,4-oxadiazol-2-yl)-3-phenoxybenzamide, N-[5-(2-chlorophenyl)-1,3,4-oxadiazol-2-yl]-3-phenoxybenzamide; 6-F2518-0248, N-(5-(2,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)-2,6-difluorobenzamide; 7-F0608-0617, 2-fluoro-N-[5-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,3,4-oxadiazol-2-yl]benzamide; 8- F2518-0218, 3-butoxy-N-(5-(2,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl)benzamide, 3-butoxy-N-[5-(2,4-dimethylphenyl)-1,3,4-oxadiazol-2-yl]benzamide; 9-F2518-0041, 5-bromo-N-(5-(2,5-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thiophene-2-carboxamide.
2.2. BstA Purification
The S. aureus bacillithiol transferase candidate gene NWMN_2591 was codon optimized for expression in E. coli and synthesized by GenScript (Piscataway, New Jersey, USA). The gene was cloned into pET28a+ vector using NdeI and XhoI cloning sites, which generated N-terminal His6-tagged protein that contained a thrombin cleavage site following the His6 tag. E. coli C41 (DE3) was used to express the His6-tagged protein. Ten liters of Luria Broth containing kanamycin (50 μg/mL) were inoculated with cells grown to late exponential phase. His6-BstA production was induced with 1 mM IPTG at 30°C for 3 hours, and cells were collected by centrifugation at 4 °C. The cells were resuspended in 400 mM NaH2PO4 pH 8.0, 2.4 M NaCl, 1 mg/mL lysozyme, 35 μM each of TPCK and TLCK. Cells were lysed by sonication and extracts were centrifuged at 15000 rpm for 30 minutes. The cell-free extract was purified on a Zn2+-liganded PrepEase column (USB) and His6-tagged protein purification kit according to the manufacturer's instructions. After elution, the buffer was exchanged to 25 mM HEPES, 0.1 M NaCl, 2 mM DTT, pH 7.0 on a Sephadex G25 column. Protein-containing fractions were pooled and BstA (55 mg) was incubated with 800 units of thrombin for 16 hours at room temperature. To monitor cleavage of the His6-tag, the protein was analyzed by SDS gel electrophoresis. The solution was concentrated using a 30 kDa Centricon ultrafilter (Millipore). Thrombin was removed using free benzamidine resin (GE Healthcare). 5 mL of resin was packed in a column and the protein solution was loaded and washed with 20 mM Tris-HCl, 0.1 M NaCl, pH 7.4. Cleaved BstA and the His6-tag were eluted with 10 mM HCl, 0.5 M NaCl, pH 2.0 into a tube containing 200 μL of 1 M Tris-HCl, pH 9.0. The protein solution was loaded onto Zn2+-liganded PrepEase columns and washed with 25 mM HEPES, 100 mM NaCl, 2 mM DTT, pH= 7.0 to remove the His6-tag peptides. Cleaved BstA was concentrated in a 30 kDa concentrator, and the protein was stored in 25 mM HEPES, 100 mM NaCl, 2 mM DTT, and 20% glycerol. Protein concentration was estimated using A280 = 3.6 mg mL−1 (http://ca.expasy.org).
2.3. Assay of BstA Activity with Monochlorobimane and HPLC
Due to the limited amounts of BSH available, the sensitive HPLC analysis of the fluorescent BSmB conjugate (2, Equation 1) of monochlorobimane proved to be a generally useful BST assay[18]. With purified BstA the samples were processed without precipitation of the protein prior to HPLC analysis. Briefly, the standard assay consisted of 50 μM each of BSH and monochlorobimane in 0.1M NaCl, 25 mM NaPO4 5% glycerol, pH= 7.0 (assay buffer) in the presence of 220 nM of S. aureus BstA. The thiol was added last and the reaction (50 μl) was incubated at room temperature (23°C) and 15 μL samples were quenched with 55 μL of 40 mM methanesulfonic acid. This was injected on a reverse phase HPLC column with fluorescent detection as previously described[12]. Reaction rates obtained from the “−enzyme” reactions were subtracted from the “+enzyme” reactions to obtain a net enzymatic reaction. Initial rates were linearly extrapolated to zero time using Kaleidagraph (Synergy Software). For determination of the thiol co-substrate and thiol transferase activity, cysteine, glutathione, coenzyme A, and mycothiol were freshly prepared in water, standardized using the DTNB assay[19] and substituted for bacillithiol in the standard assay. To determine the metal ion dependence of BstA, fresh stock solutions of 100 mM 1,10-phenanthroline and 1,7-phenanthroline were prepared in dimethyl sulfoxide (DMSO). Solutions of 220 nM BstA and 1 mM phenanthrolines were incubated in assay buffer for 5 minutes. Subsequently, 50 μM mBCl, 50 μM BSH were added, and samples were analyzed by HPLC as described above. These assays were performed at 23°C. All rates were calculated for each time point and were linearly extrapolated to time zero using Kaleidagraph.
2.4. Phylogenetic Analysis
112 proteins from the YfiT Superfamily were selected from Gram-positive and Gram-negative species of bacteria. Proteins sharing the branch with BstA were used as query sequences for BLAST searches to construct a tree of closely related BstA proteins (Figure 2A). ClustalX was used to align sequences (using the PAM series) and to construct phylogenetic trees. Trees were generated by the neighbor-joining clustering method with 1000 bootstrap trials and were visualized using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/). Similar trees were obtained regardless of the method used.
Figure 2. Phylogenetic analysis of proteins closely related to BstA.

(A) Phylogenetic tree of proteins related to BstA as determined by Superfamily and BLAST searches. Proteins closely related to BstA were identified via BLAST searches as described in the Materials and Methods. Bootstrap values are shown for selected branches and correspond to confidence levels. Main branches are labeled I–V. Outgroups are labeled #1 (S. aureus FosB), #2 (predicted BST from B. anthracis), and #3 (B. subtilis YfiT); BstA is #8. See Table S4 in Supplementary Material for accession numbers that correspond to numbers. (B) Percent identity analysis of representatives from each branch. Percent identity was calculated using pairwise alignments (see Supplementary Figure S11) using ClustalW. The list of accession numbers for both trees can be found in the Supplementary Data (Tables S3 and S4).
2.5. Saturation Kinetics
All saturation kinetics studies were performed in assay buffer at pH 7.0. BSH saturation kinetics were analyzed by saturating BstA with 200 μM mBCl and varying the BSH concentration to 10 μM, 20 μM, 50 μM, 100 μM, and 200 μM. mBCl saturation kinetics were analyzed by saturating BstA with 50 μM BSH and varying the mBCl concentration to 60 μM, 250 μM, 500 μM, 1 mM, and 2 mM. BSH and mBCl saturation kinetics were performed at 37°C with 220 nM BstA. Initial rates were obtained using HPLC analysis as previously described[12]. CDNB saturation kinetics were analyzed by saturating BstA with 50 μM BSH and varying the CDNB concentration to 50 μM, 100 μM, 250 μM, 500 μM, 1 mM, 2 mM, 3 mM, and 4 mM. CDNB saturation kinetics were performed at room temperature (23°C). Initial rates were obtained spectrophotometrically at 340 nm using an extinction coefficient of 9.6 mM−1 cm−1. The data were fit to the Michaelis-Menten equation using Kaleidagraph.
2.6. Identification of BstA Substrates
Thiol depletion assays using DTNB thiol analysis were initially used to determine the substrates of BstA. Reactions containing 0.5 mM candidate substrate and 0.5 mM BSH were mixed at 23°C in assay buffer. Samples were removed and assayed for thiol content with 0.2 mM DTNB (final concentration) at 412 nm as previously described[12]. For mBCl competition reactions, 50 μM mBCl, 50 μM BSH, and 100 μM candidate substrate were mixed in assay buffer at 23°C with and without 220 nM BstA. Samples were analyzed by HPLC as previously described[12]. Rates were calculated for each time point and were linearly extrapolated to time zero using Kaleidagraph. Candidate substrates of interest that showed inhibition in either or both mBCl and DTNB assays were further studied in direct reactions with BSH (Section 2.7).
2.7. Direct BstA Reactions
Reactions with thiol reactive compounds other than monochlorobimane were carried out at higher concentrations due to the low absorbance of substrates and products. Initial reactions contained 0.5 mM of substrate (4-hydroxynonenal, caffeic acid, cerulenin, ciprofloxacin, clindamycin, etacrynic acid, mupirocin, sulforaphane, tiamulin, or triclosan) and was incubated with 0.5 mM BSH in 20 mM NaPO4, pH 7.0 and 100 mM NaCl with and without 5.5 μM BstA. Reactions with obvious products were repeated with 200 μM substrate, etacrynic acid, 4-hydroxynonenal, and suforaphane and 200 μM BSH in 0.1 M NaCl with 25 mM NaPO4 pH 7.0, with or without 5.5 μM S. aureus BstA at 23°C unless otherwise specified. Cerulenin reactions contained 500 μM cerulenin and BSH with and without 220 nM BstA. Reactions (50 μl) were sampled by diluting 5-fold into 10 mM methanesulfonic acid and concentrated using a SpeedVac prior to HPLC[12]. Reactions were assayed on an Atlantis T3 (C18, Waters) 2.1 by 150 mm reverse phase HPLC column at 0.4 mL/min and ambient temperature. Etacrynic acid, 4-hydroxynonenal, and sulforaphane were assayed by HPLC with monitoring at 215 nm (cerulenin 220 nm) using a linear gradient from 0–70% B over 40 min (0.05% TFA-water, A buffer; 60% acetonitrile-water, B buffer). For etacrynic acid, the B solvent was 100% acetonitrile. Due to the chemical reactivity of rifamycin S with BSH, the concentrations were reduced to 50 μM each and the reaction was monitored at 331 nm by HPLC as previously described for rifamycin S conjugates of mycothiol[20]. Samples of potential adducts were purified by HPLC and high resolution mass spectra (HRMS) of each was determined by the Molecular Mass Spectrometry Facility (UC San Diego). Chromatographic separations and product analysis by HRMS are shown in Supplemental Figures S3–7.
2.8. Studies with carboxamido-1, 3, 4-oxadiazole Scaffold Kinase Inhibitors
Inhibition constants of the kinase inhibitors were determined under steady state conditions assuming simple competitive inhibition. Assays were performed using 220 nM BstA, 60 μM mBCl (the Km of mBCl), and 50 μM BSH in assay buffer. Ki values of each inhibitor were calculated using the equation R0/Ri=0.5 + 0.5(1 + I/Ki) where R0 is the uninhibited rate and Ri is the inhibited rate. A linear plot of R0/Ri versus inhibitor concentration was generated for each inhibitor and Ki was estimated as 0.5/slope. Data for the plot were generated using 3 or more concentrations for each inhibitor between 2 and 100 μM. Reported Ki values represent the mean and standard deviation of triplicate experiments.
2.9. Studies with Cell-Free Extracts
Cell-free protein extracts were prepared by growing wild type S. aureus USA 300 LAC, the bstA Tn-mutant, and the bshB Tn-mutant strains in 250 mL trypticase soy broth (TSB, BBL) until OD600 was approximately 0.4. The cells were harvested by centrifugation (9,700 × g at 4 °C) and resuspended in 2 volumes of extraction buffer containing 25 mM HEPES, pH 7.5, 100 mM NaCl, 2 mM 2-ME, and 35 μM each of the protease inhibitors TPCK and TLCK. Zirconia/silica beads (0.1 mm) were added to 2 mL of cells in extraction buffer, and a BioSpec MiniBeadBeater was used to lyse cells. Cells were subjected to three cycles at 3450 rpm for 5 minutes, with cooling on ice for 5 minutes between each cycle. The extracts were clarified by centrifugation for 30 minutes (27,000 × g 4 °C). The supernatant was dialyzed (6 kDa MWCO, Spectra/Por Biotech Membrane) once against 500 volumes of extraction buffer overnight. The protein concentrations were 7–8 mg mL−1 as determined by bicinchoninic acid protein assay (Pierce). Extracts were stored at 4°C on ice. All assays were performed on the same day. BstA activity assays were performed with 250 μg cell-free extract. Control reactions in the absence of extract contained 50 μM BSH and 50 μM mBCl and were performed in extraction buffer. All reactions were performed at 37°C and were quenched by 1:1 dilution with 40 mM methanesulfonic acid in acetonitrile. Quenched samples were then incubated at 60°C for 10 minutes, followed by 5 minutes on ice, and centrifuged (15,000 × g for 3 minutes) to pellet the precipitated protein. The supernatant was diluted 5-fold in 10 mM methanesulfonic acid and analyzed by HPLC as previously described[12]. Bacillithiol conjugate amidase (BCA) assays were performed on the same cell-free extracts by the addition of 50 μM HPLC purified BSmB to 250 μg of S. aureus crude extract. Samples were quenched (as above) and the protein was removed prior to analysis by HPLC for production of CySmB as previously described[21].
3. Results
3.1. Cloning and Purification of BstA
To determine if S. aureus Newman encoded a bacillithiol transferase in the YfiT-like Superfamily, a BLAST search was performed against the S. aureus Newman genome with B. subtilis YfiT as the input sequence. There were no hits that had an E-value above 0.01. We therefore used the structure based SUPERFAMILY search engine (http://supfam.cs.bris.ac.uk/SUPERFAMILY/), to identify proteins with structural similarity to YfiT. This program predicted that the NWMN_2591 ORF in S. aureus Newman as well the corresponding ORF in the S. aureus USA 300 LAC strain (SAUSA300_2626) encoded proteins in the YfiT structural superfamily, and thus were candidate BST enzymes. The S. aureus Newman NWMN_2591 sequence was codon optimized for expression in Escherichia coli and cloned into the pET28a+ expression vector by Genscript. BstA appeared to be toxic to many commercial BL21 (DE3) E. coli expression hosts, showing little or no BstA expression. However, a low level of BstA was produced (~5 mg/L) using E. coli C41 (DE3). After induction with IPTG, cells were lysed and BstA was purified on a Zn2+ resin. The purified protein contained an additional 20 amino acids on the N-terminus due to the His6 tag, which was removed by thrombin cleavage, leaving just 3 additional amino acids (Gly-Ser-His) on the N-terminus. Mass spectrometry indicated that the protein had the expected sequence and molecular weight.
3.2. BstA Thiol Co-substrate Specificity
The thiol transferase activity and thiol co-substrate specificity of BstA were measured using the model glutathione transferase substrate monochlorobimane (mBCl, Equation 1)[18, 22, 23]. Monochlorobimane is non-fluorescent, but the product formed with a thiol is fluorescent (BSmB; 2, Equation 1) and is readily analyzed using reverse phase HPLC separation with fluorescence detection. Cysteine, glutathione, mycothiol, coenzyme A, and bacillithiol are all major LMWTs found in bacteria[4] and were tested as possible BstA co-substrates for conjugation with mBCl (Figure 1B). Importantly, the rate of non-enzymatic product formation between these thiols and mBCl is slow, but addition of a thiol transferase with appropriate substrate specificity catalyzes the reaction. Our results indicated that BstA catalyzed BSmB product formation above the non-enzymatic rate only with BSH, but does not enhance the rate of product formation with any of the other thiol substrates (Table 1). This indicates that S. aureus BstA is an thiol transferase that specifically uses bacillithiol as a co-substrate.
Figure 1. Purification and activity of BstA, a metal-dependent bacillithiol transferase.

(A) Analysis of recombinant BstA purification with 15% Tris-HCl SDS-PAGE gel of His6 uncleaved and cleaved BstA. Lane 1, crude His6-BstA extract; lane 2, pass through from Zn2+ affinity column before imidazole elution steps; lane 3, purified recombinant His6-BstA; lane 4, purified recombinant thrombin-cleaved His6-tagged BstA; lane 5, Bio-Rad polypeptide molecular weight standards. For (B) and (C): Closed symbols represent +BstA reactions, open symbols represent −BstA reactions. Error bars show standard deviation (n=3). (B): Monochlorobimane (mBCl, 50 μM) reactions for BST activity and thiol specificity. Reactions contained 50 μM thiol; bacillithiol (circles), cysteine (right triangle), glutathione (squares), mycothiol (triangles), or coenzyme A (diamonds). (C) Effects of metal chelation on BstA activity. Samples were treated with 1 mM 1,10-phenanthroline (squares), 1 mM 1,7-phenanthroline (circles), or were untreated (right triangles).
Table 1.
BstA activity is bacillithiol-dependent
| Thiol | Specific Activity (nmoles min−1 mg−1) | |
|---|---|---|
|
| ||
| − BstA | + BstA | |
| Glutathione | 4.4 ± 0.4 | 3.9 ± 0.1 |
| Cysteine | 6.7 ± 0.3 | 8.3 ± 0.5 |
| Mycothiol | 3.0 ± 0.1 | 3.3 ± 0.1 |
| Coenzyme A | 1.2 ± 0.1 | 1.4 ± 0.1 |
| Bacillithiol | 8.4 ± 0.3 | 152 ± 8 |
All values represent mean ± standard deviation (n=3). Reactions consist of 50 μM thiol, 50 μM mBCl, pH 7.0; +BstA reactions contained 220 nM BstA.
3.3. Monomeric and Active Molecular Weight of BstA
Purified His6-tagged BstA was sequenced from a SDS PAGE gel (Figure 1A) by the University of California, San Diego Biomolecular and Proteomics Mass Spectrometry Facility with 78% sequence coverage at 95% confidence. After thrombin cleavage of the His6 tag (see Section 2.2), MALDI confirmed that the protein was not post-translationally modified by the C41 (DE3) E. coli host, and the molecular weight was estimated to be 17793 ± 100 daltons (Supplementary Figure S1). Fast-protein liquid chromatography (FPLC)-gel filtration chromatography on Superdex S-200 resin was utilized to establish the active oligomeric state of the enzyme. Samples containing 2 mg, 1 mg, 0.5 mg, or 0.05 mg BstA were injected, and all samples resulted in a single peak, which corresponded to a molecular weight of 48,000 Da, 2.7 times the monomeric molecular weight (Supplementary Figure S2A). Sedimentation velocity experiments were subsequently performed and revealed a single peak with so20,w=2.78 S. This corresponds to a molecular weight of 44,000 Da, 2.5 times the molecular weight, and a frictional ratio of 1.55 (Supplementary Figure S2B). Globular shaped proteins typically show a frictional ratio of 1.1–1.3[24], therefore the high frictional ratio observed in the SV experiments likely indicates that the active enzyme is elongated. Two of the three crystallized B. subtilis YfiT-like Superfamily members, YizA and YjoA (PDB ID: 2QE9 and 3DKA, respectively) show a dimeric structure, but little similarity in overall quaternary structure (NCBI). The data are most consistent with BstA being an elongated dimer, but another possibility is that the solution form of BstA exists in a dimer-trimer equilibrium that is fast relative to the timescale of gel filtration or sedimentation velocity separations (hours).
3.4. Metal Ion Dependence of BstA
When the Bacillus subtilis BST YfiT was crystallized, it was hypothesized that the enzyme is a metalloenzyme because it contains metal-ion coordinating residues in the active site that are arranged with a geometry similar to that of metalloproteases[25]. To determine the metal ion requirement of BstA, the divalent cation chelating agent 1,10-phenanthroline was added to a BstA -catalyzed mBCl assay to determine whether addition of this chelating agent would decrease enzyme activity. 1,10-phenanthroline was utilized because it allowed the use of a non-chelating molecule with the same chemical scaffold (1,7-phenanthroline) as a control to verify that any affect on activity was due to chelation rather than an indirect effect due to structure (Figure 1C). After correction for the background chemical rate, treatment with 1,10-phenanthroline reduced the enzymatic reaction to < 1% (12.5 ± 0.7 nmoles min−1 mg−1), which was comparable to the background chemical rate of the reaction without added BstA (12.9 ± 1.8 nmoles min−1 mg−1). This represents a reduction of >99% from the untreated +BstA rate (143 nmoles min−1 mg−1). In contrast, treatment with the non-chelating molecule 1,7-phenanthroline reduced the enzymatic rate by only 27% (105 nmoles min−1 mg−1) compared to the rate of the untreated enzymatic reaction. Collectively, these data suggest that BstA requires a divalent metal cation for activity. In keeping with this hypothesis, the activity of BstA increased significantly when the N-terminal His6 tag was cleaved (data not shown), suggesting that the His6 tag, which has been previously shown to disrupt the activity of metalloenzymes by effectively removing metals from the active site, was interfering with the activity of this metalloenzyme. Furthermore, treatment with ethylenediaminetetraacetic acid (EDTA), another divalent cation chelator, also abolished activity, reducing the enzymatic reaction rate to the same slow rate as the chemical reaction (data not shown). From these studies, we have determined that BstA activity is metal-ion dependent, although we did not perform a detailed characterization of the specific divalent metal.
3.5. Phylogenetic Analysis of BstA
After establishing bacillithiol transferase activity of the enzyme, we performed primary sequence alignments of B. subtilis YfiT and BstA and determined that they share 18% sequence identity (data not shown). To further investigate the evolutionary relationship between BstA and YfiT, we conducted a phylogenetic analysis of a subset of YfiT-like Superfamily proteins, resulting in a tree containing 112 members total (Supplementary Figure S9). As expected, YfiT and BstA clustered into two separate families. The S. aureus FosB, which shares merely 8% sequence identity with BstA, failed to group with any of the proteins, allowing FosB to serve as an outgroup for the analysis.
Proteins that cluster on the same branch as BstA were then used as input sequences for BLAST searches to identify additional proteins related to BstA (in blue, Supplementary Figure S9). These proteins were used to construct a tree of proteins closely related to BstA. S. aureus FosB, B. subtilis YfiT, and a predicted bacillithiol transferase from Bacillus anthracis were added to the tree as outgroups because of their low sequence identity to the BstA related proteins (Supplementary Figure S10). This analysis of 43 total proteins generated a tree with four main branches (I–IV, Figure 2A). Sequences from representative members of each branch were aligned to determine the percent identity between the proteins (Figure 2B, Supplementary Figure S11). We observed that proteins from branches I and II were most closely related to each other (58% sequence identity) and contain exclusively Staphylococcus species. Proteins from branches III and IV shared ≤30% sequence identity with proteins from branches I and II. Branches III and IV share 34% sequence identity and contain a variety of Bacillus species. Members of branch I share 89% sequence identity or more with BstA, except for #18, which shares 70% sequence identity with BstA (Supplementary Figure S12). This analysis emphasizes the diversity of YfiT-like proteins in Staphylococcus species and in the phylum Firmicutes.
3.6. Saturation Kinetics
Detailed saturation kinetics were performed under saturating conditions of BSH and the two model substrates mBCl and 1-chloro-2,4-dinitrobenzene (CDNB). Substantial chemical reactions at high BSH and mBCl concentrations were observed and the values for Km and Vmax were adjusted for the velocity of the chemical reaction (Figure 3A and 3B). Such rapid chemical reactions were not observed with high CDNB concentrations (Figure 3C). The catalytic efficiency of BstA (kcat/Km) for BSH (1.4 ± 0.03 × 104 M−1 s−1) is the same order of magnitude as that of FosB (5–14 × 104 M−1 s−1; [14]), the only other known S. aureus bacillithiol transferase. The Km for BSH was determined to be 16 ± 4 μM (Table 2), which is over 10 fold below the calculated BSH concentration in exponentially growing S. aureus cells (~230 μM; [7]). This indicates that BstA has high affinity for bacillithiol and is likely saturated with thiol co-substrate in vivo. The model substrate mBCl was found to have a Km of 61 ± 26 μM, which is over 10 fold below the Km of CDNB, which was determined to be 780 ± 220 μM (Table 2). Consequently, the catalytic efficiency for CDNB is 8 fold below that of mBCl. BstA appears to follow Michaelis-Menten kinetics with respect to all three substrates. From these data, we have determined that BstA has higher affinity for mBCl than CDNB, and the highest affinity for its thiol co-substrate BSH.
Figure 3. Substrate saturation kinetics.
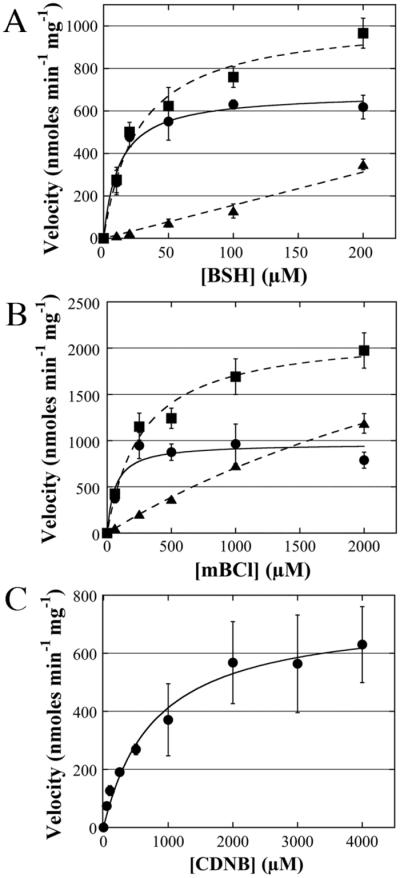
Detailed saturation kinetics of BstA using BSH, mBCl, and CDNB. Error bars show standard deviation (n=3). For (A) and (B): enzymatic velocity (circles, solid lines) were obtained by subtracting chemical velocity (triangles, dashed lines) from total velocity (squares, dashed lines). (A) BSH saturation kinetics. (B) mBCl saturation kinetics. (C) CDNB saturation kinetics.
Table 2.
Substrate saturation kinetics of BstA
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| BSHb | 16 ± 4a | 0.21 ± 0.01 | 14000 ± 290 |
| mBClb | 61 ± 26a | 0.30 ± 0.02 | 4900 ± 2100 |
| CDNBc | 780 ± 220 | 0.22 ± 0.02 | 280 ± 40 |
Values represent adjusted enzymatic rates (chemical reaction velocity subtracted from total reaction velocity).
Reactions performed at 37°C with 220 nM BstA.
Reactions performed at room temperature (23°C) with 2.8 μM BstA.
All values represent mean ± standard deviation (n=3) at pH=7.0.
3.7. Substrate Specificity of BstA
To identify potential substrates or inhibitors of BstA, antibiotics and thiol-reactive molecules were screened for inhibition of the BstA-catalyzed mBCl reaction. As a complementary assay, BSH levels were measured after reaction with candidate substrates by titration with 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB). A decrease in TNB− (anion) formed indicated that a reaction between BSH and the candidate substrate occurred. These two assays served as an initial screen for potential inhibitors or substrates reacting with BSH in a BstA-independent manner. Candidates that inhibited product formation in both assays were subsequently reacted directly with BSH to determine whether adducts were observed using HPLC with UV absorbance detection (Supplementary Figure S8). Adduct formation with the fatty acid synthesis antibiotic cerulenin was observed to be 300 nmoles min−1 mg−1, almost 2 fold above the non-enzymatic rate (170 nmoles min−1 mg−1) (Figure 4, Supplementary Table S1). Cerulenin was previously determined to be a substrate of the B. subtilis YfiT by monitoring BS-cerulenin product formation[12]. Using high resolution mass spectrometry (HRMS), the [MH]+ ion of the BS-cerulenin adduct was measured to be 622.2237 m/z, which is consistent with a formula of C25H40N3O13S with a Δppm 0.5 (Supplementary Figure S3). Thus, while cerulenin-BSH formation is catalyzed by BstA, the low rate of catalysis that is just two fold above the chemical rate indicates that cerulenin is a weak substrate and is therefore unlikely to be the natural substrate of this enzyme.
Figure 4. Cerulenin is a weak BstA substrate.

A direct reaction between 500 μM BSH and 500 μM cerulenin exhibited BstA -dependent adduct formation. +BstA reactions (closed triangles) and −BstA reactions (open triangles) are depicted. Error bars show standard deviation (n=3).
Previously, it was postulated that detoxification of rifamycin S, a bacterial RNA polymerase inhibitor, might be bacillithiol transferase dependent in S. aureus[21, 26]. We found that BSH reacted directly with rifamycin S, primarily forming the reduced conjugate, BS-rifamycin SV, in a BstA-independent manner at pH 7.0 and pH 6.0 with a rate of >35 μM min−1 (Supplementary Table S1). The [MH]− ion of the BS-rifamycin SV adduct was measured to be 1092.3865 m/z, consistent with a formula of C50H66N3O22S with a Δppm 0.1 (Supplementary Figure S4). The substrates etacrynic acid, 4-hydroxynonenal, and sulforaphane were also found to react with BSH in a BstA-independent manner (Supplementary Table S1). The [MH]+ ion of the BS-etacrynic acid adduct was measured to be 701.1180 m/z, consistent with a formula of C26H35Cl2N2O14S with a Δppm −0.1 (Supplementary Figure S5). The [MH]− ion of the BS-4-hydroxynonenal adduct was measured to be 553.2064 m/z, consistent with a formula of C22H37N2O12S with a Δppm −1.6 (Supplementary Figure S6). The [MH]+ ion of the BS-sulforaphane adduct was measured to be 576.1350 m/z, consistent with a formula of C19H34N3O11S3 with a Δppm 0 (Supplementary Figure S7). Many other antibiotics were tested in the above in vitro assays, and no additional substrates were identified (Supplementary Table S2). Thus, we determined that cerulenin is a weak substrate of BstA, while BSH dependent detoxification of rifamycin S, etacrynic acid, 4-hydroxynonenal, and sulforaphane were determined to occur without BstA catalysis.
3.8. Inhibitors of BstA
Previously, inhibitors of the YfiT-like M. smegmatis mycothiol transferase (MsMST)[12] were found while screening a small inhibitor library from the NIH Molecular Library Program (an NIH Roadmap Initiative) and the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) for mycothiol biosynthesis enzyme inhibitors. Although inhibitors of mycothiol biosynthesis enzymes were not found, ten compounds were found that inhibited the newly discovered M. smegmatis MST to varying degrees (Fahey and Newton, unpublished). Of the ten inhibitors, seven contained a common 2-carboxamido-1, 3, 4-oxadiazole scaffold (Figure 5) found to be among the most effective of 26,000 kinase inhibitors tested for killing M. tuberculosis[27]. These compounds were added to assays in competition with 50 μM BSH and 50 μM mBCl to determine whether they inhibit BstA. Five of the seven compounds inhibited BstA to varying degrees with 3 giving the lowest Ki value of ~3 μM (Figure 5). In concentration dependent inhibitor studies compound 3 was an effective inhibitor of BstA at a low concentration (5 μM). Although compound 3 could be a substrate of BstA, as such it would only consume <10% of the BSH present at the lowest inhibitor concentration used. This small loss of BSH would not substantially decrease the enzymatic rate of BSmB formation, contrary to what was observed. Further control experiments indicated that the compounds were unlikely to be chemically reactive because they had no effect at 50 μM on the chemical reaction of mBCl and BSH, and did not react directly with BSH in the absence of mBCl to form adducts (data not shown). The strong inhibition of compound 3 may provide clues to the structure of the natural substrates of BstA.
Figure 5. Carboxamido-1, 3, 4-oxadiazole scaffold kinase inhibitors inhibit BstA to varying degrees.

Structures and Ki values of inhibitors 3–9. “−BstA” reaction rates were subtracted from “+BstA” reaction rates. Values represent mean ± standard deviation (n=3).
3.9. Cell-free extracts from uninduced cells display low BstA activity
To determine the relative abundance of BstA activity in S. aureus cells, cell-free extracts were prepared from wild type USA 300 LAC cells, as well as from strains containing transposon insertions in bstA (ORF SAUSA300_2626) and bshB (ORF SAUSA300_0552) obtained from the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) program[28]. As predicted, the bstA mutant extract displayed a chemical rate similar to the minus extract control, which were 0.09 ± 0.004 nmoles min−1 mg−1 and 0.12 ± 0.003 nmoles mg−1, respectively. The bshB mutant extract similarly displayed a rate of 0.10 ± 0.014 nmoles min−1 mg−1 (Figure 6A). Surprisingly, the crude extract obtained from the wild type strain presented only a slightly elevated BstA rate with mBCl of 0.16 ± 0.009 nmoles min−1 mg−1 compared to the chemical reaction rate of the bstA mutant (0.09 ± 0.004 nmoles min−1 mg−1). When 55 nM (0.05 μg) of purified BstA was added to 250 μg of the bstA mutant crude extract, the rate increased to 0.28 ± 0.013 nmoles min−1mg−1 (Figure 6A). When rates were corrected for the chemical reaction observed in the BstA mutant extract to obtain the net rate, addition of purified BstA resulted in a rate 3 fold higher than the wild type extract rate. These results indicate that exponentially growing cells cultured in nutrient rich medium display low levels of BstA equivalent to the activity of 70 ng purified protein per mg of cell extract.
Figure 6. Extracts from growing cells exhibit low BstA activity, but high bacillithiol conjugate amidase (BCA) activity.

(A) Estimation of BstA activity in wild type cell-free extract. Extracts were made using the wild type USA 300 LAC (closed circles), the bstA Tn-mutant (closed triangles) and the bshB Tn-mutant (closed diamonds); the bstA Tn-mutant extract was supplemented with 55 nM BstA to estimate the amount of BstA in wild type cell extracts (closed squares) and all samples were subjected to monochlorobimane assays. Reactions with no extract (open circles) were also performed. (B) Determining BCA activity in the wild type cell-free extract. Extract prepared from wild type USA 300 LAC (closed circles) and the bstA Tn-mutant (closed diamonds), and the bshB Tn-mutant (closed triangles) are depicted. Error bars in (A) and (B) show standard deviation (n=3).
Based upon the sequence similarity of the B. anthracis BshB2/bacillithiol conjugate amidase (BCA) to the S. aureus BshB enzyme, it has been suggested that the S. aureus BshB possesses BCA activity[29]. Thiol analysis of the bshB Tn-mutant has shown that unlike B. subtilis bshB1 and bshB2 single mutants, the S. aureus bshB mutant does not produce BSH, indicating that there is no compensating BshB activity (data not shown and [26]). Indeed, the wild type S. aureus USA300 (JE2) dialyzed cell-free extract showed robust BCA activity (670 ± 70 nmoles min−1 mg−1) similar to that of the bstA mutant extract (570 ± 10 nmoles min−1 mg−1), whereas the bshB mutant extract showed BCA activity at just 10% of the wild type level (70 ± 13 nmoles min−1 mg−1, Figure 6B). These data indicate that BshB serves as an amidase both during the biosynthesis of BSH and during subsequent reactions with bacillithiol adducts such as BSmB and that it produces the majority of BCA activity in S. aureus USA 300 LAC.
4. Discussion
The DinB/YfiT-like Superfamily of thiol transferases is a new class thiol transferases in which members share structural similarity, but very little sequence similarity. Previous work based on structural similarity to B. subtilis YfiT as predicted by Superfamily[12] postulated that S. aureus Newman has one DinB/YfiT-like thiol transferase that is encoded by ORF Newman_2591. We have here demonstrated that the enzyme encoded by this ORF is indeed the bacillithiol transferase from the DinB/YfiT-like Superfamily and have named the enzyme BstA (Figure 7A). BstA is not related by sequence or structure to FosB, the only other bacillithiol transferase identified in S. aureus, however identical or nearly identical proteins are encoded by all S. aureus genomes, including pathogens such as S. aureus USA 300 LAC (ORF SAUSA_2626).
Figure 7. BstA catalyzes bacillithiol-dependent detoxification reactions in S. aureus.

(A) Model for BstA -dependent detoxification in S. aureus using the substrate mBCl. When mBCl enters the cell, the S. aureus BstA (NWMN_2591/SAUSA300_2626) catalyzes BSH addition, yielding BSmB. BshB, the amidase utilized in BSH biosynthesis (NWMN_0530/SAUSA300_0552), possesses bacillithiol conjugate amidase (BCA) activity and hydrolyzes BSmB to CysmB. GlcN-L-malate is recycled back to BSH biosynthesis, where BshC adds cysteine in the final step of synthesis. A cysteine conjugate N-acetyltransferase (CAT) transfers acetyl to cysteine to form AcCysmB (a mercapturic acid), which exits the cell via passive diffusion or an efflux transporter. (B) Genomic context of S. aureues bstA. bstA is in close proximity to the rarD drug translocator, an N-acetyltransferase family protein (GNAT), and a 2-oxoglutarate/malate translocator. Upstream of bstA is padR, a negative transcriptional regulator of phenolic acid stress genes.
Phylogenetic analysis of BstA revealed that although B. subtilis YfiT and BstA are both bacillithiol transferases that share the YfiT-like domain, the two enzymes cluster into separate families (Supplementary Figure S9). Further analysis revealed that BstA clusters closely with proteins from other Staphylococcus species, and less closely with proteins from other Firmicutes (Figure 2). Our analysis indicates that there are many families of YfiT proteins with several intermediate families separating BstA and B. subtilis YfiT even though S. aureus and B. subtilis are closely related organisms. One reason these enzymes may show so much sequence diversity is because the thiol transferases are expected to act on a variety of substrates with different chemical structures. Our observations allude to the exciting evolutionary history of these enzymes, and further analysis with a larger group of DinB/YfiT related proteins is needed to ascertain how many additional proteins are related to BstA and to begin to determine how many families belong in this divergent group of bacillithiol transferase-like proteins.
The thiol cofactor of FosB has been shown to be BSH [13, 14, 30] although it was previously thought to be cysteine[31]. We demonstrate here that BstA also specifically utilizes bacillithiol as its thiol cofactor. In addition, studies with chelating agents showed that BstA activity is metal-dependent, as is FosB activity[14, 15]. This is an interesting observation because the only other characterized bacillithiol transferase in the DinB/YfiT-like Superfamily, B. subtilis YfiT, is likely to also be a metalloenzyme[25]. BstA was active with the His6-tag intact, but the activity increased when the tag was removed, likely because the chelating activity of this tag affected the endogenous metal cofactor of the enzyme. The precise metal used in vivo remains unclear, but in our in vitro studies Zn2+ is likely to be the ligand since BstA, like YfiT, was purified using a Zn2+-IMAC column.
More detailed kinetic analysis demonstrates that BstA follows saturation kinetics for the bacillithiol co-substrate and two classic thiol transferase substrates, mBCl and CDNB. Our kinetic experiments required substantial corrections to account for the chemical reaction that takes place between mBCl and BSH, but this was not necessary for CDNB, which showed a low chemical reaction rate with BSH. This analysis showed that BstA has a Km for mBCl that is substantially lower than that of CDNB (61 ± 26 μM and 780 ± 220 μM, respectively), indicating that CDNB is a weaker substrate than mBCl. We found that BstA has a Km value for BSH of 16 ± 4 μM, which is 260-fold lower than that of the S. aureus FosB (4.2 ± 0.7 mM; [14]) and ~10-fold below the intracellular BSH concentration (~230 μM; [7]). This result indicates that BstA is saturated with its thiol co-substrate and is poised for catalysis in the organism.
Our results demonstrated that BstA catalyzes BSH-dependent halide displacement with both mBCl and CDNB. We also demonstrated that BstA conjugates BSH to the epoxide moiety of cerulenin in vitro, and were able to verify the product using high-resolution mass spectrometry (Supplementary Figure S4). However, there was the BstA catalyzed reaction rate with cerulenin was very slow (130 nmoles min−1 mg−1). This is likely smaller than the previously reported rate for cerulenin loss using cell-free S. aureus extracts[26], suggesting the existence of other cerulenin degradation pathways. The low reaction rate also suggests that cerulenin is a suboptimal BstA substrate and that other molecules might be more optimal BstA substrates.
In the related Gram-positive organism B. subtilis, the bshA-C operon that synthesizes bacillithiol is part of the Spx regulon, which is induced by oxidants and other stressors[11]. Interestingly, the oxidant H2O2 was found to react spontaneously with BSH ~40-fold faster than glutathione or mycothiol[12], suggesting that BSH might directly detoxify H2O2. We found that BSH also reacts with a surprisingly fast rate with the quinone-containing translational inhibitor rifamycin S. The addition of purified BstA did not enhance the rate of reaction (Supplementary Table S1), suggesting that BSH also directly detoxifies rifamycin S. This finding is in contrast to a previous report that BstA is responsible for BSH-rifamycin S adduct formation using cell-free S. aureus extracts[26], which we were unable to confirm using purified BstA. We also observed rapid, BstA-independent BSH conjugate formation with the thiol reactive molecules 4-hydroxynonenal, sulforaphane, and etacrynic acid (Supplementary Table S1). These results provide evidence for the reactive nature of BSH in conjugation reactions with halides, isothiocyanates, enals, enones and epoxides, perhaps due to the low thiol pKa (pKa 7.46) for BSH[32]. This suggests that BSH might be able to directly detoxify many molecules in living cells without requiring a thiol transferase.
It is possible that the natural BstA substrates might need to be bioactivated, perhaps by a cytochrome P450 type reaction; any such BstA substrates would not be identified in these experiments. BstA might also be involved in detoxification of endogenous metabolites that could occur when production of antibiotics or metabolites goes awry. For example, two of the metabolites made by S. aureus are synthesized via pathways that contain intermediates that could be toxic and highly thiol-reactive: the staphyloxanthin biosynthestic pathway contains intermediates with aldehydes and α/β unsaturated bonds and, indeed even the final staphyloxanthin product could be thiol-reactive[33] while the aureusimine biosynthetic pathway contains an aldehyde intermediate[34]. If noxious intermediates or off pathway reaction products accumulate, BstA could detoxify these molecules before they can damage the cell. Thus, the possibility that the natural substrate of BstA could be one of these metabolites or intermediates in the biosynthetic pathways warrants further investigation.
Although the natural BstA substrates have not been identified thus far, we have established a subset of compounds containing the 2-carboxamido-1, 3, 4-oxadiazole scaffold that can inhibit BstA activity. This scaffold is common to kinase inhibitors that have been found to be potent M. tuberculosis antibiotics[27] and target trans-translation protein degradation pathways in bacteria[35]. Many of these compounds also inhibited the structurally related DinB/YfiT-like M. smegmatis MST. Given the low Ki for some of these inhibitors, specifically compound 3, they might be structurally more similar to the natural BstA substrates than the model compounds mBCl and CDNB.
We were able to estimate the endogenous abundance of BstA activity in the absence of stress or potential exogenous substrates using cell-free extracts of wild type S. aureus USA 300 LAC. The wild type extract exhibited low bacillithiol transferase activity but accounted for >90% of the activity in this extract, while the bstA mutant extract displayed a rate similar to the chemical reaction rate. This indicates that BstA was the major mBCl utilizing bacillithiol transferase detected in S. aureus. Adding known quantities of purified enzyme allowed us to estimate the relative abundance of BstA to be about 0.007% of the total soluble protein. Thus, our observations with cell-free extracts indicate that BstA is a low abundance protein when S. aureus is growing in nutrient rich medium.
Although the relative abundance of BstA is low in rich medium, prior transcriptomic studies have demonstrated induction of bstA expression during treatment with natural products and oxidants. Treatment of S. aureus MW2 with the neutrophil microbicides hypochlorous acid, hydrogen peroxide, and azurophilic granule proteins increased bstA expression 2-, 3-, and 2-fold, respectively[36], and treatment of S. aureus COL with the growth inhibitor orange essential oil increased bstA expression 3-fold[37]. These conditions also induced additional bacillithol-related detoxification genes, including the S. aureus MW2 ortholog of B. subtilis bacilliredoxin brxB, whose product functions analogously to glutaredoxins by removing BSH from protein mixed disulfides[38] formed under conditions causing cytoplasmic oxidation. This S. aureus brxB ortholog was upregulated 2-, 6-, and 2-fold during treatment with hypochlorous acid, hydrogen peroxide, and azurophilic granule proteins. S. aureus MW2 encodes an ortholog of B. subtilis glxA, which encodes a glyoxylase that catalyzes the formation of S-lactoyl bacillithiol during methylglyoxal detoxification. This gene was upregulated 3-fold and 2-fold during hypochlorous acid and hydrogen peroxide stress[36]. Treatment of S. aureus COL with orange essential oil induced a 10-fold increase in gene expression of the glxA ortholog and the BSH biosynthesis genes bshB2 and bshC were each induced 2-fold[37]. Importantly, the fosB gene was not upregulated in these studies, demonstrating the specificity of this bacillithiol dependent enzyme for fosfomycin detoxification. These microarray studies display a concerted induction of bstA and other bacillithiol-related detoxification components during a variety of stressful conditions.
Examination of the genomic context of bstA in S. aureus revealed that it is adjacent to genes encoding proteins that could be involved in detoxification or BSH synthesis (Figure 7B). The gene downstream of bstA encodes a 2-oxoglutarate/malate translocator, which is significant because l-malate is a substrate of in the first step of BSH biosynthesis. It is conceivable that when bstA expression is upregulated, there is a need for additional BSH and its biosynthetic substrates such as l-malate. Also in the region is a gene encoding a protein with high sequence similarity to the GNAT family of N-acetyltransferases, which could acetylate the Cys-adduct cleavage product of the bacillithol conjugate amidase, and a protein in the RarD efflux transporter family, which could export acetylated Cys-adducts (mercapturic acids). Products of these reactions have been previously observed during detoxification of electrophiles in S. aureus Newman in culture[21]. Finally, the upstream and divergently transcribed gene encodes PadR, a protein that negatively regulates the phenolic acid stress response and that in many species governs expression of efflux pumps and phenolic acid decarboxylases that convert toxic phenolic acids to their vinyl phenol derivatives[39]. Further experiments are necessary to determine if the other genes in this region encode proteins involved in detoxification reactions.
As previously noted[12, 40], despite the DinB name, the DinB/YfiT-like Superfamily of proteins is not related to the type IV DNA polymerases of which E. coli DinB is the prototype[41, 42]. Of the eight proteins studied in this superfamily thus far, all have been verified to be bacillithiol transferases ([12, 43], unpublished data, and this study). Thus, we propose renaming the DinB/YfiT-like Superfamily as the S-transferase-like (STL) Superfamily to avoid confusion between the two distinct types of DinB proteins, which could lead to the improper assignment of enzyme function. Furthermore, we propose a naming system in which proteins are named based on the organism of origin, function, and order of discovery. B. subtilis YfiT is the only experimentally validated bacillithiol transferase from the STL Superfamily from this organism, so we propose renaming this protein BstA (gene name bstA). The number of STL superfamily proteins encoded by each genome varies greatly, but in the case of B. subtilis eight are encoded. In such cases we suggest that the second protein whose activity is demonstrated would be named BstB, the third BstC, etc. This nomenclature does not depend on sequence homology, because our phylogenetic analysis revealed an extraordinary diversity in these enzymes, so homologs of each enzyme often fail to exist in closely related organisms let alone distantly related ones. The diversity in these enzymes is remarkable, and might reflect a strong selective pressure to cope with an equally diverse array of endogenous or exogenous toxic metabolites and oxidants.
5. Conclusions
We have here verified the bacillithiol transferase activity of BstA and determined that the enzyme specifically uses bacillithiol as a co-substrate. The genomic context of BstA along with the low Km value of BSH point toward a detoxification function of this enzyme in the cell, although so far we demonstrated only relatively slow reaction rates with one antibiotic (cerulenin) and higher reaction rates with the model substrates mBCl and CDNB. While additional BstA substrates were not identified, a number of substrates were found to react directly with BSH in an enzyme-independent manner. One proposed function of thiol transferases is to lower the thiol pKa of the co-substrate during catalysis[44]. However, the thiol pKa of BSH is more acidic[32] than the pKa values of the other major endogenous LMWTs from S. aureus (cysteine pKa=8.6; coenzyme A pKa=9.6)[45]. Thiol transferases can also function to bring the thiol cofactor in close proximity to the substrate[44], and it is possible that this is the major role played by BstA. Thus, further studies are needed to fully understand the physiological role of BstA in S. aureus.
Supplementary Material
Highlights.
-
-
We demonstrate that S. aureus NWMN_2591 encodes a bacillithiol transferase, BstA
-
-
The Km for bacillithiol (16 ± 4 μM) indicates BstA is saturated in vivo
-
-
Cerulenin is a BstA substrate, while rifamycin S reacts directly with bacillithiol
-
-
BstA is the first DinB/YfiT-like thiol transferase identified in S. aureus
-
-
Several molecules that inhibit BstA at low μM concentration have been identified
Acknowledgements
We thank Partho Ghosh (UCSD) for helpful advice and for providing expression strains, as well as Joe Pogliano (UCSD) and Eric Allen (UCSD) for helpful advice and assistance with the phylogenetic analysis. This research was supported by the National Institutes of Health (R01-AI095125). The content is solely the responsibility of the authors and doesn't necessarily represent the official views of the National Institute of Health or the National Institute of Allergy and Infectious Disease. The following isolates were obtained through the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) Program: JE2, NE248, and NE1728, supported under NIAID/ NIH Contract No. HHSN272200700055C.
Abbreviations
- BSH
bactillithiol
- BST
bacillithiol transferase
- LMWT
low molecular weight thiol
- mBCl
monochlorobimane
- CDNB
1-chloro-2,4-dinitrobenzene
- BCA
bacillithiol conjugate amidase
- 2-ME
2-mercaptoethanol
- DTT
dithiothreitol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Josephy PD, Mannervik B. Molecular toxicology. 2nd ed Oxford University Press; Oxford; New York: 2006. pp. 333–364. [Google Scholar]
- [2].Allocati N, Federici L, Masulli M, Di Ilio C. Glutathione transferases in bacteria. FEBS J. 2009;276:58–75. doi: 10.1111/j.1742-4658.2008.06743.x. [DOI] [PubMed] [Google Scholar]
- [3].Allocati N, Federici L, Masulli M, Di Ilio C. Distribution of glutathione transferases in Gram-positive bacteria and Archaea. Biochimie. 2012;94:588–596. doi: 10.1016/j.biochi.2011.09.008. [DOI] [PubMed] [Google Scholar]
- [4].Fahey RC. Glutathione analogs in prokaryotes. Biochim. Biophys. Acta. 2013;1830:3182–3198. doi: 10.1016/j.bbagen.2012.10.006. [DOI] [PubMed] [Google Scholar]
- [5].Newton GL, Buchmeier N, Fahey RC. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008;72:471–494. doi: 10.1128/MMBR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jothivasan VK, Hamilton CJ. Mycothiol: synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008;25:1091–1117. doi: 10.1039/b616489g. [DOI] [PubMed] [Google Scholar]
- [7].Newton GL, Rawat M, La Clair JJ, Jothivasan VK, Budiarto T, Hamilton CJ, Claiborne A, Helmann JD, Fahey RC. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 2009;5:625–627. doi: 10.1038/nchembio.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gaballa A, Newton GL, Antelmann H, Parsonage D, Upton H, Rawat M, Claiborne A, Fahey RC, Helmann JD. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. U. S. A. 2010;107:6482–6486. doi: 10.1073/pnas.1000928107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee JW, Soonsanga S, Helmann JD. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8743–8748. doi: 10.1073/pnas.0702081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chi BK, Roberts AA, Huyen TT, Basell K, Becher D, Albrecht D, Hamilton CJ, Antelmann H. S-bacillithiolation protects conserved and essential proteins against hypochlorite stress in firmicutes bacteria. Antioxid Redox Signal. 2013;18:1273–1295. doi: 10.1089/ars.2012.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gaballa A, Chi BK, Roberts AA, Becher D, Hamilton CJ, Antelmann H, Helmann JD. Redox regulation in Bacillus subtilis: the bacilliredoxins BrxA (YphP) and BrxB (YqiW) function in de-bacillithiolation of S-bacillithiolated OhrR and MetE. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Newton GL, Leung SS, Wakabayashi JI, Rawat M, Fahey RC. The DinB superfamily includes novel mycothiol, bacillithiol, and glutathione S-transferases. Biochemistry. 2011;50:10751–10760. doi: 10.1021/bi201460j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sharma SV, Jothivasan VK, Newton GL, Upton H, Wakabayashi JI, Kane MG, Roberts AA, Rawat M, La Clair JJ, Hamilton CJ. Chemical and Chemoenzymatic syntheses of bacillithiol: a unique low-molecular-weight thiol amongst low G + C Gram-positive bacteria. Angew. Chem. Int. Ed. Engl. 2011;50:7101–7104. doi: 10.1002/anie.201100196. [DOI] [PubMed] [Google Scholar]
- [14].Roberts AA, Sharma SV, Strankman AW, Duran SR, Rawat M, Hamilton CJ. Mechanistic studies of FosB: a divalent-metal-dependent bacillithiol-S-transferase that mediates fosfomycin resistance in Staphylococcus aureus. Biochem. J. 2013;451:69–79. doi: 10.1042/BJ20121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thompson MK, Keithly ME, Harp J, Cook PD, Jagessar KL, Sulikowski GA, Armstrong RN. Structural and chemical aspects of resistance to the antibiotic fosfomycin conferred by FosB from Bacillus cereus. Biochemistry. 2013;52:7350–7362. doi: 10.1021/bi4009648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Taguchi T, Yabe M, Odaki H, Shinozaki M, Metsa-Ketela M, Arai T, Okamoto S, Ichinose K. Biosynthetic conclusions from the functional dissection of oxygenases for biosynthesis of actinorhodin and related Streptomyces antibiotics. Chem. Biol. 2013;20:510–520. doi: 10.1016/j.chembiol.2013.03.007. [DOI] [PubMed] [Google Scholar]
- [17].Unson MD, Newton GL, Davis C, Fahey RC. An immunoassay for the detection and quantitative determination of mycothiol. J. Immunol. Methods. 1998;214:29–39. doi: 10.1016/s0022-1759(98)00034-9. [DOI] [PubMed] [Google Scholar]
- [18].Eklund BI, Edalat M, Stenberg G, Mannervik B. Screening for recombinant glutathione transferases active with monochlorobimane. Anal. Biochem. 2002;309:102–108. doi: 10.1016/s0003-2697(02)00258-0. [DOI] [PubMed] [Google Scholar]
- [19].Ellman GL. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- [20].Steffek M, Newton GL, Av-Gay Y, Fahey RC. Characterization of Mycobacterium tuberculosis mycothiol S-conjugate amidase. Biochemistry. 2003;42:12067–12076. doi: 10.1021/bi030080u. [DOI] [PubMed] [Google Scholar]
- [21].Newton GL, Fahey RC, Rawat M. Detoxification of toxins by bacillithiol in Staphylococcus aureus. Microbiology. 2012;158:1117–1126. doi: 10.1099/mic.0.055715-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shrieve DC, Bump EA, Rice GC. Heterogeneity of cellular glutathione among cells derived from a murine fibrosarcoma or a human renal cell carcinoma detected by flow cytometric analysis. J. Biol. Chem. 1988;263:14107–14114. [PubMed] [Google Scholar]
- [23].Cook JA, Pass HI, Russo A, Iype S, Mitchell JB. Use of monochlorobimane for glutathione measurements in hamster and human tumor cell lines. Int. J. Radiat. Oncol. Biol. Phys. 1989;16:1321–1324. doi: 10.1016/0360-3016(89)90307-6. [DOI] [PubMed] [Google Scholar]
- [24].Brown PH, Balbo A, Schuck P. Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr. Protoc. Immunol. 2008;Chapter 18 doi: 10.1002/0471142735.im1815s81. Unit 18 15. [DOI] [PubMed] [Google Scholar]
- [25].Rajan SS, Yang X, Shuvalova L, Collart F, Anderson WF. YfiT from Bacillus subtilis is a probable metal-dependent hydrolase with an unusual four-helix bundle topology. Biochemistry. 2004;43:15472–15479. doi: 10.1021/bi048665r. [DOI] [PubMed] [Google Scholar]
- [26].Rajkarnikar A, Strankman A, Duran S, Vargas D, Roberts AA, Barretto K, Upton H, Hamilton CJ, Rawat M. Analysis of mutants disrupted in bacillithiol metabolism in Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2013;436:128–133. doi: 10.1016/j.bbrc.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Reynolds RC, Ananthan S, Faaleolea E, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Sosa MI, Thammasuvimol E, White EL, Zhang W, Secrist JA., 3rd High throughput screening of a library based on kinase inhibitor scaffolds against Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb) 2012;92:72–83. doi: 10.1016/j.tube.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bose JL, Fey PD, Bayles KW. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl. Environ. Microbiol. 2013;79:2218–2224. doi: 10.1128/AEM.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fang Z, Roberts AA, Weidman K, Sharma SV, Claiborne A, Hamilton CJ, Dos Santos PC. Cross-functionalities of Bacillus deacetylases involved in bacillithiol biosynthesis and bacillithiol-S-conjugate detoxification pathways. Biochem. J. 2013;454:239–247. doi: 10.1042/BJ20130415. [DOI] [PubMed] [Google Scholar]
- [30].Lamers AP, Keithly ME, Kim K, Cook PD, Stec DF, Hines KM, Sulikowski GA, Armstrong RN. Synthesis of bacillithiol and the catalytic selectivity of FosB-type fosfomycin resistance proteins. Org. Lett. 2012;14:5207–5209. doi: 10.1021/ol302327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cao M, Bernat BA, Wang Z, Armstrong RN, Helmann JD. FosB, a cysteine-dependent fosfomycin resistance protein under the control of sigma(W), an extracytoplasmic-function sigma factor in Bacillus subtilis. J. Bacteriol. 2001;183:2380–2383. doi: 10.1128/JB.183.7.2380-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sharma SV, Arbach M, Roberts AA, Macdonald CJ, Groom M, Hamilton CJ. Biophysical Features of Bacillithiol, the Glutathione Surrogate of Bacillus subtilis and other Firmicutes. Chembiochem. 2013 doi: 10.1002/cbic.201300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pelz A, Wieland KP, Putzbach K, Hentschel P, Albert K, Gotz F. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J. Biol. Chem. 2005;280:32493–32498. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- [34].Sun F, Cho H, Jeong DW, Li C, He C, Bae T. Aureusimines in Staphylococcus aureus are not involved in virulence. PLoS One. 2010;5:e15703. doi: 10.1371/journal.pone.0015703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ramadoss NS, Alumasa JN, Cheng L, Wang Y, Li S, Chambers BS, Chang H, Chatterjee AK, Brinker A, Engels IH, Keiler KC. Small molecule inhibitors of trans-translation have broad-spectrum antibiotic activity. Proc. Natl. Acad. Sci. U. S. A. 2013;110:10282–10287. doi: 10.1073/pnas.1302816110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Palazzolo-Ballance AM, Reniere ML, Braughton KR, Sturdevant DE, Otto M, Kreiswirth BN, Skaar EP, DeLeo FR. Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J. Immunol. 2008;180:500–509. doi: 10.4049/jimmunol.180.1.500. [DOI] [PubMed] [Google Scholar]
- [37].Muthaiyan A, Martin EM, Natesan S, Crandall PG, Wilkinson BJ, Ricke SC. Antimicrobial effect and mode of action of terpeneless cold-pressed Valencia orange essential oil on methicillin-resistant Staphylococcus aureus. J. Appl. Microbiol. 2012;112:1020–1033. doi: 10.1111/j.1365-2672.2012.05270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chandrangsu P, Dusi R, Hamilton CJ, Helmann JD. Methylglyoxal resistance in Bacillus subtilis: contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 2014;91:706–715. doi: 10.1111/mmi.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tran NP, Gury J, Dartois V, Nguyen TK, Seraut H, Barthelmebs L, Gervais P, Cavin JF. Phenolic acid-mediated regulation of the padC gene, encoding the phenolic acid decarboxylase of Bacillus subtilis. J. Bacteriol. 2008;190:3213–3224. doi: 10.1128/JB.01936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cooper DR, Grelewska K, Kim CY, Joachimiak A, Derewenda ZS. The structure of DinB from Geobacillus stearothermophilus: a representative of a unique four-helix-bundle superfamily. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010;66:219–224. doi: 10.1107/S1744309109053913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, Nohmi T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- [42].Sung HM, Yeamans G, Ross CA, Yasbin RE. Roles of YqjH and YqjW, homologs of the Escherichia coli UmuC/DinB or Y superfamily of DNA polymerases, in stationary-phase mutagenesis and UV-induced mutagenesis of Bacillus subtilis. J. Bacteriol. 2003;185:2153–2160. doi: 10.1128/JB.185.7.2153-2160.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Feng J, Che Y, Milse J, Yin YJ, Liu L, Ruckert C, Shen XH, Qi SW, Kalinowski J, Liu SJ. The gene ncgl2918 encodes a novel maleylpyruvate isomerase that needs mycothiol as cofactor and links mycothiol biosynthesis and gentisate assimilation in Corynebacterium glutamicum. J. Biol. Chem. 2006;281:10778–10785. doi: 10.1074/jbc.M513192200. [DOI] [PubMed] [Google Scholar]
- [44].Ketterer B. The role of nonenzymatic reactions of glutathione in xenobiotic metabolism. Drug Metab. Rev. 1982;13:161–187. doi: 10.3109/03602538209002234. [DOI] [PubMed] [Google Scholar]
- [45].Jocelyn PC. Biochemistry of the SH group; the occurrence, chemical properties, metabolism and biological function of thiols and disulphides. Academic Press; London, New York: 1972. pp. 52–55. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.