

Abstract
Lenalidomide is an immunomodulatory agent clinically active in chronic lymphocytic leukemia patients. The specific mechanism of action is still undefined, but includes modulation of the microenvironment. In chronic lymphocytic leukemia patients, nurse-like cells differentiate from CD14+ mononuclear cells and protect chronic lymphocytic leukemia cells from apoptosis. Nurse-like cells resemble M2 macrophages with potent immunosuppressive functions. Here, we examined the effect of lenalidomide on the monocyte/macrophage population in chronic lymphocytic leukemia patients. We found that lenalidomide induces high actin polymerization on CD14+ monocytes through activation of small GTPases, RhoA, Rac1 and Rap1 that correlated with increased adhesion and impaired monocyte migration in response to CCL2, CCL3 and CXCL12. We observed that lenalidomide increases the number of nurse-like cells that lost the ability to nurture chronic lymphocytic leukemia cells, acquired properties of phagocytosis and promoted T-cell proliferation. Gene expression signature, induced by lenalidomide in nurse-like cells, indicated a reduction of pivotal pro-survival signals for chronic lymphocytic leukemia, such as CCL2, IGF1, CXCL12, HGF1, and supported a modulation towards M1 phenotype with high IL2 and low IL10, IL8 and CD163. Our data provide new insights into the mechanism of action of lenalidomide that mediates a pro-inflammatory switch of nurse-like cells affecting the protective microenvironment generated by chronic lymphocytic leukemia into tissues.
Introduction
Chronic lymphocytic leukemia (CLL) patients present a progressive immunodeficiency due to the ability of CLL cells to manipulate their microenvironment, escaping immunosurveillance and inducing immunosuppression. CLL cells evade immune detection through different mechanisms involving secretion of immunosuppressive cytokines and formation of the protective niches needed to change the function of immune effector cells and to escape drug-induced apoptosis.1 In addition, alteration of different signaling molecules involved in actin polymerization influences the communication between CLL cells and effector cells.2 CLL cells are accompanied by an expanded population of regulatory and exhausted T cells, and surrounded by a macrophage population with M2 properties and dysregulated expression of molecules involved in antigen-presentation and immune response.3
Nurse-like cells (NLCs) are round or fibroblast-shaped adherent cells differentiated from peripheral blood-derived monocytes in vitro. NLCs can also be detected in lymph nodes (LN) of CLL patients.4–6 NLCs share several features with tumor-associated macrophages: secretion of IL10, IL8, but not IL12; high surface expression of CD11b, HLA-DR, CD163, CD206; and expression of indoleamine 2,3-dioxygenase (IDO).3,5,7 Furthermore, NLCs also show deregulated expression of genes involved in immunocompetence.8 NLCs protect leukemic cells from undergoing spontaneous or drug-induced apoptosis in a contact-dependent manner.9
Lenalidomide is an immunomodulatory agent (IMID) clinically active in patients with CLL.10–12 The mechanism of action of lenalidomide includes ‘re-educating’ immune cells such as T cells, monocytes and NK cells, increasing anti-tumor immunity in CLL.13 By in vitro studies and in the TCL1 mouse model for CLL, lenalidomide was shown to reverse defects in adhesion and motility functions, as well as in immunological synapse formation between CLL and T cells, by modulating several cytoskeletal molecules.14–16 Recently, lenalidomide was also shown to interfere with the mutualistic interaction between CLL and NLCs.17
Together these findings prompted us to investigate the functional effects of lenalidomide on NLCs in CLL. We found that lenalidomide modifies CLL-circulating monocytes, inducing firm adhesion to endothelium and loss of migration through modulation of small GTPases. Lenalidomide induces a pro-inflammatory profile in NLCs improving their phagocytic activity and ability to activate T-cell proliferation. Overall, our study provides new insights into the mode of action of lenalidomide that targets microenvironmental elements interfering with the supporting and protective milieu generated by CLL cells into tissues.
Methods
A detailed description of the protocols used is available in the Online Supplementary Appendix.
Patients and samples
Written informed consent was obtained in accordance with the Declaration of Helsinki with a protocol approved by the local Institutional Review Board. CD14+ monocytes were obtained by immunomagnetic selection. To generate NLCs, PBMCs from CLL patients were cultured (107/mL) as previously described.18 Lenalidomide was used at the clinically relevant doses of 0.5 μM, 1 μM and 10 μM as in previous studies.2,16,17,19
Actin polymerization
Actin polymerization was inspected in monocytes and in NLCs from CLL patients with F-actin Visualization Biochem Kit (Cytoskeleton, Denver, CO, USA).
Adhesion assays
CD14+ monocytes from 10 CLL patients were added onto the confluent HUVEC (Life Technologies) layer and allowed to adhere for 2 h at 37°C. After incubation, monocytes were treated with lenalidomide 0.5 μM for 20 min or vehicle. In some experiments, adherent monocytes (n=6) were treated with Rac1 inhibitor (50μM) or Rap1 inhibitor (10 μM) for 30 min. Then, monocytes firmly adherent to HUVEC were counted by staining with CD14 APC antibody (Miltenyi Biotec), as previously described.20 Furthermore, adhesion of CLL cells on NLCs was also evaluated. We generated NLCs from PBMCs (n=7), treated or not with lenalidomide 0.5 μM or 1 μM or vehicle. Firmly adherent CLL cells were collected and counted by CD19 staining.
Chemotaxis assays
Migration assays on CLL monocytes (n=5) treated or not with 0.5 μM lenalidomide were performed using 5-mm pore PET inserts (Millipore, Billerica, MA, USA), as previously described.21
Gene expression analysis
Gene expression profiling (GEP) was performed by hybridizing RNA of NLCs treated with lenalidomide 0.5 μM or vehicle (control) for ten days (n=4) on 4X44K Whole Human Genome Microarray (Agilent Technologies, Palo Alto, CA, USA) as previously described.22 Gene transcripts were also amplified using LightCycler 480 SYBR Green I Master Mix (Roche). Primers are listed in Online Supplementary Table S1.
Immunoblotting
CD14+ monocytes were cultured in the presence of lenalidomide or vehicle for 4 h at 37°C. Proteins (100 μg/lane) were electrophoresed and membranes were immunoblotted with primary antibodies listed in Online Supplementary Table S2.
Flow cytometry
Nurse-like cells were stained with the following antibodies and corresponding isotype controls: APC-conjugated CD14 (Miltenyi Biotec), CD163, CD11b (BD Biosciences Pharmingen, San Jose, CA, USA) and PE-conjugated CD11b CBRM 1/5 (eBioscience).
Phagocytosis assays
Phagocytosis was inspected using CytoSelect™ 96-Well Phagocytosis assay (Cell Biolabs, San Diego, CA, USA). In a separate set of experiments, NLCs were generated on coverslips with lenalidomide or vehicle and FITC-dextran particle engulfment was quantified as previously described.23
Cell activation and proliferation
Nurse-like cell activation was monitored using MTT assay (Trevigen, Gaithersburg, MD, USA). NLC proliferation was evaluated using CFSE dilution assay, Ki-67 staining and cell cycle analy sis. Allogeneic mixed lineage reactions were performed to measure T-cell proliferation.
Cytokine secretion assay
To determine secretion of IL-2, NLCs were analyzed using the Cytokine Secretion Assay (CSA) for IL-2 (CSA Detection Kit; Miltenyi Biotec).
Statistical analyses
Data were analyzed using SPSS v.20.0 (SPSS, Chicago, IL, USA). P values were calculated by Student t-test (*P<0.05, **P<0.01).
Results
Lenalidomide induces actin polymerization in monocytes derived from CLL patients
Immunomodulatory drugs (IMiDs) such as lenalidomide were shown to activate Rho family GTPases and induce cytoskeleton reorganization.24 To evaluate whether lenalidomide could affect actin cytoskeleton in monocytes from CLL patients, we treated purified CD14+ monocytes (n=9) with lenalidomide for 20 min at doses ranging from 0.5 μM to 10 μM and measured the F-actin content. Lenalidomide stimulated actin polymerization at the clinically relevant doses of 0.5 μM and 1 μM (Figure 1A). In particular, F-actin formation increased to 174% (±27%), 300% (±62%), and 350% (±131%) upon stimulation with 0.5 μM, 1 μM and 10 μM lenalidomide, respectively, compared to unstimulated control (100%) (n=9; P<0.05 in all cases) (Figure 1B).
Figure 1.
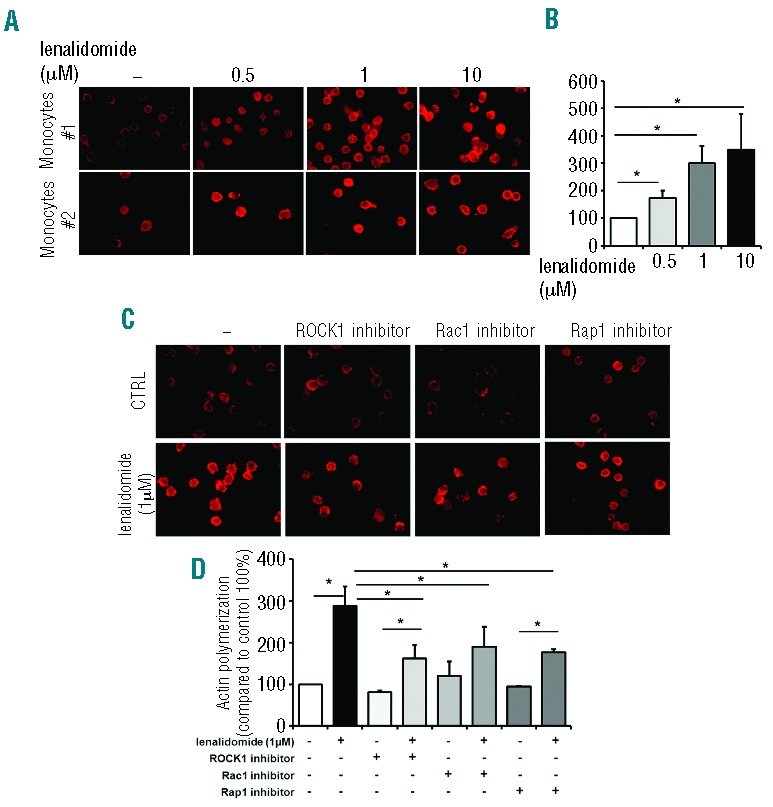
Lenalidomide induces actin polymerization in circulating monocytes from CLL patients throughout Rho and Ras family GTP-binding proteins. (A and B) Monocytes purified from peripheral blood mononuclear cells (PBMC) of 9 CLL patients were allowed to adhere and then treated with lenalidomide at the indicated concentrations or vehicle (DMSO) for 20 min. F-actin content was then inspected by rhodamine-phalloidin staining. (A) Two representative samples. (B) Quantification of cell staining, as mean value obtained from 5 different fields at 400X magnification normalized on control (100%, DMSO-treated monocytes). Lenalidomide induces actin polymerization in monocytes from CLL patients. Data represent 8 independent experiments. Columns and error bars represent mean±SEM (Student t-test, *P<0.05). (C and D) Monocytes from 8 CLL patients were treated with ROCK, Rap1 and Rac1 inhibitors for 30 min before stimulation with lenalidomide 1 μM or DMSO for an additional 20 min and measurement of the F-actin content. Quantification of staining, as mean fluorescence intensity value obtained from individual cells from 5 different fields at 400X magnification, is normalized on control (100%, inhibitors-untreated DMSO-treated monocytes). Lenalidomide mediates actin polymerization through the activation of RhoA, Rac1 and Rap1 GTPases. (C) One representative case. (D) Histograms represent mean±SEM of 7 independent experiments (Student t-test, *P<0.05).
In addition, we asked ourselves whether lenalidomide induced actin polymerization by activating specific GTPases. To investigate this, in a separate set of experiments, lenalidomide-induced actin polymerization in CLL monocytes from 8 patients was measured after blocking ROCK1, Rap1 or Rac1 kinases by using specific inhibitors. These agents invariably reduced lenalidomide-induced actin polymerization in CLL monocytes (Figure 1C). Specifically, addition of Y27632 (ROCK1 inhibitor), GGTI-268 (Rap1 inhibitor), and Rac1-specific inhibitor reduced the effect of lenalidomide from 288% (±46%) to 162% (±31%), 190% (±48%) and 177% (±7%), respectively (P<0.05 in all cases) (Figure 1D). However, the specific inhibition of each GTPase did not completely neutralize the stimulation of F-actin formation mediated by lenalidomide, suggesting that other molecular mediators are involved in actin polymerization (Figure 1D). Thus, lenalidomide promotes the activation of Rho-family small GTPases, RhoA and Rac1 and Ras-family small GTPase Rap1, stimulating actin polymerization in circulating monocytes from CLL patients.
Lenalidomide improves adhesion and impairs migration of CLL monocytes
The actin cytoskeleton supports a multitude of essential functions in adherent and migrating cells from participating in the formation of protrusions to the generation of tensile forces and cell motility. Thus, we speculated that the cytoskeleton reorganization, induced by lenalidomide treatment, might modify adhesive and migratory properties of CLL monocytes. First, we inspected the adhesive potential of monocytes by culturing CD14+ cells on HUVEC monolayers. Lenalidomide treatment strongly stimulated monocyte adhesion to the HUVEC layer, increasing to 238% (±37%) the mean relative adhesion compared to the untreated control (100%) (n=10; P<0.05) (Figure 2A and B). Accordingly, treatment with lenalidomide improved the amount of ILK (integrin linked kinase) and phosphorylated Akt in CD14+ CLL monocytes (Figure 2C). In agreement with a role for small GTPases, adhesion induced by lenalidomide (204%±46%) was significantly reduced in the presence of Rac1 (123%±34%) or Rap1 (67%±7%) inhibitors (n=6; P<0.05 in all cases) (Figure 2D). These data demonstrate that lenalidomide promotes monocyte adhesion by Rap1 and Rac1 signaling pathways. IMiDs were shown to increase migration of normal monocytes and to repair motility dysfunction of T cells from CLL patients.16,24 Conversely, IMiDs impaired migration capability of CLL cells and endothelial cells.17,25 To determine the effects exerted by lenalidomide on CLL monocytes, we performed chemotaxis assays on monocytes (n=5) treated with 0.5 μM lenalidomide or vehicle using 10 ng/mL CCL2, 10 ng/mL CCL3, and 200 ng/mL CXCL12 as chemoattractants. Stimulation of monocyte migration mediated by CCL2 (217%±37%), CCL3 (181%±10%), and CXCL12 (166%±17%) was reduced by exposure with lenalidomide to 163% (±26%), 123% (±12%) and 143% (±22%), respectively (n=5; P<0.05 in all cases) (Figure 3A). We explored the mechanisms involved in lenalidomide-induced migration impairment. We excluded the possibility that lenalidomide affects viability (data not shown) or modified the expression levels of chemokine receptors during the migration assay (n=6) (Figure 3B). Lenalidomide was reported to modulate the expression of the Rho GTPase RhoH, and the actin binding protein CORO1B in T lymphocytes and CLL cells.17,26,27
Figure 2.
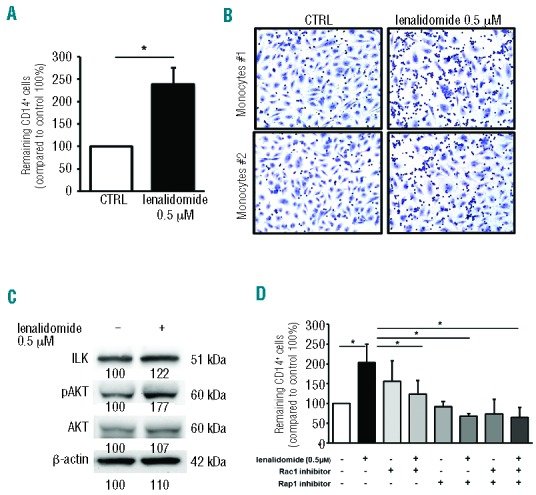
Lenalidomide promotes CLL monocytes adhesion to endothelium. (A) CD14+ monocytes from CLL patients (n=10) were allowed to adhere to HUVEC layer for 2 h and then treated with lenalidomide 0.5 μM or vehicle (DMSO). Histograms represent the mean relative adhesion of lenalidomidetreated monocytes compared to the untreated control. Data represent 8 independent experiments. Columns and error bars represent mean±SEM (Student t-test, *P<0.05). (B) Representative May-Grunwald Giemsa staining shows CLL monocytes adhesion to HUVEC in presence or absence of lenalidomide 0.5 μM. (C) Western blot analysis of CLL CD14+ monocytes was performed with ILK, anti-phospho Akt, total Akt and β-actin antibodies after 4 h of treatment with lenalidomide. The immunoblots show ILK and Akt activation in one representative case. Densitometric quantification of bands normalized to the untreated control is shown below the immunoblots. (D) Monocytes from 6 CLL patients were treated with Rac1 inhibitor or Rap1 inhibitor or both for 30 min before stimulation with lenalidomide 0.5 μM for an additional 20 min. Histograms represent the mean relative adhesion to HUVEC. (Student t-test, *P<0.05).
Figure 3.
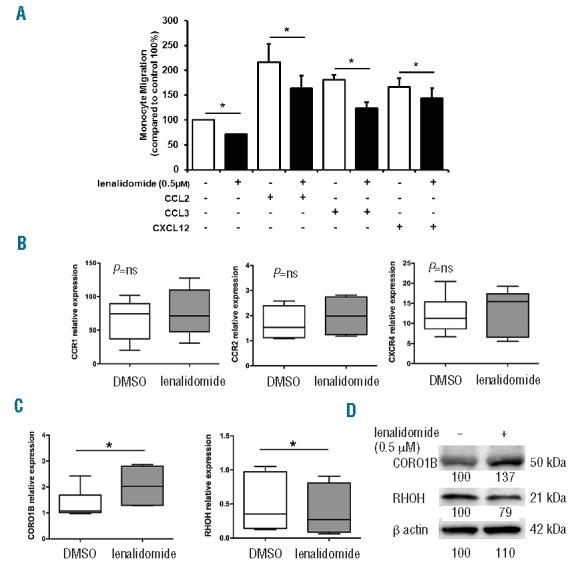
Lenalidomide reduces migration of monocytes from CLL patients. (A) Monocytes purified from 5 CLL patients were loaded in the upper chamber of 5-mm pore PET inserts with the addition of 0.5 μM lenalidomide or vehicle (DMSO). CCL2 (10 ng/mL), CCL3 (10 ng/mL), or CXCL12 (200 ng/mL) were added in the bottom chamber as chemoattractants. Monocytes were allowed to migrate for 4 h and migrated cells were quantified by fluorescence plate reader. Data are normalized on control (100%, DMSO-treated monocytes). Histograms represent mean±SEM of 5 independent experiments (Student t-test, *P<0.05). (B and C) Monocytes purified from 6 CLL patients were cultured in 24-well plates with 0.5 μM lenalidomide or vehicle (DMSO) for 4 h. (B) Gene expression of CCR1, CCR2, CXCR4, and (C) gene expression of RhoH and CORO1B, were measured by quantitative reverse-transcription PCR. Results of 2 independent experiments with 6 patient samples are presented as box plots; whiskers show min and max values. Lenalidomide does not modify CCR1, CCR2 and CXCR4 expression on CLL monocytes. Conversely, CORO1B is significantly up-regulated and RhoH down-regulated by lenalidomide treatment (Student t-test, *P<0.05). (D) Immunoblots show CORO1B and RHOH quantities after treatment with lenalidomide 0.5 μM in a representative sample of CLL monocytes. Densitometric quantification of bands normalized to the untreated control is shown below the immunoblots.
We monitored the expression levels of RhoH and CORO1B mRNA in CLL monocytes (n=6) treated for 4 h with lenalidomide 0.5 μM. Lenalidomide increased the expression levels of CORO1B, whereas it down-modulated RhoH in monocytes (P<0.05) (Figure 3C and D). These modifications may account for the inhibition of monocyte migration induced by lenalidomide.
Lenalidomide induces activation and proliferation of NLCs
Peripheral blood-monocytes from CLL patients spontaneously differentiate in vitro into large adherent cells, the so-called NLCs that deliver survival signals to leukemic cells.18,28 We confirmed that lenalidomide reduced CLL survival in contact with NLCs from 54.2% to 44.5% after ten days (n=5; P<0.05) (Online Supplementary Figure S1), as reported by Schulz et al.17
The next part of the study was dedicated to the analysis of the effect of lenalidomide treatment on NLCs. First, we cultured PBMCs from 9 CLL patients for ten days with 0.5 μM or 1 μM lenalidomide or vehicle. The number of NLCs was significantly increased upon treatment with lenalidomide to 268% (±37%) and 309% (±73%) compared to untreated control (100%) (n=9; P<0.01 and P<0.05, respectively) (Figure 4A and B). We observed an increase in NLC activation to 161% (±18%) as measured by solubilization of intracellular purple formazan, after treatment with lenalidomide for five days, compared to untreated control (100%) (n=6; P<0.05) (Figure 4C). Accordingly, lenalidomide stimulated NLC proliferation after five days of culture from 44% (±3%) to 55% (±3%) of dividing cells (n=5; P<0.01) (Figure 4D). NLC proliferation was also confirmed by Ki-67 staining and cell cycle analysis (Online Supplementary Figure S2). Lenalidomide increased the percentage of Ki-67+NLCs from 7.01%±2.09% to 9.42%±2.12% (n=6; P<0.01). It has to be considered that NLCs protect CLL from apoptosis in a contact-dependent fashion. Surprisingly, we found that CLL cells, despite a lower viability, were more adherent to NLCs treated with lenalidomide 0.5 μM and 1 μM, with a mean increase of 227% (±91%) and 212% (±54%), respectively (n=6; P<0.05) (Figure 4E).
Figure 4.
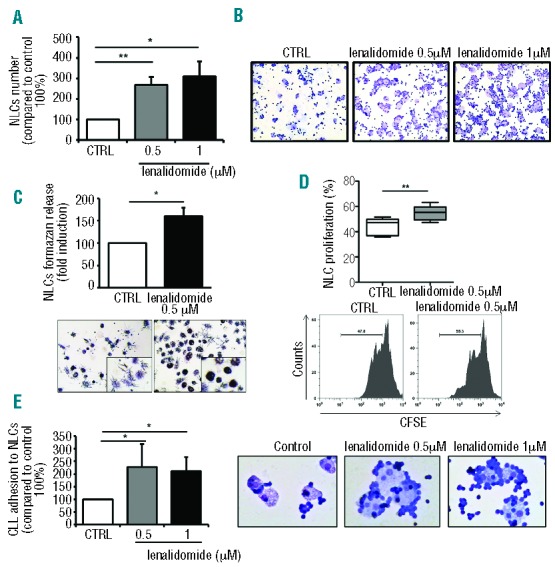
Lenalidomide modifies the number and functional properties of nurse-like cells. NLCs were generated from PBMCs isolated from CLL patients in presence or absence of lenalidomide. (A) Number of NLCs were determined by CD14 staining and analyzed by flow cytometry for each condition (n=9, Student t-test, *P<0.05, **P<0.01). (B) May-Grunwald Giemsa staining documented the high number of NLCs induced by treatment with lenalidomide 0.5 μM and 1 μM compared to untreated control in a representative case. (C) Histograms represent the formazan release by metabolically active NLCs treated with lenalidomide 0.5 μM compared to untreated control. Data are presented as mean±SEM of 6 independent experiments performed in triplicates (Student t-test, *P<0.05). (Bottom panels) Representative phase-contrast micrographs demonstrating increased NLCs activation (as insoluble formazan purple precipitate) after treatment with lenalidomide. (D) CFSE-labeled PBMCs collected from 5 CLL patients were cultured for five days either in presence or absence of lenalidomide 0.5 μM. Box plot represents the proliferative rate of NLCs in each condition, stained with CD14 antibody and analyzed by flow cytometry (Student t-test, **P<0.01). (Bottom) Histograms represent one representative case of NLCs treated or not with lenalidomide. Values indicate the percentage of proliferating cells with low expression of CFSE. (E) Histograms represent the mean relative adhesion (±SEM) of CLL cells to NLCs in the presence or absence of lenalidomide compared to control (n=6; Student t-test, *P<0.05). (Right) May-Grunwald Giemsa stainings demonstrate the increased adhesion of CLL cells to NLCs after treatment with lenalidomide.
Taken together, these findings indicate that lenalidomide supports the formation of NLCs from PBMCs of CLL patients by promoting cell activation and proliferation. However, NLCs generated in the presence of lenalidomide adhere closely to CLL cells but fail to efficiently sustain leukemic cell viability, prompting us to explore the functional and molecular features of lenalidomide-treated NLCs.
Lenalidomide-treated NLCs improve the phagocytic activity and activate T-cell proliferation
Published evidence suggests that NLCs are closely related to tumor-associated macrophages (TAM), showing M2-skewed properties.3,5,7 We then asked ourselves whether lenalidomide could interfere with the immunosuppressive profile of NLCs. In agreement with observations in monocytes, we found a strong stimulation of F-actin content in lenalidomide-treated NLCs (Figure 5A). In line with our hypothesis, NLCs (n=6) significantly increased their phagocytic activity to 141% (±16%) and 155% (±9%), respectively, after lenalidomide exposure (0.5 μM and 1μM for 4 h; n=6; P<0.05) (Figure 5B). Consistently, the same treatment increased FITC-dextran uptake by NLCs (n=8; P<0.01) (Figure 5C). Treatment with the Rap1 inhibitor before stimulation with lenalidomide was followed by a strong impairment in phagocytosis (P<0.05) (Figure 5D), suggesting that small GTPases may be a common target of lenalidomide. We then evaluated T-cell proliferation in an allogeneic mixed lineage reaction. NLCs (n=6), treated or not with lenalidomide 0.5 μM overnight, were cultured for 7 days with CFSE-labeled T lymphocytes from healthy donors (HD) in fresh medium. T-cell proliferation increased both in the presence of NLCs (P<0.05) and with aCD3/CD2/CD28-coated beads (positive control) (P<0.01) compared to unstimulated conditions. Moreover, lenalidomide-treated NLCs strongly improved T-cell proliferation from 20% (±6%) to 35% (±11%) (n=6; P<0.05) (Figure 5E and F).
Figure 5.
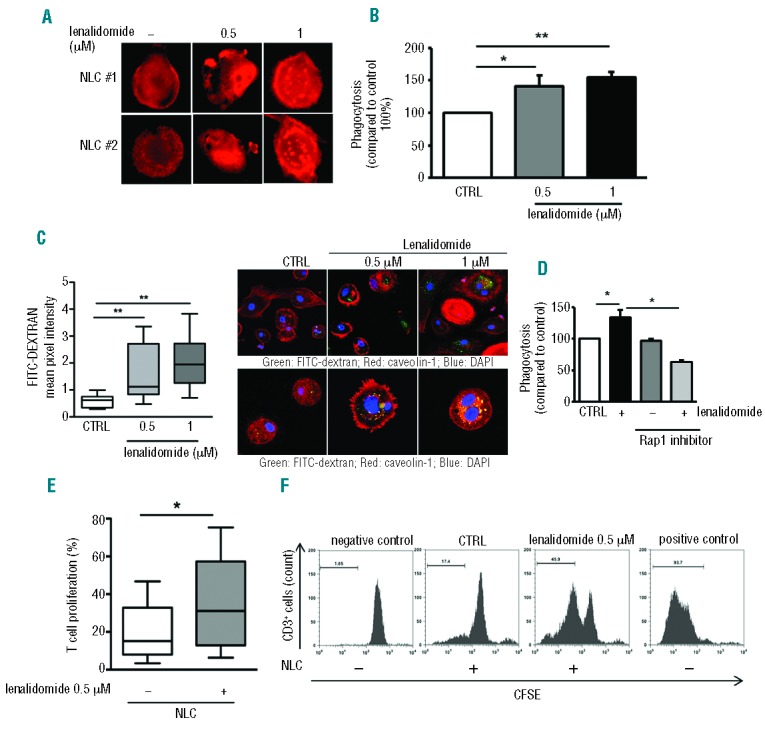
Lenalidomide enhances phagocytosis and the ability to stimulate T-cell proliferation in NLCs. (A) Actin polymerization was strongly increased in NLCs after treatment with lenalidomide in one representative CLL patient. F-actin content was inspected by rhodamine-phalloidin staining. (B) NLCs obtained from 6 CLL patients were treated or not with lenalidomide 0.5 μM and 1 μM for 4 h, followed by phagocytosis assay. Data represent mean±SEM of 6 independent experiments (Student t-test, *P<0.05, **P<0.01). (C) Box plots summarize FITC-Dextran uptake by NLCs treated or not with lenalidomide relative to 8 independent experiments; whiskers show min and max values (Student t-test, **P<0.01). (Right) First row shows confocal staining of NLCs with FITC-dextran, phalloidin and DAPI; second row shows FITC-dextran, caveolin-1 and DAPI staining. (D) NLCs generated from 3 CLL patients were treated or not with Rap1 inhibitor (GGTI) for 30 min and then stimulated with lenalidomide 1 μM for 4 h. Rap1 inhibitor affects lenalidomide-induced phagocytosis in NLCs. Histogram shows mean±SEM of 3 independent experiments (Student t-test, *P<0.05). (E and F) T cells from healthy donors were cultured on NLCs treated or not with lenalidomide 0.5 μM overnight. CD3+ cell proliferation was measured after 7 days by CFSE staining. (E) Box plot shows the improvement in T-cell proliferation (as %CFSE-low CD3+ cells) on lenalidomide-treated NLCs (Student t-test, n=6, P<0.05). (F) Representative histograms show CFSE proliferation profiles of healthy T cells cultured onto NLC layer pre-treated or not treated with lenalidomide overnight. T cells cultured alone for seven days or stimulated with αCD3/CD2/CD28-coated beads are shown as negative and positive controls, respectively.
Lenalidomide modifies gene expression profile and immunophenotype of NLCs
Gene expression profiles of NLCs (n=4), generated with lenalidomide or vehicle-control (0.5 μM for 10 days), were analyzed by microarrays (control sample vs. lenalidomidetreated sample). Supervised analysis identified 584 genes that were differentially expressed upon lenalidomide treatment: 352 up-regulated and 232 down-regulated (P<0.05). Classifying the modulated entities into biological function categories by Gene Ontology, we found that lenalidomide-induced signature was enriched in genes involved in immune response, activation/proliferation of T cells, complement activation, antigen processing and presentation as well as regulation of cellular movement, cytokine and chemokine activity (Figure 6A). In particular, modulation of several chemokines such as CXCL11, CXCL9, CCL19, XCL1 and XCL2 (up-regulated) or CCL2 and CXCL12 (down-regulated) was apparent (Figure 6B). Furthermore, NLCs generated in the presence of lenalidomide, showed upregulation of IL12B (FC=1.9), IL2 (FC=1.8), and TNFSF4 (FC=2.8), and downregulation of IL17D (FC=−2.4), ANGPT2 (FC=−2.3), IGF1 (FC=−5.4), and HGF (FC=−2.1). Among the up-regulated genes in NLCs generated with lenalidomide, we also detected IDO1 (FC=3.6) and the lysosomal-associated protein 3 (LAMP3, FC=1.5), as well as SPON2, opsonin for macrophage phagocytosis of bacteria (FC=8.8), genes coding for CD1 molecules that mediate the presentation of lipid and glycolipid antigens, and CD209 involved in pathogen-recognition and endocytosis (FC=1.7). Moreover, lenalidomide induced the downregulation of CD163 (FC=−2.0), EDNRB (FC=−2.2) and TLR5 (FC=−1.6). The upregulation of IL2 and IDO1 in NLCs (n=8) generated with/without lenalidomide was confirmed by real-time PCR (Figure 6C). Accordingly, lenalidomide increased the percentage of NLCs secreting IL-2 protein (Figure 6D). The modulation of gene expression of IL10 and IL8 (showing a borderline significance in the microarray data) was also evaluated by real-time PCR, and showed a downregulation of both genes (71% for IL10, P<0.05; 60% for IL8, P<0.01) (Figure 6C). Lastly, we quantified the surface expression levels of CD11b, the activated epitope MAC-1 and CD163 on CD14+ NLCs. Lenalidomide-treated NLCs expressed significantly higher levels of CD11b and MAC-1 (P<0.05 for CD11b; P<0.05 for MAC-1) (Figure 6E) and lower levels of CD163 (P<0.05) (Figure 6E).
Figure 6.
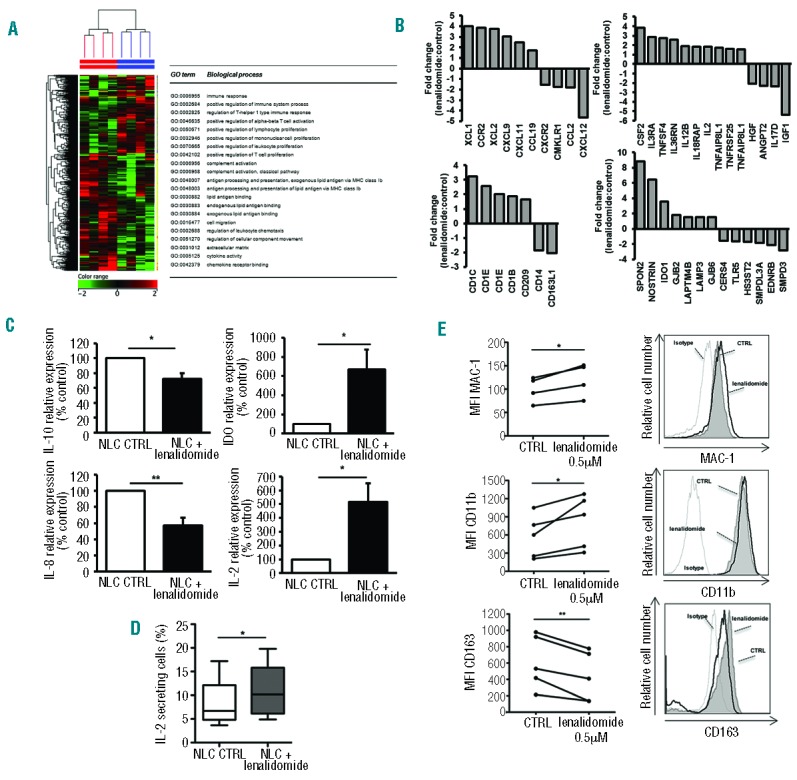
Lenalidomide induces pro-inflammatory modification in NLCs. (A) Heat map shows differentially expressed genes between NLCs treated and not treated with lenalidomide for ten days. (B) Histograms represent fold change of normalized intensity values of genes expressed in NLCs treated with lenalidomide 0.5 μM to untreated control. (C) Histograms represent the mRNA relative quantity of IL2, IL10, IDO1, IL8 evaluated by RT-PCR in NLCs obtained from 7 CLL patients (Student t-test, *P<0.05, **P<0.01). (D) Box plots show the percentage of IL-2 secreting cells in NLCs populations treated or not with lenalidomide for 10 days relative to 4 independent experiments; whiskers shown min and max values (Student t-test, *P<0.05). (E) Histograms show the fluorescence intensity of NLCs after treatment with lenalidomide stained with anti-MAC1, anti-CD11b and anti-CD163 Abs in one representative sample. (Left). Values of untreated and treated samples (n=5) are connected by lines (Student t-test, *P<0.05, **P<0.01).
Discussion
In this study, we demonstrate that lenalidomide alters the migratory and adhesive properties of monocyte/macrophage populations in CLL. Moreover, lenalidomide counteracts the ability of leukemic cells to generate a macrophage population, defined as nurse-like cells, characterized by an immunosuppressive, M2-skewed and nursing profile. Instead, lenalidomide promotes the expansion of a macrophage population with M1 phenotype, characterized by enhanced phagocytic activities and support to T-cell proliferation, with less ability to nurture leukemic cells.
We found that lenalidomide strongly induces cytoskeleton reorganization through activation of small GTPases in monocytes isolated from CLL patients. Consequently, this induction of actin polymerization is reduced, even if not completely neutralized, by using specific inhibitors for Rap1, Rac1 and RhoA, suggesting that lenalidomide exerts its mechanism of action by orchestrating the concomitant activation of several GTPases. Actin cytoskeleton reorganization is essential during immune response, regulating cell motility, migration, extravasation, antigen recognition and phagocytosis.29 In particular, Rac1 mediates the formation of lamellipodia at the leading edge and activation of integrin allowing a stable adhesion.30,31 Activation of Rap1 induces a redistribution of integrin from uropod to the leading edge necessary for immunological synapse formation, macrophage phagocytosis and migration.32,33 We demonstrate that lenalidomide stabilizes firm adhesion of monocytes to endothelium through stimulation of Rac1 and Rap1, also activating both PI3K and ILK. Stimulation of PI3K, induced by Rap1, activates Rac1, cell adhesion and pseudopod formation.34 In addition, ILK is a regulator of adhesion, cell spreading, migration through integrin activation modulating intracellular signaling pathways, and recruiting molecules involved in actin polymerization.29,35,36 Similarly, lenalidomide was reported to target Rho GTPase signaling and to promote Rap1 trafficking to the membrane, restoring the adhesion and motility function of T cells from CLL patients.16
Nevertheless, we observed that lenalidomide reduces the migratory capacity of monocytes derived from CLL patients. Since lenalidomide does not affect the viability of monocytes and the expression of chemokine receptors, its inhibitory effect on migration is most likely related to intracellular modification of cytoskeletal molecules. One possible explanation may account for the simultaneous activation of GTPases by lenalidomide. Constitutively active Rac1 inhibits growth-factor-induced migration, because lamellopodia extend all around the cells blocking polarization. Similarly, constitutively activated RhoA maintains cells unpolarized, thus abolishing the chemotactic response,37 and Rap1 suppresses cell migration.38 Other modifications of cytoskeletal molecules induced by lenalidomide in CLL-monocytes are the upregulation of the coronin CORO1B and the downregulation of RhoH GTPase. Coronins are actin-binding proteins that regulate lamellopodia protrusion, whole-cell motility and chemotaxis. CORO1B inhibits actin filament nucleation and its downmodulation is required for smooth muscle cell migration.39 Moreover, it is well known that RhoH is strictly required for cell migration in response to CXCL12. The diminished level of RhoH in monocytes treated with lenalidomide could impair the cellular distribution of phosphorylated focal adhesion kinase that fails to effectively co-ordinate the activation of the Rho GTPases RhoA and Rac, leading to defective migration.26 The capability of lenalidomide to restrain monocyte migration is in line with studies demonstrating that this drug also interferes with CLL cell chemotaxis, suggesting that CORO1B upregulation and RhoH downregulation may be involved in migration impairment induced by lenalidomide in CLL cells and monocytes.17,26 Conversely, high levels of RhoH in the presence of chemokine signals impair T-cell chemotaxis, explaining the opposite promoting effect of lenalidomide on T-cell migration. In treated CLL patients, lenalidomide would be expected to restrain monocyte migration towards chemokine gradients generated by CLL-infiltrated cells.
In CLL, in vitro, a subset of CD14+ mononuclear cells from CLL patients differentiates into large, round adherent cells called NLCs.18 Lenalidomide treatment resulted in significantly increased numbers of NLCs. We correlated this increased development of NLCs to a strong stimulation in their activation and proliferation status induced by lenalidomide. It has to be considered that the percentage of CD14+Ki67+ NLCs was approximately 7% after five days of culture without lenalidomide. Conversely, we detected less than 1% of CD14+Ki67+ cells in culture from healthy donors (data not shown), in accordance with data reported in literature;40 this means that the presence of CLL cells is able to induce the proliferation of CD14+ cells. Lenalidomide significantly improved the proliferation rate of NLCs. Despite the high number of NLCs induced by lenalidomide, NLCs maintain the ability to attract and to establish physical contact with CLL cells, but lose the ability to nurture the CLL leukemic clone. Accordingly, we demonstrated that lenalidomide induces the downregulation of genes involved in pivotal pro-survival signals for CLL, such as CCL2,41,42 CXCL1218 and IGF1.43
Previous studies demonstrated that NLCs are characterized by deregulation of genes involved in the process of phagocytosis.8 Here, we examined the ability of lenalidomide to stimulate NLCs to engulf and ingest particles. Lenalidomide improves the content of F-actin in NLCs and their phagocytic activity. Interestingly, this effect was abolished by blocking Rap1 GTPase, thus indicating that Rap1 activation is essential for lenalidomide-mediated NLCs-phagocytosis. Activation of Rap1 has been demonstrated to be essential for phagocytosis by promoting the polymerization of actin filament at the site membrane beneath the forming phagosome and by functionally activating MAC-1 (CD11b/CD18), and bolstering the integrin-dependent events necessary for effectuating the phagocytic response.33,44
It is well known that monocytes isolated from CLL patients have immunosuppressive properties characterized by a strong reduction in T-cell proliferation, and this anti-proliferative effect is caused, in part, by TGFβ, IDO1 and IL10.5,7,21 We observed that lenalidomide is able to improve the ability of NLCs to stimulate T-cell proliferation. In line with these findings, a switch of NLCs properties to a pro-inflammatory profile was supported by gene expression profiling analysis. Specifically, we found that lenalidomide seems to modulate a peculiar cluster of genes involved in the polarization of monocytes towards classical activated macrophages (M1) induced by LPS and IFN-γ, i.e IDO1 and LAMP3.45 The role of IDO1 in the immune response is still controversial, implying a possible involvement in M1 polarization or alternatively in immune tolerance.7,46,47 Lenalidomide up-regulated IDO1 expression in NLCs. One possible explanation may account for the counter regulatory role of IDO1 to restrain excessive or inappropriate immune activation during an inflammatory response mediated by lenalidomide. Alternatively, a dose-dependent mechanism of IDO1 function may be envisioned. We also observed a decreased expression of CD163, a peculiar marker of NLCs, after treatment with lenalidomide. CD163 expression is a marker of M2 macrophages; its function is carried out through scavenging of the haptoglobin-hemoglobin complex and production of anti-inflammatory metabolites.48 Upregulation of IL2 and downregulation of IL10 in lenalidomide-treated NLCs may be expected to be involved in the recovery of T-cell proliferation. IL2 is a potent inducer of T-cell proliferation and IL10 is an M2 cytokine, strongly expressed in NLCs, that is involved in immunosuppression. Finally, lenalidomide reduces the expression of IL8 that not only promotes cell invasion and increases macrophage infiltration, but also protects CLL cells from apoptosis.49
CLL patients are typically immunocompromised with defective function of T lymphocytes, NK cells and accessory cells, and this could promote a tollerogenic milieu for leukemic cells. On the other hand, there is some evidence to show that an inflammatory microenvironment is induced in survival-supporting culture of CLL cells and that the levels of some inflammatory cytokines are increased in sera of CLL patients compared to healthy controls.42,50 However, CLL patients with good outcome are characterized by higher levels of several pro-inflammatory cytokines (IL-2, IL-4, IL-15, IL-1β, IL-6, TNFα) as compared to CLL with poor outcome. This implies that a more aggressive disease is accompanied by a progressive suppression of immune cell activation and anti-tumor responses.
Our data provide new insights into the mechanism of action of lenalidomide that induces an ‘immune re-education’ of NLCs. Lenalidomide interferes with the supporting and protective microenvironment generated by CLL cells inside tissue niches and counteracts the pro-leukemia role and the immunodeficiency typical of NLCs inducing properties of pro-inflammatory cells.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC IG14376-R.Mar. and IG12754-S.D), Milan, Italy. V.A. is supported by an AIRC/FIRC Italian fellowship (#15047). Additional research support was received from Celgene (San Diego, CA, USA). Lenalidomide for in vitro studies was provided from Celgene.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Cutucache CE. Tumor-induced host immunosuppression: special focus on CLL. Int Immunopharmacol. 2013;17(1):35–41. [DOI] [PubMed] [Google Scholar]
- 2.Ramsay AG, Johnson AJ, Lee AM, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008;118(7):2427–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filip AA, Cisel B, Koczkodaj D, Wasik-Szczepanek E, Piersiak T, Dmoszynska A. Circulating microenvironment of CLL: are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol Dis. 2013; 50(4):263–270. [DOI] [PubMed] [Google Scholar]
- 4.Tsukada N, Burger JA, Zvaifler NJ, Kipps TJ. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood. 2002; 99(3):1030–1037. [DOI] [PubMed] [Google Scholar]
- 5.Ysebaert L, Fournie JJ. Genomic and phenotypic characterization of nurse-like cells that promote drug resistance in chronic lymphocytic leukemia. Leuk Lymphoma. 2011;52(7):1404–1406. [DOI] [PubMed] [Google Scholar]
- 6.Jia L, Clear A, Liu FT, Matthews J, et al. Extracellular HMGB1 promotes differentiation of nurse-like cells in chronic lymphocytic leukemia. Blood. 2014;123(11):1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giannoni P, Pietra G, Travaini G, et al. Chronic lymphocytic leukemia nurse-like cells express hepatocyte growth factor receptor (c-MET) and indoleamine 2,3-dioxygenase and display features of immunosuppressive type 2 skewed macrophages. Haematologica. 2014; 99(6):1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharya N, Diener S, Idler IS, et al. Nurse-like cells show deregulated expression of genes involved in immunocompetence. Br J Haematol. 2011;154(3):349–356. [DOI] [PubMed] [Google Scholar]
- 9.Burger JA. Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011:96–103. [DOI] [PubMed] [Google Scholar]
- 10.Badoux XC, Keating MJ, Wen S, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood. 2011;118(13):3489–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrajoli A, Lee BN, Schlette EJ, et al. Lenalidomide induces complete and partial remissions in patients with relapsed and refractory chronic lymphocytic leukemia. Blood. 2008;111(11):5291–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CI, Bergsagel PL, Paul H, et al. Single-agent lenalidomide in the treatment of previously untreated chronic lymphocytic leukemia. J Clin Oncol. 2011;29(9):1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotla V, Goel S, Nischal S, et al. Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorgun G, Ramsay AG, Holderried TA, et al. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc Natl Acad Sci USA. 2009; 106(15):6250–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120(7):1412–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood. 2013;121(14):2704–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulz A, Durr C, Zenz T, et al. Lenalidomide reduces survival of chronic lymphocytic leukemia cells in primary cocultures by altering the myeloid microenvironment. Blood. 2013;121(13):2503–2511. [DOI] [PubMed] [Google Scholar]
- 18.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 19.Chen N, Lau H, Kong L, et al. Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J Clin Pharmacol. 2007; 47(12):1466–1475. [DOI] [PubMed] [Google Scholar]
- 20.Fiorcari S, Brown WS, McIntyre BW, et al. The PI3-kinase delta inhibitor idelalisib (GS-1101) targets integrin-mediated adhesion of chronic lymphocytic leukemia (CLL) cell to endothelial and marrow stromal cells. PLoS One. 2013; 8(12):e83830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maffei R, Bulgarelli J, Fiorcari S, et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica. 2013;98(7):1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maffei R, Fiorcari S, Bulgarelli J, et al. Physical contact with endothelial cells through beta1- and beta2- integrins rescues chronic lymphocytic leukemia cells from spontaneous and drug-induced apoptosis and induces a peculiar gene expression profile in leukemic cells. Haematologica. 2012;97(6):952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arruga F, Gizdic B, Serra S, et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014; 28(5):1060–1070. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, Li J, Ferguson GD, et al. Immunomodulatory drugs reorganize cytoskeleton by modulating Rho GTPases. Blood. 2009;114(2):338–345. [DOI] [PubMed] [Google Scholar]
- 25.De Luisi A, Ferrucci A, Coluccia AM, et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin Cancer Res. 2011;17(7):1935–1946. [DOI] [PubMed] [Google Scholar]
- 26.Troeger A, Johnson AJ, Wood J, et al. RhoH is critical for cell-microenvironment interactions in chronic lymphocytic leukemia in mice and humans. Blood. 2012; 119(20):4708–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riches JC, Sangaralingam A, Kiaii S, et al. Impact of Lenalidomide on Gene Expression Profiles of Malignant and Immune Cells in Patients with Chronic Lymphocytic Leukemia. Blood. 2011; 118(21):446–452.21596854 [Google Scholar]
- 28.Deaglio S, Vaisitti T, Bergui L, et al. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood. 2005; 105(8):3042–3050. [DOI] [PubMed] [Google Scholar]
- 29.Vicente-Manzanares M, Sanchez-Madrid F. Role of the cytoskeleton during leukocyte responses. Nat Rev Immunol. 2004; 4(2):110–122. [DOI] [PubMed] [Google Scholar]
- 30.Rougerie P, Delon J. Rho GTPases: masters of T lymphocyte migration and activation. Immunol Lett. 2012;142(1–2):1–13. [DOI] [PubMed] [Google Scholar]
- 31.D’Souza-Schorey C, Boettner B, Van Aelst L. Rac regulates integrin-mediated spreading and increased adhesion of T lymphocytes. Mol Cell Biol. 1998;18(7):3936–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2(5):369–377. [DOI] [PubMed] [Google Scholar]
- 33.Hattori M, Minato N. Rap1 GTPase: functions, regulation, and malignancy. J Biochem. 2003;134(4):479–484. [DOI] [PubMed] [Google Scholar]
- 34.Kortholt A, Bolourani P, Rehmann H, et al. A Rap/phosphatidylinositol 3-kinase pathway controls pseudopod formation [corrected]. Mol Biol Cell. 2010;21(6):936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boulter E, Grall D, Cagnol S, Van Obberghen-Schilling E. Regulation of cell-matrix adhesion dynamics and Rac-1 by integrin linked kinase. FASEB J. 2006; 20(9):1489–1491. [DOI] [PubMed] [Google Scholar]
- 36.Qin J, Wu C. ILK: a pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr Opin Cell Biol. 2012;24(5):607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141(5):1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohba Y, Ikuta K, Ogura A, et al. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 2001;20(13):3333–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams HC, San Martin A, Adamo CM, et al. Role of coronin 1B in PDGF-induced migration of vascular smooth muscle cells. Circ Res. 2012;111(1):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clanchy FI, Holloway AC, Lari R, Cameron PU, Hamilton JA. Detection and properties of the human proliferative monocyte subpopulation. J Leukoc Biol. 2006;79(4):757–766. [DOI] [PubMed] [Google Scholar]
- 41.Burgess M, Cheung C, Chambers L, et al. CCL2 and CXCL2 enhance survival of primary chronic lymphocytic leukemia cells in vitro. Leuk Lymphoma. 2012;53(10):1988–1998. [DOI] [PubMed] [Google Scholar]
- 42.Schulz A, Toedt G, Zenz T, Stilgenbauer S, Lichter P, Seiffert M. Inflammatory cytokines and signaling pathways are associated with survival of primary chronic lymphocytic leukemia cells in vitro: a dominant role of CCL2. Haematologica. 2011; 96(3):408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yaktapour N, Ubelhart R, Schuler J, et al. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood. 2013; 122(9):1621–1633. [DOI] [PubMed] [Google Scholar]
- 44.Chung J, Serezani CH, Huang SK, et al. Rap1 activation is required for Fc gamma receptor-dependent phagocytosis. J Immunol. 2008; 181(8):5501–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–7311. [DOI] [PubMed] [Google Scholar]
- 46.Wang XF, Wang HS, Wang H, et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: focus on macrophage polarization of THP-1 cells. Cell Immunol. 2014;289(1–2):42–8. [DOI] [PubMed] [Google Scholar]
- 47.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117(5):1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akila P, Prashant V, Suma MN, Prashant SN, Chaitra TR. CD163 and its expanding functional repertoire. Clin Chim Acta. 2012;413(7–8):669–674. [DOI] [PubMed] [Google Scholar]
- 49.di Celle PF, Carbone A, Marchis D, et al. Cytokine gene expression in B-cell chronic lymphocytic leukemia: evidence of constitutive interleukin-8 (IL-8) mRNA expression and secretion of biologically active IL-8 protein. Blood. 1994;84(1):220–228. [PubMed] [Google Scholar]
- 50.Yan XJ, Dozmorov I, Li W, Yancopoulos S, Sison C, Centola M, et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood. 2011;118(19):5201–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]