

Abstract
Although it has been reported that mesenchymal stromal cells are unable to provide sufficient hematopoietic support in myelodysplastic syndrome, the underlying mechanisms remain elusive. In this study, we found that mesenchymal stromal cells from patients with myelodysplastic syndrome displayed a significant increase in senescence, as evidenced by their decreased proliferative capacity, flattened morphology and increased expression of SA-β-gal and p21. Senescent mesenchymal stromal cells from patients had decreased differentiation potential and decreased stem cell support capacity. Gene knockdown of Dicer1, which was down-regulated in mesenchymal stromal cells from patients, induced senescence. The differentiation and stem cell-supporting capacities were significantly inhibited by Dicer1 knockdown. Overexpression of Dicer1 in mesenchymal stromal cells from patients reversed cellular senescence and enhanced stem cell properties. Furthermore, we identified reduced expression in the microRNA-17 family (miR-17-5p, miR-20a/b, miR-106a/b and miR-93) as a potential factor responsible for increased p21 expression, a key senescence mediator, in Dicer1 knockdown cells. Moreover, we found that miR-93 and miR-20a expression levels were significantly reduced in mesenchymal stromal cells from patients and miR-93/miR-20a gain of function resulted in a decrease of cellular senescence. Collectively, the results of our study show that mesenchymal stromal cells from patients with myelodysplastic syndrome are prone to senescence and that Dicer1 down-regulation promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells. Dicer1 down-regulation seems to contribute to the insufficient hematopoietic support capacities of mesenchymal stromal cells from patients with myelodysplastic syndrome.
Introduction
Myelodysplastic syndrome (MDS) is a heterogeneous group of clonal diseases derived from hematopoietic stem cells and is characterized by ineffective bone marrow hematopoiesis and a substantial risk of progression to acute myeloid leukemia. In spite of intense research on the cellular and molecular pathophysiology of MDS1–3 over the past decade, including studies of epigenetic changes, mutations and abnormalities in cytokines and the immune system, the role of the bone marrow (BM) microenvironment in MDS remains to be characterized.
The BM microenvironment is composed of various cells and an extracellular matrix, and these components cooperate to regulate hematopoiesis. Mesenchymal stromal cells (MSC), which are undifferentiated and pluripotent, are key components of the BM microenvironment.4 It has become evident that the interaction between MSC and hematopoietic stem cells is important in inducing the quiescence of hematopoietic stem cells both in vivo and in vitro.5 The possible involvement of MSC in the pathogenesis of MDS has not been clearly studied, and existing data regarding the cytogenetics and function of MSC in patients with MDS are contradictory. Some authors have reported that MDS-derived MSC have a decreased capacity to support hematopoiesis and defective proliferative, differentiation and immunosuppressive potential.6–10 However, other authors report results indicating the opposite.11–13 The discrepancies between the results of the studies might be attributed to the use of different MDS subtypes, different passage numbers and even dissimilar cell populations. Recently, Schroeder et al. tested a large number of cell samples from MDS patients.10 They found that MSC from patients with MDS are structurally, epigenetically and functionally altered and their stromal support function is impaired. However, the precise mechanisms of insufficient stromal support in MDS remain elusive.
Evidence accumulated in recent years demonstrates that MSC from MDS are intrinsically pathological, exhibiting a continuous decline in proliferation as a result of increased senescence.10,14 Cellular senescence is usually accompanied by functional changes. Indeed, decreased differentiation has been linked to senescence and aging in MSC.15–17 Determining how and whether increased cellular senescence contributes to insufficient stromal support in patients with MDS is a topic for further investigation.
Cellular senescence is a complex process. Increasing evidence suggests that microRNA (miRNA, miR) are functionally involved in MSC senescence.18–20 The ablation of Dicer1 (an RNAse III endonuclease essential for miRNA biogenesis) and the loss of mature miRNA in embryonic fibroblasts and endothelial cells inhibited cell proliferation and induced a premature senescence phenotype.21,22 Although recent striking findings indicate that Dicer1 deletion can induce an MDS phenotype in mice23 and that Dicer1 expression is reduced in MSC from MDS patients,24 the precise mechanism by which the Dicer1 gene contributes to the pathogenesis of MDS remains elusive. We inferred that disordered expression of Dicer1 could contribute to abnormal MSC senescence and impaired stromal support in MDS.
The aims of this study were, therefore, to evaluate the senescent features of BM-MSC from patients with MDS and to explore the role of Dicer1 in the senescence of MSC and the disturbed stromal function observed in patients with MDS.
Methods
Patients and control samples
A total of 77 patients with MDS were included in this study. Their characteristics are detailed in Online Supplementary Table S1. Patients were diagnosed with MDS according to the minimum diagnostic criteria established by the Conference on MDS.25 Patients were classified for the study as “lower-risk” (LR) [International Prognostic Scoring System (IPSS) low/int-1] and as “higher-risk” (HR) (IPSS-int-2/high).26 Twenty-two healthy volunteers were used as controls (median age 62 years; age range, 43–78 years) and were matched for gender and age. All study participants signed an informed consent form. The research was approved by the ethics committee of the Sixth Hospital affiliated with Shanghai Jiao Tong University, and all patient-relevant research strictly abided by the Declaration of Helsinki.
Mesenchymal stromal cell characterization
BM-MSC were isolated and cultured; their immunophenotype, clonogenic and proliferative potential, and differentiation were studied and SA-β-Gal assay were performed. Detailed information about the experiments is provided in the Online Supplementary Material.
RNA isolation and real-time polymerase chain reaction analysis
Total RNA was isolated using TRIzol (Invitrogen, Paisley, Scotland) according to the manufacturer’s instructions. For mRNA detection, the RNA was reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Fermentas, Burlington, Canada) according to the manufacturer’s protocol, and real-time polymerase chain reaction (RT-PCR) was performed using RealMasterMix (Takara, Dalian, China). The process used was described in detail in our previous study.27 The primers are listed in Online Supplementary Table S2. RT-PCR for miRNA was performed using the PrimeScript™ miRNA qPCR Starter Kit Ver. 2.0 (Takara, Dalian, China). RNU6B was used as the housekeeping gene. Fold change was calculated using the ΔΔCT method of relative quantification.
Isolation of CD34+ cells and CD271+ cells
Mononuclear cells were obtained from BM aspirates by density gradient separation and then subjected to immunomagnetic enrichment of CD34+ cells or CD271+ cells (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s protocol.
Long-term culture
To evaluate the capacity of MSC to sustain the survival and proliferation of early hematopoietic progenitor cells, a long-term culture-initiating cell (LTC-IC) assay was performed according to the manufacturer’s protocol. Detailed information is provided in the Online Supplementary Material.
Dicer1 short-hairpin RNA and cell transductions
Three pLenti X1 Puro-shDicer1-eGFP vectors and plentiX1 puro-shcontrol (negative vector) were constructed by Guangzhou Cyagen Biosciences, Inc. For the lentiviral transduction, lentiviruses were produced in 293T cells using standard protocols and pLV/helper-SL3, pLV/helper-SL4 and pLV/helper-SL5 as packaging vectors. Detailed information about cell transductions is provided in the Online Supplementary Material.
Construction of adenoviral vectors for expression of Dicer1, production of adenovirus and cell transductions
The adenovirus expression vector that encodes human Dicer1 was generated using the Adeno-X Expression System (BD Biosciences Clontech). The recombinant virus was packaged and amplified in HEK293 cells and purified by CsCl density gradient centrifugation. To package adenovirus, the adenoviral vectors were linearized with the restriction enzyme PacI and transfected into HEK293 cells using Lipofectamine2000. After several rounds of propagation, recombinant adenovirus was purified by an AdEasy virus purification kit (Stratagene). The transfection was performed according to the manufacturer’s protocol.
Overexpression of microRNA-93/microRNA-20a
The human miR-93, miR-20a and scrambled control lentivirus were purchased from Genechem Company, Shanghai, China. MDS-MSC were transfected according to the manufacturer’s protocol. Twenty-four hours after transfection, the medium was removed and replaced with fresh medium.
Results
Senescent features of bone marrow mesenchymal stromal cells from patients with myelodysplastic syndrome
The BM-MSC cultured from each patient with MDS and from healthy controls were successfully expanded. Cells from cultures were devoid of hematopoietic cells, being negative for the hematopoietic antigens CD45 and CD34, and were positive for CD73, CD90, CD105, and CD166.28 MSC from healthy controls (HC-MSC) displayed a characteristic spindle-like morphology, whereas MDS-MSC were larger and irregular (Figure 1A). Under routine in vitro culture conditions, we observed that MDS-MSC ceased growing earlier (passages 6 to 9) than HC-MSC (passages 13 to 15) (data not shown). Colony-forming unit (CFU)-fibroblast (CFU-F) assays were performed to assess the clonogenic potential of the MSC. The BM-MSC from all MDS subtypes exhibited decreased cumulative CFU-F numbers at passages 1, 3 and 5 compared with HC-MSC (all P<0.05, Figure 1B,C). Furthermore, the doubling times of MSC at passages 1, 3 and 5 were significantly higher in MDS patients than in controls (Figure 1D). The data regarding the cell doubling time were also confirmed by CCK-8 assay. Over the 7-day culture period, the number of live cells corresponding to the optical density from both LR- and HR-MDS groups increased more slowly than that from the control group (Figure 1E).
Figure 1.
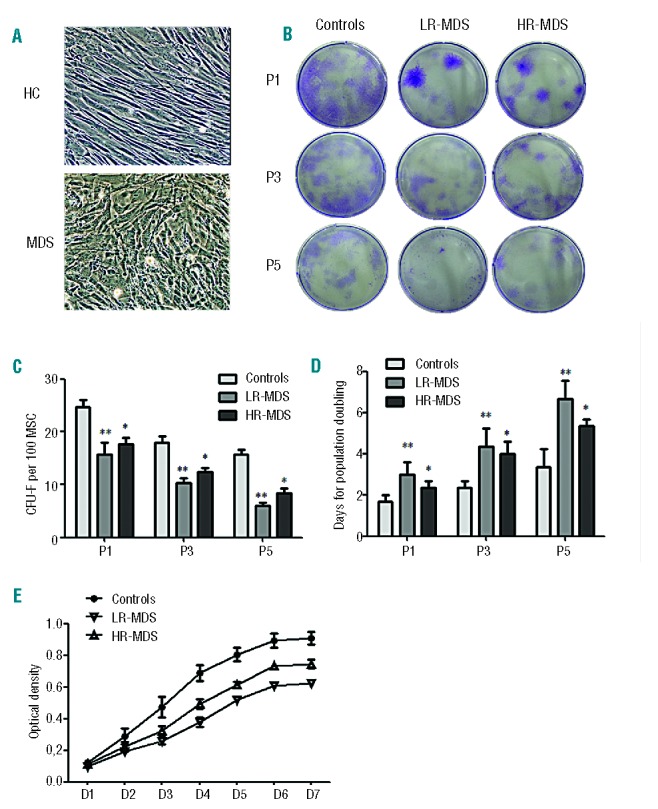
Impaired proliferative capacity of MDS-MSC. (A) Representative morphology of MDS-MSC and HC-MSC. HC-MSC displayed fibroblast-like morphology, whereas MDS-MSC were larger and appeared disorganized (100 × magnification). (B, C) CFU-F assays. Representative colonies in six-well plates are displayed (B). At each passage, the cumulative CFU-F numbers of MSC from both LR-MDS (n=15) and HR-MDS (n=12) patients decreased (C). (D) Doubling times of HC-MSC (n=9), LR-MDS-MSC (n=15) and HR-MDS-MSC (n=12) at each passage. (E) The cell proliferation ratio was detected using the CCK-8 assay. MSC from both LR-MDS (n=8) and HR-MDS (n=7) patients grew more slowly than those from the control group (n=5). Compared with HC-MSC, the significance was set as *P≤0.05; **P≤0.001.
To investigate whether the decreased cell growth of MDS-MSC was due to increased apoptosis, annexin V+ cells were measured by flow cytometry. No apoptotic cells were observed in our BM-MSC cultures (<1%, data not shown). Furthermore, SA-β-gal was used to examine the presence of senescence in MSC. The number of SA-β-gal-positive cells was notably increased in MDS-MSC at each time point assessed (Figure 2A,B). At passage 3, the median percentage of senescent cells in HC-MSC was 5.5% of the total. Using a cutoff value of 5.5%, increased senescence was observed in 82.4% (14/17) of LR-MDS-MSC and 60% (9/15) of HR-MDS-MSC. The distribution of senescence among samples is shown in Online Supplementary Figure S1A.
Figure 2.
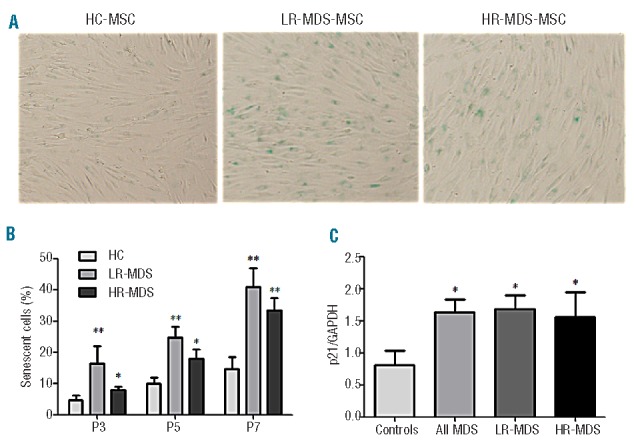
Cellular senescence in MDS-MSC. (A, B) SA-β-gal was used to examine MSC senescence. One hundred MSC per individual sample were counted using light microscopy (100 × magnification). The number of SA-β-gal-positive cells clearly increased among LR-MDS-MSC (n=17) and HR-MDS-MSC (n=15). (C) Expression of senescence-related molecular p21 in MDS-MSC, as determined by RT-PCR (n=77). The results are expressed as means ± SD. Compared with HC-MSC, the significance was set as *P≤0.05; **P≤0.001.
We then evaluated the expression of multiple key mediators, including p21, p16 and RB, which are involved in regulating cell senescence. We observed an up-regulation of p21 in MDS-MSC compared with healthy controls (Figure 2C). However, no differences were observed for p16 and RB expression between MDS and healthy controls. Furthermore, we found increased expression of p21 in senescent MDS-MSC (SA-β-gal-positive cells ≥5.5%) than in non-increased senescence MDS-MSC (NS-MDS-MSC, SA-β-gal-positive cells <5.5%) (data not shown).
In line with these findings, primary (CD271+) MSC from MDS patients also displayed increased cellular senescence, as shown by a significantly higher number of SA-β-gal-positive cells and higher expression of p21, compared with healthy controls (Online Supplementary Figure S1B-D). Taken together, these data suggest that MSC from MDS patients had decreased proliferation capacity and increased senescence.
Senescent myelodysplastic syndrome mesenchymal stromal cells exhibited decreased differentiation and stem cell-supporting capacity
Considering that cell dysfunction is associated with senescence, we evaluated the capacity of senescent MDS-MSC and HC-MSC to differentiate and to support stem cell growth. The osteoblastic and adipogenic differentiation potentials of MSC were evaluated by cytochemical staining and relative mRNA expression. Compared with HC-MSC and NS-MDS-MSC, the osteogenic differentiation potential of senescent MDS-MSC was significantly reduced, as reflected by the mineralization analyses and ALP activity measurement (Figure 3A–C). Consistent with the results of the cytochemical staining, the mRNA levels of ALP and Runx2, two key factors involved in osteogenic differentiation, were significantly reduced in senescent MDS-MSC during the process of osteogenic differentiation (Figure 3D,E). The adipogenic differentiation potential was evaluated using oil red-O staining and was 2-fold higher in MSC from healthy subjects than in senescent MDS-MSC (Figure 3F,G). The differences in the adipogenic differentiation potential between HC-MSC and senescent MDS-MSC were also reflected by the mRNA expression levels of FABP4 and C/EBPα (Figure 3H,I).
Figure 3.

Differentiation of senescent (S)-MDS-MSC. Osteogenic differentiation of S-MDS-MSC (A–E). Before MSC osteogenic differentiation, the degree of senescence was estimated by SA-β-gal staining. MDS-MSC were divided into two groups: S-MDS-MSC (SA-β-gal-positive cells ≥5.5%, n=15) and non-senescent (NS)-MDS-MSC (SA-β-gal-positive cells <5.5%, n=12). (A, B) Mineralization analysis. The mineralization was visualized by Alizarin red S staining at 21 days (100 × magnification). (C) ALP activity measurements. ALP activity was measured at 7 days after osteogenic differentiation using an alkaline phosphatase activity kit. (D, E) Relative levels of RUNX2 and ALP mRNA in cells differentiated for 21 days. Adipogenic differentiation of S-MDS-MSC (F–I). (F) Representative images of lipid droplet formation on day 21 after adipogenic differentiation (100 × magnification). (G) Quantitation of oil red-O incorporation was determined according to the amount of extracted dye at 510 nm. (H, I) Relative FABP4 and C/EBPαmRNA expression levels in cells differentiated for 21 days. The results are expressed as means ± SD. Compared with HC-MSC, the significance was set as *P≤0.05; **P≤0.001.
We estimated the capacities of senescent MDS-MSC or HC-MSC grown in layers to sustain the proliferation of CD34+ cells. For this purpose, long-term culture assays were implemented. The results showed that the growth of normal LTC-IC on expanded senescent MDS-MSC was significantly less than that of HC-MSC (Online Supplementary Figure S2A). Compared with HC-MSC, co-culture with senescent MDS-MSC resulted in depressed numbers of CFU-granuloctye, macrophage (CFU-GM) (2.32-fold), burst-forming unit-erythroid (BFU-E) (1.9-fold) and CFU-granulocyte, erythrocyte, monocyte, megakaryocyte (CFU-GEMM) (2.0-fold) (Online Supplementary Figure S2B). In summary, the differentiation and stem cell-supporting capacities of senescent MSC were reduced.
Low Dicer1 expression was observed in myelodysplastic syndrome mesenchymal stromal cells, and knockdown of Dicer1 induced senescence in mesenchymal stromal cells
Dicer1 ablation caused a rapid onset of cell senescence in various cell types.21,22 We measured the expression of Dicer1 in MDS-MSC and controls. Using RT-PCR and western blotting, we found that the expression of Dicer1 was down-regulated in the MSC from both LR-MDS and HR-MDS patients (Figure 4A,B). This finding remained significant when we looked at primary (CD271+) MDS-MSC (Figure 4C).
Figure 4.

Under-expression of Dicer1 in MDS-MSC. (A) The expression of Dicer1 in expanded LR-MDS-MSC (n=38), HR-MDS-MSC (n=34) and HC-MSC (n=22) was measured by quantitative PCR and is presented as means ± SD. (B) Protein expression of Dicer1 in MDS-MSC. (C) The expression of Dicer1 in primary MDS-MSC (n=24) and primary HC-MSC(n=10). Compared with controls, the significance was set as *P≤0.05; **P≤0.001.
To further assess the role of Dicer1 in MSC senescence, we constructed a recombinant lentivirus to generate shRNA targeting Dicer1. Nearly 95% of cells were transfected with the shNA constructs (Figure 5A). As shown in Figure 5B,C, the shRNA groups had significantly lower Dicer1 mRNA and protein expression levels compared with the control group. MSC that underwent Dicer1 knockdown (KD) proliferated far more slowly than either control MSC or the negative group (Figure 5D). Furthermore, Dicer1 KD resulted in an increased number of cells in the G1 phase without inducing apoptosis (Figure 5E).
Figure 5.

Dicer1 knockdown-induced cellular senescence of MSC. (A) Transfection efficiency under the fluorescence microscope. (B, C) Dicer1 shRNA decreased Dicer1 expression in MSC, as determined by western blotting (B) and RT-PCR analyses (C). (D) The cell proliferation capacity was measured by CCK-8 assay. (E) Cell cycle analysis of Dicer1-KD MSC by FACS. (F) Representative micrographs depict the morphologies of Dicer1-KD MSC (shRNA), negative MSC (transfected with control lentiviruses) and control-MSC (HC-MSC without transfection) and also show SA-β-gal staining (100 × magnification). (G) One hundred MSC per sample were counted using light microscopy, and the percentages of SA-β-gal-positive cells were determined. The average of three replicates is displayed. Compared with controls, the significance was set as *P≤0.05; **P≤0.001.
To determine whether these cells were becoming senescent, Dicer1-KD MSC or controls were passaged once they reached confluence. After 14 days, Dicer1-KD MSC stopped dividing and assumed a large, flattened morphology (Figure 5F). Harvesting and re-plating cells in fresh medium at higher or lower densities failed to stimulate their growth. To confirm that Dicer1-KD MSC were undergoing senescence, the cells were stained in a SA-β-gal assay. Small amounts of staining were detected in the control and negative MSC, whereas SA-β-gal activity was readily apparent in the Dicer1-KD MSC (Figure 5G). Over 50% of the Dicer1-KD MSC underwent senescence by day 7, compared with less than 15% of senescence in the control and negative MSC. These results suggest that down-regulation of Dicer1 is sufficient to induce MSC senescence.
Dicer1 knockdown inhibited osteogenic and adipogenic differentiation of mesenchymal stromal cells
We then tested the effect of Dicer1-KD on the capacity of MSC to differentiate into the osteogenic and adipogenic lineages. After osteogenic induction, Dicer1-KD MSC showed undetectable mineralization when stained with Alizarin red (Online Supplementary Figure S3A). Moreover, the Dicer1-KD MSC exhibited more than a 90% reduction (P<0.001) in ALP activity, and a 95% reduction (P<0.001) in incorporation of Alizarin red stain compared with negative MSC and control cells (Online Supplementary Figure S3B,C). During the course of the osteogenic induction, mRNA levels of RUNX2 and ALP increased strikingly in control MSC. In contrast, the levels of these gene transcripts only slightly increased in Dicer1-KD MSC (Online Supplementary Figure S3D,E).
A similar method was used to assess the capacity of MSC to differentiate into an adipogenic lineage after Dicer1-KD. After 21 days, oil red-O-stained cultures demonstrated that more than 90% control MSC could differentiate into adipocytes, showing abundant intracellular lipid droplets. In contrast, the intracellular lipid accumulation in Dicer1-KD MSC was sparse and undetectable, often consisting of small droplets that did not coalesce (Online Supplementary Figure S4A). During adipogenic induction, the expression of C/EBPα and FABP4 gradually increased. After 14 days, control MSC exhibited a 4.5-fold increase in C/EBPα with a 5.2-fold increase in FABP4 when compared with undifferentiated cells. In contrast, Dicer1-KD MSC exhibited a mild increase in both mRNA levels, with a 1.8-fold increase in adiponectin and a 0.8-fold increase in FABP4 compared with undifferentiated cells (Online Supplementary Figure S4B,C).
Dicer1 knockdown decreased the hematopoietic-support properties of mesenchymal stromal cells
Given that senescent MSC exhibited a decrease in stem cell-support capacity, we speculated that Dicer1-KD could impair the hematopoietic-support properties of MSC. We assessed the hematopoietic support of Dicer1-KD MSC using long-term culture assays. The results showed that Dicer1-KD MSC have a 6-fold reduced capacity to support healthy CD34+ cells compared with control MSC [LTC-IC frequency on control MSC (mean±s.e.): 1.61%±0.42%; on Dicer1-KD MSC: 0.24%±0.09%]. By counting the CFU, we found that the numbers of CFU-GM, BFU-E and CFU-GEMM were significantly lower after co-culture with Dicer1-KD MSC than with control MSC (15.4±4.8 and 98.6±9.8 CFU-GM, 8.4±3.5 and 42.2±6.6, BFU-E, and 6.8±2.2 and 38.5±8.0 CFU-GEMM per 104 CD34+ cells in the presence of Dicer1-KD MSC and control MSC, respectively) (Figure 6A). Our results suggest that Dicer1-KD MSC layers were inefficient in their hematopoietic-supporting capacity.
Figure 6.
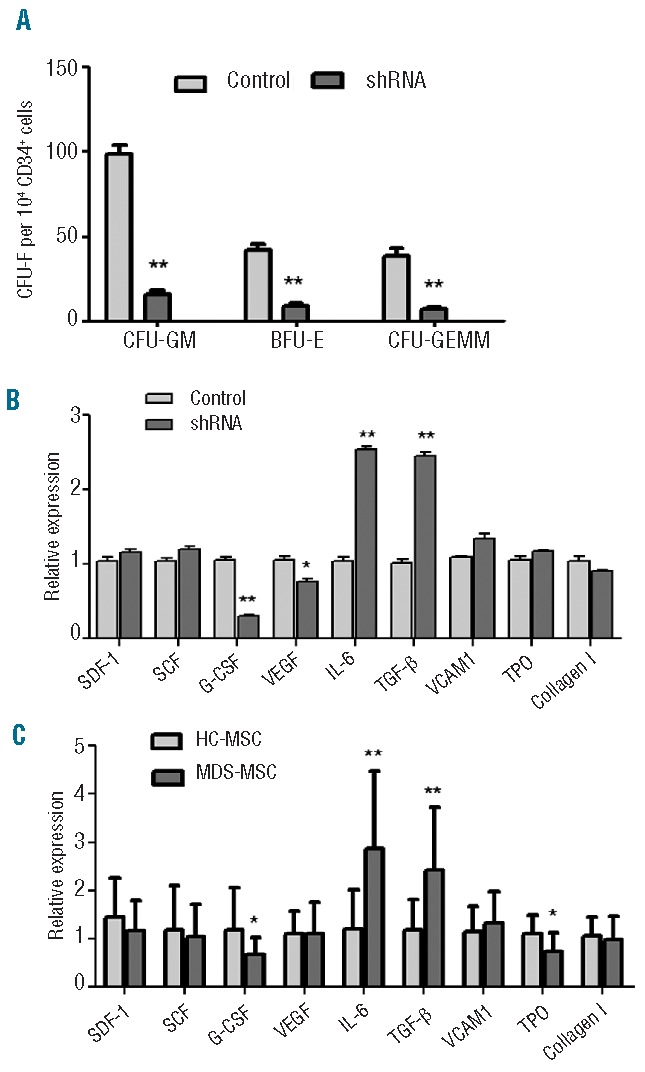
Dicer1-KD decreased the hematopoietic-supporting properties of MSC. (A) CD34+cells from four healthy controls were cultured on Dicer1-KD MSC (shRNA), negative MSC or control MSC feeder layers for 7 weeks. The numbers of CFU-GM, CFU-E and CFU-GEMM were counted. (B,C) Hematopoietic niche cytokine expression levels were measured by RT-PCR in Dicer1-KD MSC (B) and primary MDS-MSC (C). The results are expressed as means ± SD. The average of three replicates is displayed. Compared with controls, the significance was set as *P≤0.05; **P≤0.001.
MSC interact with hematopoietic stem cells, secreting chemokines that support the long-term growth of the hematopoietic stem cells. Alterations in chemokine levels might contribute to the defective support of hematopoiesis characteristic of Dicer1-KD MSC. We further tested the expression of hematopoietic cytokines in Dicer1-KD MSC and found that Dicer1-KD MSC exhibited decreased expression of granulocyte colony-stimulating factor (G-CSF) and vascular endothelial growth factor compared with control MSC. However, the levels of interleukin-6 and transforming growth factor-β expression in the Dicer1-KD MSC were significantly higher than in control MSC. The expression levels of stromal cell-derived factor-1, stem cell factor, vascular cell adhesion molecule 1, thrombopoietin and collagen I were largely unchanged (Figure 6B).
The levels of expression of these cytokines were also measured in primary MDS-MSC. We found that primary MDS–MSC (n=24) exhibited decreased expression of G-CSF and thrombopoietin, and increased expression of interleukin-6 and transforming growth factor-β, compared with primary HC-MSC (n=10) (Figure 6C).
Overexpression of Dicer1 reversed the senescent features of mesenchymal stromal cells from patients with myelodysplastic syndrome, and enhanced stem cell properties
In the following experiment, MSC from four MDS patients were considered. We used MDS-MSC transfected with adenovirus carrying the Dicer1 gene (AD-Dicer1) or green fluorescent protein (AD-GFP). Dicer1 expression was considerably increased in the AD-Dicer1 MSC at both the mRNA (Figure 7A) and protein (Figure 7B) levels. We found that cell viability was not affected in the AD-Dicer1 MSC. There were less SA-β-gal-positive cells when Dicer1 expression was up-regulated in MDS-MSC (Figure 7C). Cell-cycle analysis revealed that G1 phase arrest was reversed in AD-Dicer1 MSC (from 94.34±3.6% to 84.48±2.8%). In addition, AD-Dicer1 MSC exhibited increased osteoblastic (Online Supplementary Figure S5A) and adipogenic differentiation potential (data not shown) compared with MSC transfected with GFP vectors. Proliferation, as measured by CFSE staining of healthy CD34+ cells, was increased markedly when co-cultured with AD-Dicer1 MSC compared with AD-GFP MSC (Online Supplementary Figure S5B). Taken together, overexpression of Dicer1 reversed cellular senescence and enhanced stem cell properties in MDS-MSC.
Figure 7.
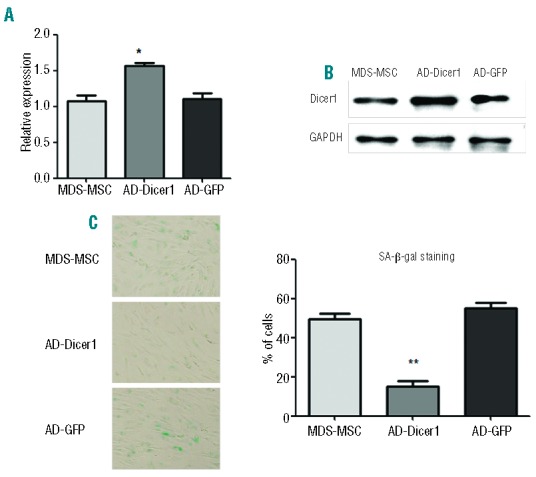
Overexpression of Dicer1 reversed cellular senescence in MDS-MSC (n=4). The mRNA (A) and protein expression (B) of Dicer1 after adenovirus transfection. (C) The percentages of SA-β-gal-positive cells. The results are expressed as means ± SD. The average of three replicates is displayed. Compared with MDS-MSC, the significance was set as **P≤0.001.
The microRNA-17 family, which targets p21, is associated with Dicer1 knockdown-induced cellular senescence
To determine whether down-regulation of Dicer1 in MSC could affect miRNA biogenesis and participate in cellular senescence, we tested a set of specific miRNA that were predicted to target senescence-related molecules, such as p21 and p16. Three miRNA target databases, miRBase, PicTar and Targetscans, were used for target analysis. Dicer1-KD resulted in decreased expression of miR-17, miR-93, miR-106a, miR-106b, miR-20a and miR-20b, which target p21and are known as the miR-17 family (Figure 8A). In comparison, miR-125b was overexpressed in Dicer1-KD MSC. We did not detect statistically significant changes in miR-212, miR-132, miR-301b, miR-22, miR-134, miR-146b or miR-128 expression. These data revealed that Dicer1-KD-induced senescence may be mainly implemented by the regulation of p21-related miRNA. To confirm the results, we measured the expression levels of some cell cycle markers in Dicer1-KD MSC. As expected, p21 expression levels were increased in the Dicer1-KD MSC. However, little difference was observed in the levels of the p16 or RB (Figure 8B).
Figure 8.
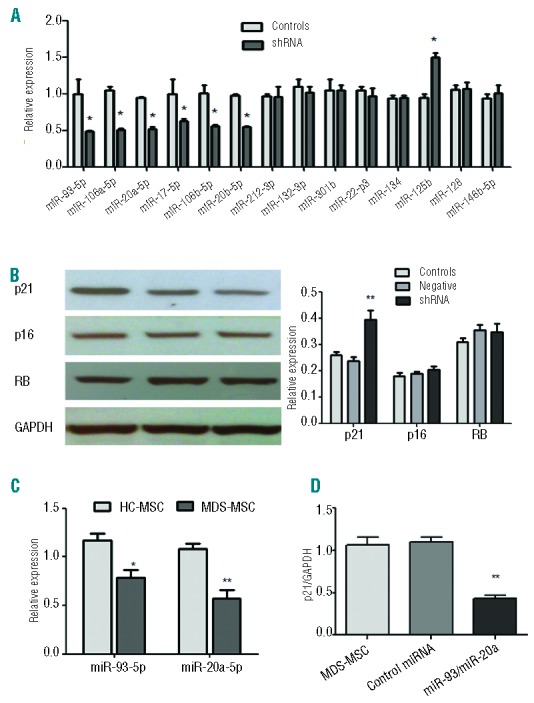
The miR-17 family is associated with Dicer1-KD-induced cellular senescence. (A) Effect of Dicer1-KD on miRNA expression. Real-time PCR was performed on RNA isolated from Dicer1-KD MSC (shRNA) and controls. (B) Protein expression of p21, p16 and RB in Dicer1-KD MSC. (C) miR-93 and miR-20a expression in MDS-MSC (n=56) and healthy controls (n=20). (D)Overexpression of miR-93 and miR-20a in MDS-MSC (n=3). The expression of p21 in MDS-MSC, miR-93/miR-20a group (MDS-MSCs transfected with miR-93/miR-20a adenovirus) and miRNA control group (transfected with scrambled control lentivirus). These experiments were performed three times. Compared with controls, the significance was set as *P≤0.05; **P≤0.001.
We further verified the expression of these disordered miRNA in MDS-MSC. Our results showed that miR-93 and miR-20a expression levels were significantly reduced in MDS-MSC compared with controls (Figure 8C). A similar down-regulation of miR-93 and miR-20a was also observed in senescent MDS-MSC,when compared with NS-MDS-MSC (data not shown).
To show that p21 is a target of miR-93 and miR-20a in MDS-MSC, experiments of miR-93/miR-20a overexpression were performed. We transfected MDS-MSC with miR-93 and miR-20a lentivirus, and assessed p21 expression levels 48 h after transfection. The degree of miRNA overexpression was monitored by RT-PCR 2 days after transfection and showed satisfactory results (Online Supplementary Figure S6A). In three analyzed samples of MDS-MSC, this resulted in a 2.2-fold reduction of p21 mRNA levels (Figure 8D) and a decrease of cellular senescence (Online Supplementary Figure S6B-D) when compared with transfection with scrambled control lentivirus. However, transfection of cells with miR-93/miR-20a did not reveal statistically significant changes in osteogenic differentiation of MDS-MSC (data not shown), suggesting other miRNA may be involved.
Discussion
MSC are key components of the hematopoietic microenvironment. The abnormal production of hematopoietic cytokines and the insufficient stromal support by MSC in patients with MDS are widely accepted.10 However, little is known about the underlying mechanisms of these defects.
In this study, we show that MDS-MSC were more prone to cellular senescence than HC-MSC and that senescent MDS-MSC exhibited decreased differentiation potential and stem cell support capacity. Interestingly, knockdown of Dicer1, which is under-expressed in MDS-MSC, promoted cellular senescence and decreased the differentiation and stem cell-supporting capacities of MSC. Overexpression of Dicer1 in MDS-MSC reversed cellular senescence and enhanced stem cell properties. The miR-17 family members were involved in the Dicer1 KD-induced senescence.
Although MSC from all MDS patients were expanded successfully in the current study, MDS-MSC showed reduced expansion potential in vitro when compared to HC-MSC. These observations agree with the findings of several previously published studies,7,10,12,29,30 but contrast with the data from others6,9,13 in which MDS-MSC had normal growth. The contradiction might be attributed to the heterogeneity of MDS patients. To overcome this barrier, we recruited a large sample of MDS patients. This finding remained significant when MDS subtypes were considered separately. Because the decreased cell growth of MDS-MSC was not due to an increase in apoptosis, the senescence of MDS-MSC was studied. The classic characteristics of cellular senescence include growth arrest, enlarged/flattened morphology and increased SA-β-gal expression.31,32 Just as predicted, most of the cultured and primary MDS-MSC were larger and irregular and expressed significantly higher amounts of SA-β-gal and the senescence-related molecule, p21. These findings are similar to those of previous studies,10,14 but the investigators of those studies did not provide a detailed analysis of the different MDS subsets and the functional changes related to senescence in MDS pathology. In our study, not all of the cases exhibited an increase in senescence (22/32, 68.7%). Cellular senescence was more common in LR-MDS-MSC than in HR-MDS-MSC (14/17 versus 9/15), suggesting biological differences between the two subsets. In fact, the immunoregulatory role of MSC has been reported to be different between LR-MDS and HR-MDS.9,33 It remains to be investigated whether different primary defects are present in MSC from LR and HR patients with MDS, which may contribute to the different biological characteristics of LR-MDS and HR-MDS.
Senescent MSC are also characterized by altered cell function. The loss of MSC osteogenic and adipogenic potential has been demonstrated in vitro.15,16 By quantitatively and dynamically evaluating the differentiation potential of MSC, we identified significantly decreased osteoblastic and adipogenic potentials in senescent MDS-MSC. In addition, senescent MDS-MSC exhibited decreased stem cell-supporting capacity in long-term culture. Thus, the senescence of MSC may be a contributing factor in the ineffective hematopoiesis of patients with MDS. In keeping with our findings, some studies demonstrated that MDS-MSC displayed insufficient hematopoietic support capability compared with normal controls.10,13,14 However, other reports suggested that MDS-MSC may have normal support capacity.11,12,34 The discrepancies in these results might be attributed to differences between patients, cellular populations and experimental protocols in the various studies. Our study also suggested that differences in the cellular senescence of MDS-MSC could contribute to these discrepant results.
Recently, a prominent group of regulatory factors (miRNA) has also been considered to be key modulators of cellular senescence.18,20 Raaijmakers et al. reported that the deletion of Dicer1, an RNAse III endonuclease essential for microRNA biogenesis, in murine MSC-derived osteoprogenitors induces an MDS phenotype in mice.23 Based on these findings, we analyzed Dicer1 expression in MDS-MSC. We noted reduced expression levels of Dicer1 in expanded and primary MDS-MSC. To explore the role of Dicer1 under-expression in MDS-MSC, we generated a stable knockdown of Dicer1 in MSC. Our data support a role for Dicer1 in the appropriate proliferation and senescence of MSC. Dicer1-KD accelerated cellar senescence, which manifested as G1 growth arrest, flat cell morphology and increased SA-β-gal staining. Experiments of Dicer1 overexpression further confirmed the results. Although the loss of miRNA biogenesis by the disruption of Dicer1 has been associated with cellular senescence in primary cells and endothelial cells,21,22,35 the mechanisms of this event in BM-MSC remain elusive. Senescent cells usually display up-regulation of cell cycle inhibitors such as p53/p21 or p16/Rb,36 which have been reported to be regulated by miRNA. In the present study, we identified reduced expression of the miR-17 family (miR-17-5p, miR-20a/b, miR-106a/b and miR-93) as a potential factor responsible for increased p21 expression in Dicer1-KD MSC. In fact, many studies have used luciferase assays to indicate that miRNA in the miR-17 family (also called the miR-106b family) can regulate cell cycle progression by targeting p21.37–40 Our study and previous studies suggested that loss of expression of the miR-17 family is responsible for the Dicer1-KD-induced cellular senescence in MSC.
Recently, a global down-regulation of miRNA expression was reported in MDS-MSC, suggesting that impaired miRNA expression might be involved in the pathogenesis of MDS.24 We found that miR-93 and miR-20a expression levels were significantly reduced in MDS-MSC. What is more, overexpression of miR-93/miR-20a reversed cellular senescence in MDS-MSC by targeting p21. These results demonstrated that the two repressed miRNA had a key role in the increased cellular senescence of MDS-MSC.
In line with the reports of Raaijmakers et al.,23 Dicer1-KD MSC exhibited reduced osteoblastic differentiation potential, characterized by a reduction in ALP activity, calcium deposits and the expression of osteoblastic maker genes. Moreover, quantification of lipid formation revealed a 90% reduction in Dicer1-KD MSC. Overexpression of Dicer1 reversed the impaired differentiation potential of MDS-MSC. These results indicated that the disruption of miRNA processing by Dicer1-KD was sufficient to greatly reduce the differentiation capability of MSC, suggesting that precise levels of mature miRNA are critical for MSC differentiation.41–43
MSC senescence influences not only differentiation potential but also the support of hematopoiesis.44 To date, knowledge regarding the role of MSC senescence in hematopoietic support is limited. In this study, other than the classic feature of cellular senescence, Dicer1-KD MSC showed a significantly reduced capacity to support CD34+ cells in LTC-IC assays. Accordingly, the expression of two cytokines, G-CSF and vascular endothelial growth factor were significantly down-regulated in Dicer1-KD MSC. These molecules are involved in the regulation of hematopoiesis,45,46 suggesting that their deregulation contributes to the impaired support of hematopoiesis characteristic of Dicer1-KD MSC. It is worth mentioning that the level of G-CSF expression has been reported to be decreased in MDS-MSC.13,29 We found that G-CSF and thrombopoietin expression levels were significantly reduced in primary MDS-MSC. These results suggest that the disruption of Dicer1 may represent a mechanism responsible for the abnormal cytokine expression in MDS-MSC. Interleukin-6 and transforming growth factor-β were up-regulated in the Dicer1-KD MSC, in line with the senescent phenotype of MSC, in which the two molecules were up-regulated.47,48
In conclusion, we investigated the characteristics and molecular mechanisms of MSC senescence in patients with MDS. Our data demonstrate that Dicer1 plays an essential role in the aging process of MSC from MDS patients by regulating miRNA and causing impaired stromal support of hematopoietic stem and progenitor cells. Our findings are in agreement with the notion that a primary stromal defect causes MDS.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This study was supported by the National Nature Science Foundation of China (NNSFC81170463, NNSFC81400090).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia. 2014;28(3):497–506. [DOI] [PubMed] [Google Scholar]
- 2.Tothova Z, Steensma DP, Ebert BL. New strategies in myelodysplastic syndromes: application of molecular diagnostics to clinical practice. Clin Cancer Res. 2013;19(7): 1637–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warlick ED, Miller JS. Myelodysplastic syndromes: the role of the immune system in pathogenesis. Leuk Lymphoma. 2011;52(11):2045–2049. [DOI] [PubMed] [Google Scholar]
- 4.Sacchetti B, Funari A, Michienzi S, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131(2):324–336. [DOI] [PubMed] [Google Scholar]
- 5.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6(2):93–106. [DOI] [PubMed] [Google Scholar]
- 6.Flores-Figueroa E, Arana-Trejo RM, Gutiérrez-Espíndola G, Pérez-Cabrera A, Mayani H. Mesenchymal stem cells in myelodysplastic syndromes: phenotypic and cytogenetic characterization. Leuk Res. 2005;29(2):215–224. [DOI] [PubMed] [Google Scholar]
- 7.Aanei CM, Flandrin P, Zugun Eloae F, et al. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2011;21(10):1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aanei CM, Eloae FZ, Flandrin-Gresta P, et al. Focal adhesion protein abnormalities in myelodysplastic mesenchymal stromal cells. Exp Cell Res. 2011;317(18):2616–2629. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Z, Wang Z, Li Q, Li W, You Y, Zou P. The different immunoregulatory functions of mesenchymal stem cells in patients with low-risk or high-risk myelodysplastic syndromes. PloS one. 2012;7(9):e45675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geyh S, Öz S, Cadeddu R, et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia. 2013;27(9):1841–1851. [DOI] [PubMed] [Google Scholar]
- 11.Flores-Figueroa E, Montesinos JJ, Flores-Guzmán P, et al. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk Res. 2008;32(9): 1407–1416. [DOI] [PubMed] [Google Scholar]
- 12.Klaus M, Stavroulaki E, Kastrinaki M-C, et al. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev. 2009;19(7):1043–1054. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Z-G, Xu W, Yu H-P, et al. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett. 2012;317(2):136–143. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer RA, Wobus M, List C, et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica. 2013;98(11):1677–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonab MM, Alimoghaddam K, Talebian F, Ghaffari SH, Ghavamzadeh A, Nikbin B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006;7(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003;33(6):919–926. [DOI] [PubMed] [Google Scholar]
- 17.Zhou S, Greenberger JS, Epperly MW, et al. Age related intrinsic changes in human bone marrow derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell. 2008;7(3):335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inukai S, Slack F. MicroRNAs and the genetic network in aging. J Mol Biol. 2013;425(19):3601–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez I, Almstead LL, DiMaio D. MicroRNAs and senescence. Aging (Albany NY). 2011;3(2):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato M, Slack FJ. Ageing and the small, non-coding RNA world. Ageing Res Rev. 2013;12(1):429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mudhasani R, Zhu Z, Hutvagner G, et al. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008;181(7):1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ungvari Z, Tucsek Z, Sosnowska D, et al. Aging-induced dysregulation of dicer1-dependent microrna expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci. 2013;68(8):877–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raaijmakers MH, Mukherjee S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santamaría C, Muntión S, Rosón B, et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica. 2012;97(8): 1218–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valent P, Horny H-P, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from a working conference. Leuk Res. 2007;31(6):727–736. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–2088. [PubMed] [Google Scholar]
- 27.Zhao Y, Zhang X, Guo J, et al. Downregulation of p21 in myelodysplastic syndrome is associated with p73 promoter hypermethylation and indicates poor prognosis. Am J Clin Pathol. 2013;140(6):819–827. [DOI] [PubMed] [Google Scholar]
- 28.Fei C, Zhao Y, Gu S, et al. Impaired osteogenic differentiation of mesenchymal stem cells derived from bone marrow of patients with lower-risk myelodysplastic syndromes. Tumour Biol. 2014;35(5): 4307–4316. [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Villar O, Garcia JL, Sanchez-Guijo FM, et al. Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q− syndrome. Leukemia. 2009;23(4):664–672. [DOI] [PubMed] [Google Scholar]
- 30.Varga G, Kiss J, Várkonyi J, et al. Inappropriate Notch activity and limited mesenchymal stem cell plasticity in the bone marrow of patients with myelodysplastic syndromes. Pathol Oncol Res. 2007;13(4):311–319. [DOI] [PubMed] [Google Scholar]
- 31.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. [DOI] [PubMed] [Google Scholar]
- 32.Krtolica A, Campisi J. Cancer and aging: a model for the cancer promoting effects of the aging stroma. Int J Biochem Cell Biol. 2002;34(11):1401–1414. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Tang X, Xu W, et al. The different immunoregulatory functions on dendritic cells between mesenchymal stem cells derived from bone marrow of patients with low-risk or high-risk myelodysplastic syndromes. PloS one. 2013;8(3):e57470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soenen-Cornu V, Tourino C, Bonnet M-L, et al. Mesenchymal cells generated from patients with myelodysplastic syndromes are devoid of chromosomal clonal markers and support short-and long-term hematopoiesis in vitro. Oncogene. 2005;24(15):2441–2448. [DOI] [PubMed] [Google Scholar]
- 35.Andl T, Murchison EP, Liu F, et al. The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr Biol. 2006;16(10):1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ksiazek K. A comprehensive review on mesenchymal stem cell growth and senescence. Rejuvenation Res. 2009;12(2):105–116. [DOI] [PubMed] [Google Scholar]
- 37.Ivanovska I, Ball AS, Diaz RL, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28(7):2167–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibcus JH, Kroesen BJ, Koster R, et al. MiR17/106b seed family regulates p21 in Hodgkin’s lymphoma. J Pathol. 2011;225(4):609–617. [DOI] [PubMed] [Google Scholar]
- 39.Kim Y-K, Yu J, Han TS, et al. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37(5):1672–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Liu M, Zhu H, et al. Suppression of p21 by c-Myc through members of miR-17 family at the post-transcriptional level. Int J Oncol. 2010;37(5):1315–1321. [PubMed] [Google Scholar]
- 41.Gao J, Yang T, Han J, et al. MicroRNA expression during osteogenic differentiation of human multipotent mesenchymal stromal cells from bone marrow. J Cell Biochem. 2011;112(7):1844–1856. [DOI] [PubMed] [Google Scholar]
- 42.Baglìo SR, Devescovi V, Granchi D, Baldini N. MicroRNA expression profiling of human bone marrow mesenchymal stem cells during osteogenic differentiation reveals Osterix regulation by miR-31. Gene. 2013;527(1):321–331. [DOI] [PubMed] [Google Scholar]
- 43.Clark EA, Kalomoiris S, Nolta JA, Fierro FA. Concise Review: MicroRNA function in multipotent mesenchymal stromal cells. Stem Cells. 2014;32(5):1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walenda T, Bork S, Horn P, et al. Co culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J Cell Mol Med. 2010;14(12):337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li T, Wu Y. Paracrine molecules of mesenchymal stem cells for hematopoietic stem cell niche. Bone Marrow Res. 2011;2011: 353878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiozawa Y, Havens A, Pienta K, Taichman R. The bone marrow niche: habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia. 2008;22(5):941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lafferty-Whyte K, Bilsland A, Cairney CJ, et al. Scoring of senescence signalling in multiple human tumour gene expression datasets, identification of a correlation between senescence score and drug toxicity in the NCI60 panel and a pro-inflammatory signature correlating with survival advantage in peritoneal mesothelioma. BMC Genomics. 2010;11(1):532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren J, Stroncek DF, Zhao Y, et al. Intra-subject variability in human bone marrow stromal cell (BMSC) replicative senescence: Molecular changes associated with BMSC senescence. Stem Cell Res. 2013;11(3):1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]