

Facilitates chromatin transcription (FACT) plays essential roles in chromatin remodeling during DNA transcription, replication, and repair. Tsunaka et al. studied human FACT–histone interactions that present precise views of nucleosome reorganization, conducted by the FACT-SPT16 Mid domain and its adjacent acidic AID segment.
Keywords: chromatin remodeling, nucleosome reorganization, FACT, transcription, histone chaperone, H2A–H2B displacement
Abstract
Facilitates chromatin transcription (FACT) plays essential roles in chromatin remodeling during DNA transcription, replication, and repair. Our structural and biochemical studies of human FACT–histone interactions present precise views of nucleosome reorganization, conducted by the FACT-SPT16 (suppressor of Ty 16) Mid domain and its adjacent acidic AID segment. AID accesses the H2B N-terminal basic region exposed by partial unwrapping of the nucleosomal DNA, thereby triggering the invasion of FACT into the nucleosome. The crystal structure of the Mid domain complexed with an H3–H4 tetramer exhibits two separate contact sites; the Mid domain forms a novel intermolecular β structure with H4. At the other site, the Mid–H2A steric collision on the H2A-docking surface of the H3–H4 tetramer within the nucleosome induces H2A–H2B displacement. This integrated mechanism results in disrupting the H3 αN helix, which is essential for retaining the nucleosomal DNA ends, and hence facilitates DNA stripping from histone.
In the eukaryotic nucleus, genomic DNA is organized into densely packed chromatin, whose higher-order architectures are dominated by arrays of the basic repeating units termed nucleosomes (Luger et al. 1997; Tremethick 2007; Woodcock and Ghosh 2010). The orchestrated actions of chromatin remodeling factors, such as histone chaperones, ATP-dependent chromatin remodelers, and histone modifiers, rearrange dynamic chromatin architectures at nucleosome or higher-order levels, thereby playing critical roles in gene regulation linked to epigenetics (Suganuma and Workman 2011; Narlikar et al. 2013; Gurard-Levin et al. 2014). Facilitates chromatin transcription (FACT), commonly classified as a histone chaperone protein, is crucial for gene expression processes from yeast to humans, since this protein complex reorganizes individual nucleosomes to generate accessible DNA templates (Belotserkovskaya et al. 2003; Nakayama et al. 2007; Winkler et al. 2011; Kwak and Lis 2013; Kemble et al. 2015). In the chromatin-based transcription, the human FACT (hFACT) in fact facilitates RNA polymerase II-driven elongation by helping to displace a histone H2A–H2B dimer from the nucleosome (Belotserkovskaya et al. 2003). On the other hand, Drosophila FACT counteracts the spreading of silent chromatin at the boundary between heterochromatin and euchromatin, highlighting its important roles in epigenetic regulation (Nakayama et al. 2007).
The heterodimeric FACT molecule, a highly conserved eukaryotic protein, consists of a structure-specific recognition protein-1 (SSRP1) and a larger subunit, SPT16 (suppressor of Ty 16). Together, these subunits have five structural domains: the heterodimerization domain, the middle domain and HMG domain of SSRP1, and the amino-peptidase-like domain and middle domain (Mid, residues 644–930 in humans) of SPT16 (Fig. 1A; VanDemark et al. 2006, 2008; Tsunaka et al. 2009; Hondele et al. 2013; Kemble et al. 2013). The individual domains are connected by several intrinsically disordered regions (IDRs). Genetic and structural studies indicated that the SPT16-Mid domain from fungi plays key roles in histone recognition by FACT (Myers et al. 2011; Hondele et al. 2013; Kemble et al. 2013). The C-terminal region of SPT16, which includes a highly acidic segment (AID, residues 931–988 in humans) (Fig. 1A), is also critical for the histone chaperone activity of FACT (Belotserkovskaya et al. 2003; Winkler et al. 2011; Kemble et al. 2015). Notably, the FACT subunits display an intriguingly broad range of physical and genetic interactions with other factors involved in DNA transcription, replication, and repair (VanDemark et al. 2006; Heo et al. 2008; Ransom et al. 2010; Kwak and Lis 2013). Therefore, it appears to universally play key roles in nucleosome dynamics during different processes within nuclei. In spite of this broad functional spectrum, remarkably, every eukaryotic species contains only one ortholog of the FACT complex, suggesting that FACT should conduct universal actions in terms of chromatin remodeling. In this context, the functional behaviors of FACT appear to be somewhat distinct from those of other conventional histone chaperones, while the precise mechanism for actions of FACT on nucleosomes remains elusive.
Figure 1.
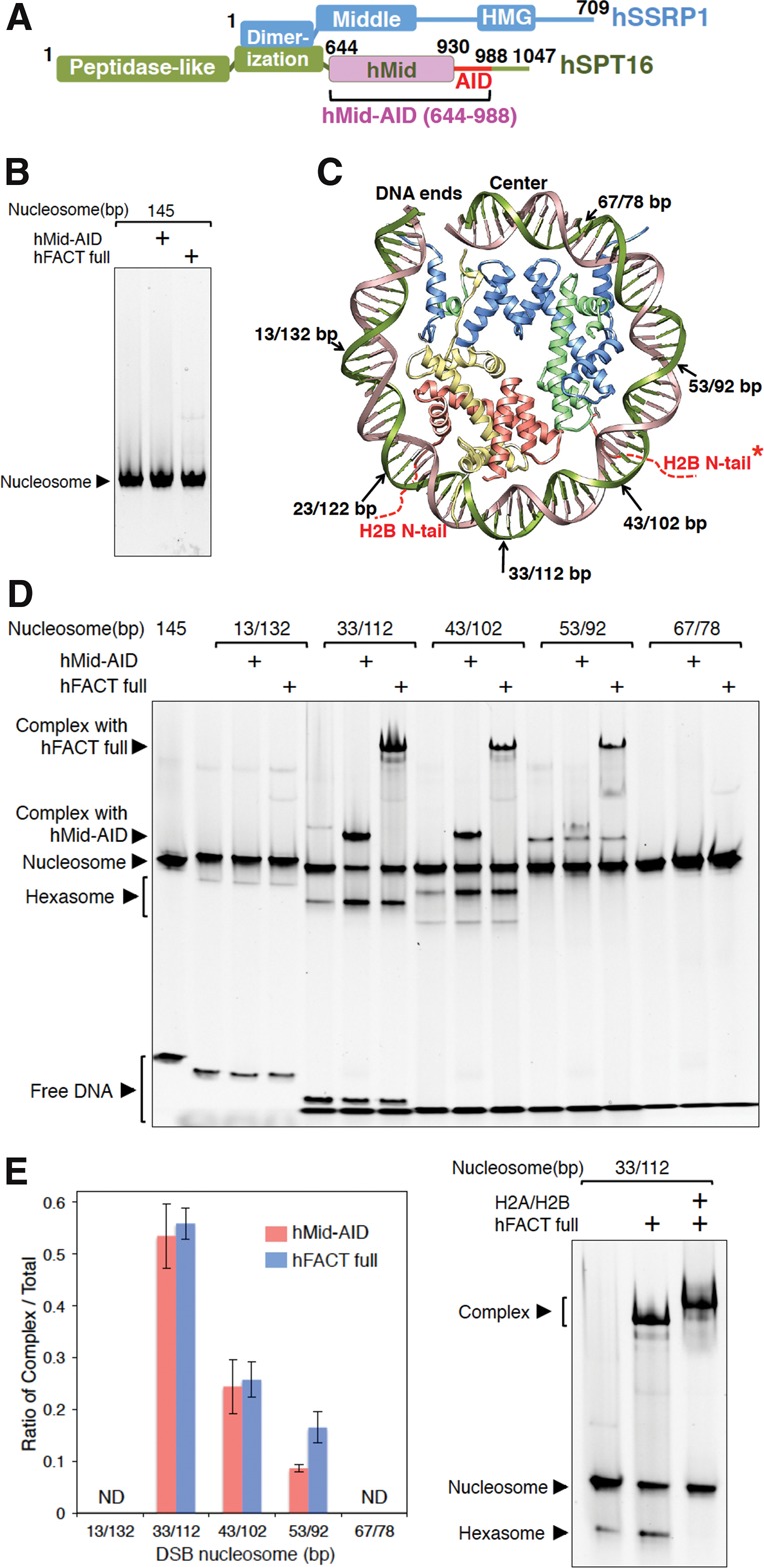
hFACT interacts with nucleosomes at double-strand break (DSB) sites in contact with H2A–H2B dimers. (A) Domain organization of hFACT. Human Mid (hMid)-AID, hMid, and AID proteins were used in this study. (B) EMSAs (electrophoretic mobility shift assays) detected no complex of hFACT or hMid–AID with intact nucleosomes. (C) DSB sites are marked by arrows on the crystal structure of one-half of a human nucleosome (Protein Data Bank [PDB] code 2CV5). Red dotted lines denote two H2B N-terminal tails (N-tails) passing between DNA gyres. An asterisk indicates the other H2B N-tail of H2A–H2B in one-half of a nucleosome lacking DSB. (D) EMSAs detected complexes of hFACT or hMid–AID with DSB nucleosomes at the positions represented in C as well as hexasomes upon addition of hFACT or hMid–AID. (E) Ratios of the complexes of hFACT (blue) or hMid–AID (pink) with DSB nucleosomes in the EMSAs represented in D. hFACT and hMid–AID most efficiently form the complexes at the DSB sites (33 base pairs [bp] and 112 bp [33/112 bp]) in the close vicinity of the H2B N-tail. Data are mean and SD for each data point. n = 3. (ND) Not determined; band intensities of complexes were below our detection limit. (F) Confirmation of hexasome formation by hFACT. (Middle lane) Upon addition of hFACT, the hexasome band appears. (Right lane) Upon further addition of H2A–H2B, the hexasome band disappears, as the nucleosome band recovers. Simultaneously, the H2A–H2B addition results in a supershift of the complex, suggesting that the complex reacquires free H2A–H2B. Experiments were repeated at least three times.
Here, we reveal the precise molecular mechanism for nucleosome reorganization by hFACT. The introduction of a double-strand break (DSB) into the nucleosomal DNA in the close vicinity of the H2B N-terminal tail (N-tail) allows hFACT to efficiently form a complex with nucleosomes. Notably, the interaction of the H2B N-tail with AID facilitates the invasion of the adjacent hFACT Mid domain (hMid) into nucleosomes. Thus, the hMid domain associates with the H2A-docking surface of the H3–H4 tetramer [(H3–H4)2] and thereby displaces an H2A–H2B dimer (H2A–H2B) from nucleosomes. The displacement generates disorder of the H3 αN-helix structure and results in the stripping of the nucleosomal DNA ends from the histone octamer. This scenario for nucleosome reorganization could be generalized for various nuclear processes whose progressions are impeded by nucleosomes.
Results
FACT binds to DSB nucleosomes in the close vicinity of the H2B N-tail
hFACT binds to a histone H2A–H2B dimer and a histone H3–H4 complex (Winkler et al. 2011). In addition, we revealed that hFACT interacts with a histone octamer to form a complex with a histone hexamer, which lacks one H2A–H2B dimer from the octamer (Supplemental Fig. S1A). In contrast, hFACT phosphorylated in insect cells never interacts with the intact nucleosome reconstituted from recombinant human histones and a 145-base-pair (bp) Widom 601 DNA (Fig. 1B; Lowary and Widom 1998), indicating that the nucleosomal DNA blocks the interaction of hFACT with histone proteins. On the other hand, the dephosphorylated FACT nonspecifically interacts with the nucleosomal DNA (Tsunaka et al. 2009; Winkler et al. 2011). Thus, we used the phosphorylated hFACT to avoid the nonspecific DNA binding by the dephosphorylated hFACT.
Transcription generates positive superhelical torsion in DNA ahead of RNA polymerase (Liu and Wang 1987). In the chromatin-based transcription, the superhelical torsion locally disrupts DNA–histone contacts within the nucleosome (Sheinin et al. 2013). In addition, the previous structural reports indicate competitive relationships between the nucleosomal DNA and several histone chaperones, including FACT, during the association with internal histones (Hu et al. 2011; Winkler et al. 2011; Elsässer et al. 2012; Liu et al. 2012; Chen et al. 2015; Huang et al. 2015; Kemble et al. 2015; Richet et al. 2015). Thus, we assumed that the interaction of hFACT with nucleosomes would involve local detachments of the nucleosomal DNA from histones (Winkler et al. 2011; Hsieh et al. 2013; Kemble et al. 2015). Our preliminary analysis revealed that hFACT forms stable complexes with nucleosomes treated with DNase I (data not shown). This result implies that DNA cleavages on nucleosomes would cause partial unwrapping of the nucleosomal DNA in the vicinity of the cleavage sites upon contacting hFACT, which subsequently allows direct interactions with internal histones. This tempted us to systematically analyze the DSB sites, where hFACT most efficiently forms a complex with nucleosomes. We produced a series of nucleosomes containing one DSB site at different positions (Fig. 1C; Supplemental Fig. S1B). Notably, when comparing ratios of successfully reconstituted DSB nucleosomes between the left half of the 601 DNA segment (601L) and the right half of 601 (601R), 601L showed more efficient reconstitution than 601R (Supplemental Fig. S1B). This agrees with the previous report, which found that 601L has a higher affinity for the histone octamer than 601R (Chua et al. 2012). In addition, as described later, nucleosomes containing DSB in 601L were almost completely protected from nuclease digestion, suggesting that DSBs in 601L retain the nucleosome structure. EMSAs (electrophoretic mobility shift assays) using these DSB nucleosomes revealed that hFACT more efficiently forms complexes at DSB sites in contact with H2A–H2B dimers, whereas the complex formation is hardly observed near the entry–exit and center sites (Fig. 1C–E; Supplemental Fig. S1C,D). The DSB sites interacting with hFACT are consistent with the region cross-linked with histone H2B, as recently identified by a chromatin immunoprecipitation (ChIP) exonuclease (ChIP-exo) assay (Rhee et al. 2014). Most notably, the N-tails of H2B pass between two gyres of the nucleosomal DNA near these sites (Supplemental Fig. S1E; Luger et al. 1997; Tsunaka et al. 2005), implying that hFACT may interact with the H2B tail exposed by DSBs. Furthermore, the interaction between FACT and nucleosomes does not occur at entry–exit sites of nucleosomes, since the complex formations with FACT are not observed for 13-bp and 132-bp DNAs (13/132-bp DSBs) or intact nucleosomes (Fig. 1B–E).
The interaction between hFACT and DSB nucleosomes produced much faster mobility bands, corresponding to hexasome, which lacks one H2A–H2B dimer in nucleosomes (Fig. 1D; Belotserkovskaya et al. 2003). This interpretation was validated by adding extra H2A–H2B dimers to the reaction mixtures of hFACT and nucleosomes (Fig. 1F); upon the addition of H2A–H2B, the hexasome band disappeared, as the nucleosome band recovered. In fact, the quantified intensity of the nucleosome band in the last lane of Figure 1F is increased by ∼30% in comparison with that in the middle lane. Simultaneously, the H2A–H2B addition results in a supershift of the complex (Fig. 1F, last lane), suggesting that the complex reacquires free H2A–H2B. On the other hand, the displaced H2A–H2B upon the addition of FACT would interact with FACT free from the complex in this assay. In a gel filtration (GF) assay, hFACT mixed with a histone octamer was eluted as a complex with a histone hexamer, indicating that one H2A–H2B dimer was evicted from the octamer by hFACT (the band intensity of H2B was 45.7% relative to the H4 band intensity) (Supplemental Fig. S1A). Based on these results from our original assay, we suggest that the introduction of DSBs in the close vicinity of the H2B N-tail allows hFACT to efficiently interact with histones in nucleosomes, thereby inducing the conversion of nucleosomes into hexasomes.
We next investigated which regions of hFACT are responsible for binding to DSB nucleosomes. The hMid domain plus its C-terminal AID segment (hMid–AID) (Fig. 1A) showed the same tendency as the full-length complex in the above assay using DSB nucleosomes (Fig. 1D,E). Similarly to hFACT, hMid–AID formed stable complexes with the DSB nucleosomes and increased the rate of hexasome production (Fig. 1D). These observations led us to the conclusion that the hMid domain and the AID segment play dominant roles in the interaction of FACT with DSB nucleosomes and the subsequent conversion to hexasomes. In contrast, in the same assay using hMid–AID in place of FACT in Figure 1F, we failed to observe a supershift of the complex as well as nucleosome recovery upon the H2A–H2B addition due to smeared gel patterns (data not shown). This indicates that hMid–AID has no obvious nucleosome reassembly activity, in contrast to FACT.
Crystal structure of the hMid–AID/(H3–H4)2 complex
To clarify how the hMid domain and the AID segment behave to histones in nucleosome, we determined two X-ray crystal structures: the hMid domain alone at 1.92 Å resolution (Supplemental Table S1; Supplemental Figs. S2A, S3A) and the complex of hMid–AID with a nearly intact human H3Δ34–H4 tetramer (H3 residues 35–135 and H4 full length) at 2.98 Å resolution (Fig. 2A; Supplemental Table S1; Supplemental Fig. S3B). The comparison of the hMid backbone structure with the two Mid structures from Saccharomyces cerevisiae and Chaetomium thermophilum exhibited high similarity, with 1.16 and 1.13 Å root mean square deviation (RMSD) values, respectively (Supplemental Fig. S2A; Hondele et al. 2013; Kemble et al. 2013). The complex structure showed that hMid–AID contacts one entire molecule of (H3–H4)2 through interactions with each molecule of four histone proteins (H3, H4, H3′, and H4′) at two separate sites. The binding sites are entirely different from those in the previous binding assay using histone H3 peptides (Hondele et al. 2013) located within the globular domain of (H3–H4)2. In addition, this binding scheme is quite novel in comparison with other structures of the complexes so far determined between histone chaperones and an H3–H4 dimer (Supplemental Fig. S2B; English et al. 2006; Natsume et al. 2007; Hu et al. 2011; Elsässer et al. 2012; Liu et al. 2012). Importantly, the hMid–AID/(H3–H4)2 complex retains the original (H3–H4)2 subunit structure composed of two copies of an H3–H4 dimer, whereas other histone chaperones disrupt the structure of an H3–H4 tetramer to form complexes with an H3–H4 dimer (English et al. 2006; Natsume et al. 2007; Hu et al. 2011; Elsässer et al. 2012; Liu et al. 2012). Recently, the crystal structures of the MCM2/(H3–H4)2 and Spt2/(H3–H4)2 complexes have been determined (Chen et al. 2015; Huang et al. 2015; Richet et al. 2015). These complexes retain the original (H3–H4)2 structure in the same manner as that of the hMid–AID/(H3–H4)2 complex. However, the binding sites of these three complexes are entirely dissimilar to each other, except for partial overlapping among the three structures, as described in detail later (Supplemental Fig. S2C).
Figure 2.
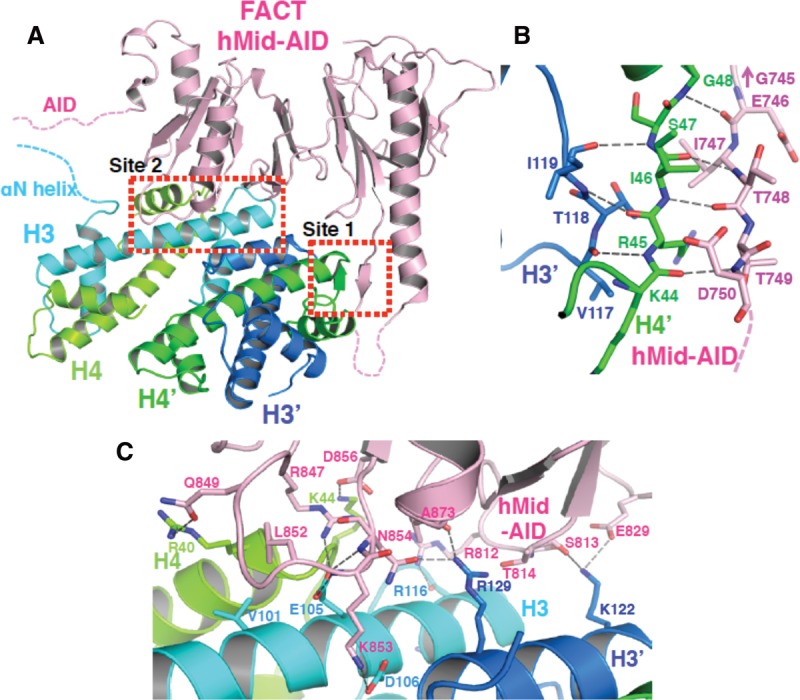
hMid–AID interacts with a histone H3–H4 tetramer [(H3–H4)2] at two separate sites. (A) Crystal structure of the complex between hMid–AID (pink) and (H3–H4)2 (H3 [cyan], H3′ [blue], H4 [yellow green], and H4′ [green]). Dotted lines indicate disordered regions of hMid–AID and histones. Red dotted boxes show the two binding sites (site 1 and site 2). (B,C) Close-up views showing the three-stranded anti-parallel β-sheet structure in site 1 (B) and the mixture of hydrophobic and hydrophilic interactions in site 2 (C). Hydrogen bonds are indicated by gray dashed lines, and contact residues are depicted. The representation of G745 in B indicates the adjacent residue of E746 on hMid–AID.
In the complex structure, one binding site constitutes a three-stranded anti-parallel β sheet by combining one strand from hMid–AID and the original β structure between H4′ and H3′, thus establishing a 479 Å2 interface (site 1) (Fig. 2B; Supplemental Fig. S3C). Residues at site 1 of hMid–AID are highly conserved in a sequence alignment among metazoans but are diverged in fungi (Supplemental Fig. S4A). The comparison of the free and histone-bound hMid domains reveals few differences in main chain structures, except for the alteration of a flexible loop toward the β structure at this binding site (Supplemental Fig. S2D). This binding site is quite similar to those in the MCM2/(H3–H4)2 and Spt2/(H3–H4)2 complexes, both of which form the β structure at the same position as site 1 (Supplemental Fig. S2C; Chen et al. 2015; Huang et al. 2015; Richet et al. 2015). However, these amino acid sequences are completely different from each other (Supplemental Fig. S2C). Therefore, the structural aspect rather than the sequence at site 1 may be important.
The other binding site contains a mixture of hydrophobic and hydrophilic interactions with H3, H4, and H3′ to construct a broad interface of 923 Å2 (site 2) (Fig. 2C; Supplemental Fig. S3B,C). Residues at site 2 of hMid are evolutionarily conserved across species from yeast to humans (Supplemental Fig. S4A), supporting their functional importance. Residues (Arg847, Gln849, Leu852, Lys853, Asn854, and Asp856) of hMid–AID bound to (H3–H4)2 are mostly concentrated in a loop, which we named the (H3–H4)2-binding loop. Notably, the side chain of Leu852 in hMid–AID is flipped out from hydrophobic core within hMid so as to interact with the Val101 side chain of H3 (Supplemental Fig. S3D). Hydrophilic interactions mainly accompany the rearrangements of polar side chains of (H3–H4)2, such as Glu105 of H3, Lys122 and Arg129 of H3′, and Arg40 and Lys44 of H4 (Supplemental Fig. S3E). These extensive interactions result in a 7° rotation of one H3–H4 dimer half in the complex, as compared with (H3–H4)2 within the nucleosome (Supplemental Fig. S2E).
The interactions between hMid–AID and (H3–H4)2 were validated by a combined approach of biochemical and thermodynamic assays, including site-directed mutagenesis. Wild-type hMid–AID forms a 1:1 complex with intact (H3–H4)2 in GF chromatography (Fig. 3A). In contrast, the mutations at the two binding sites of hMid–AID (hMidm1m2–AID; Δ746–751/S813A/E829A/R847A/Q849A/L852A/K853A/N854A/D856A) abolish the complex formation with (H3–H4)2 (Fig. 3A). Isothermal titration calorimetry (ITC) measurements of the binding between hMid–AID and (H3–H4)2 revealed endothermic binding with a 2 µM dissociation constant (Kd) and a 1:1 stoichiometry, whereas the Kd value of hMidm1m2–AID is increased by approximately fivefold (Fig. 3B; Supplemental Fig. S5). Notably, the GF and ITC assays exhibited the 1:1 stoichiometry even in solution, excluding the possibility that a 2:1 complex is formed between hMid–AID and (H3–H4)2. In GF and ITC assays on the histone side, mutations of the polar residues on H3 (H3m2; E105A/K122A/R129A) prevented the complex formation with hMid–AID and also reduced the affinity by fivefold (Fig. 3A,B; Supplemental Fig. S5). Taken together, these results demonstrate that the mutated residues are important for association between hMid–AID and the H3–H4 tetramer. Furthermore, the previous report showed that the S765P (the corresponding mutation at G745, the adjacent residue of E746 in humans) (Fig. 2B; Supplemental Fig. S4A) and E857K (the corresponding mutation at E829 in humans) (Fig. 2C; Supplemental Fig. S4A) mutations in yeast SPT16 result in reduced recruitments of FACT to highly transcribed regions of the S. cerevisiae genome (Hainer et al. 2012), supporting the biological significance of the hMid–AID/(H3–H4)2 complex structure.
Figure 3.
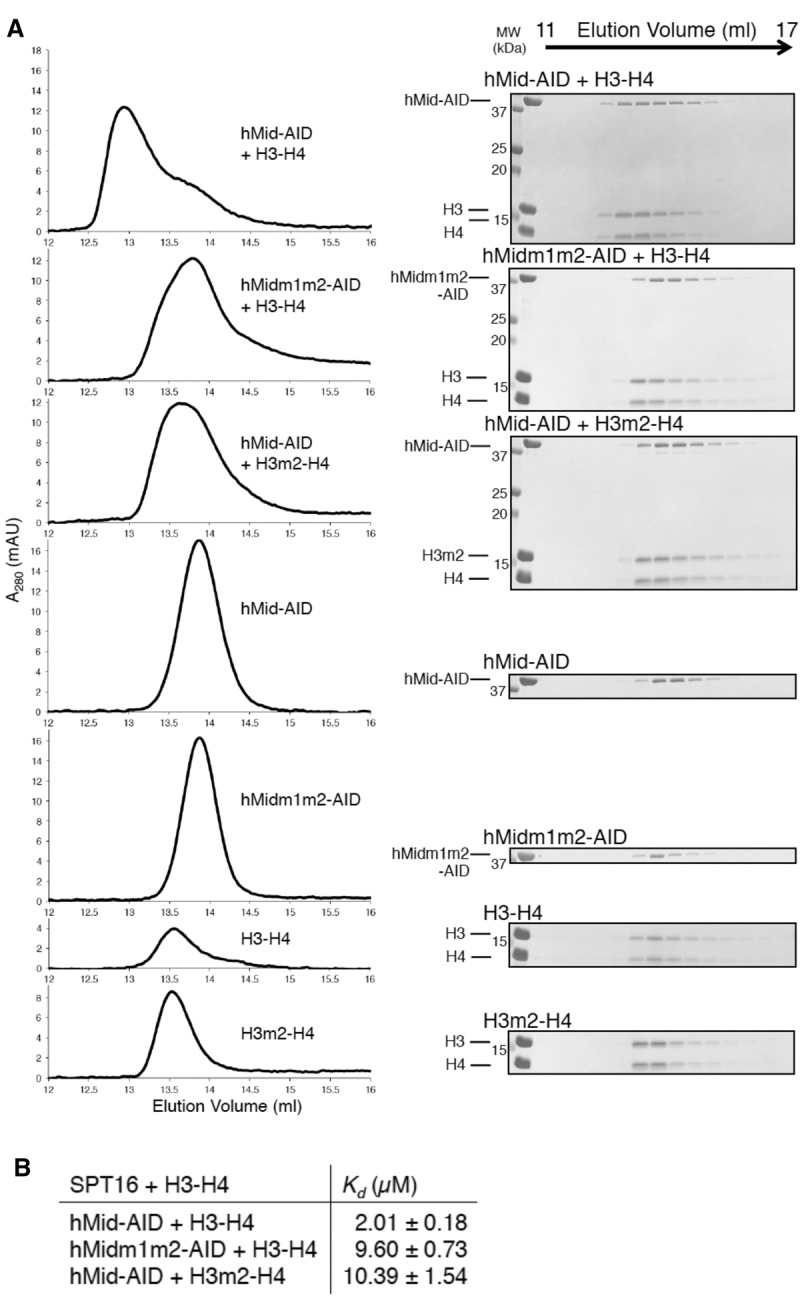
Biochemical analyses of the interaction between hMid–AID and (H3–H4)2. (A) GF analyses show that mutations of residues at site 1 and site 2 disrupt the complex between hMid–AID and (H3–H4)2. Protein mixtures with stoichiometric amounts were eluted from a Superdex200 10/300 column in 750 mM NaCl. Elution profiles and the corresponding SDS-PAGE are shown at the left and right sides, respectively. (B) ITC measurements between various hFACT and (H3–H4)2 constructs in 750 mM NaCl at 25°C. ITC profiles and fitting data are shown in Supplemental Figure S5. Data are mean ± SD for each data point. n = 3.
Meanwhile, in the GF assay, the deletion of the AID segment from hMid–AID completely abolished the interaction with (H3–H4)2 (Supplemental Fig. S6). In addition, AID alone exhibits the significant thermal transition upon binding to (H3–H4)2 in the ITC measurements (Supplemental Fig. S5). This implies that the bindings observed in ITC measurements of hMidm1m2–AID and H3m1–H4 mutants are ascribed to the interaction between AID and H3–H4. Therefore, AID may play a crucial role in the affinity for an isolated H3–H4 tetramer, although AID was disordered in the hMid–AID/(H3–H4)2 complex structure at the atomic level (Fig. 2A). As described later, AID makes functionally critical contributions to nucleosome reorganization.
Structural basis for H2A–H2B displacement and DNA stripping by hFACT
To investigate the mechanism for H2A–H2B displacement from nucleosomes by hFACT, we constructed a model in which hMid was docked to the human nucleosome structure (Tsunaka et al. 2005) by making the best superposition of the (H3–H4)2 structures between hMid–AID/(H3–H4)2 and nucleosomes (Fig. 4A; Supplemental Fig. S7A). Overall, this model is stereochemically reasonable; the histone regions generate few collisions, and a broad basic surface of hMid contacts the nucleosomal DNA around the central region (Supplemental Fig. S7A). However, the acidic surfaces of hMid cause steric hindrance with the nucleosomal DNA at one end and at a position ∼10 bp distant from the dyad axis (Supplemental Fig. S7A,B). Notably, at the latter position, hMid forms an intermolecular β structure with H4 (Fig. 2B), which is important for retaining the acidic surface structure (Supplemental Fig. S7A,B). These features imply that electrostatically repulsive forces between hMid and DNA contribute to the peeling of the nucleosomal DNA.
Figure 4.

Structural model of H2A–H2B displacement from nucleosomes by hMid–AID. (A) Docking model constructed by making the best superposition of the (H3–H4)2 structure between hMid–AID/(H3–H4)2 and the nucleosome (PDB code 2CV5). hMid is colored deep pink. The red box represents a close-up view shown in the middle panel of B. (B) H2A–H2B is displaced from nucleosomes by steric hindrances. (Left panel) The (H3–H4)2 binding loop on hMid–AID (deep pink) interacts with (H3–H4)2 in the hMid–AID/(H3–H4)2 structure (black arrow). (Middle panel) The clashing loop on hMid–AID collides with the H3 αN helix in the docking model (black arrow). (Middle and right panels) Both the (H3–H4)2-binding loop and the clashing loop on hMid–AID cause steric hindrances (red arrows in the middle panel) around the H2A-docking segment (yellow) and the H3 αN helix (cyan), which form hydrophobic interactions to tether H2A–H2B to (H3–H4)2 in the nucleosome (right panel). (C) Detailed views of hMid–AID in the hMid–AID/(H3–H4)2 complex (left panel) and the H2A-docking segment in nucleosomes (right panel) on the contact surface of (H3–H4)2 (colored as in Fig. 2A). They share interactions with key residues (Val101 and Glu105 of H3, Arg129 of H3′, and Lys44 of H4). Hydrogen bonds are indicated by gray dashed lines, and contact residues are depicted. Water molecules are shown as orange balls.
Remarkably, in site 2, the C-terminal docking segment of H2A (residues 107–119), which makes extensive contacts to traverse the surface of (H3–H4)2 within nucleosomes, is replaced by the (H3–H4)2-binding loop of hMid (Fig. 4B). In more detail, hMid–AID and the H2A-docking segment in nucleosomes share interactions with key residues (Val101 and Glu105 of H3, Arg129 of H3′, and Lys44 of H4) on the contact surface of (H3–H4)2 (Fig. 4C). The (H3–H4)2-binding loop of hMid also clashes with more residues of the H2A-docking segment, such as Ile87, Arg88, and residues of Gly105–Leu116. In addition, the previous report indicates that the extensive lack of the H2A-docking segment leads to prominent structural perturbations within nucleosomes (Shukla et al. 2011). Taken together, it is convincing that the H2A-docking segment, which occupies the same site as the (H3–H4)2-binding loop, is detached from histones H3–H4 upon invasion of hMid–AID into nucleosomes, thereby inducing the conversion of nucleosomes into hexasomes. In good agreement with this scenario, the interaction of hMid–AID with DSB nucleosomes facilitated the conversion from nucleosomes to hexasomes, whereas the hexasome formation of hMidm1m2–AID was reduced by approximately twofold in comparison with the wild type (Fig. 5A).
Figure 5.
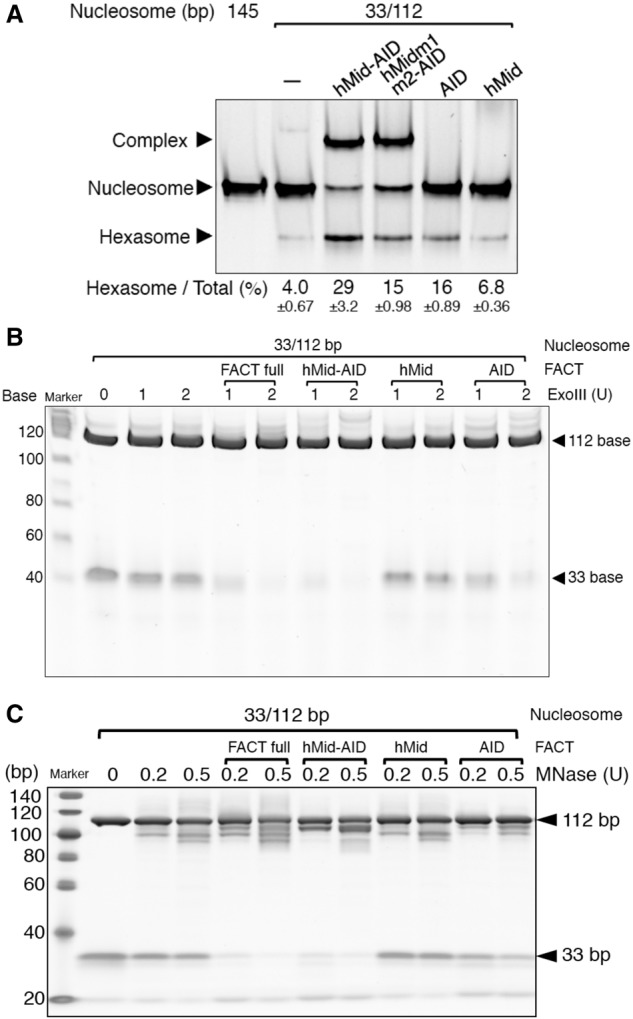
Biochemical analyses of H2A–H2B displacement from nucleosomes by hMid–AID. (A) EMSAs detected hexasome formation upon binding of hMid–AID and its mutants to DSB nucleosomes. Indicated values for ratios (percentages) of hexasome bands are mean ± SD for each data point. n = 3. Hexasome formation by hMidm1m2–AID, AID, and hMid is reduced in comparison with the wild type. (B,C) Exonuclease III (ExoIII) (B) and micrococcal nuclease (MNase) (C) treatment assays of the complex between DSB nucleosomes and various hFACT constructs. Similarly to the ExoIII assays, MNase completely digests a 33-bp DNA in the complexes with hFACT and hMid–AID but not those with hMid and AID. Experiments were repeated at least three times.
As observed in both the human nucleosome structure and chicken erythrocyte histone octameric structure (Wood et al. 2005), the H2A-docking segment forms hydrophobic interactions with the αN helix of H3 (residues 45–56), thereby fixing this helix to H2A and H4. The detachment of this docking segment would destabilize the αN helix of H3. One loop of hMid (Val897–Ser899) also clashes with the αN helix of H3 in the docking model (Fig. 4B). As observed in the hMid–AID/(H3–H4)2 complex structure (Fig. 2A), these two factors appear to cause the disorder of the αN helices of H3. Importantly, the structural disorder of the αN helix would result in the detachment of the nucleosomal DNA ends, fixed by basic residues (Arg49, Arg53, and Lys 56) of the αN helix (Fig. 4B). The docking model also shows that the steric hindrance with the acidic moiety of hMid at the corresponding DNA end may facilitate the peeling of DNA from the histone surface (Supplemental Fig. S7A,B). Taken together, we propose that H2A–H2B displacement by hMid–AID leads to DNA stripping. In agreement with this interpretation, the nucleosomal DNA ends are detached from nucleosomes, which contain H2A lacking the docking segment, similar to hexasomes (Shukla et al. 2011; Arimura et al. 2012).
To obtain definite evidence, we examined the nuclease susceptibility of the nucleosomal DNA using mixtures of hMid–AID and a DSB nucleosome reconstituted with 33-bp and 112-bp DNAs (33/112-bp DSB nucleosome) (Fig. 5B,C). In 33/112-bp DSB nucleosomes alone, both DNA fragments were completely protected from exonuclease III (ExoIII) digestion. In the mixture, only 112-bp DNA was protected, whereas 33-bp DNA was completely digested with ExoIII (Fig. 5B). Similar digestion patterns were also observed in micrococcal nuclease (MNase) assays (Fig. 5C). These results indicate that the 33-bp DNA segment is stripped from the nucleosomal end upon the invasion of hMid–AID into DSB nucleosomes. However, similar to the previous report (Arimura et al. 2012), the 112-bp DNA segment was protected from nuclease digestion. We thus conclude that hMid–AID and hexasomes lacking one H2A–H2B dimer are able to form the nuclease-protected stable complex. The same results were observed for the full-length hFACT molecule (Fig. 5B,C), indicating that hMid–AID alone completely reflects nucleosome reorganization by hFACT.
AID simultaneously interacts with the H2B N-tail and (H3–H4)2
The docking model revealed the critical roles of hMid in nucleosome reorganization. However, hMid alone, without AID, lacks the ability to conduct H2A–H2B displacement and DNA stripping from nucleosomes (Fig. 5A–C). In contrast, using a series of AID-truncated mutants, the binding assay revealed that hMid–AID with >34 residues of AID is essential for the complex formation with DSB nucleosomes and the simultaneous increase in hexasome production (Fig. 6A). These results suggest that AID plays a critical role in correctly bringing hMid to the H2A-docking surface of (H3–H4)2. Nevertheless, AID alone was less efficient in hexasome production and DNA stripping than hMid–AID (Fig. 5A–C), indicating that their cooperative action is required to accomplish nucleosome reorganization.
Figure 6.
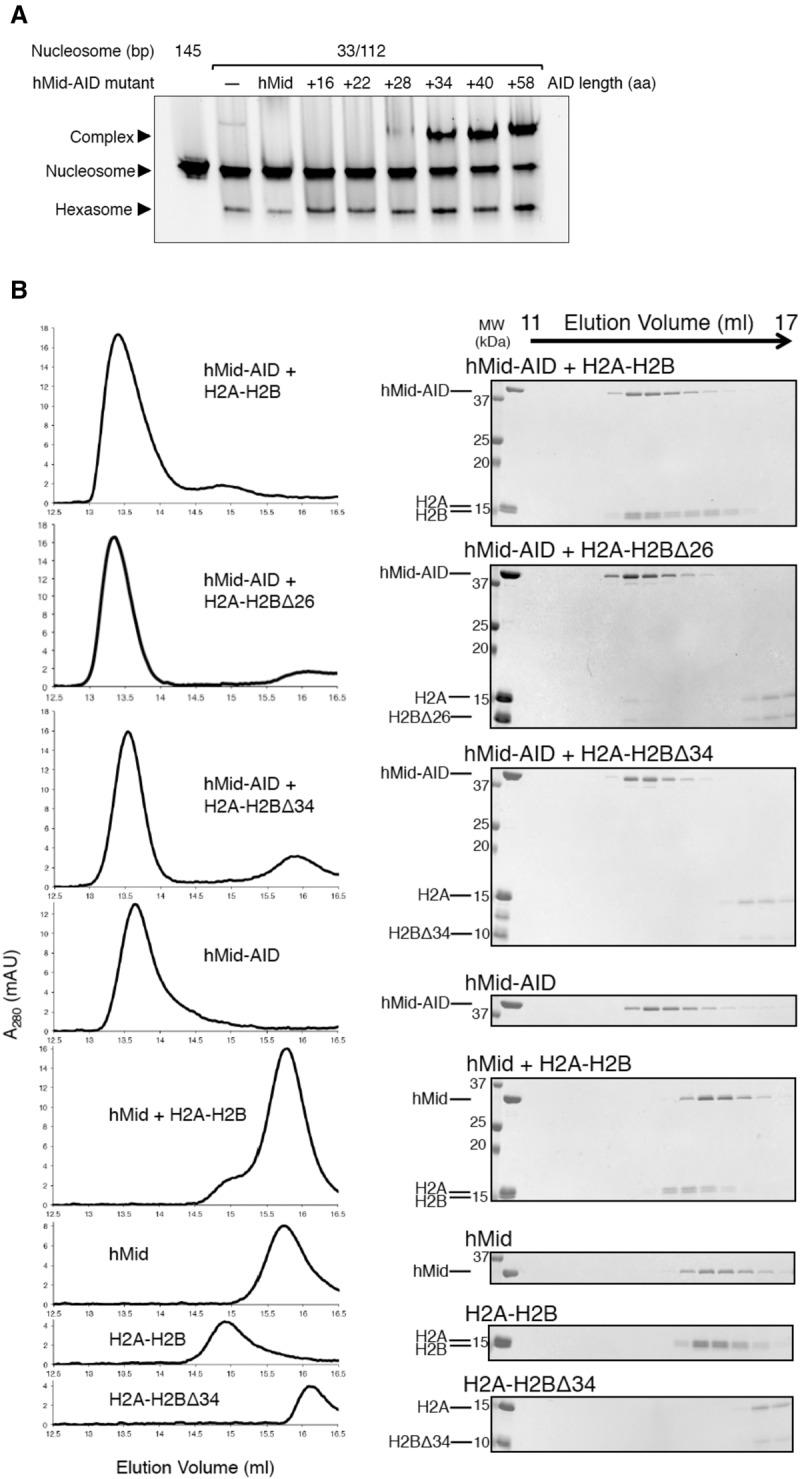
AID interacts with the H2B N-tail. (A) EMSAs show that the AID lengths of hMid–AID are critical for complex formation with DSB nucleosomes and the simultaneous increase in hexasome production. Experiments were repeated at least twice. (B) GF analyses show that AID and the H2B N-tail are required for interaction between hMid–AID and H2A–H2B. Protein mixtures with stoichiometric amounts were eluted from a Superdex200 10/300 column in 500 mM NaCl. Elution profiles and the corresponding SDS-PAGE are shown at the left and right sides, respectively.
Meanwhile, hMid–AID exhibited the most efficient accessibility to DSB nucleosomes in the close vicinity of the basic H2B N-tail (Fig. 1C–E). Thus, GF analyses were carried out to observe the direct interactions between AID and the H2B N-tail using truncated mutants of hMid–AID and the isolated H2A–H2B dimer. The full-length H2A–H2B and the mutant lacking the partial H2B N-tail (deletion of residues 1–26, H2A–H2BΔ26) formed a stable complex with hMid–AID (Fig. 6B). However, H2A–H2B lacking the entire H2B N-tail (deletion of residues 1–34, H2A–H2BΔ34) lost this binding ability (Fig. 6B). These results indicate that the proximal segment in the H2B N-tail (resides 27–34 in humans) is important for the interaction. Similarly, hMid lacking AID did not bind to the full-length H2A–H2B (Fig. 6B). The ITC analyses provided overall consistent results for the same mutant proteins (Supplemental Fig. S8A). Notably, the ITC analyses also showed that both AID alone and hMid–AID interact with the full-length H2A–H2B to a similar extent (Supplemental Fig. S8A). Collectively, these findings indicate that AID and the H2B N-tail are important for the interaction between FACT and H2A–H2B.
To further clarify the functional role of AID, whose structure is not observed in the crystal (Fig. 2A), we performed a GF assay of AID mixed with a histone octamer (Supplemental Fig. S8B). The histone octamer disintegrated into the H2A–H2B dimer and H3–H4 tetramer in 500 mM NaCl buffer. In contrast, the mixture of AID and the histone octamer in the same buffer was eluted at the stoichiometry of the histone octamer, indicating that AID can promote the formation of the histone octameric complex. Thus, AID is likely to make simultaneous interactions with the H2A–H2B dimer and H3–H4 tetramer.
Discussion
Our structural and biochemical studies reveal the integrated molecular mechanism of nucleosome reorganization involving H2A–H2B displacement by hFACT (Fig. 7). Initially, the AID segment interacts with the H2B N-tail detached from the nucleosomal DNA, possibly due to a local disruption of DNA–histone contacts by actions of other factors. Next, the hMid domain correctly binds to the H2A-docking surface of the nucleosome. Thus, hMid displaces an H2A–H2B dimer from the nucleosome through steric collisions on the H2A-docking surface of (H3–H4)2. Simultaneously, this results in inducing the disorder of the H3 αN helix. Consequently, the complex between FACT and the hexasome strips ∼30 bp of the nucleosomal DNA from histones. The DNA stripping may facilitate the invasion of other remodeling factors and polymerases, which must surmount the barriers against their progress (Kulaeva et al. 2013). On the other hand, the H2A–H2B dimer displaced from (H3–H4)2 may remain tethered to the nucleosome via FACT, for instance, through transient interactions possibly retained between FACT–AID and the H2B N-tail.
Figure 7.
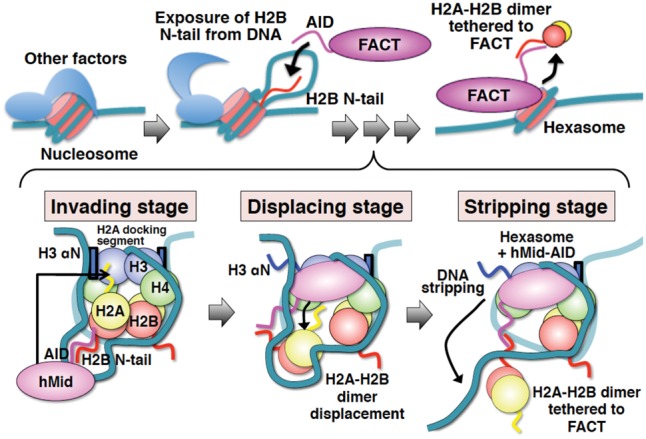
Schematic diagrams showing the integrated mechanism for nucleosome reorganization by hFACT. Histone proteins and DNA in nucleosomes are indicated by different colors as follows: H2B (red), H2A (yellow), H3 (blue), H4 (green), and DNA (light sea green). FACT, hMid, and AID are each colored in magenta. Blue boxes show the two αN helices of H3. Wavy lines represent the disordered regions of proteins. The invading stage involves speculation, because the corroborative assays (Fig. 1) are based on the assumption that DSB nucleosomes mimic the exposure of internal histones from DNA by other factors. However, the interaction of AID with the H2B N-tail was revealed by our biochemical data (Fig. 6). The displacing stage is entirely supported by our crystal structure and the related biochemical data (Figs. 2, 3, 4, and 5A). At the stripping stage, the DNA stripping is supported by our nuclease susceptibility assays (Fig. 5B,C). On the other hand, the anchoring of H2A–H2B via AID is based on the interpretation that AID simultaneously binds to one H3–H4 tetramer and one H2A–H2B dimer. This interpretation is supported by our biochemical data (Fig. 1F; Supplemental Fig. S8B).
The significance of a space created between DNA and the H2B N-tail
In the H2B N-terminal-proximal segment, a highly basic and conserved H2B repression region (HBR) (residues 27–34 in Supplemental Fig. S4B) is buried by DNA in nucleosomes, passing through the two gyres of DNA (Supplemental Fig. S1E; Luger et al. 1997; Tsunaka et al. 2005). The HBR of H2B appears to lock histone octamers into position on the DNA and thus may mediate the structural and dynamic polymorphism of nucleosomes (Sivolob et al. 2003). In fact, DSBs at the direct contact site with the HBR (122/23-bp DSB) were found to cause nucleosome deformation (Supplemental Fig. S1B), suggesting that the HBR is crucial to stabilize the nucleosome structure (Parra et al. 2006; Nag et al. 2010). On the other hand, the HBR is necessary for yeast FACT (yFACT) to bind to H2A–H2B (Zheng et al. 2014). Furthermore, hFACT and DNA compete with each other for nonnucleosomal H2A–H2B binding (Winkler et al. 2011). Naturally, FACT binding and DNA binding to the HBR are mutually competitive; the HBR should be detached from DNA before FACT can engage in the interaction with the H2B N-tail. Hence, we assume that DSBs in the close vicinity of the H2B N-tail would generate specific unwrapping of the nucleosomal DNA by an unknown mechanism, possibly through electrostatic AID actions. Consistently, in response to addition of AID, the DSB nucleosome becomes more susceptible to digestion by MNase and ExoIII (Fig. 5B,C).
On the other hand, hMid–AID addition results in more efficient digestion of DSB nucleosomes than that of AID (Fig. 5B,C). In addition, hMid–AID is more efficient in hexasome production than AID (Fig. 5A). This suggests that AID binding to DSB nucleosomes facilitates the local DNA detachment from the HBR near DSBs but cannot accomplish more extensive DNA stripping, which requires H2A–H2B displacement by hMid invasion into the nucleosome. Therefore, the DNA stripping coupled with H2A–H2B displacement should be discriminated from the local DNA detachment from histones to expose the H2B N-tail. The DNA stripping coupled with H2A–H2B displacement would be an irreversible alteration of nucleosome structure unless H2A–H2B is reassembled. Thus, this may be followed by the invasion of other factors, such as histone chaperones and remodeling factors, thereby leading to further disruption of nucleosome and histone replacement. In contrast, the local DNA detachment from the H2B N-tail is a dynamic process that possibly involves a temporal alteration of two DNA gyres within nucleosomes. This detachment is crucial for FACT to subsequently invade into nucleosomes and cause larger alterations of the nucleosome structure.
What factors induce the detachment of the nucleosomal DNA from the HBR during the DNA transaction processes within nuclei? First, the DNA torsional strain, generated by elongating polymerases, locally disrupts DNA–histone contacts within nucleosomes (Sheinin et al. 2013; Teves and Henikoff 2014). Second, ATP-dependent chromatin remodelers, such as ISWI and SWI/SNF, also disrupt local DNA–histone contacts, creating DNA loops or bulges on nucleosomes during DNA translocation (Mueller-Planitz et al. 2013; Bartholomew 2014). Such disruption of local DNA–histone contacts may promote the exposure of the HBR, which triggers the FACT invasion into nucleosomes. Indeed, an ATP-dependent remodeler is required for the FACT-dependent histone H3.3 replacement at chromatin boundaries (Nakayama et al. 2012). Third, DNA lesions may be removed through complex pathways involving disruptions of local DNA–histone contacts. In fact, FACT-dependent H2A–H2B exchange is accelerated at UV-induced DNA damage sites in mammalian cells (Heo et al. 2008; Dinant et al. 2013).
General implications for nucleosome reorganization by hFACT
Several mechanisms have been proposed so far for nucleosome reorganization by FACT. In all of these mechanisms, FACT and DNA exhibit mutually competitive binding to H2A–H2B within nucleosomes (Winkler et al. 2011; Hsieh et al. 2013; Zheng et al. 2014; Kemble et al. 2015). Thus, FACT shields DNA-binding surfaces of H2A–H2B within nucleosomes and partially peels DNA from histones. In agreement with this interpretation, we suggest that hFACT interacts with the H2B N-tail detached from the nucleosomal DNA, thereby striping ∼30 bp of the nucleosomal DNA from histones after H2A–H2B displacement at the final step (Fig. 7). However, our findings contradict the previous study, suggesting that the FACT Mid domain binds to an H2A–H2B dimer (Hondele et al. 2013). This previous study relies on the crystal structure of a complex in which a mismatched pair of a C. thermophilum FACT Mid domain and a Xenopus H2A–H2B dimer were fused to each other. Such fusion complexes may sometimes exhibit nonfunctional contacts in crystal structures. In fact, we could not reproduce the corresponding interactions when using the cognate pair of hMid domain and human H2A–H2B (Fig. 6B; Supplemental Fig. S8A). We found that, instead of the Mid domain, its adjacent acidic AID segment interacts with H2A–H2B. The consistent result was also obtained from the study using the cognate pair of yFACT and yeast H2A–H2B (Kemble et al. 2015).
The structure of the hMid–AID/(H3–H4)2 complex indicates that H2A and FACT are mutually exclusive on the H2A-docking surface within nucleosomes, thus causing H2A–H2B displacement from nucleosomes (Fig. 4). However, in the previously reported mechanisms for nucleosome reorganization by FACT, H2A–H2B displacement is not obligatory (Xin et al. 2009; Winkler et al. 2011; Hsieh et al. 2013; Zheng et al. 2014). This scientific disagreement could be explained by the alternative interpretation of the previous data. In addition to its role in nucleosome disassembly, FACT is involved in nucleosome reassembly in cells (Formosa et al. 2002), suggesting that FACT maintains a reversible equilibrium between stable and partially dissociated nucleosomes in vivo; namely, FACT displaces an H2A–H2B dimer from nucleosomes for a time through steric collisions on the H2A-docking surface of (H3–H4)2, but the H2A–H2B dimer may remain tethered to nucleosomes via FACT, presumably through transient interaction retained between the FACT AID segment and the H2B N-tail. This interpretation is supported by our result that AID simultaneously interacts with the H3–H4 tetramer and H2A–H2B dimer (Supplemental Fig. S8B). In addition, the recent report has proposed a similar mechanism in which yFACT retains nucleosomes in partially loosened structures by interactions between acidic IDRs of FACT and H2A–H2B (Kemble et al. 2015). Thus, H2A–H2B displacement would not necessarily be observed in nuclear processes, such as transcription. After the relevant processes have concluded, FACT immediately executes nucleosome reassembly. In agreement with this interpretation, we indeed showed that the FACT–hexasome complex reacquires H2A–H2B upon their further addition (Fig. 1F).
Meanwhile, our results from human cognate proteins are different from those from yeast (Kemble et al. 2015) in two aspects. First, the acidic C terminus of the yeast SSRP1 homolog Pob3 exhibits domain architectures distinct from those of metazoan, such as an HMG domain and mixed acidic and basic IDRs. Corresponding to this difference, the C terminus of Pob3 interacts with H2A–H2B (Kemble et al. 2015), while the previous study of human SSRP1 failed to detect interactions with them (Winkler et al. 2011). This result may be rationalized by our previous study, in which the phosphorylated acidic IDR of Drosophila SSRP1 makes strong intramolecular contacts with its adjacent basic IDR and the HMG domain (Tsunaka et al. 2009; Hashimoto et al. 2013). Second, the major interaction between yeast H2A–H2B and AID of yeast SPT16 is specific: An aromatic residue on AID contacts hydrophobic residues in a globular domain of H2B (Tyr45 and Met62 in yeast), although electrostatic interactions also contribute to their binding (Kemble et al. 2015). On the other hand, we found that human H2A–H2B lacking the entire H2B N-tail (residues 1–34 in humans) (Supplemental Fig. S4B) loses the ability to bind to hMid–AID (Fig. 6B; Supplemental Fig. S8A), although the recent report has not confirmed whether AID interacts with H2A–H2B lacking the corresponding H2B N-tail in yeast (resides 1–37 in yeast) (Supplemental Fig. S4B; Kemble et al. 2015). Thus, the corresponding hydrophobic residues of human H2B (Tyr42 and Met59 in humans) do not make significant contributions to AID binding. Instead, electrostatic interactions between AID and the H2B N-tail appear to be more important for the binding between human proteins. This minor difference may not underestimate the importance of the interaction between FACT–AID and H2B.
Our complex structure may serve for understanding how FACT causes further disruption of nucleosome and histone replacements through cooperation with histone chaperones, such as Asf1, HIRA, and MCM2 (Tan et al. 2006; Nakayama et al. 2007; Takahata et al. 2009; Foltman et al. 2013). For instance, hMid–AID and one MCM2 histone-binding domain could simultaneously bind to (H3–H4)2 without the steric hindrance in the superimposed model (Supplemental Fig. S9; Huang et al. 2015; Richet et al. 2015). This structural model is consistent with the previous reports, which suggest the functional cooperation between FACT and MCM2 (Tan et al. 2006; Foltman et al. 2013). In addition, several conventional H3–H4 chaperones disrupt the dimer interface of H3–H4 (English et al. 2006; Natsume et al. 2007; Hu et al. 2011; Elsässer et al. 2012; Liu et al. 2012), in contrast to hFACT, which retains their original interface (Supplemental Fig. S2B). This highlights the mechanistic difference in nucleosome reorganization between FACT and these chaperones while implying their functional complementarity. The hFACT–hexasome complex was also found to be remarkably stable against nuclease digestions (Fig. 5B,C). The maintenance of this complex may be connected with epigenetic preservation of histones throughout RNA polymerase II elongation (Hsieh et al. 2013).
In conclusion, the integrated mechanism reported here promises to universally rationalize how FACT operates to create a transient nucleosome structure, thus assisting the passage of polymerases without entire nucleosome disassembly so as to ensure chromatin integrity.
Materials and methods
Protein expression and purification
Recombinant human histone proteins were produced in Escherichia coli and purified as reported previously (Tsunaka et al. 2005). Truncated mutants of histone complexes contained the following residues: an H2A–H2BΔ26 dimer (H2A full length and H2B residues 27–125), an H2A–H2BΔ34 dimer (H2A full length and H2B residues 35–125), and an H3Δ34–H4 tetramer (H3 residues 35–135 and H4 full length). All DNA segments are based on the 601 nucleosome positioning sequence (Lowary and Widom 1998). DNA fragments of 13, 23, 33, 43, 53, 67, and 78 bp were synthesized as two complementary oligomers and annealed. DNA fragments of 92, 102, 112, 122, 132, and 145 bp were constructed and purified as described previously (Han et al. 2014). A 145-bp mononucleosome and DSB nucleosomes were reconstituted from histones and DNAs by the salt dialysis method (Tsunaka et al. 2005). The 33/112-bp DSB nucleosome was crystallized under essentially the same conditions as those for the human nucleosome (Tsunaka et al. 2005). The X-ray diffraction study revealed similar crystal data between the DSB and intact nucleosomes. The preliminary electron density map at 4 Å resolution allowed us to trace continuous DNA strands, indicating that the 33- and 112-bp DNA fragments are retained in the DSB nucleosome (data not shown). The reconstituted nucleosomes were also subjected to ExoIII and MNase digestion analyses.
Baculovirus-driven expression of the hFACT complex (coexpression of N-terminal His-tagged SPT16 and nontagged SSRP1) and the mutants (N-terminal His-tagged hMid–AID and hMid) in Sf9 insect cells was carried out as reported previously (Tsunaka et al. 2009). The infected cells were collected by centrifugation and suspended in lysis buffer containing 150 mM NaCl, 20 mM Tris-HCl (pH 8.5), 0.1% Nonidet P-40, and a protease inhibitor mixture (Nacalai-Tesque). The hFACT proteins were purified according to previously published protocols (Tsunaka et al. 2009) with a few modifications. The hFACT proteins were partially purified on a HisTrap column (GE Healthcare) and then subjected to a Superdex200 GF column (GE Healthcare). Fractions containing hFACT proteins were further purified on a HiTrap Q anion exchange column (GE Healthcare). For the purification of the AID protein, we expressed the hMid–AID protein with a TEV protease cleavage site between residues 930 and 931 in Sf9 cells and then treated it with TEV protease for 16 h at 4°C. AID separated from hMid was finally purified on a Superdex200 GF column.
For crystallization, the His tags of hMid and hMid–AID were removed by TEV digestion during purification. The purified hMid–AID was mixed with an equimolar amount of the H3Δ34–H4 tetramer. After an incubation for 5 min at 20°C, the hMid–AID/(H3–H4)2 complex was purified on a Superdex200 column in the purification buffer (750 mM NaCl, 20 mM Tris-HCl at pH 7.5, 1 mM DTT).
Crystallization and data collection
For hMid alone, monoclinic crystals belonging to space group C2 (Supplemental Table S1) were grown at 4°C from hanging drops composed of 1.2 µL of protein solution (3.5 mg mL−1 protein in 150 mM NaCl, 20 mM Tris-HCl at pH 8.5, 1 mM DTT, 10% glycerol) and 1.5 µL of reservoir solution (5% PEG1000, 100 mM citrate at pH 4.4) suspended over 1 mL of the reservoir. Crystals were cryoprotected in the reservoir supplemented with 30% glycerol and flash-frozen in liquid nitrogen. For the hMid–AID/(H3–H4)2 complex, orthorhombic crystals belonging to space group P21221 (Supplemental Table S1) were grown at 4°C from hanging drops composed of 2 µL of protein solution (4.5 mg mL−1 protein in 750 mM NaCl, 20 mM Tris-HCl at pH 7.6, 1 mM DTT, 10% glycerol) and 1 µL of reservoir solution (10% PEG400, 0.9–1.1 M imidazole at pH 7.0, 100–150 mM L-histidine) suspended over 1 mL of the reservoir. Crystals were transferred into a cryoprotection buffer (30% PEG400, 0.925 M imidazole at pH 7.0, and 58.5 mM L-histidine) and flash-frozen in liquid nitrogen. All X-ray diffraction data were collected at the synchrotron radiation source at BL44XU (SPring-8) at 100 K using a nitrogen stream. Data processing and scaling were performed with HKL2000 (Otwinowski and Minor 1997).
Structure determination and refinement
The structures of hMid alone and the hMid–AID complex with (H3–H4)2 were determined by molecular replacement using PHASER. The hMid alone structure was solved using the yeast Mid structure (Kemble et al. 2013) as a probe, and the complex structure was solved using the hMid structure determined here and the histone (H3–H4)2 structure from the human nucleosome core particle (Tsunaka et al. 2005). Refinement of the structures was performed by iterative cycles of model adjustment in COOT (Emsley and Cowtan 2004) and refinement in PHENIX (Adams et al. 2010). The structures were drawn using PyMOL (http://www.pymol.org) and University of California at San Francisco Chimera (Pettersen et al. 2004). Electrostatic surface potentials were calculated using APBS (Baker et al. 2001). Structural superposition was calculated with the SSM superpose of COOT.
EMSAs
Nucleosomes (1.5 pmol) and various hFACT constructs (hFACT full, 3.6 pmol; others, 10 pmol) were mixed in a reaction buffer containing 400 mM NaCl, 20 mM Tris-HCl (pH 8.5), 10% glycerol, and 1 mM DTT and incubated for 15 min at 30°C. In the assay of Figure 1F, 5 pmol of the H2A–H2B dimer was additionally mixed in the above reaction mixture. The samples were electrophoresed at 4°C on a 7.5% native PAGE in 1× Tris-glycine buffer and then visualized by SYBR Gold nucleic acid gel stain. Each band was quantified using Image Lab software (Bio-Rad). n = 3 was found to be sufficient for reproducible data with low standard deviations (Figs. 1E, 5A; Supplemental Fig. S1C).
GF
Histones and various hFACT constructs were mixed at equimolar ratios (1.5 nmol) and incubated for 5 min at 20°C. Proteins were separated on a Superdex200 10/300 GL column in buffer containing 20 mM Tris-HCl (pH 7.5), 1 mM DTT, and either 500 mM NaCl (for histone octamer and H2A–H2B) or 750 mM NaCl [for (H3–H4)2]. The eluted fractions were analyzed by 15% SDS-PAGE and stained with Coomassie brilliant blue. Each band was quantified by Image Lab.
ITC
Binding affinities were determined at 25°C by using an iTC200 calorimeter (GE Life Science, MicroCal). Proteins were dialyzed against ITC buffer {25 mM Tris-HCl at pH 7.5, containing either 200 mM NaCl [for H2A–H2B] or 750 mM NaCl [for (H3–H4)2]} prior to measurements. Injections consisted of 1 µL of 250–600 µM hMid–AID into 20–35 µM histones at 5-min intervals at 25°C. The heat of dilution was obtained by injecting the hMid–AID solution into ITC buffer and was subtracted from the heat of the binding reaction before the fitting process. Data were analyzed using Origin software (version 7.0). A single binding site model for (H3–H4)2 gave the best fit to the data. n = 3 was found to be sufficient for reproducible data with low standard deviations (Fig. 3B).
Nuclease susceptibility assays
The nuclease susceptibility assays were performed as described previously (Arimura et al. 2012). The reaction mixtures of 33/112-bp DSB nucleosomes (1.5 pmol) with various hFACT constructs (hFACT full, 3.6 pmol; others, 10 pmol) were incubated for 15 min at 30°C in 10 µL of 150 mM NaCl, 20 mM Tris-HCl (pH 8.5), 10% glycerol, and 1 mM DTT. The reaction mixtures were then treated with 1 or 2 U of E. coli ExoIII (Takara) in 20 µL of 75 mM NaCl, 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 5% glycerol, and 1 mM DTT. The reaction was continued for 5 min at 30°C and was stopped by the addition of 55 µL of proteinase K solution (20 mM Tris-HCl at pH 8.0, 80 mM EDTA, 0.25% SDS, 0.5 mg mL−1 proteinase K [Roche]). After a 20-min treatment at 30°C, the DNA samples were extracted and then analyzed by 10% urea-PAGE containing 7 M urea in 0.5× TBE buffer. The reaction mixtures were also treated with 0.2 or 0.5 U of MNase (Takara) in 20 µL of 77.5 mM NaCl, 20 mM Tris-HCl (pH 8.0), 5% glycerol, and 2.5 mM CaCl2. After a 5-min incubation at 30°C, the reaction was stopped by the addition of 15 µL of proteinase K solution. After a 30-min incubation at 30°C, the DNA samples were analyzed by 15% PAGE in 1× TBE. The samples were stained with SYBR Gold.
Accession numbers
Atomic coordinates and structure factors for the reported crystal structures have been deposited in the PDB under accession codes 4Z2N (hMid domain) and 4Z2M [hMid–AID/(H3–H4)2 complex].
Supplementary Material
Acknowledgments
We are grateful to Henri-Obadja Kumada and Miyako Natsume for technical assistance. We thank Kouta Mayanagi and Kazuhiro Yamada for helpful discussions. We also thank Mariko Ariyoshi, Masahiro Shirakawa, and Masayuki Oda for help in ITC measurements. The synchrotron radiation experiments were performed at the BL44XU of SPring-8 under the Cooperative Research Program of the Institute for Protein Research, Osaka University (proposal nos. 2012B6727, 2013A6830, 2013B6830, 2014A6931, and 2014B6931). This work was supported by Grants-in-Aid for Scientific Research (A) (Japan Society for the Promotion of Science KAKENHI grant nos. 23247016 and 26251008) and Precursory Research for Embryonic Science and Technology, Japan Science and Technology Agency.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.274183.115.
Freely available online through the Genes & Development Open Access option.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H. 2012. Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry 51: 3302–3309. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci 98: 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew B. 2014. Regulating the chromatin landscape: structural and mechanistic perspectives. Annu Rev Biochem 83: 671–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090–1093. [DOI] [PubMed] [Google Scholar]
- Chen S, Rufiange A, Huang H, Rajashankar KR, Nourani A, Patel DJ. 2015. Structure–function studies of histone H3/H4 tetramer maintenance during transcription by chaperone Spt2. Genes Dev 29: 1326–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua EYD, Vasudevan D, Davey GE, Wu B, Davey CA. 2012. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res 40: 6338–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C, Ampatziadis-Michailidis G, Lans H, Tresini M, Lagarou A, Grosbart M, Theil AF, van Cappellen WA, Kimura H, Bartek J, et al. 2013. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol Cell 51: 469–479. [DOI] [PubMed] [Google Scholar]
- Elsässer SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. 2012. DAXX envelops an H3.3–H4 dimer for H3.3-specific recognition. Nature 491: 560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- English CM, Adkins MW, Carson JJ, Churchill MEA, Tyler JK. 2006. Structural basis for the histone chaperone activity of Asf1. Cell 127: 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, Labib K. 2013. Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep 3: 892–904. [DOI] [PubMed] [Google Scholar]
- Formosa T, Ruone S, Adams M, Olsen A, Eriksson P, Yu Y, Rhoades AR, Kaufman P, Stillman DJ. 2002. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics 162: 1557–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin ZA, Quivy J-P, Almouzni G. 2014. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem 83: 487–517. [DOI] [PubMed] [Google Scholar]
- Hainer SJ, Charsar BA, Cohen SB, Martens JA. 2012. Identification of mutant versions of the Spt16 histone chaperone that are defective for transcription-coupled nucleosome occupancy in Saccharomyces cerevisiae. G3 (Bethesda) 2: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y-W, Tsunaka Y, Yokota H, Matsumoto T, Kashiwazaki G, Morinaga H, Hashiya K, Bando T, Sugiyama H, Harada Y. 2014. Construction and characterization of Cy3- or Cy5-conjugated hairpin pyrrole–imidazole polyamides binding to DNA in the nucleosome. Biomater Sci 2: 297–307. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Kodera N, Tsunaka Y, Oda M, Tanimoto M, Ando T, Morikawa K, Tate S-I. 2013. Phosphorylation-coupled intramolecular dynamics of unstructured regions in chromatin remodeler FACT. Biophys J 104: 2222–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo K, Kim H, Choi S, Choi J, Kim K, Gu J, Lieber M, Yang A, An W. 2008. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell 30: 86–97. [DOI] [PubMed] [Google Scholar]
- Hondele M, Stuwe T, Hassler M, Halbach F, Bowman A, Zhang ET, Nijmeijer B, Kotthoff C, Rybin V, Amlacher S, et al. 2013. Structural basis of histone H2A–H2B recognition by the essential chaperone FACT. Nature 499: 111–114. [DOI] [PubMed] [Google Scholar]
- Hsieh F-K, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, Studitsky VM. 2013. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci 110: 7654–7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Liu Y, Wang M, Fang J, Huang H, Yang N, Li Y, Wang J, Yao X, Shi Y, et al. 2011. Structure of a CENP-A–histone H4 heterodimer in complex with chaperone HJURP. Genes Dev 25: 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Strømme CB, Saredi G, Hödl M, Strandsby A, González-Aguilera C, Chen S, Groth A, Patel DJ. 2015. A unique binding mode enables MCM2 to chaperone histones H3–H4 at replication forks. Nat Struct Mol Biol 22: 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble DJ, Whitby FG, Robinson H, McCullough L, Formosa T, Hill CP. 2013. Structure of the Spt16 middle domain reveals functional features of the histone chaperone FACT. J Biol Chem 288: 10188–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble DJ, McCullough L, Whitby FG, Formosa T, Hill CP. 2015. FACT disrupts nucleosome structure by binding H2A–H2B with conserved peptide motifs. Mol Cell 60: 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Hsieh F-K, Chang H-W, Luse DS, Studitsky VM. 2013. Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim Biophys Acta 1829: 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Lis JT. 2013. Control of transcriptional elongation. Annu Rev Genet 47: 483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, Wang JC. 1987. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci 84: 7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-P, Xiong C, Wang M, Yu Z, Yang N, Chen P, Zhang Z, Li G, Xu R-M. 2012. Structure of the variant histone H3.3–H4 heterodimer in complex with its chaperone DAXX. Nat Struct Mol Biol 19: 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary P, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276: 19–42. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader A, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Mueller-Planitz F, Klinker H, Becker PB. 2013. Nucleosome sliding mechanisms: new twists in a looped history. Nat Struct Mol Biol 20: 1026–1032. [DOI] [PubMed] [Google Scholar]
- Myers CN, Berner GB, Holthoff JH, Martinez-Fonts K, Harper JA, Alford S, Taylor MN, Duina AA. 2011. Mutant versions of the S. cerevisiae transcription elongation factor Spt16 define regions of Spt16 that functionally interact with histone H3. PLoS One 6: e20847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag R, Kyriss M, Smerdon JW, Wyrick JJ, Smerdon MJ. 2010. A cassette of N-terminal amino acids of histone H2B are required for efficient cell survival, DNA repair and Swi/Snf binding in UV irradiated yeast. Nucleic Acids Res 38: 1450–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Nishioka K, Dong Y-X, Shimojima T, Hirose S. 2007. Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes Dev 21: 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Shimojima T, Hirose S. 2012. The PBAP remodeling complex is required for histone H3.3 replacement at chromatin boundaries and for boundary functions. Development 139: 4582–4590. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. 2013. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154: 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume R, Eitoku M, Akai Y, Sano N, Horikoshi M, Senda T. 2007. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature 446: 338–341. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Parra MA, Kerr D, Fahy D, Pouchnik DJ, Wyrick JJ. 2006. Deciphering the roles of the histone H2B N-terminal domain in genome-wide transcription. Mol Cell Biol 26: 3842–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- Ransom M, Dennehey BK, Tyler JK. 2010. Chaperoning histones during DNA replication and repair. Cell 140: 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Bataille AR, Zhang L, Pugh BF. 2014. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159: 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet N, Liu D, Legrand P, Velours C, Corpet A, Gaubert A, Bakail M, Moal-Raisin G, Guerois R, Compper C, et al. 2015. Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res 43: 1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinin MY, Li M, Soltani M, Luger K, Wang MD. 2013. Torque modulates nucleosome stability and facilitates H2A/H2B dimer loss. Nat Commun 4: 2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla MS, Syed SH, Goutte-Gattat D, Richard JLC, Montel F, Hamiche A, Travers A, Faivre-Moskalenko C, Bednar J, Hayes JJ, et al. 2011. The docking domain of histone H2A is required for H1 binding and RSC-mediated nucleosome remodeling. Nucleic Acids Res 39: 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivolob A, Lavelle C, Prunell A. 2003. Sequence-dependent nucleosome structural and dynamic polymorphism. Potential involvement of histone H2B N-terminal tail proximal domain. J Mol Biol 326: 49–63. [DOI] [PubMed] [Google Scholar]
- Suganuma T, Workman JL. 2011. Signals and combinatorial functions of histone modifications. Annu Rev Biochem 80: 473–499. [DOI] [PubMed] [Google Scholar]
- Takahata S, Yu Y, Stillman DJ. 2009. FACT and Asf1 regulate nucleosome dynamics and coactivator binding at the HO promoter. Mol Cell 34: 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BC-M, Chien C-T, Hirose S, Lee S-C. 2006. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J 25: 3975–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, Henikoff S. 2014. Transcription-generated torsional stress destabilizes nucleosomes. Nat Struct Mol Biol 21: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremethick DJ. 2007. Higher-order structures of chromatin: the elusive 30 nm fiber. Cell 128: 651–654. [DOI] [PubMed] [Google Scholar]
- Tsunaka Y, Kajimura N, Tate S, Morikawa K. 2005. Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res 33: 3424–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunaka Y, Toga J, Yamaguchi H, Tate S-I, Hirose S, Morikawa K. 2009. Phosphorylated intrinsically disordered region of FACT masks its nucleosomal DNA binding elements. J Biol Chem 284: 24610–24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDemark A, Blanksma M, Ferris E, Heroux A, Hill CP, Formosa T. 2006. The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol Cell 22: 363–374. [DOI] [PubMed] [Google Scholar]
- VanDemark AP, Xin H, McCullough L, Rawlins R, Bentley S, Heroux A, Stillman DJ, Hill CP, Formosa T. 2008. Structural and functional analysis of the Spt16p N-terminal domain reveals overlapping roles of yFACT subunits. J Biol Chem 283: 5058–5068. [DOI] [PubMed] [Google Scholar]
- Winkler DD, Muthurajan UM, Hieb AR, Luger K. 2011. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem 286: 41883–41892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CM, Nicholson JM, Lambert SJ, Chantalat L, Reynolds CD, Baldwin JP. 2005. High-resolution structure of the native histone octamer. Acta Crystallogr Sect F Struct Biol Cryst Commun 61: 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock CL, Ghosh RP. 2010. Chromatin higher-order structure and dynamics. Cold Spring Harb Perspect Biol 2: a000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Takahata S, Blanksma M, McCullough L, Stillman DJ, Formosa T. 2009. yFACT induces global accessibility of nucleosomal DNA without H2A–H2B displacement. Mol Cell 35: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Crickard JB, Srikanth A, Reese JC. 2014. A highly conserved region within H2B is important for FACT to act on nucleosomes. Mol Cell Biol 34: 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.