

Until 15 years ago, inherited thrombocytopenias (ITs) were quite an indistinct group of disorders, only a few forms of which had been clearly defined. Moreover, the genetic defect was known for only two disorders: Bernard-Soulier syndrome (BSS) and Wiskott Aldrich syndrome (WAS).
Since then, our knowledge of ITs has greatly advanced and we currently know at least 21 genes whose mutations result in 19 disorders (Table 1). The study of large series of patients identified the particular characteristics of the different forms and revealed that they have different degrees of clinical complexity and a great variation in prognosis. Furthermore, we realized that different mutations in the same gene may cause from different phenotypes. Finally, specific treatments for specific disorders have been identified, and given this, we are now truly in an era in which personalized medicine can play a role in the treatment of ITs.
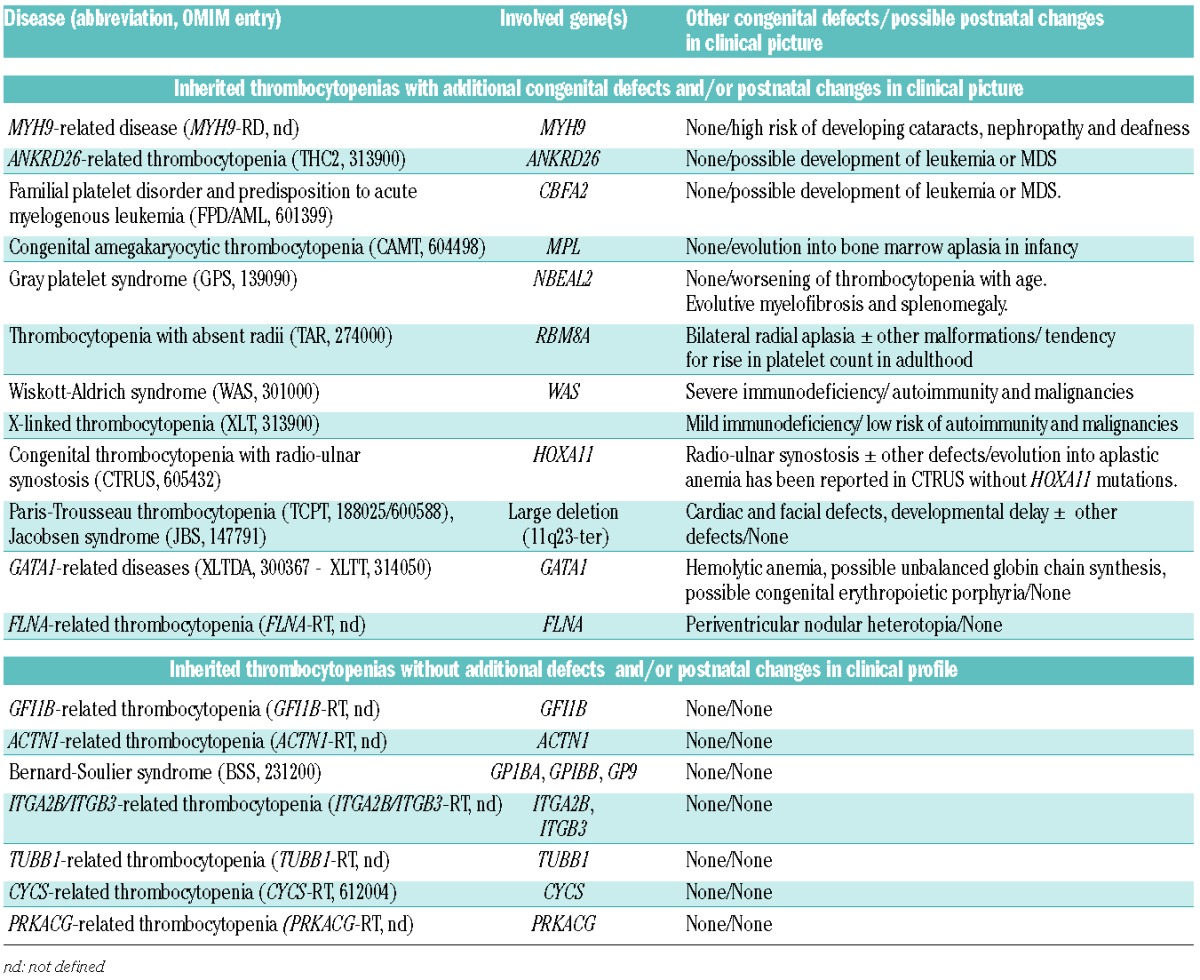
Table 1.
Inherited thrombocytopenias classified accordingly to complexity of the clinical profile.
Molecular characterization for defining prognosis
For a long time, the most frequently diagnosed form of IT was BSS, which typically presents from birth with recurrent hemorrhage.1 Bleeding tendency is usually severe also in the other well-known ITs, such as WAS, congenital amegakaryocytic thrombocytopenia (CAMT), and gray platelet syndrome (GPS). A major concern in these patients has always been to prevent bleedings from hemostatic challenge and to stop spontaneous hemorrhage.
In the last few years, our understanding of ITs has changed because the discovery of several ‘new’ ITs revealed that they are much more frequent than the ‘old’ ones, and that most patients with these ‘new’ disorders have only mild or moderate thrombocytopenia, with trivial bleeding episodes or no bleeding at all. Thus, the bleeding risk is no longer a major concern for most patients with ITs.2 However, this improved knowledge of ITs revealed that many of them expose affected subjects to another threat, that of acquiring additional defects that worsen the quality of life or that can even prove fatal (Table 1).
The typical disorder with this feature is MYH9-related disease (MYH9-RD), the most frequent form of IT.3 It derives from monoallelic mutations in the gene MYH9 for the heavy chain of myosin IIA and usually presents with mild bleeding tendency. However, it exposes patients to a 30% risk of developing a glomerulonephritis that evolves into end stage renal failure and requires dialysis or kidney transplantation. Moreover, 16% of patients develop presenile cataracts and 60% acquire a sensorineural hearing defect that may lead to deafness at a young age. Importantly, genotype-phenotype studies revealed that the molecular defect predicts the risk of anomalies other than thrombocytopenia.4 For instance all subjects with mutations affecting the R702 residue in the head of myosin IIA develop glomerulonephritis by the fourth decade of life, while those with mutations in the non-helical portion of the tail almost never suffer from this problem. Also the risk of deafness and cataracts can be defined on the basis of patient genotype.
Patients with ITs also risk developing another category of disorders: hematologic malignancies. Subjects with germline mutations in RUNX1 (familial platelet disorders with predisposition to acute myeloid leukemia, FDP/AML) or ANKRD26 (ANKRD26-related thrombocytopenia, ANKRD6-RT) have mild thrombocytopenia and negligible bleeding tendency, but often acquire myelodysplastic syndromes and acute leukemias. This has been reported in 40% of subjects with FDP/AML5 and in 8% of those with ANKRD6-RT.6,7
Acquired bone marrow aplasia is a further possible consequence of mutations resulting in ITs. Subjects with MPL mutations (CAMT) have a very low platelet count from birth that, in approximately 25% of cases, induces intracranial hemorrhage. Nonetheless, the major problem for subjects with CAMT is not bleeding, but is instead the bone marrow aplasia that invariably develops before adulthood and leads to death if left untreated.8 Evolution into bone marrow aplasia has been reported also in patients with a poorly defined form of IT associated with congenital skeleton defects.9
It is worth remembering that also WAS mutations result in both congenital defects and the risk of acquiring additional disorders. More recently, it has been shown that different WAS mutations result in very different phenotypes. In general, mutations that abolish WAS protein expression cause a very low platelet count associated with severe immunodeficiency and a high risk of autoimmunity and malignancies (WAS). At variance with this, mutations that only reduce WAS protein levels cause a much less critical clinical picture [X-linked thrombocytopenia (XLT)].10
Finally, the time course of platelet count can be predicted by the genotype. Platelet count tends to remain stable in most ITs, but in a few cases, thrombocytopenia improves or worsens. Subjects with mutations in the gene RBM8A [thrombocytopenia with absent radii (TAR)] present at birth with skeletal defects combined with a severe thrombocytopenia that hinders surgical correction of bone abnormalities.11 However, platelet count usually improves or even normalizes with aging.12 In contrast, in subjects with NBEAL2 mutations (GPS), the degree of thrombocytopenia worsens with aging, probably because, over time, they develop myelofibrosis and splenomegaly.13
Molecular characterization for guiding patient management
It was previously thought that patients with ITs need medical attention after hemostatic challenge or bleeding events. But the finding that some of them are at risk of developing additional disorders has changed this and indicated the need for personalized follow-up regimens. Subjects with RUNX1 and ANKRD26 mutations require close hematologic surveillance so the emergence of hematologic malignancies can be identified as soon as possible, and, if appropriate, a search can begin for a compatible bone marrow donor. Close monitoring of renal function is recommended for subjects with mutations of MYH9 that predispose to glomerulonephritis because angiotensin receptor blockers and/or angiotensin-converting enzyme inhibitors may reduce or even abolish proteinuria,14 and it is, therefore, expected that their early administration counteracts the progression to kidney failure. Moreover, ototoxic drugs and agents that may damage renal function have to be used sparingly in patients with MYH9-RD at risk of non-hematologic complications.3 Finally, cochlear implant should be offered early to patients with MYH9-RD who develop deafness because the timely application of this device greatly improves their hearing capacity.15
As far as management of bleeding tendency is concerned, the important news is that thrombopoietin mimetic eltrombopag increases platelet count in most patients with MYH9 mutations.16 As a matter of fact, this drug is beginning to be used in place of platelet transfusions in order to prepare subjects with MYH9-RD for elective surgery.17 Clinical trials are required to verify whether TPO mimetics also work in other ITs and whether they can be used as a long-term treatment for subjects with severe bleeding episodes.
Because it is still not known whether TPO mimetics are effective in conditions other than MYH9-RD, advances in hematopoietic stem cell transplantation (HSCT) have made it an attractive possibility for patients with life-threatening bleeding episodes. HSCT is currently being used as first-line treatment for subjects with WAS and CAMT, also because it not only benefits thrombocytopenia, but also cures immunodeficiency in WAS10 and prevents bone marrow aplasia in CAMT.8 HSCT has been used successfully also in deeply thrombocytopenic patients with BSS who developed refractoriness to platelet transfusions,18 and in some patients with IT and skeletal defects associated or not with HOXA11 mutations.9
Gene therapy represents a potential alternative to HSTC in patients with known mutations whenever HSTC is not feasible, and encouraging results have been obtained in a few subjects with WAS mutations.9
Finally, progress in the knowledge of IT etiology provides a rational basis for the use of splenectomy, an intervention that in the past has sometimes been used empirically in the hope of increasing platelet count. Indeed, it has been shown that splenectomy reduces thrombocytopenia (but increases the risk of infections) in patients with WAS mutations,10 while it does not provide a durable benefit in all other known conditions.
What remains to be done
There are two main problems that remain to be solved before the potential of personalized medicine in ITs can be fully exploited. The first obstacle is that nearly half the patients have disorders that have not yet been identified2 and, therefore, we cannot define their prognosis or personalize their management. This flaw also hampers effective genetic counseling, as well as pre-implantation selection and prenatal diagnosis. Application of next generation sequencing techniques to large series of patients with yet unknown disorders is potentially able to fill this gap in the short term, and such an attempt should, therefore, be encouraged.
The second obstacle is the difficulty in diagnosing known ITs. According to the diagnostic algorithm proposed several years ago by the Italian Platelet Study Group,19 specific sequences of tests are used to raise a diagnostic suspicion, and then sequencing of candidate genes is required to confirm diagnosis. However, preliminary tests are expensive and are only available in a few centers, and, in fact, diagnosis is often missed.20 Targeted sequencing of all known IT genes as an initial diagnostic approach could be advantageous with respect to the traditional multi-step methodology both in terms of cost and of efficacy. Sequencing technologies are becoming increasingly effective and affordable, and such a test for ITs is expected to cost less than 1000 euros, a sum that compares favorably with that required for the complex and time-consuming methodology currently in use.
Achieving these goals would allow us to rationalize the clinical approach towards all cases of IT, with benefits to both patients and the health care system.
Footnotes
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Savona A, Kunishima S, De Rocco D, et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat. 2014;35(9):1033–1045. [DOI] [PubMed] [Google Scholar]
- 2.Balduini CL, Pecci A, Noris P. Inherited thrombocytopenias: the evolving spectrum. Hamostaseologie. 2012;32(4):259–270. [DOI] [PubMed] [Google Scholar]
- 3.Balduini CL, Pecci A, Savoia A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br J Haematol. 2011;154(2):161–174. [DOI] [PubMed] [Google Scholar]
- 4.Pecci A, Klersy C, Gresele P, et al. MYH9-related disease: a novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum Mutat. 2014;35(2):236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liew E, Owen C. Familial myelodysplastic syndromes: a review of the literature. Haematologica. 2011;96(10):1536–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noris P, Perrotta S, Seri M, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117(24):6673–6680. [DOI] [PubMed] [Google Scholar]
- 7.Noris P, Favier R, Alessi MC, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood. 2013;122(11):1987–1989. [DOI] [PubMed] [Google Scholar]
- 8.Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Semin Thromb Hemost. 2011;37(6):673–681. [DOI] [PubMed] [Google Scholar]
- 9.Castillo-Caro P, Dhanraj S, Haut P, Robertson K, Dror Y, Sharathkumar AA. Proximal radio-ulnar synostosis with bone marrow failure syndrome in an infant without a HOXA11 mutation. J Pediatr Hematol Oncol. 2010;32(6):479–485. [DOI] [PubMed] [Google Scholar]
- 10.Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285:26–43. [DOI] [PubMed] [Google Scholar]
- 11.Albers CA, Paul DS, Schulze H, et al C. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44(4):435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geddis AE. Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Hematol Oncol Clin North Am. 2009;23(2):321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunay-Aygun M, Zivony-Elboum Y, Gumruk F, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood. 2010;116(23):4990–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pecci A, Granata A, Fiore CE, Balduini CL. Renin-angiotensin system blockade is effective in reducing proteinuria of patients with progressive nephropathy caused by MYH9 mutations (Fechtner-Epstein syndrome). Nephrol Dial Transplant. 2008;23(8):2690–2692. [DOI] [PubMed] [Google Scholar]
- 15.Pecci A, Verver EJ, Schlegel N, et al. Cochlear implantation is safe and effective in patients with MYH9-related disease. Orphanet J Rare Dis. 2014;9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pecci A, Gresele P, Klersy C, et al. Eltrombopag for the treatment of the inherited thrombocytopenia deriving from MYH9 mutations. Blood. 2010;116(26):5832–5837. [DOI] [PubMed] [Google Scholar]
- 17.Balduini CL, Pecci A, Noris P. Diagnosis and management of inherited thrombocytopenias. Semin Thromb Hemost. 2013;39(2):161–171. [DOI] [PubMed] [Google Scholar]
- 18.Pecci A, Barozzi S, d’Amico S, Balduini CL. Short-term eltrombopag for surgical preparation of a patient with inherited thrombocytopenia deriving from MYH9 mutation. Thromb Haemost. 2012;107(6):1188–1189. [DOI] [PubMed] [Google Scholar]
- 19.Noris P, Pecci A, Di Bari F, et al. Application of a diagnostic algorithm for inherited thrombocytopenias to 46 consecutive patients. Haematologica. 2004;89(10):1219–1225. [PubMed] [Google Scholar]
- 20.Noris P, Schlegel N, Klersy C, et al. Analysis of 339 pregnancies in 181 women with 13 different forms of inherited thrombocytopenia. Haematologica. 2014;99(8):1387–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]